Introduction
Hepatocellular carcinoma (HCC), the most common
primary malignancy of the liver, is the leading cause of
cancer-associated mortality, the incidence of which has been
increasing exponentially worldwide in recent decades (1–3).
Surgical resection and liver transplantation are recognized as
potentially curative therapeutic strategies, however, the five-year
survival rate of HCC remains at <7%. For the majority of
patients, HCC is diagnosed in the advanced tumor stages due to the
asymptomatic nature of HCC. At this point, the scarcity of donated
livers for transplantation severely limits the options for patients
with HCC (4,5). For advanced and unresectable HCC,
chemotherapy, transarterial chemoembolization, ablation
(radiofrequency ablation, microwave, laser-induced interstitial
thermotherapy, high-intensity focused ultrasound ablation,
cryoablation and chemical ablation), and stereotactic body
radiation therapy (proton beam therapy) at present are often the
only measures for treating the disease. However, an ideal treatment
strategy is yet to be elucidated, as outcomes associated with the
existing treatments are generally poor due to low tolerance, low
efficacy and a high recurrence rate (6). Therefore, a novel therapeutic
modality is required.
MicroRNA (miR)-122, a liver-specific miR, comprises
70% of the total miR population of the liver. It is downregulated
in human HCC and is associated with hepatocarcinogenesis. miR-122
inhibits hepatoma cell proliferation and promotes apoptosis of HCC
cells (7). Gramantieri et
al (8) found that decreased
expression of miR-122a promoted the expression of cyclin G1 and
enabled more cells to enter into S phase, thereby contributing to
the proliferation of malignant liver cancer cells. Another study
found that the Bcl-2 anti-apoptotic family member Bcl-w, also
termed BCL2L2 was a direct target of miR-122. Bcl-w expression may
be downregulated by miR-122 and subsequently inhibit cancer cell
proliferation via caspase-3 apoptotic activation in HCC cells
(9).
The above-mentioned findings indicate that
hepatocarcinogenesis may be inhibited via exogenous miR-122 mimic
transfection into a pathogenic carcinoma or HCC. The interception
of a pathogenic cell survival pathway via mimic miR-122
transfection may be an important step toward a novel therapeutic
treatment for patients with HCC. In the present study, a
liposome-mediated synthetic miR-122 analog (an miR-122 mimic) was
transfected into HepG2 cells. Subsequently, the apoptotic rates of
HepG2-transfected cells were analyzed, and the protein and mRNA
levels of Bcl-w, caspase-9 and caspase-3 were investigated to
examine the possible pathway by which miR-122 is contributing to
apoptosis in HepG2 cells.
Materials and methods
Cell culture
HepG2 cells were obtained from Professor Weng
Shengmei of the College of Pharmacy, Fujian Medical University
(Fuzhou, China) and maintained in RPMI-1640 medium (Gibco Life
Technologies, Grand Island NY, USA) supplemented with 10% fetal
calf serum (Gibco Life Technologies) at 37°C with 5% CO2
(no antibiotics). HepG2 cells in the logarithmic growth phase were
acquired and the cellular concentration was adjusted to
1×106 cells/ml. The cells were then incubated at a
density of 2 ml/well in 6-well plates for 24 h. HepG2 cells were
randomly divided into four groups, normal cultured HepG2 cells, the
transfection reagent control group, the negative control group
[transfected with non-specific sequence control short interfering
(si)RNA] and miR-122 mimic transfection group. Biological
duplicates were included for each group and all experimental
procedures were repeated three times.
miR-122 transfection
A commercial miR-122 mimic was used for transfection
with the following sequence: 5′-UGGAGUGUGACAAUGGUGUUUG-3′
(synthesized by Qiagen; Germantown, MD, USA). The AllStars Negative
Control siRNA (Qiagen; cat. no. 1027280) served as a negative
control and an AllStars Hs Cell Death Control siRNA (cat. no.
1027298) served as a positive control. Transfection was achieved
using the HiPerFect transfection reagent (Qiagen) according to the
manufacturer's instructions. Briefly, 5 µl transfection
reagent was mixed with 2.5 µl 2-µM control siRNA,
AllStars Hs Cell Death Control siRNA, or miR-122 mimic and 100 ml
serum-free media, then incubated for 10 min.
Serum-supplemented media was removed from the cell
cultures and cells were washed with serum-free RPMI-1640. Fresh,
serum-free media (900 µl) was then added to the mono-layer
of cells. Subsequently, 100 µl siRNA/mimic-HiPerFect mixture
was added into each well and mixed gently. The cells were further
incubated for 24, 48 and 72 h prior to harvesting.
Transfection efficiency
AllStars Hs Cell Death Control siRNA transfection
induces cellular apoptosis. The targets of the siRNA are
predominantly genes, which are associated with cell apoptosis and
caspase-3 or -8 activation. The cells that were successfully
transfected with AllStars Hs Cell Death Control siRNA were expected
to undergo apoptosis within 24–72 h. AllStars Hs Cell Death
Control-transfected HepG2 cells and the non-transfected control
cells were cultivated for 24, 48 and 72 h. At each of these time
points, cultures were analyzed for apoptosis under a (DM 2500B
Leica optical microscope, at a magnification of ×500 (Leica
Microsystems GmbH, Wetzlar, Germany). The transfection ratio (TR)
was calculated according to the formula: TR (%) = (apoptotic cell
number in the AllStars Hs Cell Death Control siRNA transfection
group / total cells in this group - the apoptotic cell number in
the non-transfected control group / total cells in the
non-transfected group) × 100.
