Introduction
Brain ischemia results in the deprivation of oxygen
and glucose in brain tissue, which may lead to permanent brain
damage. Several clinical conditions can give rise to global
cerebral ischemia; a frequent cause is cardiac arrest, which is a
significant worldwide health problem (1–3).
Mongolian gerbils are widely used as an animal model of chronic or
transient cerebral ischemia, as ~90% of the gerbils lack the
communicating vessels between the carotid and vertebral arteries
(4). An important feature of
cerebral ischemic damage is the vulnerability of specific neuronal
populations. In particular, pyramidal neurons in the hippocampal
CA1 region do not die immediately but survive for several days,
which is referred to as 'delayed neuronal death' (5). It is well known that the delayed
neuronal death is associated with multiple mechanisms, including
changes in energy metabolism, oxidative stress, neurotoxicity and
neuroinflammation (6–9). However, the exact mechanisms
underlying neuronal damage in ischemia and delayed neuronal death
remain to be fully elucidated.
Ischemic preconditioning (IPC) represents an
important adaptation of the central nervous system to sub-lethal
ischemia, which results in an increased tolerance to ischemia in
the central nervous system to a subsequent longer or lethal period
of ischemia (10,11). IPC induces the expression of a
diverse family of genes involved in cytoprotection, which, in turn,
encode proteins that enhance brain resistance to ischemia (12). This mechanism has been termed
'ischemic tolerance,' and the basic mechanisms underlying ischemic
tolerance remain to be fully elucidated (13).
Glucokinase (GK), a member of the hexokinase family,
catalyzes the ATP-dependent phosphorylation of glucose to produce
glucose-6-phosphate. GK is expressed predominantly in the liver,
pancreatic β-cells and brain (14–17),
and its activity is generally modulated by glucokinase regulatory
protein (GKRP), which is stimulated by binding fructose-6-phosphate
and suppressed by binding fructose-1-phosphate (18–20).
GK and GKRP are important in the regulation of glucose homeostasis,
as glucose sensors involved in the control of food intake (18,21).
Certain studies have suggested an association between neuronal cell
death and glucose dysregulation (22,23).
In addition, our previous study reported that changes in the
expression of GK and GKRP, which are expressed in the hippocampus,
were associated with neuronal loss (24). However, the expression patterns of
GK and GKRP in the brain of IPC-mediated animals following
transient cerebral ischemia remain to be fully elucidated.
Therefore, the present study was performed to
examine whether GK and GKRP are associated with IPC-induced
neuroprotection, which has been widely accepted by a number of
previous studies (11,12), following transient cerebral
ischemia in the hippocampus of the gerbil, which is considered a
suitable animal model of transient cerebral ischemia (25,26).
Materials and methods
Experimental animals
The present study used male Mongolian gerbils
(Meriones unguiculatus), obtained from the Experimental
Animal Center, Kangwon National University (Chuncheon, South
Korea). The gerbils used were 6 months of age (body weight, 65–75
g). The animals were housed in a conventional state under suitable
temperature (23°C) and humidity (60%) control, with a 12-h
light/12-h dark cycle, and were provided with free access to food
and water. All experimental protocols were approved by the
Institutional Animal Care and Use Committee at Kangwon University
(approval no. KW-130424-1) and adhered to guidelines that are in
compliance with the current international laws and policies (Guide
for the Care and Use of Laboratory Animals). All the experiments
were performed in a manner to minimize the number of animals used
and the suffering caused by the procedures used in the present
study.
Induction of transient cerebral ischemia
in animal groups
The animals were divided into four groups: i)
sham-operated group (sham group; n=7 at each time point), in which
the bilateral common carotid arteries were exposed, but no ischemia
was induced (sham-surgery) in the animals; ii) ischemia-operated
group (ischemia group; n=7 at each time-point), in which the
animals were exposed to 5 min of transient ischemia; iii) IPC +
sham-operated group (IPC + sham group; n=7 at each time-point), in
which the animals were subjected to a 2 min sublethal ischemic
insult prior to sham surgery; and iv) IPC + ischemia-operated group
(IPC + ischemia group; n=7 at each time-point), in which the
animals were subjected to a 2 min sublethal ischemic insult prior
to 5 min of transient ischemia. The preconditioning paradigm has
been previously confirmed to be effective at protecting neurons
against ischemia in this ischemic model (27). The animals in the ischemia group
and the IPC + ischemia groups were assigned recovery durations of
1, 2 and 5 days, as pyramidal neurons in the hippocampal CA1 region
do not die until 3 days and begin to die 4 days after
ischemia-reperfusion (5).
Transient cerebral ischemia was developed according
to our previously described method (9). The experimental animals were
anesthetized using a mixture of 2.5% isoflurane (Baxter, Deerfield,
IL, USA) in 33% oxygen and 67% nitrous oxide (Chil-Seung Gas Co.,
Gyeonggi-do, Korea). Under an operating microscope (Boom Stand Zoom
trinocular microscope, Shanghai Optical Instrument Factory,
Shanghai, China), a ventral neck incision was made and the
bilateral common carotid arteries were gently exposed. Ischemia was
induced by occluding the arteries using non-traumatic aneurysm
clips (Yasargil FE 723 K; Aesculap, Tuttlingen, Germany). The
complete interruption of blood flow was confirmed by observing the
central artery in the retinae using an ophthalmoscope (HEINE
K180®; Heine Optotechnik, Herrsching, Germany).
Following occlusion for 2 or 5 min, the aneurysm clips were removed
from the common carotid arteries. The body (rectal) temperature
under free-regulating or normothermic (37±0.5°C) conditions was
monitored using a rectal temperature probe (TR-100; Fine Science
Tools, Foster City, CA) and maintained using a thermometric blanket
prior to, during and following surgery, until the animals had
completely recovered from anesthesia. Subsequently, the animals
were maintained on the thermal incubator (temperature, 23°C;
humidity, 60%; Mirae Medical Industry, Seoul, South Korea) to
maintain the body temperature of animals until the animals were
sacrificed.
