Introduction
Diabetes mellitus is the leading cause of end-stage
renal disease (ESRD), based on the United States renal data system
(1). Glycemic control and
currently available pharmacotherapies delay, but cannot prevent,
the progression of diabetic nephropathy (DN) towards ESRD (2,3).
However, in order to develop novel therapies, a full understanding
of the etiology of DN is required. Proteinuria, specifically
microalbuminuria, is currently one of the earliest clinically
identifiable markers of diabetes-induced renal damage, and the
likelihood of progression to ESRD is significantly correlated with
the level of albuminuria (4–7).
Proteinuria not only predicts the speed of development in ESRD, but
also correlates with renal decline. In addition, patients with DN
with low levels of proteinuria have a markedly slower rate of
disease progression, compared with those with a high rate of
urinary protein excretion (8–11).
The selection of targets and the timing of intervention are, thus
critical for effectively preventing DN. Therefore, identifying the
appropriate cellular targets for therapeutic intervention is
crucial to enable the prevention of proteinuria. The progression of
DN frequently begins with injury to podocytes, and there is a close
association between the onset of albuminuria and podocytopathies
(12,13), including foot process effacement,
podocyte hypertrophy, detachment, apoptosis and
epithelial-to-mesenchymal transition (EMT) (14). Based on various human and
experimental models, DN is associated with a decreased number of
podocytes per glomerulus and foot process effacement upon biopsy
(15). Previous studies in OVE26
transgenic mice, a model of type-1 DN, demonstrated that reduced
podocyte numbers and density frequently follow the onset of
micro-albuminuria and more subtle podocyte injuries (16,17).
Therefore, podocyte effacement and loss are critical events in the
early progression of DN.
The EMT can also result in podocyte loss, during
which epithelial cells undergo morphological changes. Podocyte EMT
can be caused by the loss of epithelial P-cadherin, zonula
occludens-1 or nephrin, and additional stimuli include the
acquisition of mesenchymal FspI, desmin, collagen I and fibronectin
(18). Podocytes can also undergo
EMT under conditions of high glucose (19), which is associated with increased
podocyte detachment, microalbuminuria and more severe glomerular
pathology. Several intracellular signaling pathways regulate EMT,
including transforming growth factor-β/small mothers against
decapentaplegic, Wnt/β-catenin and integrin-linked kinase (ILK)
(20). Glycogen synthase kinase 3β
(GSK-3β) is involved in all these pathways, and thus may be pivotal
in podocyte EMT (20). Therefore,
GSK-3β inhibitors have been investigated in mesangial proliferative
glomerulonephritis, crescent glomerulonephritis and lupus nephritis
(21). In these studies, the
GSK-3β inhibitor, (2′Z, 3′E)-6-bromoindirubin-3′-oxime (BIO), was
found to inhibit high glucose-stimulated apoptosis in mesangial
cells.
The mechanisms involved in glomerular injury and
proteinuria during diabetes mellitus require additional
investigation. The present study investigated the association
between high glucose and EMT in podocytes and in the kidneys of
db/db diabetic mice. In addition, the present study investigated
the therapeutic potential of modulating GSK-3β and EMT, in the
presence of high glucose, for DN.
Materials and methods
Animals
The Committee for the Care and Use of Laboratory
Animals of Xinxiang Medical College (Xinxiang, China) approved all
animal experiments. A total of 48 male db/db mice and 24
age-matched db/+ control mice were selected (Model Animal Research
Center of Nanjing University, Nanjing, China). At an age of 12
weeks, the db/+ mice (normal control group; NC) and 24 db/db mice
(diabetic nephropathy group; DN) were subcutaneously injected with
dimethyl sulfoxide used as a diluent (Sigma-Aldrich, St. Louis, MO,
USA), whereas the remaining 24 db/db mice (BIO intervention group;
BIO) were injected with 320 µg/kg per day BIO
(Sigma-Aldrich). All mice were housed in temperature- and
humidity-controlled IVC-II independent supply isolation cages
(Jinan Aonuo CNC Equipment Co., Ltd., Jinan, China), with free
access to pure water and standard laboratory chow. When the mice
were 12, 15 and 18 weeks old, eight mice in each group were
sacrificed. On the day prior to sacrifice, the mice were
individually housed in metabolic cages for the collection of urine
over 24 h. The levels of urinary albumin excretion (UAE) were
quantified using an albumin ELISA kit (Abcam, Cambridge, MA, USA).
Heart blood (1 ml) was drawn and centrifuged at 625 × g at 4°C for
4 min. Serum glucose was measured at the time of sacrifice using an
automatic analyzer (SPOTCHEM EZ SP-4430; ARKRAY, Inc., Kyoto,
Japan). Following sacrifice of the mice, the kidneys were rapidly
dissected and weighed, and the cortices were separated. Renal
tissue sections (4 µm) were prepared and stained with
hematoxylin and eosin (HE; Fuzhou Maixin Biotechnology Development
Co., Ltd., Fuzhou, China), followed by assessment using light
microscopy (Leica Microsystems GmbH, Wetzlar, Germany). In
addition, electron microscopy was used to observe the
micro-morphological changes in the renal tissues. A total of 20
glomeruli were evaluated for each mouse. The present study was
approved by the ethics committee of the First Affiliated Hospital
of Zhengzhou University (Zhengzhou, China).