Flow cytometric analysis of apoptotic
rate
Cell samples from all four experimental groups were
stained using an Annexin V apoptosis detection kit (MP Biomedicals,
Shanghai, China). The apoptotic rate of the cells was detected by
flow cytometry (BD FACSVerse flow cytometer; BD Biosciences, San
Jose, CA, USA). The cells were collected at a 1×106
cells/ml density per sample after culturing for 24, 48 and 72 h.
The cells were then centrifuged at 27.75–111 x g for 5 min, and the
supernatant was removed and washed with phosphate-buffered saline
(PBS). A total of 100 µl 1X Annexin-binding buffer was added
to resuspend the cells. Subsequently, 5 µl Alexa fluor 488
(green) Annexin V and 1 µl 100-µg/ml propidium iodide
(PI; red) was added to the cells. The cells were incubated at room
temperature in the dark for 15 min. Following incubation, 400
µl 1X Annexin-binding buffer was added to the cells and the
mixture was gently mixed. The cellular concentration was adjusted
to 1×106 cells/ml. The time periods that exhibited the
highest TRs from the previous screen were selected and assessed via
flow cytometry. In the bivariate flow cytometry scatter plots, the
lower left quadrant exhibits living cells [fluorescein
isothiocyanate (FITC)−/PI−]; the upper right quadrant indicates
non-living cells/necrotic cells (FITC+/PI+); the lower right
quadrant demonstrates apoptotic cells (FITC+/PI−); and the upper
left quadrant suggested early apoptotic cells (FITC+/Pl−)..
Reverse transcription-polymerase chain
reaction (RT-PCR)
Total mRNA was extracted from cells using TRIzol
reagent and reverse transcribed into cDNA using an RT-PCR kit
(Invitrogen Life Technologies, Grand Island, NY, USA). The mRNA
levels of Bcl-w, Bcl-2-associated X protein (Bax), caspase-9 and
caspase-3 were then detected with RT-PCR using gene-specific probes
(Table I; Shanghai Dinghan
Biotechnology Co. Ltd., Shanghai, China) and β-actin served as an
internal control. The PCR cycle parameters were as follows:
Denaturation at 4°C for 5 min, amplification at 9°C for 30 sec,
57°C for 30 sec and 72°C for 45 sec sequentially for 30 cycles in
total, and extension at 7°C for 7 min. The PCR products were
separated by 1.2–1.3% agarose gel electrophoresis. A total of 5
µl of each product was added into five combs, and run twice
at 110 mV for 40 min, prior to being visualized with grayscale
scanning (Gel DocTM EZ Imager system; Bio-Rad Laboratories, Inc.,
Hercules, CA, USA).
 | Table IPolymerase chain reaction primer
sequences. |
Table I
Polymerase chain reaction primer
sequences.
| Gene | Fragment, bp | Upstream | Downstream |
|---|
| β-actin | 556 | 5′-AAA GAC CTG TAC
GCC AAC ACA G-3′ | 5′-TTT TAG GAT GGC
AAG GGA CTT C-3′ |
| Bcl-w | 237 | 5′-GCA GCT GGA GATGAG
TTC G-3′ | 5′-CCA TCC ACT CCT
GCA CTT G-3′ |
| Bax | 223 | 5′-GTT TCA TCC AGG
ATC GAG C-3′ | 5′-GCC GTC AGA AAA
CAT GTC AG-3′ |
| Caspase-9 | 240 | 5′-CAG ACC AGA GAT
TCG CAA AC-3′ | 5′-CGC AAC TTC TCA
CAG TCG AT-3′ |
| Caspase-3 | 200 | 5′-ATG AGA GGC AAT
GAT TGT TAA T-3′ | 5′-CCC ACA GAT GCC
TAA GTT CT-3′ |
Immunocytochemsitry
Bcl-w, Bax, caspase-9 and caspase-3 were selected as
target proteins. Glass coverslips (1.8×20×20 mm) were soaked in
dilute hydrochloric acid overnight and washed with double distilled
water prior to drying and autoclaving. The coverslips were placed
into 6-well plates (1 coverslip/well) and HepG2 cells were
inoculated in these wells at a density of 1×105
cells/ml. After 24 h cultivation, the cell densities were of
40–50%. The coverslips recovered by cells were divided into four
experimental groups for each target protein, two coverslips per
group. Each group was transfected with miR-122 mimic, negative
control siRNA, transfection reagent and blank control,
respectively, then cultivation was continued for 24 h. In addition,
two coverslips were randomly selected from each experimental group
for immunocytochemical negative controls to verify true positive
rather than false positive. For each target protein (Bcl-w, Bax,
caspase 9 and caspase 3), both immuno-histochemical negative and
immunohistochemical positive controls were used for contrast
analysis in each experimental groups in order to verify true
positive rather than false positive immunohistochemistry. These
coverslips were retrieved from the wells and fixed with ice-cold
acetone. The cells were next prepared for immunocytochemistry using
a commercial streptavidin-peroxidase kit according to the
manufacturer's instructions (Fuzhou Maixin Biotech Co., Ltd.,
Fuzhou, China). Compatible primary antibodies (Santa Cruz
Biotechnology, Inc., Santa Cruz, CA, USA) included monoclonal mouse
anti-human Bax (1:200; cat. no. sc-20067), caspase-9 (1:200; cat.
