Introduction
Ischemic cerebrovascular disease has high rates of
morbidity, mortality and recurrence, and may result in disability
(1). A previous study has
demonstrated that oxidative stress is key in pathophysiologic
processes (2). Oxidative stress
occurs due to an imbalance between production of reactive oxygen
species and the cell's capacity to neutralize them via its
intrinsic anti-oxidant defense (3). The endogenous thioredoxin
(Trx)-peroxiredoxin (Prdx) system is important in fighting
oxidative stress damage due to its high anti-oxidant capacity
within the body (4).
Thioredoxin reductase (TrxR) catalyzes the reduction
of thioredoxin with simultaneous oxidation of NADPH. Trx1 is
crucial for maintaining and increasing the anti-oxidant activity of
the Prdxs. The 2-Cys Prdxs are the predominant Prdx sub-family and
comprise Prdx1, -2, -3 and -4 (5).
All 2-Cys Prdxs contain a cysteine residue that is oxidized to
cysteine sulfenic acid (-SOH) by peroxides. Cys-SOH forms a
disulfide bond with the remaining cysteine, which is, in turn,
reduced by Trx1 (6). Previous
studies have demonstrated that Trx1 returns Prdxs to their reduced
forms to maintain their normal biological functions (7,8).
Increasing the transcriptional activity of Trx1 may be an effective
way to reduce cell damage following ischemia/reperfusion. In 2009,
Soriano et al (6) reported
that the transcription factor activator protein-1 (AP-1) may be
associated with the expression of Trx1.
AP-1 is a redox-sensitive intracellular
transcription factor composed of proteins belonging to the c-FBJ
murine osteosarcoma viral oncogene homolog, c-Jun proto-oncogene
and activating transcription factor families (9). AP-1, a cytosolic protein, responds to
a variety of stimuli by regulating gene expression. Activated AP-1
translocates to the nucleus, recognizes
12-O-tetradecanoylphorbol-13-acetate response elements (TRE;
5′-TGAG/CTCA-3′) and induces the expression of a variety of genes
(10). A previous study observed
TRE sequences in the promoter areas of the Trx1 gene, while the
mechanism of intracellular signaling remains to be elucidated
(11).
The present study focused on the interaction between
Trx1 and Prdxs to determine the underlying mechanisms of the
neuroprotective effects of Trx1 following stroke. For this purpose,
an in vitro cell model of oxygen glucose
deprivation/reperfusion (OGD/R) was used. Furthermore, the
association between AP-1 and Trx1 expression was investigated using
a luciferase reporter assay to determine whether AP-1 directly
upregulates Trx1 expression.
Materials and methods
Reagents and cell culture
A total of 72 neonatal Sprague-Dawley rats (weight,
male, 300–600 g; female, 250–500 g) were obtained from the Animal
Experiment Center of Chongqing Medical University (Chongqing,
China) and sacrificed by decapitation 1 day after birth. Six rats
were used per experimental group. They were maintained in an
atmosphere of 40–70% humidity at 18–26°C. The animal protocol was
approved by the Institutional Animal Care and Use Committee of
Chongqing Medical University (Chongqing, China) and all
experimental procedures were approved by Chongqing Medical
University Biomedical Ethics Committee (Chongqing, China). The
procedures complied with the current national and international
laws and recommendations.
High-glucose Dulbecco's modified Eagle's Medium
(DMEM) and glucose-free DMEM were purchased from Gibco (Thermo
Fisher Scientific, Inc., Waltham, MA, USA). Fetal bovine serum
(FBS), Dulbecco's phosphate-buffered saline (PBS),
3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxyme
thoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, inner salt (MTS),
lactate dehydrogenase (LDH) as well as penicillin and streptomycin
were purchased from Gibco (Thermo Fisher Scientific, Inc.).
Dimethylsulfoxide was purchased from Merck & Co., Inc.
(Whitehouse Station, NJ, USA).
Primary astrocytes were isolated from one-day-old
Sprague-Dawley rats as described previously () and cultured in 10%
FBS/DMEM medium. Sub-culturing was performed after 6–7 days, in a
humidified atmosphere containing 5% CO2, the cells were
then cultured until they reached 90% confluence.
Groups and cell treatment
Astrocytes were divided into three groups: i)
Control group, untreated astrocytes; ii) negative control group,
astrocytes transfected with negative lentivirus (LV3-NC); and iii)
siTrx1 group, astrocytes transfected with the most effective of
four lentiviruses designed to carry small interfering RNA (siRNA)
targeting Trx1 LV3-54: 5′-GGGAGACAAGCTTGTGGTAGT-3′, LV3-135:
5′-CTGTGACAAGTATTCCAATGT-3′, LV3-222: 5′-GCCGACCTTCCAGTTCTATAA-3′,
LV3-288:5′-GCTCGAAGCCACTATTACGGA-3′ and LV3-NC:
5′-TTCTCCGAACGTGTCACGT-3′, as indicated by Trx1 knockdown.
SiTrx1 lentivirus construction and cell
transfection
The lentiviruses were constructed by Shanghai
GenePharma Co., Ltd. (Shanghai, China). These Trx1 constructs were
LV3-54, LV3-135, LV3-222 and LV3-288. LV3-NC served as the negative
control.
The astrocytes were seeded into six-well plates at a
concentration of 2×105 cells/well. When the astrocytes
approached 80–90% confluency, they were transfected with 10
µl LV3-54, LV3-135, LV3-222, LV3-288 and LV3-NC
(multiplicity of infection, 50). The media was changed on the
second day. After 72 h, the efficiency of transfection was observed
using a fluorescent microscope. Trx1 siRNA knockdown efficiency was
confirmed by reverse-transcription quantitative polymerase chain
reaction (RT-qPCR).