Cell culture and treatment
Cells of the MPC-5 clonal cell line of conditionally
immortalized mouse podocytes, cultured in vitro from
H-2Kb-tsA58 mice, were provided by American Mount Sinai Medical
College, and were cultured, as previously described (22). For propagation, the cells were
cultured at 33°C in RPMI 1640 medium (Gibco; Thermo Fisher
Scientific, Inc., Waltham, MA, USA) supplemented with 10 U/ml mouse
recombinant interferon-γ (Shanghai Sangon Biological Engineering
Technology and Services, Co., Ltd., Shanghai, China) and 10% fetal
bovine serum (Invitrogen; Thermo Fisher Scientific, Inc.) to
enhance the expression of a thermosensitive T antigen. The cells
were then grown under non-permissive conditions at 37°C, without
interferon-γ, for 14 days to induce differentiation. The podocytes
were then cultured with or without high glucose (25 mmol/l;
Sigma-Aldrich), in the presence or absence of BIO (10
µmol/l) for 36 h.
Electron microscopy (EM)
Samples from 24 mice were used for the EM. Following
anesthesia with ketamine (100 mg/kg; Sigma-Aldrich), the mice were
sacrificed by cervical dislocation and 2% glutaraldehyde was
perfused into the kidneys, which were then excised and immersed in
the same fixative overnight. The tissue blocks were then fixed in
2% osmium tetroxide (Sigma-Aldrich) for 2 h at 4°C, dehydrated
using an ethanol gradient and Epon-embedded (Fuzhou Maixin
Biotechnology Development Co., Ltd.). Ultrathin sections (80 nm)
stained with 4% uranyl acetate (Sigma-Aldrich) and with 1% lead
citrate (Sigma-Aldrich), were cut using an ultra-microtome (EM UC7;
Leica Microsystems GmbH) and examined by EM (JEM-100SX; JEOL, Ltd.,
Tokyo, Japan).
Western blotting
Western blotting was performed using established
protocols to assess specific protein expression levels, as
previously described (23).
Radioimmunoprecipitation assay lysis buffer (Santa Cruz
Biotechnology, Inc., Dallas, TX, USA) according to the
manufacturer's protocols, and protein concentration was determined
using the Coomassie brilliant blue method. Proteins (30 µg)
were separated on 10% SDS-PAGE gels for 3 h, then transferred to
polyvinylidene fluoride membranes. Following washing with
phosphate-buffered saline three times for 14 min, 1% bovine serum
albumin (Fuzhou Maixin Biotechnology Development Co., Ltd.) was
used to block the membrane for 2 h. Following blocking, the
membranes were incubated with primary antibodies overnight at 4°C
as follows: Rabbit polyclonal anti-podocin (dilution, 1:1,000;
Bioss, Inc., Woburn, MA, USA; cat. no. bs6597R) rabbit polyclonal
anti-nephrin (dilution, 1:1,000; cat. no. ab58968), rabbit
polyclonal anti-synaptopodin (dilution, 1:200; cat. no. ab109560),
rabbit polyclonal anti-α-SMA (dilution, 1:400; cat. no. ab5694),
rabbit polyclonal anti-fibronectin (dilution, 1:2,000; cat. no.
ab2413), rabbit polyclonal
anti-phosphorylated-Tyr216-GSK-3β (dilution, 1:1,000;
cat. no. ab75745), mouse polyclonal anti-vitamin D receptor (VDR;
dilution, 1:1,000; cat. no. ab3508), and mouse monoclonal
anti-β-actin (dilution, 1:1,000; cat. no. ab6276), mouse monoclonal
anti-GSK-3β (dilution, 1:1,000; cat. no. ab75745; all from Abcam),
rabbit monoclonal anti-phosphorylated-Ser9-GSK-3β
(dilution, 1:1,000; cat. no. 5558), rabbit monoclonal
anti-β-catenin (dilution, 1:1,000; cat. no. 8480), rabbit
monoclonal anti-Snail (dilution, 1:1,000; cat. no. 3879; all from
Cell Signaling Technology, Inc., Danvers, MA, USA). The secondary
antibodies used incubated with the membrane for 2 h at room
temperature and were as follows: alkaline phosphatase-labeled goat
anti-rabbit IgG (dilution l:200; Abcam; cat. no ab97048) and
alkaline phosphatase-labeled horse anti-mouse IgG (dilution l:200;
Vector Laboratories, Inc., Burlingame, CA, USA; cat. no. AP-2000),
goat anti-rabbit IgG (dilution l:200; Abcam; cat. no. ab6721) and
donkey anti-mouse IgG (dilution l:300; Abcam; cat. no. ab150105).
Visualization was performed using nitro-blue
tetrazolium/5-bromo-4-chloro-3′-indolyphosphate coloration (Wuhan
Boster Biological Technology, Ltd., Wuhan, China). The intensities
of the bands were measured and quantified using ImageJ analysis
software (version 1.46; imagej.nih.gov/ij/).