no. sc-56073), and caspase-3 (1:200; cat. no. sc-65497), and
polyclonal rabbit anti-human Bcl-w (1:200; cat. no. sc-130701),
were incubated at 37°C for 1 h. The primary antibody of the
immunocytochemical negative control group was replaced with an
equal amount of PBS buffer The secondary antibody used was
horseradish peroxidase-labeled goat anti-mouse IgG (1:400; cat. no.
sc-2004; Santa Cruz Biotechnology, Inc.), incubated at 37°C for 2
h. Optical detection was achieved using the 3,3′-diaminobenzidine
without chromogen (Beijing Zhongshan Golden Bridge Biotechnology
Co., Ltd., Beijing, China). A total of two coverslips were
collected from each of the experimental groups. The experiment was
repeated three times for all groups. Finally, images
(magnification, ×400) were captured using a Leica Microscope (DM
2500B) and analyzed with Image-ProPlus software version 6.0 (Media
Cybernetics, Inc., Rockville, MD, USA).
Statistical analysis
Data are expressed as the mean ± standard deviation
for each group. One-way analysis of variance was used to analyze
the difference between groups and the least significant difference
test was used for comparisons between two groups. Statistical
analysis was performed using SPSS 18.0 (SPSS, Inc., Chicago, IL,
USA). P<0.01 and P<0.05 were considered to indicate a
statistically significant difference.
Results
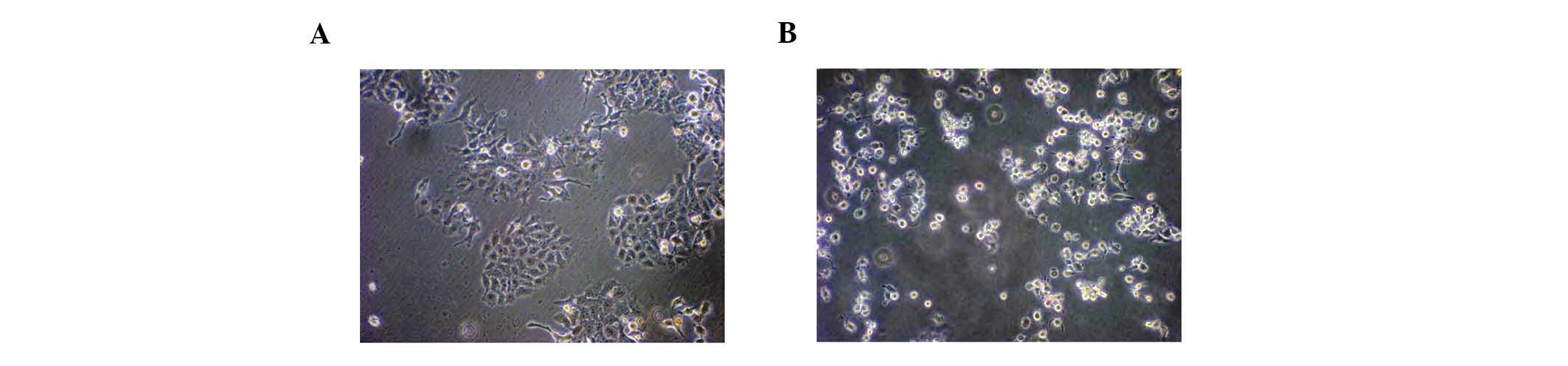
Required experimental transfection
efficiency was met
AllStars Hs Cell Death Control siRNA was used to
measure the transfection efficiency. The majority of HepG2 cells
underwent visible apoptosis 72 h after HiPerFect-mediated
transfection. The transfection efficiency was calculated at 80%
(Fig. 1), which met the
experimental requirements.
miR-122 mimic transfection increases
apoptotic rate of HepG2 cells
To investigate the role of miR-122 in HCC cell death
pathway regulation, HepG2 cells were transfected with an miR-122
mimic. The apoptotic rate of cells transfected with the mimic was
28.68±2.48%. By contrast, the negative control, positive control,
and non-transfection control groups exhibited apoptotic rates of
2.48±1.58%, 2.20±1.53% and 1.77±0.731%, respectively. The apoptotic
rate was significantly higher in the miR-122 mimic group than in
any of the control groups (Fig. 2;
P<0.01), while the differences among the control groups were not
statistically significant. Additionally, the apoptotic rate of
HepG2 cells in the miR-122 mimic group was higher at 24 h
post-transfection than at 48 h and 72 h (P<0.05; data not
shown).