Establishment of oxygen glucose
deprivation/reperfusion (OGD/R model
At three days following transfection, astrocytes
were rinsed twice with PBS and cultured in glucose-free DMEM in a
hypoxic atmosphere using a hypoxic chamber (8000DH, Thermo Fisher
Scientific, Inc.) containing 5% O2, 94.5% N2
and 0.5% CO2 at 37°C for 4 h. Following OGD, the cells
were placed in normal medium and immediately transferred to an
incubator containing 5% CO2 in air for 24 h of recovery
at 37°C. The cells in the control group were treated identically
except for the OGD treatment.
Cell viability assay
An MTS assay was used to determine the number of
surviving cells following OGD/R. Astrocytes were seeded in 96-well
plates at 1×103 cells/well with glucose-free media.
Following OGD/R, cells were incubated in normal fresh medium
containing 1 mg/ml MTS at 37°C for 3 h in the dark. The absorption
of the wells was then directly measured at a wavelength of 490 nm
after 20 min using a microplate reader (Multiskan FC; Thermo Fisher
Scientific, Inc.). A blank control was established for each
treatment group by adding by adding 200 µl PBS.
LDH assay
LDH activity was quantitatively detected by
measuring the LDH release from the damaged cells into the culture
medium using a LDH assay kit (Nanjing Jancheng Bioengineering
Institute, Nanjing, China). Astrocytes were seeded in six-well
culture plates at 5×103/well prior to the experiment.
The supernatant was collected and transferred to a 1.5-ml
centrifuge tube following OGD/R. Cellular release of LDH was
determined according to the manufacturer's instructions (Nanjing
Jiancheng Bioengineering Institute). The absorbance (A) was
measured using a Multiskan FC microplate reader at a wavelength of
450 nm and LDH release was calculated as follows: LDH release
(%)=[(Aassay group)−(Acontrol
group)]/[(Astandard group)−(Ablank
group)]×200.
RNA isolation and RT-qPCR
The expression levels of Trx1 and Prdx genes were
examined using RT-qPCR. RNA was isolated following cell lysis using
an RNAiso Plus kit (Takara Biotechnology Co., Ltd., Dalian, China).
Total RNA was reverse-transcribed into cDNA using a cDNA reverse
transcription kit (Takara Biotechnology Co., Ltd.) at 37°C for 15
min, 85°C for 5 sec, and stored at −20°C. The products were used
for two-step qPCR. The primer sequences are listed in Table I. PCR reactions were performed with
iTaq™ Universal SYBR Green (Bio-Rad Laboratories, Inc., Hercules,
CA, USA). The thermocycling program was set as follows: Initial
denaturation at 94°C for 10 min; 35 cycles of denaturation at 94°C
for 1 min, annealing at 55°C for 15 sec and extension at 70°C for
15 sec; and a final extension at 70°C for 5 min. The quantification
cycle (Cq) data were collected using a CFX manager (Bio-Rad
Laboratories, Inc.). Expression of Trx1 was normalized to β-actin.
The relative quantification of gene expression was analyzed using
the 2−ΔΔCq method (12,13).
The fold change in target gene cDNA relative to the internal
control β-actin was calculated as follows: Fold change =
2−ΔΔCq, ΔΔCq = (CqSample −
Cqβ-actin) − (CqControl −
Cqβ-actin).
 | Table IPrimer sequences used for
quantitative polymerase chain reaction. |
Table I
Primer sequences used for
quantitative polymerase chain reaction.
| Gene | Gene ID | Primer sequence
(5′-3′) | Product length
(bp) |
|---|
| Trx1 | NM_053800.3 | F:
CCTTCTTTCATTCCCTCTGTGA
R: CCCAACCTTTTGACCCTTTTTA | 143 |
| Prdx1 | NM_057114.1 | F:
CATTGCTCAGGATTATGGAGTC
R: CATTTATTGTTATCTGGCGAAGG | 104 |
| Prdx2 | NM_017169.1 | F:
CGTGGTCCTCTTTTTCTATCCA
R: CTTTTAGTCACATCAGCAAGCA | 219 |
| Prdx3 | NM_022540.1 | F:
TGCTTTTCTTCTACCCTTTGGA
R: CATTCTTTCTTGGCGTGTTGAT | 165 |
| Prdx4 | NM_053512.2 | F:
CCTCTGCTGCTGTTCCTGTTAC
R: AAATCTTGGCTTTGCTTAGGTG | 175 |
| β-actin | NM_031144 | F:
CACCCGCGAGTACAACCTTC
R: CCCATACCCACCATCACACC | 207 |
Western blot analysis
Cultured cells were washed twice using PBS and the
proteins were extracted and collected in lysis buffer containing a
protease inhibitor cocktail (Beyotime Institute of Biotechnology,
Shanghai, China). Equal quantities of total cellular protein
extracts (40 µg) in each group were separated using 16%
sodium dodecyl sulfate-polyacrylamide gel electrophoresis
(Beyontime Institute of Biotechnology). The proteins were
transferred to 0.22-µm polyvinylidene fluoride membranes
(Beijing Dingguo Changsheng Biotechnology Co., Ltd., Beijing,
China). These membranes were blocked with 5% non-fat milk in
Tris-buffered saline with Tween 20 (TBST; Invitrogen; Thermo Fisher
Scientific, Inc.) at 37°C for 2 h. Blots were incubated overnight
at 4°C with the following primary antibodies obtained from Abcam
(Cambridge, MA, USA): polyclonal rabbit anti-rat Prdx1 (1:2,000;
cat. no. ab59539), polyclonal rabbit anti-rat Prdx2 (1:2,000; cat.
no. ab59539), polyclonal rabbit anti-rat Prdx3 (1:2,000; cat. no.
ab73349), monoclonal mouse anit-rat Prdx4 (1:2,000; cat. no.