Tissue processing for histology
All the animals were anesthetized with pentobarbital
sodium and perfused transcardially with 0.1 M phosphate-buffered
saline (PBS; pH 7.4) followed by 4% paraformaldehyde (Samchun Pure
Chemical Co., Ltd., Gyeonggi-do, Korea) in 0.1 M phosphate-buffer
(PB; pH 7.4). The brains were removed and post-fixed in the same
fixative for 6 h. The brain tissues were cryoprotected by
infiltration with 30% sucrose overnight. The frozen tissues were
then serially sectioned on a cryostat (CM1900 UV; Leica, Wetzlar,
Germany) into 30 µm coronal sections, and were collected
into six-well plates containing PBS.
Cresyl violet (CV) staining
The levels of neuronal death in the hippocampal CA1
region in each group were examined using CV staining. The sections
were mounted on gelatin-coated microscope slides. Cresyl violet
acetate (Sigma-Aldrich, St. Louis, MO) was dissolved at 1.0% (w/v)
in distilled water, and glacial acetic acid (Marienfeld-Superior,
Lauda-Königshofen, Germany) was added to this solution. The
sections were stained and dehydrated by immersion in serial ethanol
baths, and were then mounted using Canada balsam (Kanto Chemical,
Co., Inc., Tokyo, Japan).
Neuronal nuclei (NeuN)
immunohistochemistry
To examine the neuronal changes in the hippocampal
CA1 region following transient cerebral ischemia using anti-NeuN, a
marker for neurons, the sections were sequentially treated with
0.3% hydrogen peroxide (H2O2) in PBS for 30
min and 10% normal goat serum in 0.05 M PBS for 30 min. The
sections were then incubated with diluted mouse anti-NeuN, a
neuron-specific soluble nuclear antigen, (1:1,000; Chemicon
International, Temecula, CA) overnight at at 4°C. Subsequently, the
tissues were exposed to biotinylated horse anti-mouse IgG and
streptavidin peroxidase complex (Vector Laboratories, Inc.,
Burlingame, CA). The sections were then visualized by staining with
0.05% 3,3′-diaminobenzidine in 0.1 M Tris-HCl buffer and mounted on
the gelatin-coated slides. Following dehydration, the sections were
mounted using Canada balsam (Kanto Chemical, Co., Inc.).
Fluoro-Jade B (F-J B)
histofluorescence
To examine neuronal death in the CA1 region at each
time-point following ischemia, F-J B, a high affinity fluorescent
marker for the localization of neuronal degeneration,
histofluorescence was performed (28). The sections were first immersed in
a solution containing 1% sodium hydroxide in 80% alcohol, followed
by immersion in 70% alcohol. The sections were then transferred
into a solution of 0.06% potassium permanganate, followed by
transfer to a 0.0004% F-J B (Histochem, Jefferson, AR, USA)
staining solution. Following washing with PBS three times, the
sections were placed on a slide warmer at ~50°C, and then examined
using an epifluorescent microscope (LSM510 META NLO; Carl Zeiss,
Göttingen, Germany) with blue (450–490 nm) excitation light and a
barrier filter. Using this method, neurons that undergo
degeneration fluoresce brightly, compared with the background
(10).
Cell counts
All measurements were performed in a blinded-manner,
to ensure objectivity, by three observers for each experiment, with
measurements of experimental samples under the same conditions.
According to anatomical landmarks corresponding to AP, between −1.4
and −1.9 mm of the gerbil brain atlas, the tissue sections were
selected with a 300-µm interval, and cell counts were
obtained by averaging the total numbers of cells in 15 sections
from each animal in each group. The numbers of NeuN- and F-J
B-positive cells were counted in a 200×200 µm square, which
was centred approximately at the center of the CA1 region using an
image analyzing system (Optimas 6.5; CyberMetrics, Scottsdale, AZ,
USA). The cell counts were obtained by averaging the total number
from each animal per group.
Immunohistochemistry for GK and GKRP
To obtain accurate data for immunoreactivity, the
sections from the sham-operated, IPC + sham, ischemia-operated and
IPC + ischemia groups (n=7 at each time-point) were stained,
according to the above-mentioned NeuN immunohistochemical staining.
The sections were incubated with diluted rabbit anti-GK (1:200;
Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA) and goat
anti-GKRP (1:200; Santa Cruz Biotechnology, Inc.), followed by
incubation with and subsequently biotinylated horse anti-rabbit or
anti-goat IgG and streptavidin peroxidase complex (1:200; Vector
Laboratories, Inc.). In order to establish the specificity of the
immunostaining, a negative control was included, in which
pre-immune serum was used in place of the primary antibody. The
negative control resulted in the absence of immunoreactivity in any
structures.
A total of 20 sections per animal were selected to
quantitatively analyze the immunoreactivities of GK and GKRP. The
cellular immunoreactivities of GK and GKRP were graded in the
hippocampal CA1 region. Digital images of the CA1 region, including
the strata oriens, pyramidale and radiatum in the hippocampus
proper, were captured using an AxioM1 light microscope (Carl Zeiss)
equipped with a digital camera (MRc5; Axiocam, Carl Zeiss)
connected to a PC monitor. Semi-quantification of the intensity of
GK+ and GKRP+ structures were evaluated using digital image
analysis software (MetaMorph 4.01; Universal Imaging Corp.). The
level of immunoreactivity was scaled as −, ±, + or ++, representing
no staining (gray scale value, ≥200), weakly positive staining
(gray scale value, 150–199), moderate staining (gray scale value,
100–149), or marked staining (gray scale value, ≤99) (29).
Statistical analysis
All data are presented as the mean ± standard error
of the mean. A multiple-sample comparison was performed to compared
the differences between groups, using analysis of variance. Tukey's
multiple range test was used as a post-hoc test, using the
criterion of the least significant differences. SAS version 9.2
(SAS Institute Inc., Cary, NC, USA) was used to perform the
analyses. P<0.05 was considered to indicate a statistically
significant difference.
Results
Determination of neuronal damage using
cresyl violet-positive (CV+) cells
The present study examined whether IPC was
associated with an increase in neuronal survival in the hippocampal
CA1 region following ischemia. In the sham group, the
CV+ cells were readily observed in all subregions of the
hippocampus (Fig. 1A and B).