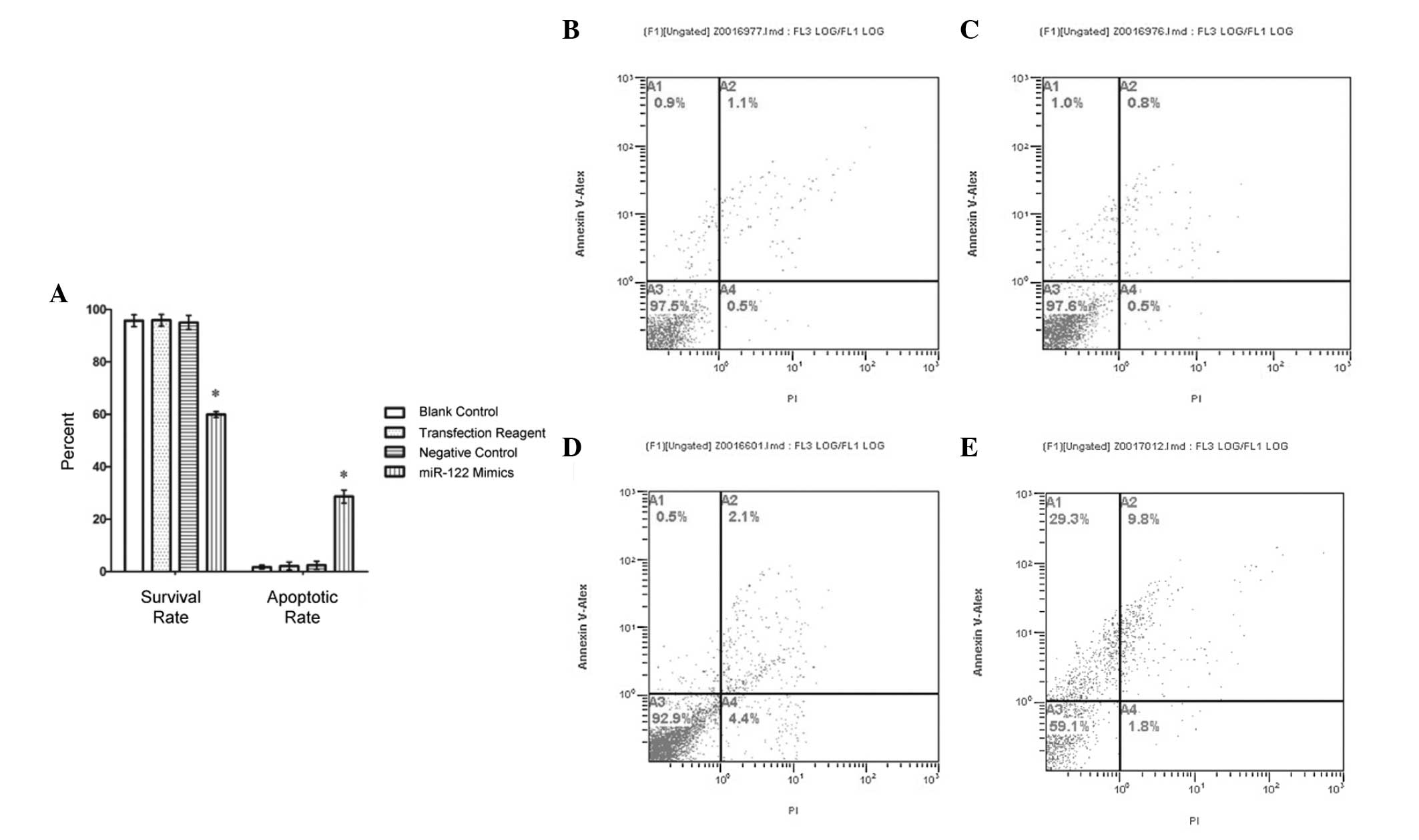 | Figure 2Survival rates decrease and apoptotic
rates increase in miR-122 mimic-transfected cells. Annexin V
staining was utilized to assess survival and apoptotic rates of
HepG2 cells. (A) Survival rate of the miR-122 mimic-transfected
cells fell below the rates of all experimental controls. The
apoptotic rates of the miR-122 mimic-transfected group were
significantly higher than control levels (*P<0.01).
No statistical significance was detected between the control
groups, which included non-transfected 'blank' control cells, a
transfection reagent positive control group and a non-sequence
specific transfected negative control group. Flow cytometry was
used as a secondary assay of apoptosis. (B–E) A1 quadrant, early
apoptosis cells (FITC+/Pl−); A3 quadrant, living cells (FITC−/PI−);
A2 quadrant, non-living cells, which may also express necrotic
cells (FITC+/PI+); A4 quadrant, apoptotic cells (FITC+/PI−). All
data are expressed as the mean ± standard deviation. miR, microRNA;
FITC, fluorescein isothiocyanate; PI, propidium iodide. |
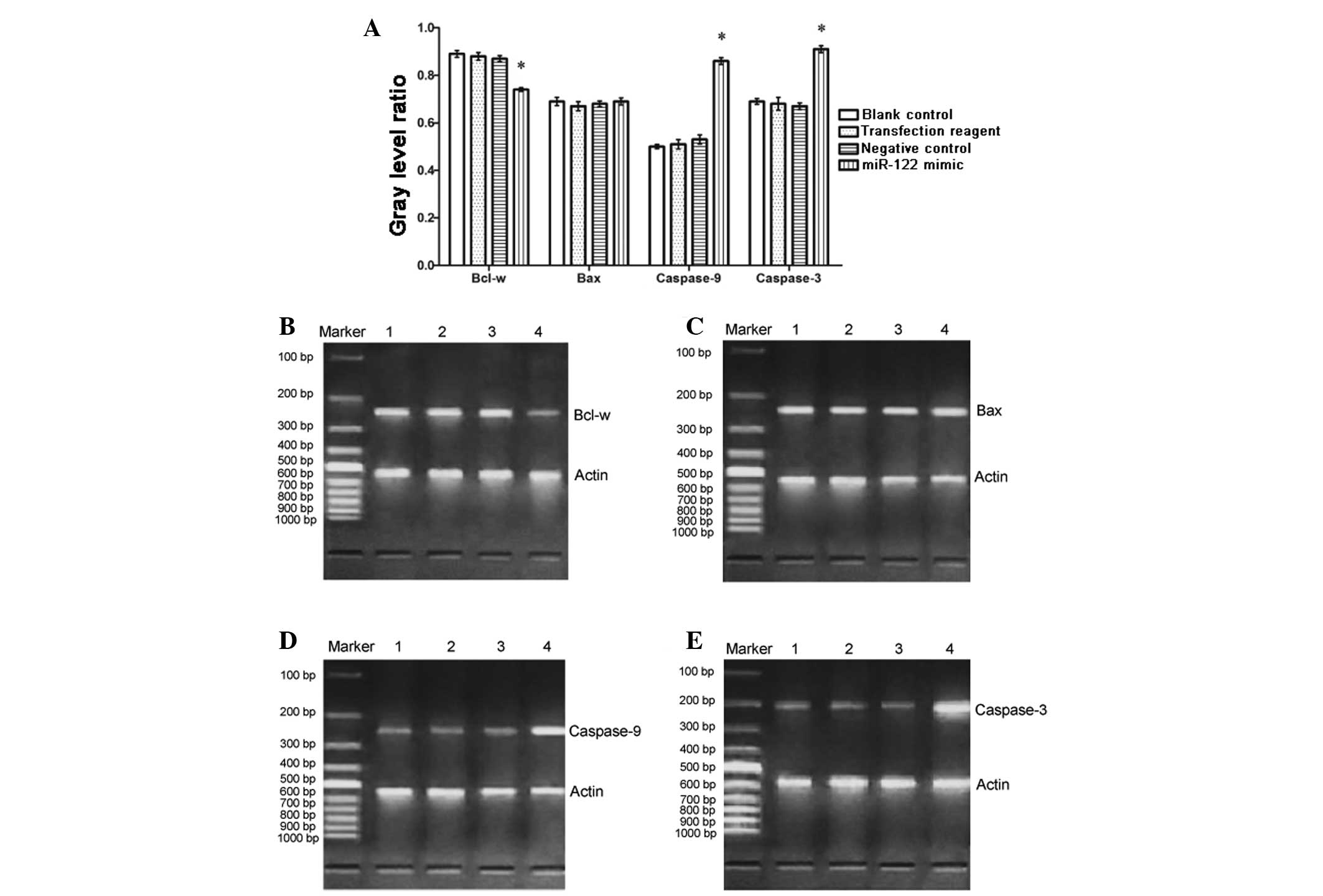
Bcl-w is downregulated, whereas caspase-9
and caspase-3 are upregulated following miR-122 mimic
transfection
HepG2 cells may express Bcl-w, Bax, caspase-9 and
caspase-3 mRNA under common medium culture conditions (37°C, 5%
CO2, 95% O2, 100% humidity). To assess
whether miR-122 mimic transfection affects the basal levels of
transcription in these genes, mRNA was measured via RT-PCR. It was
identified that the expression of Bcl-w mRNA was downregulated in
the miR-122 mimic group than in the negative, positive and
non-transfected control groups (Fig.
3; P<0.01). Conversely, caspase-9 and caspase-3 mRNA were