16943), polyclonal rabbit anti-rat Prdx-SO3 (1:2,000; cat. no.
ab16830) and β-actin (1:2,000; cat. no. A5441; Sigma-Aldrich).
Following three washes in TBST, membranes were incubated with
horseradish peroxidase-conjugated secondary antibodies (goat
anti-rabbit IgG; 1:3,000; cat. no. BA1054; rabbit anti-rat IgG,
1:3,000; cat. no. BA1058), obtained from Wuhan Boster Biological
Technology, Ltd. (Wuhan, China), at room temperature for 2 h,
followed by another three washes. The protein concentration was
determined using an Enhanced BCA Protein Assay kit (Beyotime
Institute of Biotechnology). The bands were scanned using an
imaging densitometer (ChemDoc XRS; Bio-Rad Laboratories, Inc.), and
the results were quantified using Quantity One software (version
24.0; Bio-Rad Laboratories, Inc.).
Dual-luciferase reporter assays
The binding sites of AP-1 and Trx1 were analyzed by
National Center for Biotechnology Information (NCBI; www.ncbi.nlm.nih.gov/genome). A luciferase
reporter vector driven by a fragment from the promoter of Trx1
(pGLO-Trx1-Luc) was subjected to site-directed mutagenesis of its
TRE (Biomed Gene Technology Co., Ltd., Wuhan, China). Exponentially
growing HEK293 cells (Type Culture Collection of the Chinese
Academy of Sciences, Shanghai, China) were seeded in 24-well plates
at a density of 3×104/well, and the cells were divided
into three groups: i) Control group; ii) Trx1
(WtTRE)-Luc group; and iii) Trx1(MtTRE)-Luc
group. Each group was transfected with 0.8 µg pGLO-Trx1-Luc
reporter and 0.8 µg AP-1 overexpression plasmid (Biomed Gene
Technology Co., Ltd.) using Lipofectamine 2000 (Invitrogen; Thermo
Fisher Scientific, Inc.). The control group remained untreated. The
medium was replaced after 4–6 h of transfection. Following
treatment (24 h), the cells were lysed using 100 µl of
Passive Lysis Buffer (Promega Corporation, Madison, WI, USA) and
the lysates were analyzed using a Dual-Luciferase Reporter Assay
kit according to the manufacturer's protocol (Promega Corporation).
Firefly-based reporter gene activity was normalized to the
Renilla control in all cases.
Statistical analysis
All experiments were performed at least three times
and values are expressed as the mean ± standard error of the mean.
Data were analyzed using one-way analysis of variance, followed by
a post-hoc Tukey's test. For comparative analysis between two
groups, a Student's t-test was used. Statistical analysis was
performed using SPSS, version 17.0 (SPSS, Inc., Chicago, IL, USA)
and P<0.05 was considered to indicate a statistically
significant difference.
Results
Effects of Trx1 knockdown in
astrocytes
To select the most effective lentivirus fragment for
knockdown of Trx1, the efficiency of lentiviral transfection was
examined by fluorescent microscopy (results not shown). In order to
detect the level of Trx1 knockdown, RT-qPCR was performed to
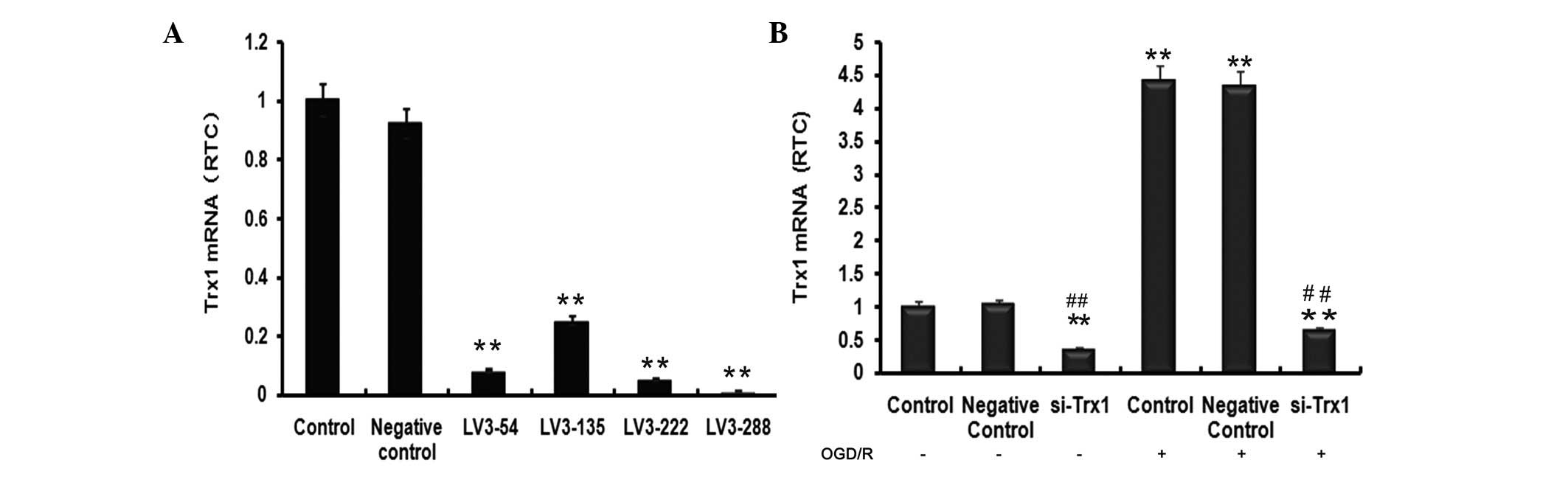
evaluate Trx1 mRNA levels (Fig.