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR) analysis
Total RNA was isolated using TRIzol reagent
(Invitrogen; Thermo Fisher Scientific, Inc.), according to the
manufacturer's protocol. Synthesis of the first strand of cDNA was
performed in 20 µl reaction buffer with 2 µg (13
µl) RNA, 4 µl 5X PrimeScript Buffer (Takara
Biotechnology, Co., Ltd., Dalian, China), 1 µl PrimeScript
RT Enzyme mix (Takara Biotechnology, Co., Ltd.), 1 µl
Oligo-dT primer (50 µmol/l) and 1 µl random hexamers
(100 µmol/l) at 37°C for 15 min, followed by 85°C for 5 sec.
qPCR was performed, according to standard protocols with 3
µl aliquots of cDNA (5 ng/µl) using specific primer
pairs, the sequences of which are shown in Table I. The reaction mixture (15
µl) contained 7.5 µl 2X Premix Ex Taq (Takara
Biotechnology, Co., Ltd.), 0.25 µl (10 µmol/l)
forward primer, 0.25 µl (10 µmol/l) reverse primer, 3
µl cDNA and 4 µl distilled water and the PCR reaction
was performed in a Veriti® 96-Well Thermal Cycler
(Applied Biosystems; Thermo Fisher Scientific, Inc.). Following an
initial denaturation at 95°C for 3 min, the thermocycling
conditions were as follows: For nephrin, 40 cycles of 95°C for 30
sec, 56°C for 30 sec and 72°C for 1 min, followed by 72°C for 5
min; for α-SMA, 40 cycles of 95°C for 30 sec, 52°C for 30 sec and
72°C for 1 min, followed by 72°C for 5 min; for GSK-3β, 40 cycles
of 95°C for 30 sec, 59°C for 30 sec and 72°C for 1 min, followed by
72°C for 5 min; for Snail, 40 cycles of 95°C for 30 sec, 55°C for
30 sec and 72°C for 1 min, followed by 72°C for 5 min; for
β-catenin, 45 cycles of 95°C for 30 sec, 51°C for 30 sec and 72°C
for 1 min, followed by 72°C for 5 min; and for GAPDH, 40 cycles of
95°C for 30 sec, 60°C for 30 sec and 72°C for 1 min, followed by
72°C for 5 min. ImageJ software was used to visualize the qPCR
product sizes fractionated on a 1.0% agarose gel
(Sigma-Aldrich).
 | Table IPrimer sequences, product sizes and
annealing temperatures/durations for reverse
transcription-quantitative polymerase chain reaction analysis. |
Table I
Primer sequences, product sizes and
annealing temperatures/durations for reverse
transcription-quantitative polymerase chain reaction analysis.
| Gene | Primer
sequence | Product (bp) | Duration (sec) | Temp (°C) |
|---|
| GAPDH | F:
5′-CCTGCACCACCAACTGCTTAGC | | | |
| R:
5′-CCAGTGAGCTTCCCGTTCAGC | 238 | 30 | 56 |
| Nephrin | F:
5′-CCCAGGTACACAGAGCACAA | | | |
| R:
5′-CTCACGCTCACAACCTTCAG | 200 | 30 | 55 |
| α-SMA | F:
5′-TACTGCCGAGCGTGAGA | | | |
| R:
5′-GCTTCGTCGTATTCCTGTTT | 489 | 30 | 51 |
| GSK-3β | F:
5′-TTCAGGCCGCTGTTCACCG | | | |
| R:
5′-GTGCTGGTCTTTCCCGCGCA | 152 | 30 | 60 |
| β-catenin | F:
5′-CTCATTCCACCAACTGCTTGGC | | | |
| R:
5′-TAAGTGAGCTTTCGGTTCAGC | 298 | 30 | 51 |
| Snail | F:
5′-CCCTAGGTACACAGAGCACG | | | |
| R:
5′-GCCACGCTCACAACCTTACG | 408 | 30 | 54 |
GSK-3β kinase activity
The activity of GSK-3β was assayed using a GSK-3β
Activity assay kit (Sigma-Aldrich), according to the manufacturer's
protocol and as previously described (24). Activity was calculated following
determining optical densities (OD) using the following formula:
GSK-3β activity = [(OD sample − OD blank) × 0.1 × gradient
concentration] / 0.005 × 6.22 × 5.
Statistical analysis
All data are presented as the means ± standard
deviation of three independent experiments and statistical analysis
was conducted using SPSS 17.0 software (SPSS, Inc., Chicago, IL,
USA). The physiological variables were analyzed using one-way
analysis of variance followed by least significant difference
multiple comparison post-hoc analysis. Other variables were
compared using a Kruskal-Wallis non-parametric test followed by
multiple comparisons using Duncan's method. For all comparisons,
P<0.05 was considered to indicate a statistically significant
difference.
Results
Effects of BIO on blood glucose, UAE,
body weight and kidney weight
As summarized in Table
II, blood glucose remained significantly elevated in the DN and
BIO groups, compared with the NC group throughout the experiment.
No significant differences were found between the DN and BIO
groups, suggesting that BIO had no effect on blood glucose. The
levels of UAE increased from the age of 12 weeks, and continued to
increase until the age of 18 weeks. The increases in UAE were
predominantly attenuated in the BIO-treated mice, compared with the
DN mice, suggesting that BIO inhibited damage to the glomerular
filtration membrane. The body and kidney weights were also
increased by 12 weeks of age, and continued to increase at 15 and
18 weeks. The increases in body weight and kidney weight were
markedly lower in the BIO group, compared with the DN group. These
data suggested that BIO significantly attenuated DN.
| Table IIEffects of BIO on blood glucose, UAE,
bodyweight, and kidney weight. |
Table II
Effects of BIO on blood glucose, UAE,
bodyweight, and kidney weight.