upregulated in the miR-122 mimic group compared with the controls
(Fig. 3; P<0.01). The level of
Bax mRNA did not significantly differ among the four groups.
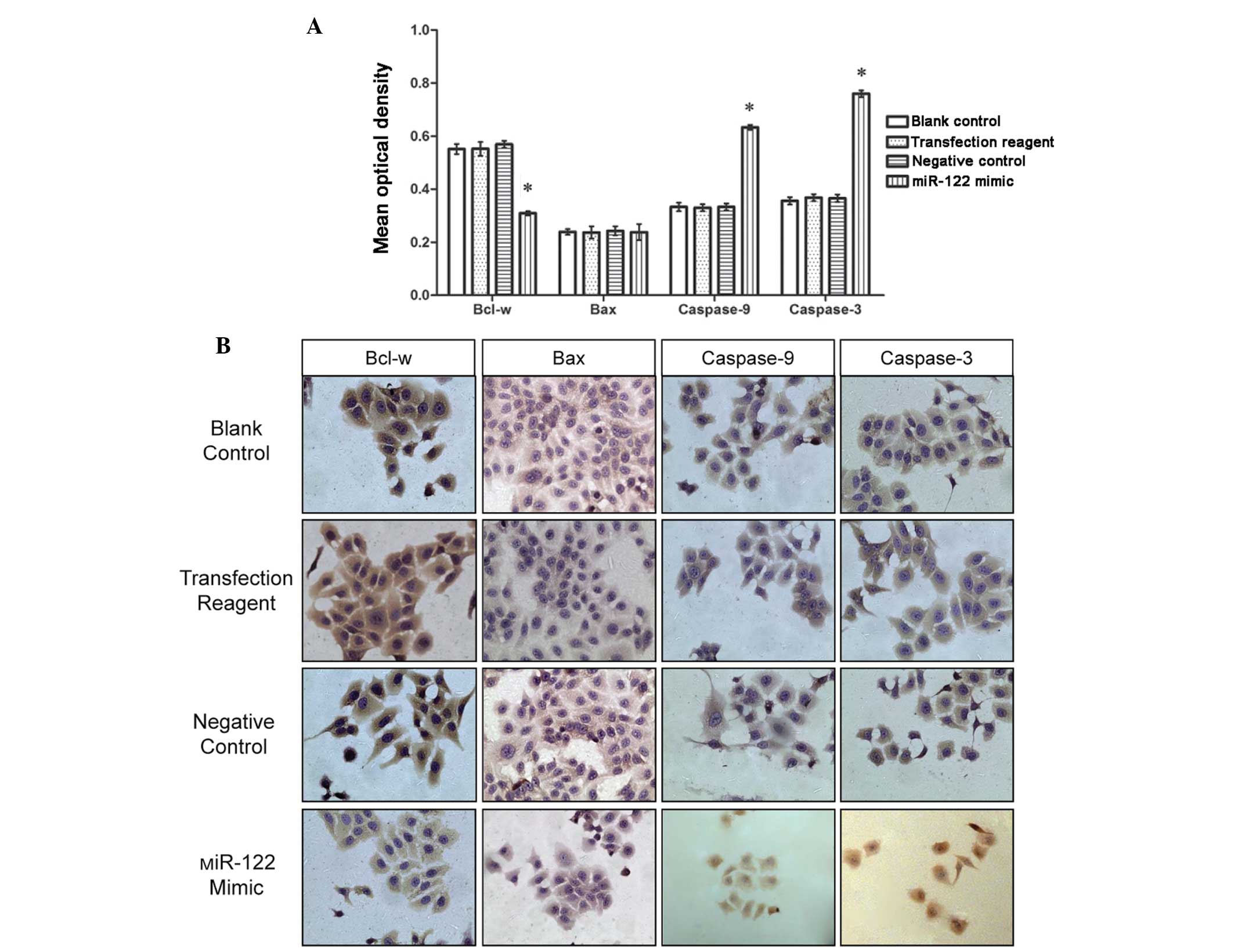
To corroborate the mRNA trends, immunocytochemistry
was performed on samples from each of the experimental groups. The
immunopositive reaction of Bcl-w and Bax was localized to the
cytoplasm, while caspase-9 and caspase-3 immunoreactivities were
traced to the nucleus and (or) the cytoplasm. The positive staining
of the cells was detected as either reddish-brown or dark claybank
chromogenic coloration. Image-ProPlus software allowed analysis of
the images and revealed that the distribution of Bcl-w, Bax,
caspase-9 and caspase-3 occurred at different degrees of intensity
in each group. The regions of the highest cellular density were
selected and the average optical density of these regions was
calculated under 5X high-power field magnification.
The Image-ProPlus analysis revealed that Bcl-w
immunoreactivity was less intense in the miR-122 mimic transfection
group compared with the negative, positive, and the non-transfected
control groups; however, the opposite was observed for caspase-9
and caspase-3 reactivity (Fig. 4;
P<0.01). No differences were observed in the level of Bax
immunoreactivity among the four groups (Fig. 4).
Discussion
To confirm whether miR-122 is responsible for
regulating human HCC cell apoptosis and to examine a possible
therapeutic intervention for HCC, HepG2 cells transfected with an
miR-122 mimic were produced via cationic liposomes and the
apoptotic rates of the cells were observed. Bcl-w, an
anti-apoptotic gene of the Bcl-2 family (significant in the
endogenous apoptotic pathway) and caspase-3 and -9 were also