Neurons in the stratum pyramidale (SP) were relatively large and
pyramid-like or round in shape. In the IPC + sham group, the CA1
pyramidal cells were well stained with CV (Fig. 1C and D). At 5 days
post-ischemia-reperfusion, the number of CV+ cells in
the sham group was significantly decreased in the SP of the CA1
region, but not the CA2/3 region, compared with those of the
ischemia group (Fig. 1E). The
damaged cells were shrunken and contained dark and polygonal nuclei
(Fig. 1F).
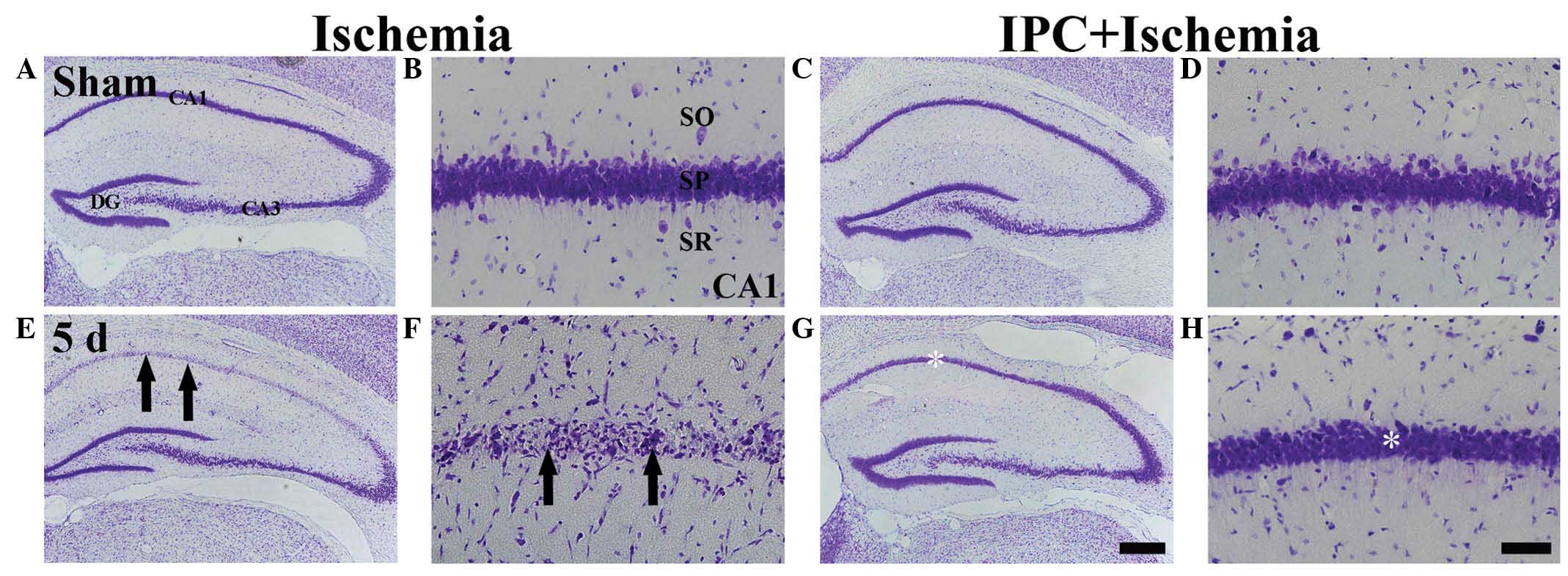 | Figure 1CV histochemical staining in the CA1
region of the ischemia (left two columns) and IPC + ischemia (right
two columns) groups at (A–D) sham and (E–H) 5 days
post-ischemia-reperfusion. In the ischemia group, few
CV+ cells (arrows) were observed in the SP 5 days
post-ischemia, whereas CV+ cells were abundant (white
asterisk) in the SP of the IPC + ischemia group. Scale bar=800
µm. (A, C, E, G) and 50 µm. (B, D, F, H). CV, Cresyl
violet; SP, stratum pyramidale; DG, dentate gyrus; SO, stratum
oriens; SR, stratum radiatum. |
In the IPC + ischemia group, the distribution
pattern of CV+ cells in the SP was similar to that
observed in the IPC + sham group 5 days after ischemia-reperfusion
(Fig. 1G and H).
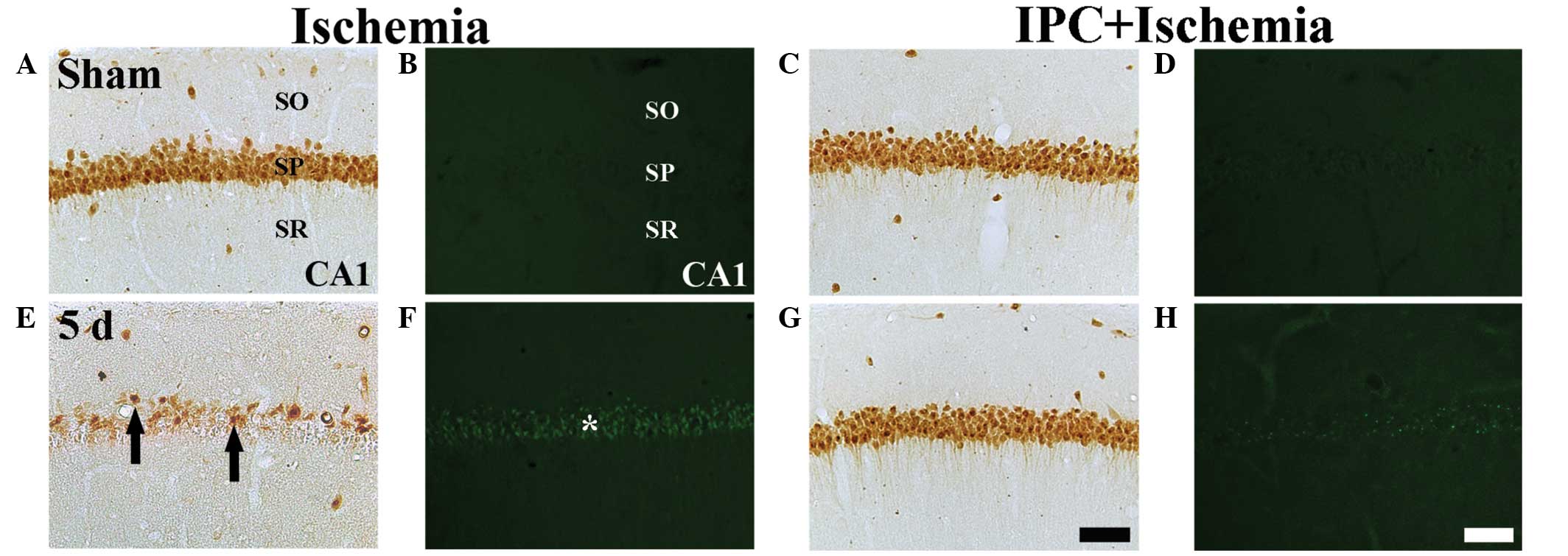
NeuN+ and F-J B+
neurons
The protection afforded by IPC in the CA1 region was
assessed using NeuN immunohisto-chemistry and F-J B
histofluorescence staining (Fig.