| Time point (age of
mice)
| F-value | P-value |
|---|
| 12 weeks | 15 weeks | 18 weeks |
|---|
| Blood glucose
(mmol/l) |
| NC group | 6.11±1.02 | 6.18±0.99 | 6.29±0.54 | 0.129 | 0.880 |
| DN group | 27.50±7.78a | 28.03±7.91a | 28.90±7.18a | 0.222 | 0.804 |
| BIO group | 27.24±7.08a | 28.29±4.57a | 28.77±2.71a | 0.502 | 0.523 |
| F value | 32.368 | 45.797 | 68.606 | – | |
| P-value | <0.05 | <0.05 | <0.05 | – | – |
| Urinary albumin
excretion (µg/24 h) |
| NC group | 36.15±7.18 | 38.39±7.24 | 39.39±4.46 | 1.466 | 0.264 |
| DN group |
239.24±37.80a |
244.66±35.30a,c |
285.41±28.41a,c | 44.826 | <0.05 |
| BIO group |
239.54±24.08a |
182.51±19.05a–c |
135.65±18.96a–d | 182.992 | <0.05 |
| F value | 160.420 | 161.764 | 310.919 | – | – |
| P-value | <0.05 | <0.05 | <0.05 | – | – |
| Body weight
(g) |
| NC group | 11.96±1.02 | 14.54±1.19 | 16.06±1.27 | 62.437 | <0.05 |
| DN group | 29.91±4.71a | 34.24±4.08a | 36.95±5.23a | 30.326 | <0.05 |
| BIO group | 29.93±4.85a | 32.09±4.93a,b | 32.49±4.77a,b | 3.664 | 0.052 |
| F value | 55.200 | 66.077 | 56.106 | – | – |
| P-value | <0.05 | <0.05 | <0.05 | – | – |
| Kidney weight
(mg) |
| NC group | 107.32±8.77 | 116.02±8.82 | 123.92±7.24 | 96.946 | <0.05 |
| DN group |
180.00±25.66a |
189.95±19.27a,c |
197.41±17.53a,c | 8.318 | <0.05 |
| BIO group |
180.78±14.97a |
174.58±16.28a,b,c |
159.90±22.53a–d | 16.383 | <0.05 |
| F value | 44.514 | 51.124 | 37.360 | – | – |
| P-value | <0.05 | <0.05 | <0.05 | – | – |
Kidney morphology
Following staining with HE, the renal cortices were
observed under a light microscope. Compared with the NC group
(Fig. 1A–C), increases in the
mesangial matrix, occlusion of kidney tubules, sclerosis of the
glomerulus, adhesion of balloon loops and Baumann's capsule walls
were observed when the DN mice were 12 weeks old, and became more
prominent by 18 weeks (Fig. 1D–F).
These changes were significantly attenuated in the BIO group,
compared with the DN group (Fig.
1G–I). Compared with the NC group (Fig. 2A–D), fusion and loss of foot
processes, increased thickness of the glomerular basement membrane
(GBM), increased mesangial matrix and collagen fiber, and hyaline
degeneration of afferent and efferent vessels were detected in the
DN mice (Fig. 2E–H). These changes