analyzed for their role in miR-122-mediated HCC cell survival.
While numerous miRs are expressed in the liver, only
miR-122 is expressed specifically and abundantly. Landgraf et
al (10) found that
liver-specific miR-122 is expressed from the early implantation
stages and reaches half of its peak levels on approximately the
17th embryonic day. Before birth, miR-122 begins to approach peak
levels (50,000 copies of miR-122 per liver cell) although a
marginal and steady rise continues even after birth, indicating
that miR-122 may be involved in the regulation of the
differentiation and development of the liver (10). miR-122 expression decreases in all
cells in HCC indicating that it may be closely-associated with
hepatic function and certain diseases. Chang et al (11) found that miR-122 antisense
oligonucleotides damage the liver and inhibit the synthesis of
cholesterol, further supporting the importance of endogenous
miR-122 for normal hepatic function.
miR-122 may inhibit the proliferation of HCC cells
and induce apoptosis via two of its targets in HCC cells, cyclin G1
and Bcl-w (9,12). Bcl-w is widely expressed in certain
transformed tumor cell lines of epithelial origin, such as colon,
cervical and breast cancer (13).
It has been suggested that Bcl-w may suppress gastric cancer cell
death by blocking stress-activated protein kinase/c-Jun N-terminal
kinase activation, and inducing the migratory and invasive
potential of cancer cells via increased matrix metalloproteinase-2
expression (14,15). Bcl-w expression is also modulated
by the Met/hepatocyte growth factor (c-Met) receptor and inhibits
apoptosis in colorectal tumors. Additionally, a binding site of
miR-122 may exist in the 3′-untranslated region (UTR) of Bcl-w
(16,17).