1A). Trx1 mRNA levels indicated a significant reduction of Trx1
following treatment with the four siRNAs compared with the control
group (P<0.01). The largest decrease was observed in the LV3-288
group (P<0.01), approaching a knockdown efficiency of 90±2.21%.
LV3-288 (target sequence, 5′-GCTCGAAGCCACTATTACGGA-3′) was
therefore selected for use in all subsequent experiments.
OGD-induced oxidative stress damage
increases Trx1 mRNA expression levels
To investigate the impact of OGD/R on Trx1 mRNA
expression levels and the efficiency of the siRNA used in
inhibiting the upregulation of Trx1 under OGD/R conditions in
astrocytes, RT-qPCR analysis was performed. As presented in
Fig. 1B, Trx1 mRNA expression in
the si-Trx1 group was reduced by 66±3.61% and 86±2.93% compared
with the controls, respectively (P<0.01). Of note, Trx1 mRNA
levels in astrocytes subjected to OGD/R were significantly elevated
compared with those in the control groups (P<0.01). However,
treatment with LV3-288 completely abrogated these increases
(P<0.01). These results indicated that OGD/R markedly increased
Trx1 mRNA expression levels, which was efficiently blocked by
LV3-288.
Trx1 knockdown decreases astrocyte
viability following OGD/R
To determine the effects of Trx1 on cell viability
and cell damage in astrocytes following OGD/R, MTS and LDH assays
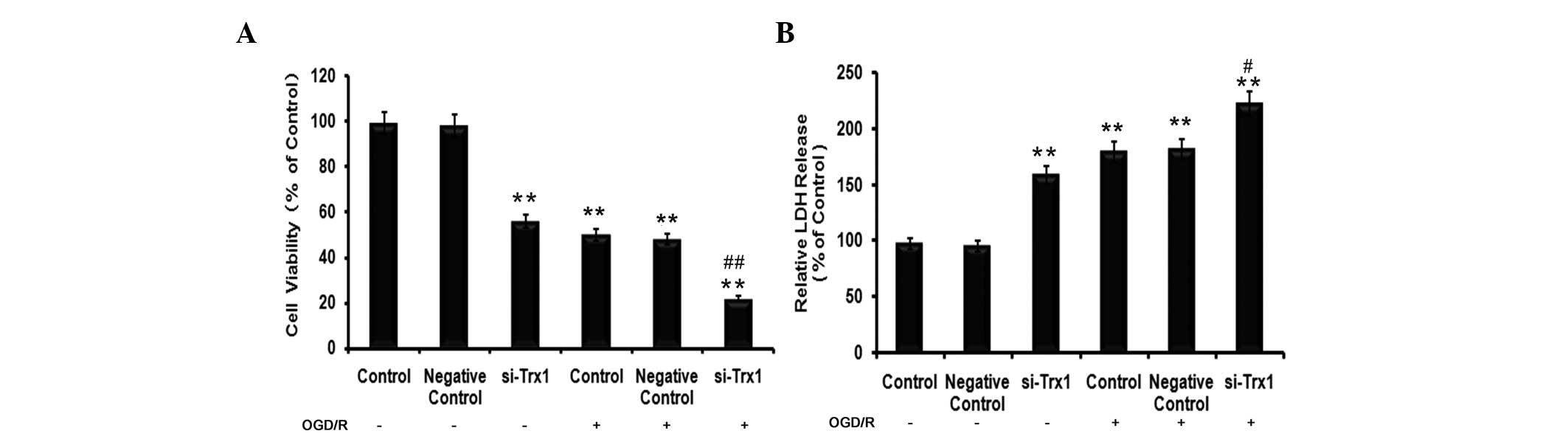
were performed. The results of the MTS assay demonstrated that,
compared with the control group, the cell viability of astrocytes
decreased by 49±1.64% (P<0.05) in the OGD/R group (Fig. 2A). However, the viability of cells
pre-treated with siRNA against Trx1 in the OGD/R group decreased to
77±1.83% of that of the control cells (P<0.01). LDH is a stable
cytoplasmic enzyme present in the majority of cells. It is rapidly
released into the cell culture supernatant upon damage to the
plasma membrane (14). As
presented in Fig. 2B, treatment of
astrocytes with siRNA against Trx1 significantly increased their
LDH release (P<0.01). Furthermore, cells in the OGD/R groups
released significantly more LDH than the control cells (P<0.01),
which was further enhanced by knockdown of Trx1 (P<0.05 vs.
OGD/R group).