2). In the sham group, the CA1 pyramidal neurons were
well-stained with NeuN (Table I;
Fig. 2A); however, no F-J
B+ neurons were identified (Table I; Fig.
2B). In the IPC + sham group, pyramidal neurons in the CA1
region were also well-stained with NeuN (Fig. 2C), and no F-J B+ cells
were observed (Table I; Fig. 2C and D).
 | Table IChanges in the mean number of
pyramidal neurons of the hippocampal CA1 region of gerbils in the
ischemia and IPC + ischemia groups. |
Table I
Changes in the mean number of
pyramidal neurons of the hippocampal CA1 region of gerbils in the
ischemia and IPC + ischemia groups.
| Time post-I-R
(days) | Ischemia-operated
group
| IPC +
ischemia-operated group
|
|---|
|
NeuN+ | F-J
B+ |
NeuN+ | F-J
B+ |
|---|
| Sham (no I-R) | 346±13.54 | 0 | 352±16.28 | 0 |
| 1 | 352±11.12 | 0 | 349±18.36 | 0 |
| 2 | 349±12.98 | 4±4.36a | 355±17.34 | 0 |
| 5 |
41±8.35a,b | 134±17.69a,b | 324±15.85a,b | 24±5.54a,b |
At 5 days post-ischemia, the number of
NeuN+ neurons were markedly decreased in the SP of the
CA1 region (Table I; Fig. 2E) and several F-J B+
cells were detected in the SP of the CA1 region (Table I, Fig.
2F).
In the IPC + ischemia group, no significant
difference was observed in the distribution patterns of the
NeuN+ and F-J B+ neurons in the SP 5 day
after ischemia-reperfusion, compared with those in the IPC + sham
group (Table I; Fig. 2G and H).
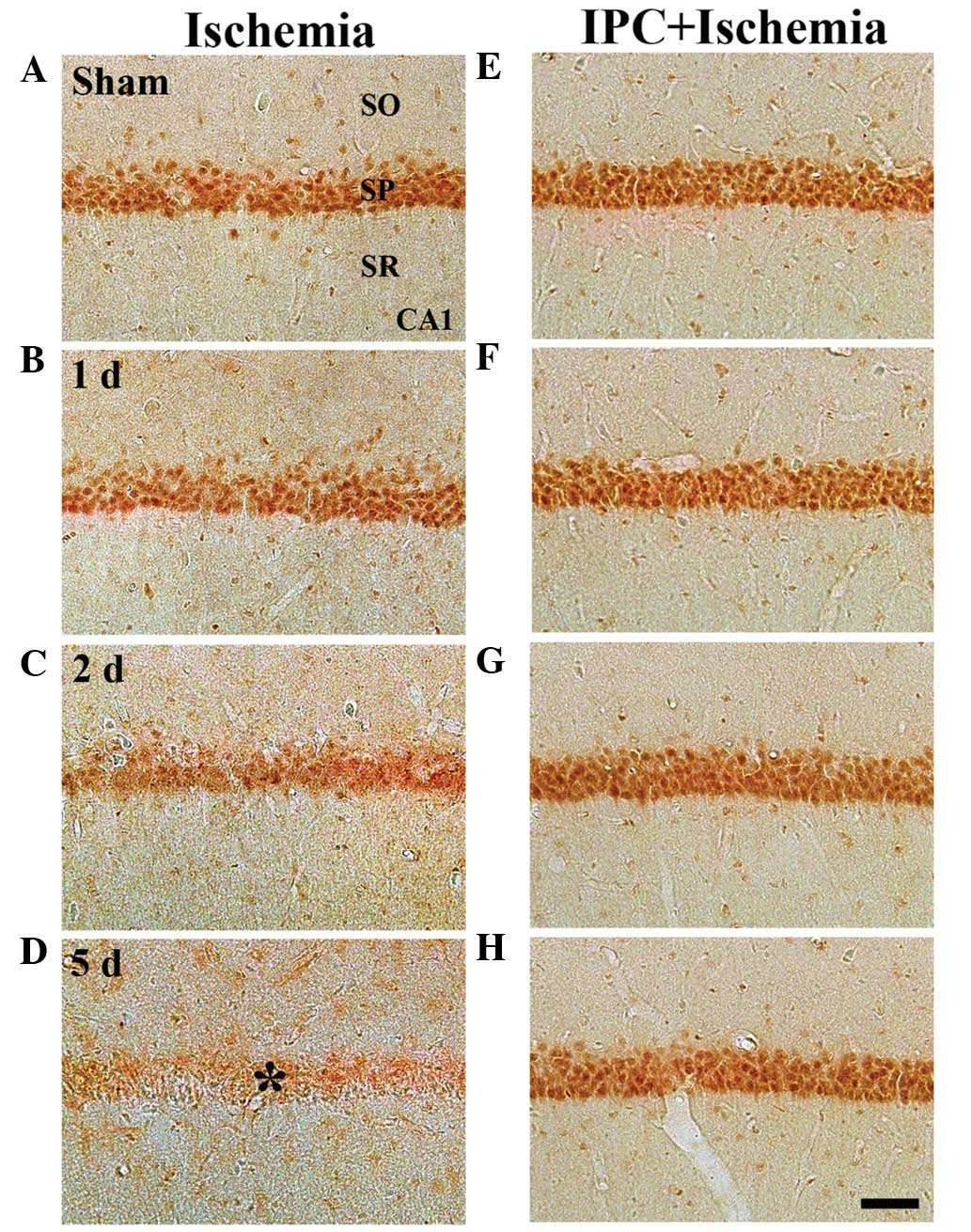
GK immunoreactivity
In the sham group, marked GK immunoreactivity was
detected in neurons of the SP of the CA1-3 regions (Table II; Fig. 3A). In the ischemia group, GK
immunoreactivity was altered in the CA1 region, but not in the
CA2/3 region. At 1 day-post ischemia-reperfusion, GK
immunoreactivity in the SP was not significantly different,
compared with that in the sham group (Table II; Fig. 3B). However, GK immunoreactivity in
the SP was distinctively decreased 2 days after
ischemia-reperfusion (Table II;
Fig. 3C). At 5 days-post
ischemia-reperfusion, GK immunoreactivity was almost undetected in
the pyramidal neurons of the SP (Table II; Fig. 3D). However, in the CA2/3 region, no
significant changes in GK immunoreactivity were observed following
ischemia-reperfusion (Table
II).