Previous studies have identified two promoters of
the miR-122 transcript in chromosome 18 of hepatocytes, however
HepG2 cells are largely miR-122-deficient. Ma et al
(18) expressed functional miR-122
via adenoviral vectors targeted to tumor cells originating from the
liver (HepG2, Hep3B, Huh7 and PLC/PRF/5), lung (NCI-H460) and
uterine cervix (HeLa) at high levels, and induce apoptosis and/or
cell cycle arrest by decreasing the expression of Bcl-W and cyclin
G1. Wu et al (19)
transfected Huh-7 and HepG2 cells with miR-122 and an miR-122
antisense strand, respectively, and no significant difference was
identified between the viabilities of the transfected HepG2 cells
and the mock-transfected HepG2 cells. However, the viability of
Huh-7 cells transfected with anti-miR-122 was significantly
elevated 48 h after transfection. While this implies that the
aberrant expression of miR-122 may contribute to
hepatocarcinogenesis, whether the transfection of HCC cells with
miR-122 mimics alleviates HCC remains controversial (19). In the present study, the apoptotic
rate was significantly increased in miR-122 mimic-transfected
cells, demonstrating that miR-122 mimic transfection induces
apoptosis in HCC cells.
Bcl-w, an anti-apoptotic Bcl-2 family member, has
been closely associated with cancer formation and progression, and
identified as a target of miR-122. Its expression is downregulated
by the binding of miR-122 to the 3′-UTR of its transcript (9,20).
It has been reported that activators of the liver-specific microRNA
in liver cancer cells are able to selectively induce apoptosis
through caspase activation (21).
Generally, there are three pathways of apoptosis: The death
receptor pathway, the endoplasmic reticulum pathway and the
mitochondrial pathway. During apoptosis, various pre-apoptotic
molecules, including reactive oxygen species (ROS) and cytochrome
c, are released by the mitochondria into the cytosol via the
mitochondrial (mt) permeability transition pore (PTP). The opening
and closing of the mtPTP is regulated by proteins of the Bcl-2
family (22). Among them, Bax is
responsible for opening of the PTP, while Bcl-w is hypothesized to
inhibit the channel from opening. Bax is a gene homologous to
Bcl-w, however, they possess different 3′-UTRs. Bax is dynamic and
able to form heterodimers with Bcl-w to inhibit apoptosis, however,
it can also homodimerize and induce apoptosis. When Bax is
overexpressed, it is easier for Bax to form homodimers, which could
antagonize the protective tendency of Bcl-w to induce cell
apoptosis. Therefore, the ratio of Bax/Bcl-w is theorized to be the
lynchpin of apoptotic regulation. Activators of miR-122 may
downregulate the expression of Bcl-w, thereby increasing the ratio
of Bax/Bcl-w and increasing the opening capacity of the mtPTP
channel, consequently generating more ROS and less antioxidant
enzymes. ROS, important apoptosis-associated molecules, are able to
further increase the rate of mtPTP opening and stimulate cytochrome
c outflow leading to apoptosis through cytochrome
c-dependent caspase activation. The release of cytochrome
c activates a cascade of caspases through complex formation
with apoptotic protease activating factor 1. The complex activates
procaspase-9 and subsequently caspase-3, a critical and
irreversible point in the progression of apoptosis.
In the present study, it is reported that Bcl-w mRNA
decreased as the levels of caspase-9 and caspase-3 mRNA increased
markedly. Furthermore, the apoptotic rate of miR-122
mimic-transfected cells is increased. miR-122 may be downregulating
Bcl-w expression and contributing to the activation of
caspase-9/caspase-3. It is likely that miR-122 is inhibiting the
translation of the anti-apoptotic gene, Bcl-w and inadvertently
activating the caspase-9/caspase-3 mitochondrial pathway of
apoptosis. The observation of these results in HepG2 cells suggests
that endogenous miR-122 in HCC may be acting to inhibit the
proliferation of HCC cells and promoting apoptosis. Further
investigation into the ability of miR-122 mimics to counter HepG2
growth and viability are warranted considering the results
presented in the current study. Stabilizing the plasmid or viral
vector target RNA fragments may optimize the gene silencing effect
and thus improve the effectiveness of the gene interference.
Additionally, Bai et al (23) validated A distintegrin and
metalloprotease family 10, serum response factor and insulin-like
growth factor 1 receptor as tumorigenic targets of miR-122, which
require further investigation (23). Finally, the growth rate of HCC
cells expressing miR-122 significantly decreased following exposure
to a multikinase inhibitor. This suggests that there are various
signaling pathways for the miR-122 mimic-associated downregulation
of HCC, in addition to the Bcl-w/caspase-3 pathway described in the
present study. While all these points require further
investigation, the case for miR-122 manipulation in the regulation
of liver cancer cell growth is apparent and promising as a novel
therapeutic approach for HCC.
Acknowledgments
The authors would like to acknowledge Dr. Marisol
Resendiz of Clarity Manuscript Consultants LLC for her assistance
in editing the manuscript.
References
|
1
|
McGlynn KA and London WT: Epidemiology and
natural history of hepatocellular carcinoma. Best Pract Res Clin
Gastroenterol. 19:3–23. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Parkin DM, Bray F, Ferlay J and Pisani P:
Global cancer statistics, 2002. CA Cancer J Clin. 55:74–108. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Altekruse SF, McGlynn KA and Reichman ME:
Hepatocellular carcinoma incidence, mortality, and survival trends
in the United States from 1975 to 2005. J Clin Oncol. 27:1485–1491.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Azam F and Koulaouzidis A: Hepatitis B
virus and hepatocarcinogenesis. Ann Hepatol. 7:125–129.