Trx1 knockdown inhibits increases in Prdx
expression following OGD/R
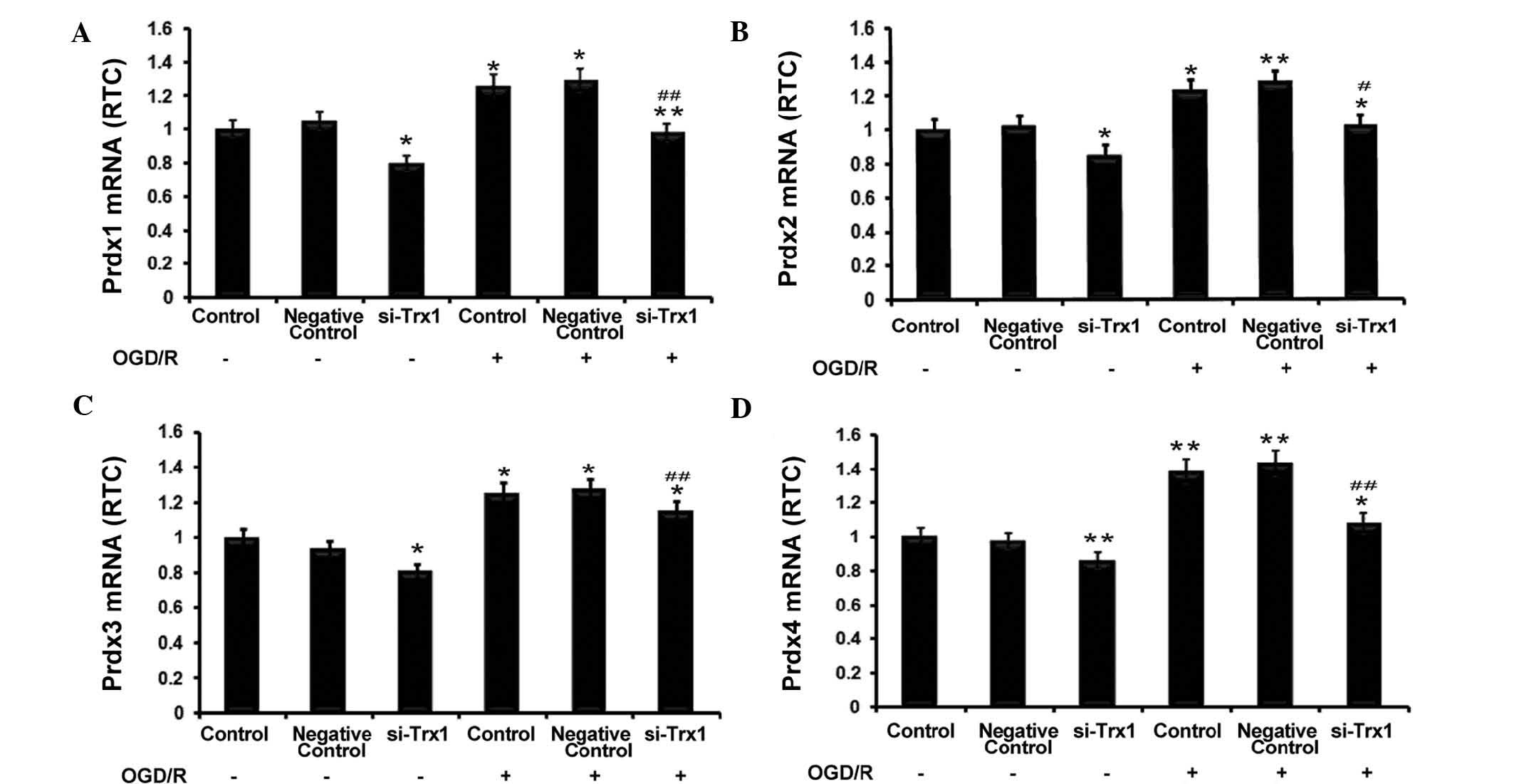
RT-qPCR was performed to investigate the association
between Trx1 and Prdxs. The results showed that Trx-1 knockdown
significantly reduced Prdx1-4 expression in astrocytes (P<0.05
or P<0.01) (Fig. 3).
Furthermore, OGD/R significantly enhanced the expression of Prdx1-4
(P<0.05 or P<0.01 vs. no OGD/R), which was partly abrogated
by knockdown of Trx1 (P<0.05 or P<0.01 vs. OGD/R control
groups). These observations indicated that enhanced expression of
Trx1 during OGD/R increases the expression of the anti-oxidant
Prdxs.
Trx1 knockdown decreases Prdx protein
expression but increases Prdx-SO3 protein
expression
Western blot analysis demonstrated that the protein
expression of Prdx1-4 and Prdx-SO3 was significantly
elevated following OGD/R (P<0.05 or P<0.01) (Fig. 4), which was consistent with the
RT-qPCR results. However, the levels of Prdx1-4 protein were
reduced following Trx1 knockdown regardless of OGD/R. Furthermore,
Prdx-SO3 protein expression was increased following Trx1
knockdown (P<0.01) (Fig. 4).
The results demonstrated that, under oxidative stress, the decrease
in Prdx expression levels was accompanied by an increase in
Prdx-SO3. This results in an imbalance of the redox
equilibrium and leads to damaged cells and tissue. These results
indicated that Trx1 enhances Prdx activity and drives the response
to oxidative stress via increasing the expression of Prdxs and
inhibiting Prdx-SO3.