were also significantly attenuated in the BIO group, compared with
the DN group, suggesting that BIO slowed down the pathological
changes in the glomerulus stimulated by DN (Fig. 2I–L).
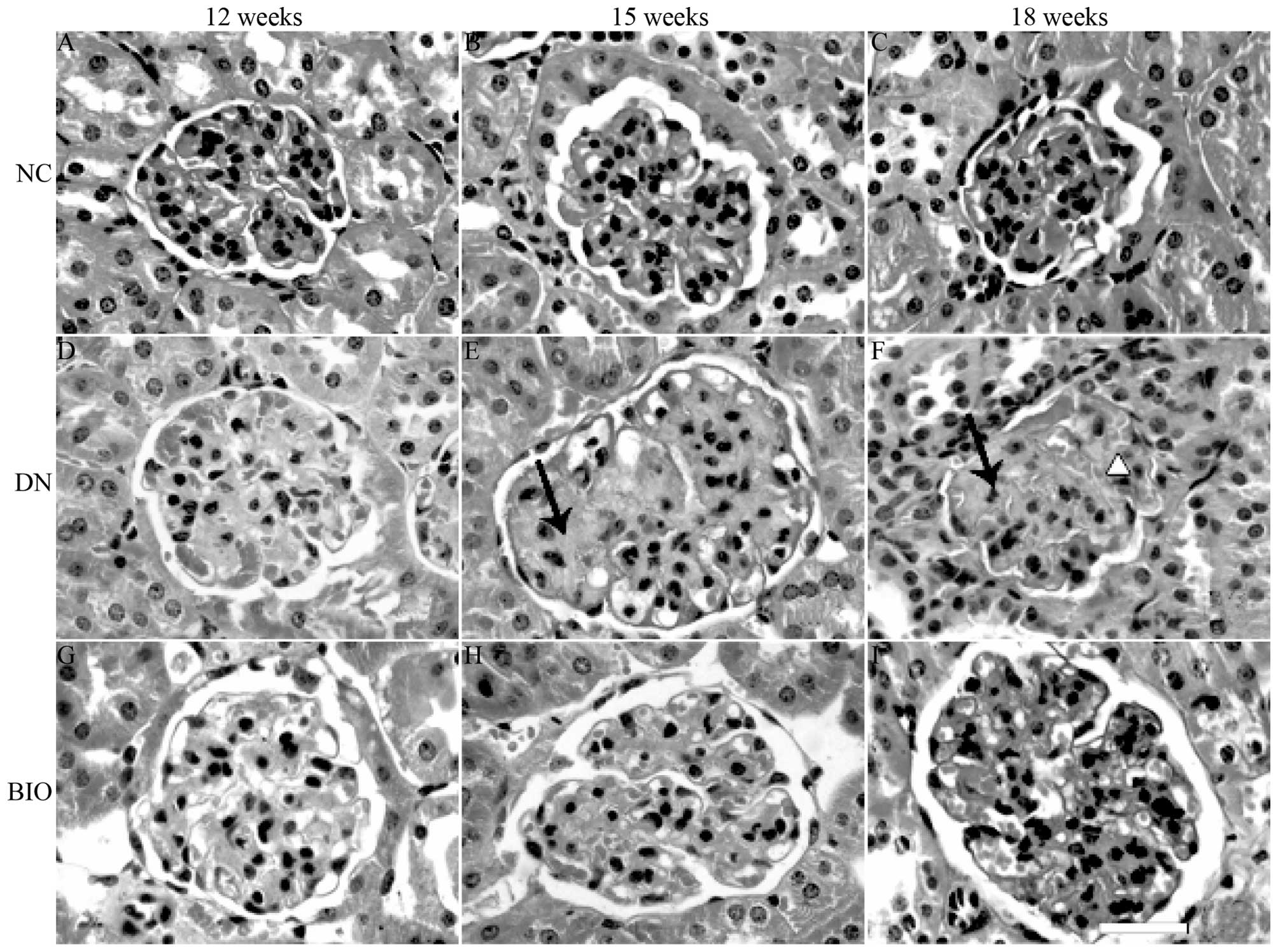 | Figure 1HE staining of mouse kidney sections
in the NC, DN and BIO groups at 12, 15 and 18 weeks of age. Kidney
sections were harvested from mice in the NC, DN and BIO groups at
12, 15 and 18 weeks of age, and were stained with HE. The DN group
exhibited increased mesangial matrix, adhesion of balloon loops and
Baumann's capsule walls at 12 weeks, and the effects were more
prominent by 18 weeks-of-age, compared with the mice in NC group.
Following treatment with BIO for 6 weeks (from 12 weeks of age),
there was a significant decrease in mesangial matrix in and the
number and extent of glomerular sclerosis, compared with the
untreated DN mice. Arrows indicate Kimmelstiel-Wilson nodules.
Triangles indicate adhesion of balloon loops and Baumann's
capsules. Scale bar=40 µm (n=8). HE, hematoxylin and eosin;
NC, normal control (db/+); DN, diabetic neuropathy (db/db); BIO,
(2′Z, 3′E)-6-bromoindirubin-3′-oxime-treated. |
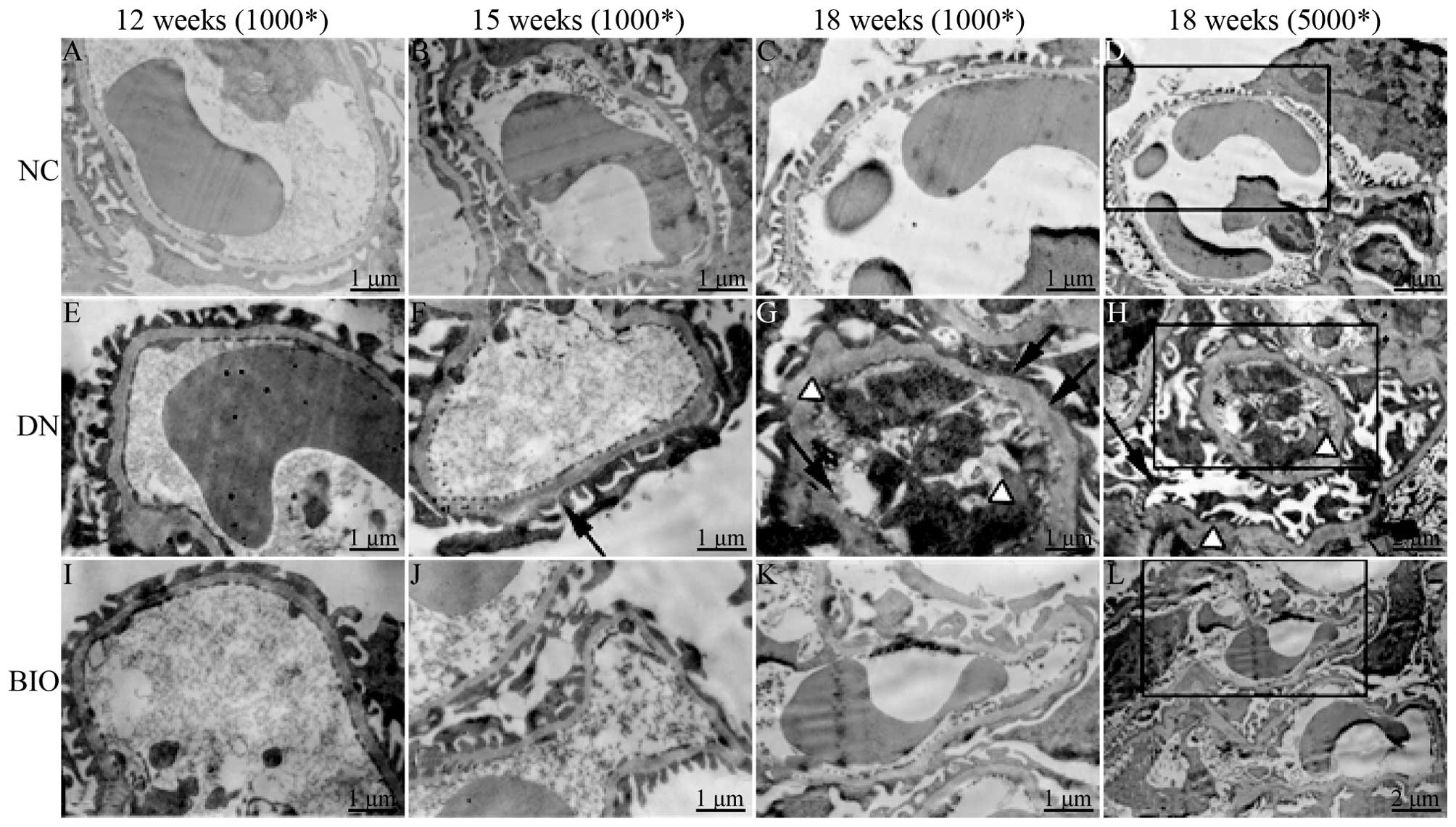 | Figure 2Ultrastructural examination using
transmission electron microscopy in the NC, DN and BIO groups at
12, 15 and 18 weeks-of-age. Ultrathin sections (80 nm) were
harvested from mice in the NC, DN and BIO groups at 12, 15 and 18
weeks of age, and were stained with 4% uranyl acetate and 1% lead
citrate. Fusion and loss of foot processes, and increase in the
thickness of the GBM were found in the DN group, compared with the
NC group. Following treatment with BIO for 6 weeks (from 12 weeks
of age), there was a significant decrease in foot process fusion
and GBM thickness, compared with the DN group. The black boxes on
the images at 18 weeks show the area of the image, which was
magnified by 200% and shown to its left. White arrowheads indicate
foot process effacement of podocytes; triangle indicate thickening
of the GBM (n=8). NC, normal control group (db/+); DN, diabetic
neuropathy group (db/db); BIO, (2′Z,
3′E)-6-bromoindirubin-3′-oxime-treated DN group; GBM, glomerular
basement membrane. |
Effects of BIO on the expression of
epithelial cell markers