| Table IISemi-quantifications of the
immunoreactivities of GK and GKRP in the hippocampal CA1 and CA2/3
regions in the ischemia and IPC + ischemia groups. |
Table II
Semi-quantifications of the
immunoreactivities of GK and GKRP in the hippocampal CA1 and CA2/3
regions in the ischemia and IPC + ischemia groups.
| Antibody | Region | Group | Category | Time following
ischemia-reperfusion
|
|---|
| Sham | 1 day | 2 days | 5 days |
|---|
| GK | CA1 | Ischemia | CSP | ++ | ++ | + | ± |
| | | CSOR | + | ± | ± | ± |
| | IPC+ischemia | CSP | ++ | ++ | ++ | ++ |
| | | CSOR | ± | ± | ± | ± |
| CA2/3 | Ischemia | CSP | ++ | ++ | ++ | ++ |
| | | CSOR | ± | ± | ± | ± |
| | IPC+ischemia | CSP | ++ | ++ | ++ | ++ |
| | | CSOR | ± | ± | ± | ± |
| GKRP | CA1 | Ischemia | CSP | ++ | ++ | + | ± |
| | | CSOR | + | + | + | ++ |
| | IPC+ischemia | CSP | ++ | ++ | ++ | ++ |
| | | CSOR | + | + | + | + |
| CA2/3 | Ischemia | CSP | ++ | ++ | ++ | ++ |
| | | CSOR | + | + | + | + |
| | IPC+ischemia | CSP | ++ | ++ | ++ | ++ |
| | | CSOR | + | + | + | + |
In the IPC + sham group, marked GK immunoreactivity
was readily detected in the SP of the CA1-3 regions (Table II; Fig. 3E). In the IPC + ischemia-group, GK
immunoreactivity in the SP of the CA1-3 regions was well-maintained
following ischemia-reperfusion (Table
II; Fig. 3F–H).
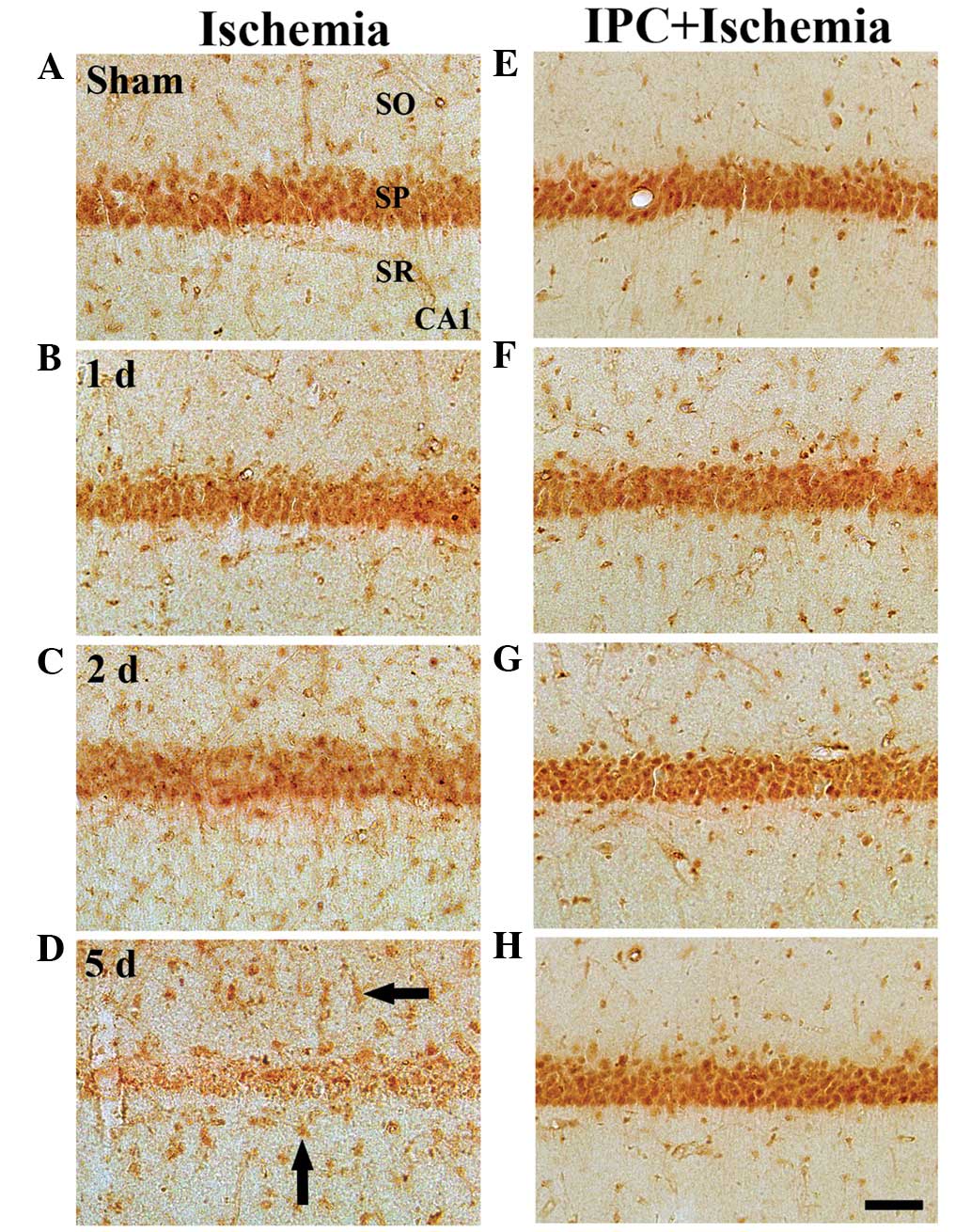
GKRP immunoreactivity
In the sham group, marked GKRP immunoreactivity was
detected, predominantly in the SP of the CA1-3 regions. In this
group, moderate GKRP immunoreactivity was observed in cells of the
strata oriens and radiatum (Table
II; Fig. 4A). In the ischemia
groups, GKRP immunoreactivity was altered in the CA1 region, but
notin the CA2/3 region. At 1 day post-ischemia-reperfusion, no
change in GKRP immunoreactivity was observed in the SP of the CA1
region, compared with that in the sham group (Table II; Fig. 4B). However, GKRP immunoreactivity
in the SP was marginally decreased 2 days after
ischemia-reperfusion (Table II;
Fig. 4C). At 5 days
post-ischemia-reperfusion, GKRP immunoreactivity was notably
decreased in the SP of the CA1 region; however, at this time-point,
marked GKRP immunoreactivity was observed in several non-pyramidal
cells in the strata oriens and radiatum (Table II; Fig. 4D). In the CA2/3 region, no
significant changes were identified in GKRP immunoreactivity
following ischemia-reperfusion (Table
II).
In the IPC + sham group, marked GKRP
immunoreactivity in the CA1-3 regions was well-detected in the
neurons of the SP, and moderate GKRP immunoreactivity was observed
in the cells of the strata oriens and radiatum (Table II; Fig. 4E). In the IPC + ischemia group, no
significant changes in GKRP immunoreactivity were observed in the
CA1-3 regions following ischemia-reperfusion (Table II; Fig. 4F–H).
Discussion
GK is important in the control of blood glucose
homeostasis. In the present study, the effect of IPC on the
immunoreactivities of GK and its regulatory protein GKRP were
examined, following 5 min of transient cerebral ischemia in
gerbils. The gerbils were randomly assigned into a sham-operated
group, ischemia-operated group, IPC + sham-operated group and IPC +
ischemia-operated group. IPC was induced by subjecting the gerbils
to 2 min of ischemia followed by 1 day of recovery. In the