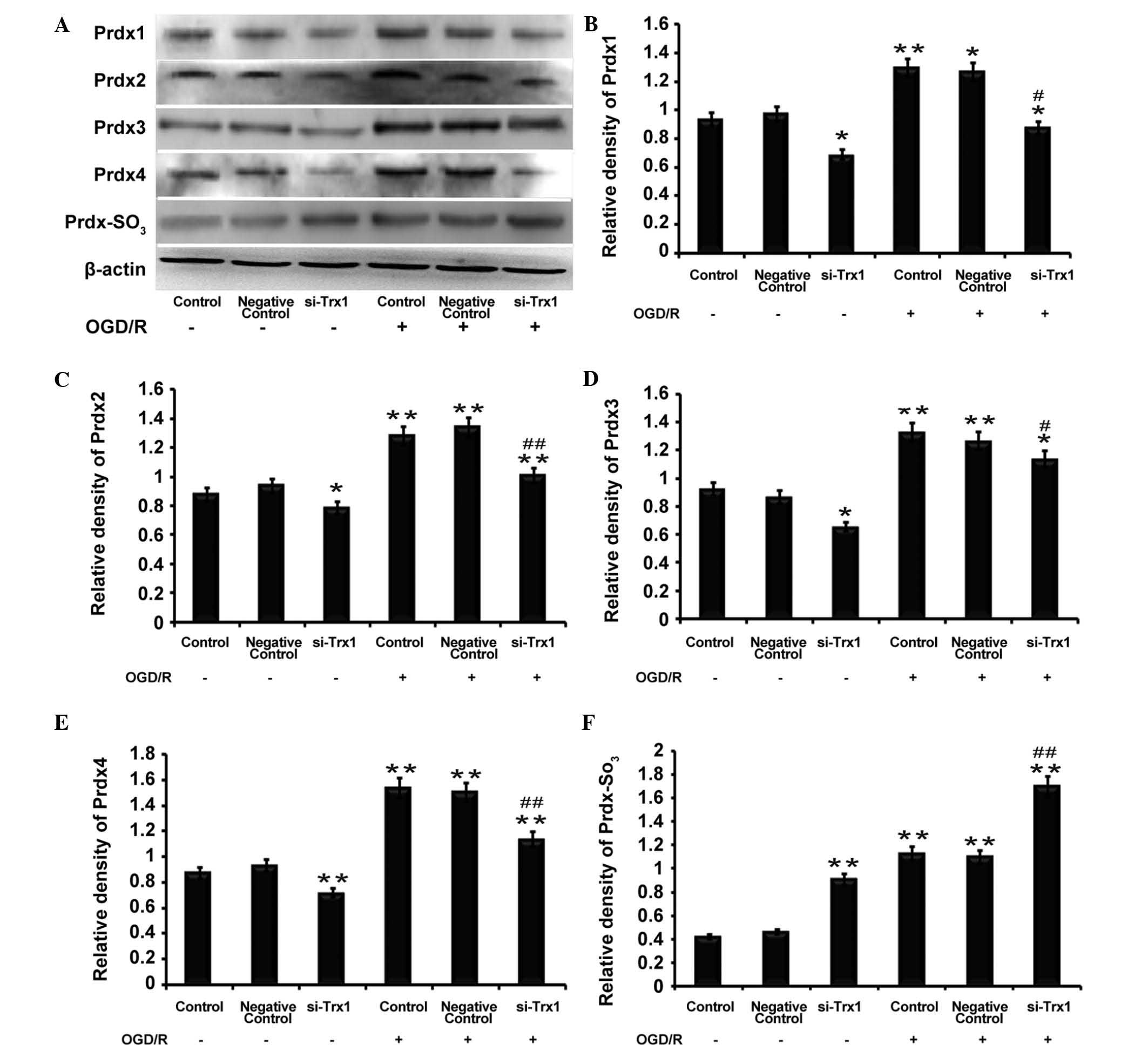 | Figure 4Trx1 knockdown attenuates 2-Cys Prdxs
protein expression and promotes Prdx-SO3 formation. (A)
Representative western blot of Prdx1-4 and Prdx-SO3 in
astrocytes following Trx1 knockdown. Quantification of (B) Prdx1,
(C) Prdx2, (D) Prdx3, (E) Prdx4 and (F) Prdx-SO3 was
performed by densitometric analysis. Following Trx1 knockdown,
expression levels of Prdx1-4 were decreased, while PRDX-SO3 was
elevated. In addition, PRDXs1-4 and PRDX-SO3 protein
expression was increased following OGD/R treatment. Values are
expressed as the mean ± standard error of the mean (n=4).
*P<0.05, **P<0.01 vs. the control;
#P<0.05, ##P<0.01 vs. the si-Trx1
group. Trx1, thioredoxin 1; si, small interfering; OGD/R, oxygen
glucose deprivation/reperfusion; Prdx, peroxiredoxin. |
AP-1 mediates Trx1 expression
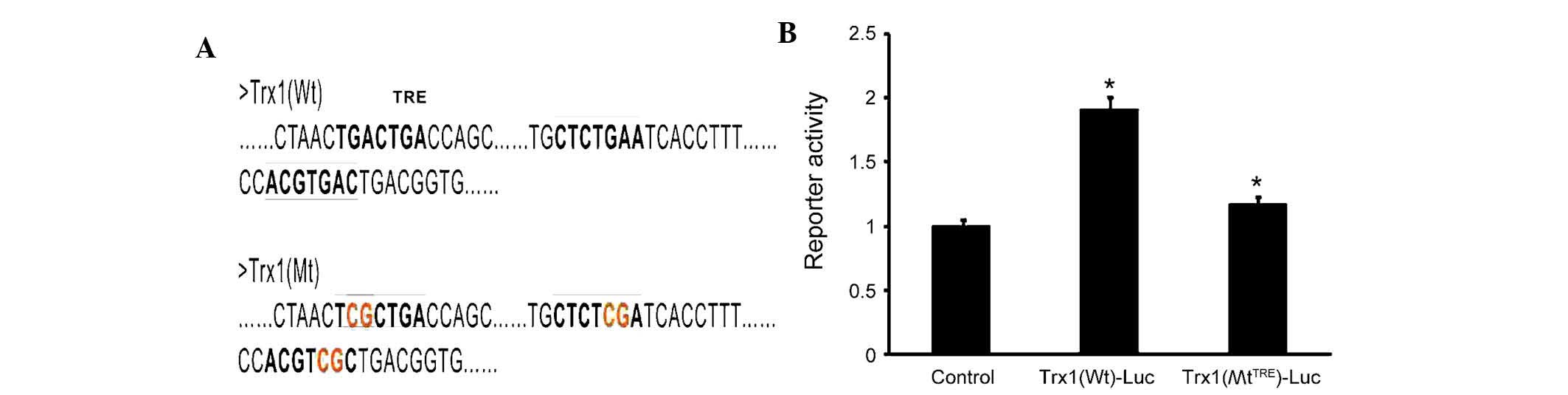
To determine whether AP-1 directly regulates the
expression of Trx1, three tandem AP-1 binding sites were predicted
in the Trx1 promoter using bioinformatics (Fig. 5A). The sequences were verified
using the NCBI database. The AP-1 plasmid as well as the luciferase
reporter plasmids PGLO-Trx1(Wt) and PGLO-Trx1(MtTRE)
were constructed. The luciferase activity in cells co-transfected
with PGLO-Trx1(Wt)-Luc and AP-1-transfected was significantly
increased by 60±4.35% compared with the control group and the
PGLO-Trx1(MtTRE)-Luc group(Fig. 5B). These results demonstrated that
site-directed mutagenesis of the TRE sequences inhibited the
specific binding between AP-1 and TRE (P<0.05), leading to a
marked reduction of luciferase activity. These results demonstrated
that the expression of Trx1 is directly and positively regulated by
AP-1.