2008.PubMed/NCBI
|
|
5
|
Kaihara S, Kiuchi T, Ueda M, Oike F,
Fujimoto Y, Ogawa K, Kozaki K and Tanaka K: Living-donor liver
transplantation for hepatocellular carcinoma. Transplantation. 75(3
Suppl): S37–S40. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lopez PM, Villanueva A and Llovet JM:
Systematic review: Evidence-based management of hepatocellular
carcinoma-an updated analysis of randomized controlled trials.
Aliment Pharmacol Ther. 23:1535–1547. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Nakao K, Miyaaki H and Ichikawa T:
Antitumor function of microRNA-122 against hepatocellular
carcinoma. J Gastroenterol. 49:589–593. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Gramantieri L, Ferracin M, Fornari F,
Veronese A, Sabbioni S, Liu CG, Calin GA, Giovannini C, Ferrazzi E,
Grazi GL, et al: Cyclin G1 is a target of miR-122a, a microRNA
frequently down-regulated in human hepatocellular carcinoma. Cancer
Res. 67:6092–6099. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lin CJ, Gong HY, Tseng HC, Wang WL and Wu
JL: miR-122 targets an anti-apoptotic gene, Bcl-w, in human
hepatocellular carcinoma cell lines. Biochem Biophys Res Commun.
375:315–320. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Landgraf P, Rusu M, Sheridan R, Sewer A,
Iovino N, Aravin A, Pfeffer S, Rice A, Kamphorst AO, Landthaler M,
et al: A mammalian microRNA expression atlas based on small RNA
library sequencing. Cell. 129:1401–1414. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Chang J, Nicolas E, Marks D, Sander C,
Lerro A, Buendia MA, Xu C, Mason WS, Moloshok T, Bort R, et al:
miR-122, a mammalian liver-specific microRNA, is processed from hcr
mRNA and may downregulate the high affinity cationic amino acid
transporter CAT-1. RNA Biol. 1:106–113. 2004. View Article : Google Scholar
|
|
12
|
Xu T, Zhu Y, Xiong Y, Ge YY, Yun JP and
Zhuang SM: MicroRNA-195 suppresses tumorigenicity and regulates
G1/S transition of human hepatocellular carcinoma cells.
Hepatology. 50:113–121. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
O'Reilly LA, Print C, Hausmann G, Moriishi
K, Cory S, Huang DC and Strasser A: Tissue expression and
subcellular localization of the pro-survival molecule Bcl-w. Cell
Death Differ. 8:486–494. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lee HW, Lee SS, Lee SJ and Um HD: Bcl-w is
expressed in a majority of infiltrative gastric adenocarcinomas and
suppresses the cancer cell death by blocking stress-activated
protein kinase/c-Jun NH2-terminal kinase activation. Cancer Res.
63:1093–1100. 2003.PubMed/NCBI
|
|
15
|
Bae IH, Park MJ, Yoon SH, Kang SW, Lee SS,
Choi KM and Um HD: Bcl-w promotes gastric cancer cell invasion by
inducing matrix metalloproteinase-2 expression via phosphoinositide
3-kinase, Akt and Sp1. Cancer Res. 66:4991–4995. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kitamura S, Kondo S, Shinomura Y, Kanayama
S, Miyazaki Y, Kiyohara T, Hiraoka S and Matsuzawa Y: Met/HGF
receptor modulates bcl-w expression and inhibits apoptosis in human
colorectal cancers. Br J Cancer. 83:668–673. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Gibson L, Holmgreen SP, Huang DC, Bernard
O, Copeland NG, Jenkins NA, Sutherland GR, Baker E, Adams JM and
Cory S: bcl-w, a novel member of the bcl-2 family, promotes cell
survival. Oncogene. 13:665–675. 1996.PubMed/NCBI
|
|
18
|
Ma L, Liu J, Shen J, Liu L, Wu J, Li W,
Luo J, Chen Q and Qian C: Expression of miR-122 mediated by
adenoviral vector induces apoptosis and cell cycle arrest of cancer
cells. Cancer Biol Ther. 9:554–561. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wu X, Wu S, Tong L, Luan T, Lin L, Lu S,
Zhao W, Ma Q, Liu H and Zhong Z: miR-122 affects the viability and
apoptosis of hepatocellular carcinoma cells. Scand J Gastroenterol.
44:1332–1339. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Shen L, Li J, Xu L, Ma J, Li H, Xiao X,
Zhao J and Fang L: miR-497 induces apoptosis of breast cancer cells
by targeting Bcl-w. Exp Ther Med. 3:475–480. 2012.PubMed/NCBI
|
|
21
|
Young DD, Connelly CM, Grohmann C and
Deiters A: Small molecule modifiers of microRNA miR-122 function
for the treatment of hepatitis C virus infection and hepatocellular
carcinoma. J Am Chem Soc. 132:7976–7981. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Martinou JC, Desagher S and Antonsson B:
Cytochrome c release from mitochondria: All or nothing. Nat Cell
Biol. 2:E41–E43. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
23
|
Bai S, Nasser MW, Wang B, Hsu SH, Datta J,
Kutay H, Yadav A, Nuovo G, Kumar P and Ghoshal K: MicroRNA-122
inhibits tumorigenic properties of hepatocellular carcinoma cells
and sensitizes these cells to sorafenib. J Biol Chem.
284:32015–32027. 2009. View Article : Google Scholar : PubMed/NCBI
|