immunohistochemical investigation, the immunoreactivities of the GK
and GKRP in neurons of the SP were distinctively decreased in the
CA1, but not the CA2/3, from 2 days post-ischemia, and weer almost
undetectable in the SP 5 days post-ischemia. In the IPC +
ischemia-operated group, the immunoreactivities of GK and GKRP in
the SP of the CA1 were similar to those in the sham group.
Therefore, these findings demonstrated that IPC maintains
immunoreactivities of GK and GKRP in neurons of the SP of the CA1
following ischemia-reperfusion, and indicated that GK and GKRP may
be necessary for neuronal survival against transient cerebral
ischemia.
The concept of IPC is defined as a brief,
non-injurious episode of ischemia, which can protect the brain
cells from a subsequent longer ischemic insult (10,11).
The first description of IPC in the brain was reported in a study
by Kitagawa et al (30) in
a gerbil model of global ischemia. Further studies have been
performed in other animal models, including global and focal
cerebral ischemia (12,31–35).
The preconditioning time-period used in the present study was
determined by that described in a previous report (27), which indicated that at least a 1
day interval between the sublethal 2 min ischemia and lethal 5 min
ischemia is necessary for induction of the neuroprotection of the
CA1 pyramidal cells. This protective mechanism has been termed
'ischemic tolerance,', however, the molecular mechanisms underlying
ischemic tolerance remain to be fully elucidated. In the present
study, a significantly protective effect of IPC against delayed
neuronal death was observed in the hippocampal CA1 region following
5 min of transient cerebral ischemia in gerbils. At 5 days
post-ischemic insult, pyramidal neurons in the CA1 region, but not
the CA2/3, region exhibited features of neuronal death, and
significant reductions in the numbers of CV+ and
NeuN+ neurons and the appearance of F-J B+
neurons were observed.
In the present study, the endogenous expression
levels of GK and GKRP in the CA1 region were compared between the
ischemia group and the IPC + ischemia group and found that the
immunoreactivities of GK and GKRP were well-detected in the
pyramidal neurons of the CA1-3 regions, although the
immunoreactivities were significantly decreased in the ischemic CA1
region following ischemia-reperfusion. Therefore, it is likely that
GK and GKRP in the pyramidal neurons, which are the principal cells
of the hippocampus, may be important in glucose metabolism. This
finding is supported by a previous study that reported that the
immunoreactivity of glucagon-like peptide-1 receptor (GLP-1R) is
decreased in the gerbil hippocampal CA1 region 2 days after
ischemia-reperfusion (36). It has
also been observed that GLP-1R stimulation with exendin-4 leads to
a protective effect against ischemic damage in the hippocampal CA1
region following transient cerebral ischemia (36). On the basis of the previous and
present findings, it is likely that the ischemia-induced alteration
of glucose metabolism may be associated with delayed neuronal death
in the hippocampal CA1 region following transient cerebral
ischemia. In addition, the maintained GK and GKRP
immunoreactivities following IPC in the present study reflected
potential survival mechanisms of the CA1 pyramidal neurons
following transient ischemic injury.