The expression levels of phenotypic markers for
epithelial cells in mouse kidney tissues were compared, with
β-actin as a loading control, using Western blotting and RT-qPCR
analysis. With the progression of DN, the expression of nephrin
decreased significantly in the DN group (Fig. 3). However, nephrin levels were
significantly attenuated at 15 and 18 weeks of age in the BIO
group, compared with the DN group (Fig. 3A). The results of the RT-qPCR
analysis of mRNA levels of nephrin in mouse kidney tissue were
analyzed using ImageJ software, which confirmed these observations
(Fig. 3B). Similar expression
patterns were observed in the protein expression levels of podocin
and synaptopodin, two other epithelial markers (Fig. 3C and D).
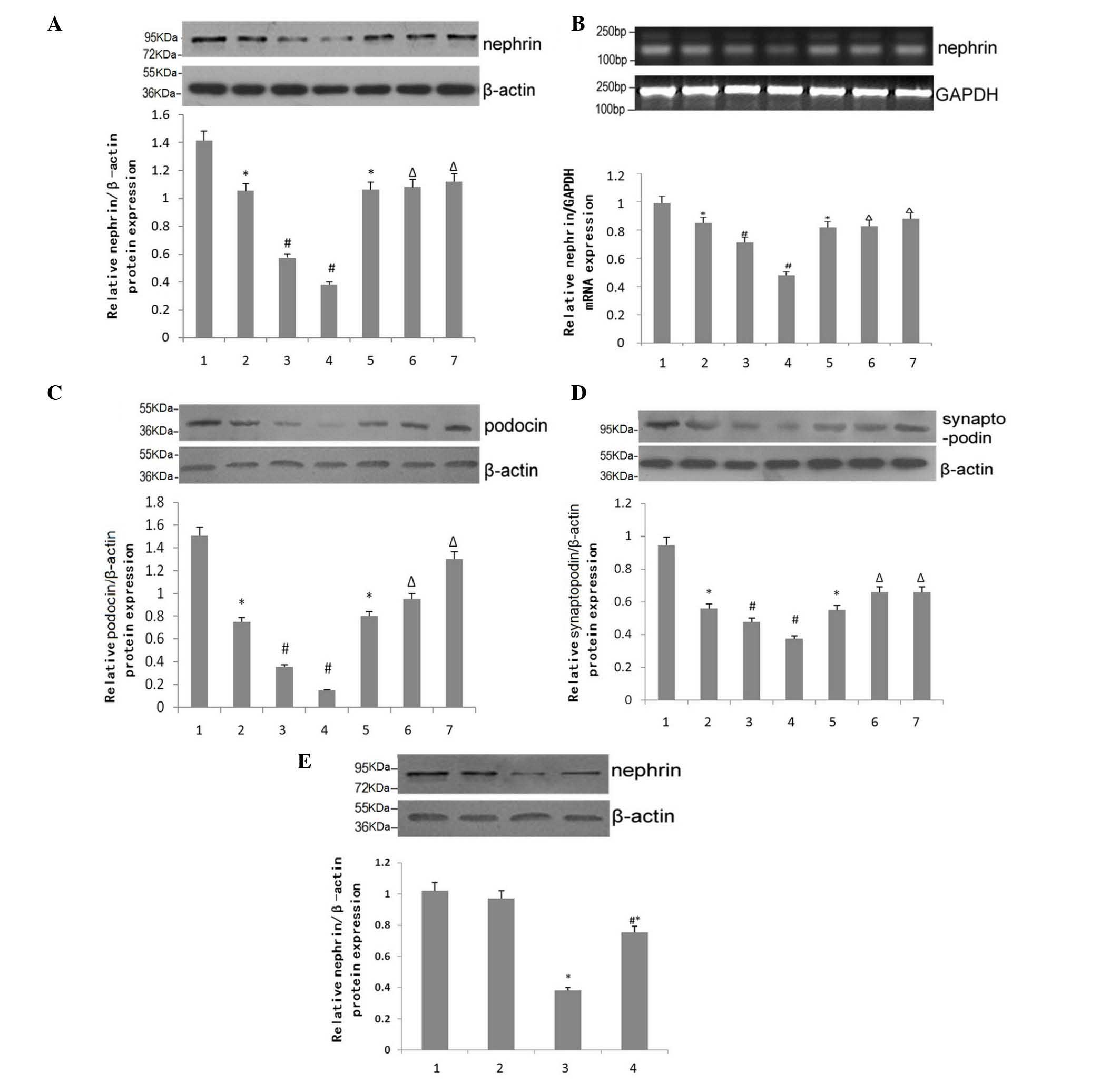 | Figure 3Effect of BIO on the expression of
epithelial cell markers. (A) Western blotting for the expression of
nephrin in kidneys of NC, DN and DN+BIO mice. Data are presented as
the mean ± standard error of the mean (n=6). (B) Reverse
transcription-quantitative polymerase chain reaction analysis for
the mRNA expression of nephrin in the kidneys of NC, DN and DN+BIO
mice. Data are presented as the mean ± standard error of the mean
(n=6). (C and D) Western blotting for the protein expression of
podocin in the kidneys of NC, DN and DN+BIO mice. Data are
presented as the mean ± standard error of the mean (n=3). 1, NC
group; 2, DN group; 3, 15 week DN group; 4, 18 week DN group; 5, 12
week BIO group; 6, 15 week BIO group; 7, 18 week BIO group. (E)
Western blotting for the protein expression of nephrin in the
kidneys of NC, DN and DN+BIO mice. Data are presented as the mean ±
standard error of the mean (n=4). 1–4: 1, normal glucose without
BIO; 2, normal glucose with BIO; 3, high glucose without BIO; 4,
high glucose with BIO. *P<0.05. vs. mice in NC group,
or podocytes in the normal glucose group without BIO;
#P<0.05, vs. 12-week-old mice in the DN group, or
podocytes in the high glucose group without BIO treatment;
ΔP<0.05, vs. 12-week-old mice in the DN+BIO group.
NC, normal control group (db/+); DN, diabetic neuropathy group
(db/db); BIO, (2′Z, 3′E)-6-bromoindirubin-3′-oxime. |
The expression levels of nephrin in podocytes
exposed to high or normal glucose, treated with or without BIO were