Discussion
The Trx system is composed of Trx, Trx receptor
(TrxR) and NADPH, and exerts vital anti-oxidant effects depending
on the activity of TrxR (15,16).
A previous study has demonstrated that in thioredoxin transgenic
mice, cell damage and the outbreak index of cerebral
ischemia/reperfusion injury were markedly attenuated in the
presence of high expression levels of Trx1 (17). Other previous studies further
indicated that Trx1 was induced in response to oxidative stress
resulting from cerebral ischemia in anaerobic environments
(18–20). The present study also suggested
that the Trx1 expression was significantly increased following
OGD/R, and the MTS and LDH results demonstrated that OGD/R
decreased the viability of astrocytes, which was markedly
aggravated by Trx1 knockdown. These findings suggested that Trx1
may protect astrocytes from damage following OGD/R and that it
protects from oxidative stress damage by exerting an anti-oxidant
effects.
In addition to its contribution to redox processes
by reducing inter- and intra-chain protein disulfide bonds, the
Trx1 system also maintains the activity of important anti-oxidant
enzymes, such as peroxiredoxins (Prdxs) (20–23).
In 2010, Hwang et al (24)
demonstrated that Trx1 markedly improves the neuroprotection of
Prdxs in Mongolian gerbils. In the present study, the expression of
Prdx1-4 was observed to decrease following Trx1 knockdown, while
Prdx-SO3 expression increased. The expression of Prdxs
was demonstrated to be closely associated with Trx1. These results
suggested that Trx1 is the cofactor of Prdxs and that Trx1
maintains Prdx activity in response to oxidative stress damage.
According to a previous study, the upregulation of
Trx1 expression is an important step of neuronal responses to
oxidative stress (25). A series
of transcription factor binding sites have been identified in the
promoter region of Trx1, including SP-1 transcription factor, AP-1,
nuclear factor-κB and nuclear factor erythroid 2-like 2 (26–28).
Among these, AP-1 has received increasing attention in recent
years. Previous studies have demonstrated that sulfiredoxin
expression is regulated by the transcription factor AP-1, which
mediates its upregulation by synaptic activity in neurons (29–31).
In the present study, AP-1 was observed to be directly activate the
expression of Trx1 by specific binding to the TRE. Thus, increasing
AP-1 levels may be an effective strategy for enhancing Trx-1
expression and achieving anti-oxidant effects for the treatment of
stroke.
In conclusion, the results of the present study
indicated that Trx1 may positively regulate the expression of
anti-oxidant Prdxs following OGD/R and that its upregulation may be
a suitable method for the prevention and treatment of cerebral
disease. Further exploration using in vitro and in
vivo studies with siRNA and overexpression technology are
required to verify the underlying mechanisms of action of Trx1 and
the interaction between Trx1 and Prdxs.
Acknowledgments
The present study was supported by grants from the
National Natural Science Foundation of China (grant nos. 81171090
and 81271460), the Natural Science Youth Foundation of China (grant
no. 81301125) and the Natural Science Foundation of Chongqing
Education Committee, China (grant no. KJ110313).
References
|
1
|
Liu R, Liu H, Ha Y, Tilton RG and Zhang W:
Oxidative stress induces endothelial cell senescence via
downregulation of Sirt6. Biomed Res Int. 9028422014.PubMed/NCBI
|
|
2
|
Silva DG, Belini Junior E, de Almeida EA
and Bonini-Domingos CR: Oxidative stress in sickle cell disease: An
overview of erythrocyte redox metabolism and current antioxidant
therapeutic strategies. Free Radic Biol Med. 65:1101–1109. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Calabrese V, Cornelius C, Mancuso C,
Pennisi G, Calafato S, Bellia F, Bates TE, Giuffrida Stella AM,
Schapira T, Dinkova Kostova AT and Rizzarelli E: Cellular stress
response: A novel target for chemoprevention and nutritional
neuroprotection in aging, neurodegenerative disorders and
longevity. Neurochem Res. 33:2444–2471. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Wood ZA, Poole LB and Karplus PA:
Peroxiredoxin evolution and the regulation of hydrogen peroxide
signaling. Science. 300:650–653. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wood ZA, Schröder E, Robin Harris J and
Poole LB: Structure, mechanism and regulation of peroxiredoxins.
Trends Biochem Sci. 28:32–40. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Soriano FX, Léveillé F, Papadia S, Higgins
LG, Varley J, Baxter P, Hayes JD and Hardingham GE: Induction of
sulfiredoxin expression and reduction of peroxiredoxin
hyperoxidation by the neuroprotective Nrf2 activator
3H-1,2-dithiole-3-thione. J Neurochem. 107:533–543. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Das KC and Das CK: Thioredoxin, a singlet
oxygen quencher and hydroxyl radical scavenger: Redox independent
functions. Biochem Biophys Res Commun. 277:443–447. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lu J and Holmgren A: Thioredoxin system in
cell death progression. Antioxid Redox Signal. 17:1738–1747. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Pronk TE, van der Veen JW, Vandebriel RJ,
van Loveren H, de Vink EP and Pennings JL: Comparison of the
molecular topologies of stress-activated transcription factors
HSF1, AP-1, NRF2 and NF-κB in their induction kinetics of HMOX1.