Cerebral ischemia leads to the alteration of energy
metabolism in the brain (37). It
is well known that transient cerebral ischemia induces the
modification of glucose metabolism in the hippocampal CA1 region,
and it is associated with the development of neuronal cell damage
and death (38). As neurons
require a constant supply of glucose for normal function, glucose
is a major source of energy metabolism for the central nervous
system (39). Several studies have
documented the effect of glucose-sensing neuropeptides on the
development of post-ischemic glucose dysfunction and neuronal
damage (22,23), and evidence from a previous in
vitro study revealed that glucose itself promotes the
development of caspase-dependent apoptosis during reoxygenation
following oxygen and glucose deprivation, and promotes the
production of reactive oxygen species, including superoxide anion,
hydrogen peroxide and peroxynitrite (40). GK and GKRP have important
regulatory roles in glucose metabolism as a glucose sensor. Alvarez
et al (18) reported that
the presence of GK interactions with GKRP in the rat hypothalamus
may be involved in glucose sensing process and metabolic
regulation. It has also been reported that GK modulates the
activity of ATP-sensitive K+ channels and determines the rate of
cell firing (41). In addition,
Mies et al (42)
demonstrated that ATP levels in the CA1 region are unchanged
between 6 h and 2 days post-ischemia, but reduce to ~70% 4 days
post-ischemia. Therefore, GK can control glycolysis and may be a
major candidate as a regulator of ATP production and
KATP channel activity (41).
In conclusion, the present study demonstrated that
IPC maintained the immunoreactivities of GK and GKRP in the CA1
pyramidal neurons following 5 min of transient ischemia, and that
CA1 pyramidal neurons death did not occur following
ischemia-reperfusion. These results suggested that GK and GKRP may
be involved in protecting neurons from ischemia-reperfusion injury
and that maintained GK and GKRP expression by IPC in the ischemic
CA1 region may be a fundamental mechanism for survival of neurons
after transient cerebral ischemia.
Acknowledgments
The authors would like to thank Mr. Seung Uk Lee for
their technical assistance. This work was supported by the Basic
Science Research Program through the National Research Foundation
(NRF) of Korea, funded by the Ministry of Science, ICT and future
Planning (grant. no. NRF-2014R1A2A2A0100 5307) and by the NRF of
Korea, funded by the Ministry of Education, Science and Technology
(grant. no. 2010-0010580).
References
|
1
|
Neigh GN, Glasper ER, Kofler J, Traystman
RJ, Mervis RF, Bachstetter A and DeVries AC: Cardiac arrest with
cardiopulmonary resuscitation reduces dendritic spine density in
CA1 pyramidal cells and selectively alters acquisition of spatial
memory. Eur J Neurosci. 20:1865–1872. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Schneider A, Böttiger BW and Popp E:
Cerebral resuscitation after cardiocirculatory arrest. Anesth
Analg. 108:971–979. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Harukuni I and Bhardwaj A: Mechanisms of
brain injury after global cerebral ischemia. Neurol Clin. 24:1–21.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kirino T and Sano K: Selective
vulnerability in the gerbil hippo-campus following transient
ischemia. Acta Neuropathol. 62:201–208. 1984. View Article : Google Scholar
|
|
5
|
Kirino T: Delayed neuronal death in the
gerbil hippocampus following ischemia. Brain Res. 239:57–69. 1982.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Chan PH: Reactive oxygen radicals in
signaling and damage in the ischemic brain. J Cereb Blood Flow
Metab. 21:2–14. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Won MH, Kang TC, Jeon GS, Lee JC, Kim DY,
Choi EM, Lee KH, Choi CD, Chung MH and Cho SS: Immunohistochemical
detection of oxidative DNA damage induced by ischemia-reperfusion
insults in gerbil hippocampus in vivo. Brain Res. 836:70–78. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Won MH, Kang T, Park S, Jeon G, Kim Y, Seo
JH, Choi E, Chung M and Cho SS: The alterations of
N-Methyl-D-aspartate receptor expressions and oxidative DNA damage
in the CA1 area at the early time after ischemia-reperfusion
insult. Neurosci Lett. 301:139–142. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lee CH, Park JH, Yoo KY, Choi JH, Hwang
IK, Ryu PD, Kim DH, Kwon YG, Kim YM and Won MH: Pre- and
post-treatments with escitalopram protect against experimental
ischemic neuronal damage via regulation of BDNF expression and
oxidative stress. Exp Neurol. 229:450–459. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Schmued LC and Hopkins KJ: Fluoro-Jade B:
A high affinity fluorescent marker for the localization of neuronal
degeneration. Brain Res. 874:123–130. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lehotský J, Burda J, Danielisová V,
Gottlieb M, Kaplan P and Saniová B: Ischemic tolerance: The
mechanisms of neuroprotective strategy. Anatomical Rec (Hoboken).
292:2002–2012. 2009. View
Article : Google Scholar
|
|
12
|
Gidday JM: Cerebral preconditioning and
ischaemic tolerance. Nat Rev Neurosci. 7:437–448. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kardesoglu E, Isilak Z, Uz O and Yiginer
O: Ischemic conditioning: A current concept in reducing reperfusion
injury. Chin Med J (Engl). 124:4802011.
|
|
14
|
Liang Y, Jetton TL, Zimmerman EC, Najafi
H, Matschinsky FM and Magnuson MA: Effects of alternate RNA
splicing on glucokinase isoform activities in the pancreatic islet,
liver and pituitary. J Biol Chem. 266:6999–7007. 1991.PubMed/NCBI
|
|
15
|
Maekawa F, Toyoda Y, Torii N, Miwa I,
Thompson RC, Foster DL, Tsukahara S, Tsukamura H and Maeda K:
Localization of glucokinase-like immunoreactivity in the rat lower
brain stem: For possible location of brain glucose-sensing
mechanisms. Endocrinology. 141:375–384. 2000.
|
|
16
|
Roncero I, Sanz C, Alvarez E, Vázquez P,
Barrio PA and Blázquez E: Glucokinase and glucokinase regulatory
proteins are functionally coexpressed before birth in the rat
brain. J Neuroendocrinol. 21:973–981. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Roncero I, Alvarez E, Vázquez P and
Blázquez E: Functional glucokinase isoforms are expressed in rat
brain. J Neurochem. 74:1848–1857. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Alvarez E, Roncero I, Chowen JA, Vázquez P
and Blázquez E: Evidence that glucokinase regulatory protein is
expressed and interacts with glucokinase in rat brain. J Neurochem.