compared. BIO had no effect under normal glucose conditions
(Fig. 3A; P>0.05). Under
conditions of high glucose, the expression of nephrin in the
podocytes was significantly reduced, compared with the normal
glucose group (P<0.05). However, treatment with BIO partially
alleviated the high glucose-mediated reduction in the expression of
nephrin (P<0.05), but did not restore expression levels to
normal (P<0.05).
Effects of BIO on the expression of
mesenchymal cell markers
The protein expression of mesenchymal cell
phenotypic markers in the mouse kidney were normalized to β-actin,
as an internal reference, for RT-qPCR analysis. As DN progressed,
the expression of α-SMA in the DN group increased significantly in
a time-dependent matter. However, these effects were attenuated at
15 and 18 weeks of age in the BIO group, compared with the DN group
(Fig. 4A). RT-qPCR analysis of
α-SMA in the kidneys of 15-week-old mice quantitatively confirmed
these observations, as analyzed using ImageJ software (Fig. 4B). Similar expression patterns of
fibronectin, another mesenchymal cell marker, were observed
(Fig. 4C).
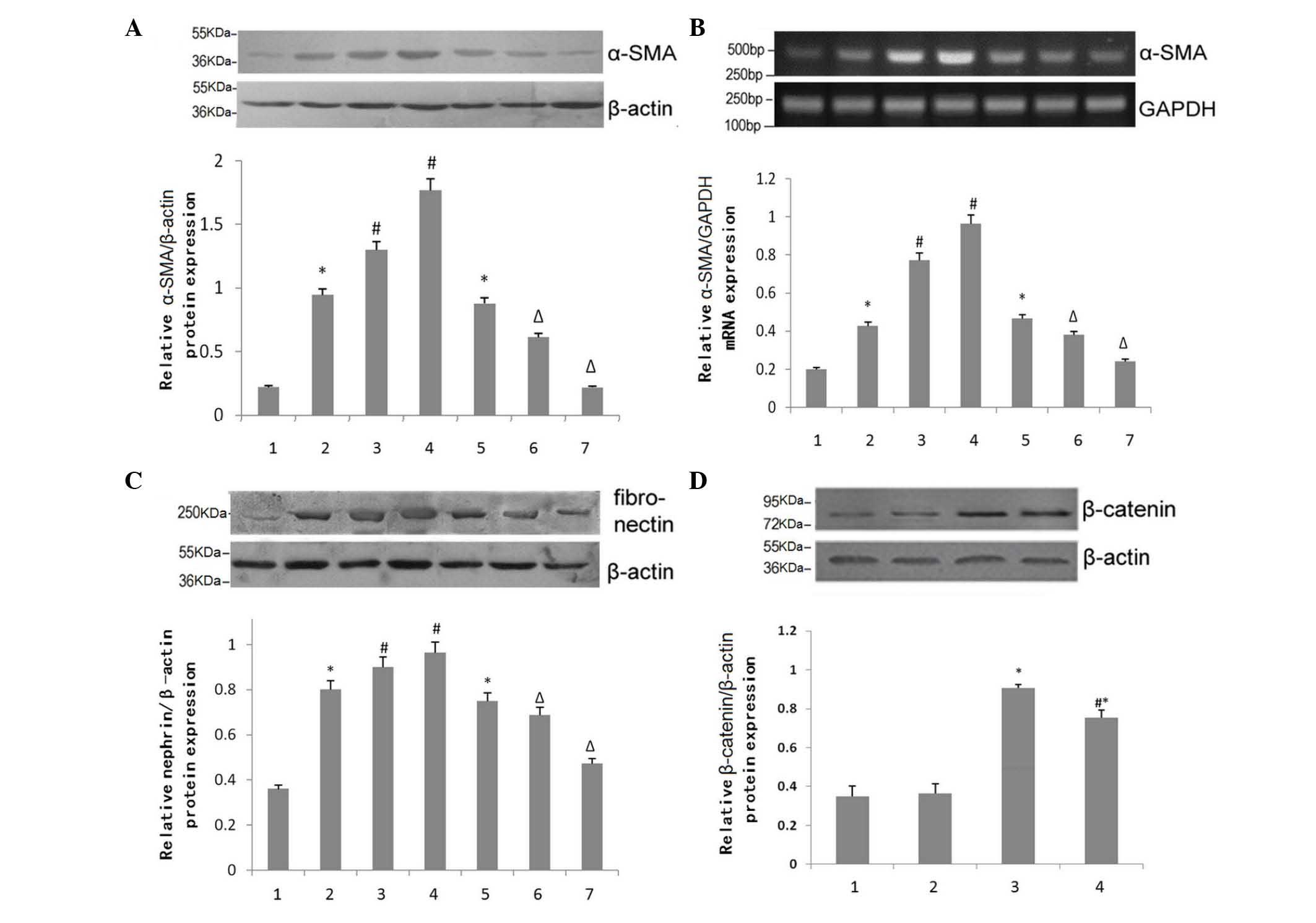 | Figure 4Effect of BIO on the expression
levels of mesenchymal cell markers. (A) Western blotting for the
expression of α-SMA in the kidneys of NC, DN and DN+BIO mice. Data
are presented as the mean ± standard error of the mean (n=6). (B)
Reverse transcription-quantitative polymerase chain reaction
analysis for the mRNA expression of α-SMA in the kidneys of NC, DN
and DN+BIO mice. Data are presented as the mean ± standard error of
the mean (n=6). (C) Western blotting for the expression of
fibronectin in the kidneys of NC, DN and DN+BIO mice. Data are
presented as the mean ± standard error of the mean (n=4). 1, NC
group; 2, DN group; 3, 15 week DN group; 4, 18 week DN group; 5, 12
week BIO group; 6, 15 week BIO group; 7, 18 week BIO group. (D)
Western blotting for the expression of β-catenin in podocytes in
high or normal glucose, with and without BIO treatment. Data are
presented as the mean ± standard error of the mean (n=5). 1–4: 1,
normal glucose without BIO; 2, normal glucose with BIO; 3, high
glucose without BIO; 4, high glucose with BIO.