Biosystems. 124:75–85. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Choi WJ: The Heterochromatin-1
phosphorylation contributes to TPA-Induced AP-1 expression. Biomol
Ther (Seoul). 22:308–313. 2014. View Article : Google Scholar
|
|
11
|
Chen B, Guan D, Cui ZJ, Wang X and Shen X:
Thioredoxin 1 downregulates MCP-1 secretion and expression in human
endothelial cells by suppressing nucleartranslocation of activator
protein 1 and redox factor-1. Am J Physiol Cell Physiol.
298:C1170–1179. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data expression data using real-time
quantitative PCR and the 2(-Delta Delta C (T)) Method. Methods.
25:402–408. 2001. View Article : Google Scholar
|
|
13
|
Xing G, Dong M, Li X, Zou Y, Fan L, Wang
X, Cai D, Li C, Zhou L, Liu J and Niu Y: Neuroprotective effects of
puerarin against beta-amyloid-induced neurotoxicity in PC12 cells
via a PI3K-dependent signaling pathway. Brain Res Bull. 85:212–218.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Guo H, Kong S, Chen W, Dai Z, Lin T, Su J,
Li S, Xie Q, Su Z, Xu Y and Lai X: Apigenin mediated protection of
OGD-evoked neuron-like injury in differentiated PC12 cells.
Neurochem Res. 39:2197–2210. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Sengupta R and Holmgren A: Thioredoxin and
thioredoxin reductase in relation to reversible S-nitrosylation.
Antioxid Redox Signal. 18:259–269. 2013. View Article : Google Scholar
|
|
16
|
Nickel C, Rahlfs S, Deponte M, Koncarevic
S and Becker K: Thioredoxin networks in the malarial parasite
Plasmodium falciparum. Antioxid Redox Signal. 8:1227–1239. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Das DK: Thioredoxin regulation of ischemic
preconditioning. Antioxid Redox Signal. 26:405–412. 2004.
View Article : Google Scholar
|
|
18
|
Taksgi Y, Hatori I, Nozaki K, Mitsui A,
Ishikawa M, Hashimoto N and Yodoi J: Excitotoxic hippocampal injury
is atenuated in thioredoxin transgenic mice. J Cereb Blood Flow
Metab. 20:829–833. 2000.
|
|
19
|
Takagi Y, Horikawa F, Nozaki K, Sugino T,
Hashimoto N and Yodoi J: Expression and distribution of redox
regulatory protein, thioredoxin during transient focal brain
ischemia in the rat. Neurosci Lett. 251:25–28. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Mansur K, Iwashashi Y, Kiryu-Seo S, Su Q,
Namikawa K, Yodoi J and Kiyama H: Up-regulation of thioredoxin
expression in motor neurons after nerve injury. Brain Res Mol Brain
Res. 62:86–91. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Rhee SG, Kang SW, Jeong W, Chang TS, Yang
KS and Woo HA: Intracellular messenger function of hydrogen
peroxide and its regulation by peroxiredoxins. Curr Opin Cell Biol.
17:183–189. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Masutani H, Ueda S and Yodoi J: The
thioredoxin system in retroviral infection and apoptosis. Cell
Death Differ. 12(Suppl 1): 991–998. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kondo N, Nakamura H, Masutani H and Yodoi
J: Redox regulation of human thioredoxin network. Antioxid Redox
Signal. 8:1881–1890. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Hwang IK, Yoo KY, Lee CH, Kim DW, Choi JH,
Kwon YG, Kim YM, Choi SY and Won MH: Changes in the expression of
mitochondrial peroxiredoxin and thioredoxin in neurons and glia and
their protective effects in experimental cerebral ischemic damage.
Free Radic Biol Med. 48:1242–1251. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Liu M, Wang Y, Zheng L, Zheng W, Dong K,
Chen S, Zhang B and Li Z: Fasudil reversed MCT-induced and chronic
hypoxia-induced pulmonary hypertension by attenuating oxidative
stress and inhibiting the expression of Trx1 and HIF-1α. Respir
Physiol Neurobiol. 201:38–46. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Bloomfield KL, Osborne SA, Kennedy DD,
Clarke FM and Tonissen KF: Thioredoxin-mediated redox control of
the transcription factor Sp1 and regulation of the thioredoxin gene
promoter. Gene. 319:107–116. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kim YC, Yamaguchi Y, Kondo N, Masutani H
and Yodoi J: Thioredoxin-dependent redox regulation of the
antioxidant responsive element (ARE) in electrophile response.
Oncogene. 22:1860–1865. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Tanito M, Masutani H, Kim YC, Nishikawa M,
Ohira A and Yodoi J: Sulforaphane induces thioredoxin through the
antioxidant-responsive element and attenuates retinal light damage
in mice. Invest Ophthalmol Vis Sci. 46:979–987. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Papadia S, Soriano FX, Léveillé F, Martel
MA, Dakin KA, Hansen HH, Kaindl A, Sifringer M, Fowler J, Stefovska
V, et al: Synaptic NMDA receptor activity boosts intrinsic
antioxidant defenses. Nat Neurosci. 11:476–487. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Soriano FX, Baxter P, Murray LM, Sporn MB,
Gillingwater TH and Hardingham GE: Transcriptional regulation of
the AP-1 and Nrf2 target gene sulfiredoxin. Mol Cells. 27:279–282.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wei Q, Jiang H, Matthews CP and Colburn
NH: Sulfiredoxin is an AP-1 target gene that is required for
transformation and shows elevated expression in human skin
malignancies. Proc Natl Acad Sci USA. 105:19738–19743. 2008.
View Article : Google Scholar : PubMed/NCBI
|