80:45–53. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Malaisse WJ, Malaisse-Lagae F, Davies DR,
Vandercammen A and Van Schaftingen E: Regulation of glucokinase by
a fructose-1-phosphate-sensitive protein in pancreatic islets. Eur
J Biochem. 190:539–545. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Van Schaftingen E: Short-term regulation
of glucokinase. Diabetologia. 37(Suppl 2): S43–S47. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Vandercammen A and Van Schaftingen E: The
mechanism by which rat liver glucokinase is inhibited by the
regulatory protein. Eur J Biochem. 191:483–489. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Harada S, Fujita-Hamabe W and Tokuyama S:
Ischemic stroke and glucose intolerance: A review of the evidence
and exploration of novel therapeutic targets. J Pharmacol Sci.
118:1–13. 2012. View Article : Google Scholar
|
|
23
|
Wang YY, Lin SY, Chuang YH, Chen CJ, Tung
KC and Sheu WH: Adipose proinflammatory cytokine expression through
sympathetic system is associated with hyperglycemia and insulin
resistance in a rat ischemic stroke model. Am J Physiol Endocrinol
Metab. 300:E155–E163. 2011. View Article : Google Scholar
|
|
24
|
Park JH, Lee CH, Kim IH, Ahn JH, Cho JH,
Yan BC, Lee JC, Lee TH, Seo JY, Cho JH, et al: Time-course changes
in immunoreactivities of glucokinase and glucokinase regulatory
protein in the gerbil hippocampus following transient cerebral
ischemia. Neurochem Res. 38:2640–2649. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Liu YR, Lei RY, Wang CE, Zhang BA, Lu H,
Zhu HC and Zhang GB: Effects of catalpol on ATPase and amino acids
in gerbils with cerebral ischemia/reperfusion injury. Neurol Sci.
35:1229–1233. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wang L, Zhu QL, Wang GZ, Deng TZ, Chen R,
Liu MH and Wang SW: The protective roles of mitochondrial
ATP-sensitive potassium channels during
hypoxia-ischemia-reperfusion in brain. Neurosci Lett. 491:63–67.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Nakamura H, Katsumata T, Nishiyama Y,
Otori T, Katsura K and Katayama Y: Effect of ischemic
preconditioning on cerebral blood flow after subsequent lethal
ischemia in gerbils. Life Sci. 78:1713–1719. 2006. View Article : Google Scholar
|
|
28
|
Candelario-Jalil E, Alvarez D, Merino N
and León OS: Delayed treatment with nimesulide reduces measures of
oxidative stress following global ischemic brain injury in gerbils.
Neurosci Res. 47:245–253. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Lee CH, Park JH, Choi JH, Yoo KY, Ryu PD
and Won MH: Heat shock protein 90 and its cochaperone, p23, are
markedly increased in the aged gerbil hippocampus. Exp Gerontol.
46:768–772. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kitagawa K, Matsumoto M, Kuwabara K,
Tagaya M, Ohtsuki T, Hata R, Ueda H, Handa N, Kimura K and Kamada
T: 'Ischemic tolerance' phenomenon detected in various brain
regions. Brain Res. 561:203–211. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Liu Y, Kato H, Nakata N and Kogure K:
Protection of rat hippo-campus against ischemic neuronal damage by
pretreatment with sublethal ischemia. Brain Res. 586:121–124. 1992.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Kirino T, Tsujita Y and Tamura A: Induced
tolerance to ischemia in gerbil hippocampal neurons. J Cereb Blood
Flow Metab. 11:299–307. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Nishi S, Taki W, Uemura Y, Higashi T,
Kikuchi H, Kudoh H, Satoh M and Nagata K: Ischemic tolerance due to
the induction of HSP70 in a rat ischemic recirculation model. Brain
Res. 615:281–288. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Toyoda T, Kassell NF and Lee KS: Induction
of ischemic tolerance and antioxidant activity by brief focal
ischemia. Neuroreport. 8:847–851. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Stagliano NE, Pérez-Pinzón MA, Moskowitz
MA and Huang PL: Focal ischemic preconditioning induces rapid
tolerance to middle cerebral artery occlusion in mice. J Cereb
Blood Flow Metab. 19:757–761. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Lee CH, Yan B, Yoo KY, Choi JH, Kwon SH,
Her S, Sohn Y, Hwang IK, Cho JH, Kim YM, et al: Ischemia-induced
changes in glucagon-like peptide-1 receptor and neuroprotective
effect of its agonist, exendin-4, in experimental transient
cerebral ischemia. J Neurosci Res. 89:1103–1113. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Dirnagl U, Iadecola C and Moskowitz MA:
Pathobiology of ischaemic stroke: An integrated view. Trends
Neurosci. 22:391–397. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Jorgensen MB, Wright DC and Diemer NH:
Postischemic glucose metabolism is modified in the hippocampal CA1
region depleted of excitatory input or pyramidal cells. J Cereb
Blood Flow Metab. 10:243–251. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Levin BE: Glucosensing neurons do more
than just sense glucose. Int J Obes Relat Metab Disord. 25(Suppl
5): S68–S72. 2001. View Article : Google Scholar
|
|
40
|
Serra-Perez A, Verdaguer E, Planas AM and
Santalucia T: Glucose promotes caspase-dependent delayed cell death
after a transient episode of oxygen and glucose deprivation in
SH-SY5Y cells. J Neurochem. 106:1237–1247. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Dunn-Meynell AA, Routh VH, Kang L, Gaspers
L and Levin BE: Glucokinase is the likely mediator of glucosensing
in both glucose-excited and glucose-inhibited central neurons.
Diabetes. 51:2056–2065. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Mies G, Paschen W and Hossmann KA:
Cerebral blood flow, glucose utilization, regional glucose and ATP
content during the maturation period of delayed ischemic injury in
gerbil brain. J Cereb Blood Flow Metab. 10:638–645. 1990.
View Article : Google Scholar : PubMed/NCBI
|