*P<0.05, vs. mice or podocytes in NC group.
#P<0.05, vs. 12-week-old mice in the DN group or
podocytes in the high glucose group without BIO treatment;
ΔP<0.05, vs. 12-week-old mice in the BIO group.
α-SMA, α-smooth muscle actin; NC, normal control group (db/+); DN,
diabetic neuropathy group (db/db); BIO, (2′Z,
3′E)-6-bromoindirubin-3′-oxime. |
The expression levels of α-SMA in podocytes under
conditions of high or normal glucose, with or without BIO treatment
were then compared (Fig. 4D).
Treatment with BIO had no effect on the expression of α-SMA under
normal glucose concentrations (P>0.05). However, the expression
of α-SMA was increased significantly under high-glucose conditions,
compared with normal glucose (P<0.05). Treatment with BIO
partially alleviated the high glucose-induced increase in the
expression of α-SMA (P<0.05), but did not restore expression
levels to normal (P<0.05).
Effects of BIO on the expression and
activity of GSK-3β
GSK-3β, which is constitutively expressed in all
eukaryotic cells, is a serine/threonine kinase. The important
regulatory amino acids are Tyr-216 for activation and Ser-9 for
inhibition. The expression levels of total-GSK-3β in the mouse
kidney tissues were compared, with β-actin as a loading control,
using RT-qPCR analysis. As DN progressed, the expression of
total-GSK-3β in the DN group increased significantly in a
time-dependent matter (P<0.05). However, total-GSK-3β levels
were substantially attenuated at 15 and 18 weeks of age in the BIO
group, compared with the DN group (P<0.05; Fig. 5A). ImageJ software was used to
quantify the mRNA expression of GSK-3β in the mouse kidney
following RT-qPCR analysis. The data from the animals at 15 weeks
of age were consistent with the observations in the protein levels
(P<0.05; Fig. 5B). The
expression levels of pSer-9-GSK-3β increased significantly at 15
and 18 weeks in the BIO group (P<0.05; Fig. 5C), whereas the patterns observed in
the expression of pTyr-216-GSK-3β were similar to those of
total-GSK-3β (P<0.05; Fig. 5D).
To confirm these observations, the kinase activity of GSK-3β was
assessed using a kinase assay to determine the glomerular activity
of GSK-3β (Fig. 5E). The activity
of GSK-3β in the DN group increased with disease progression, in a
time-dependent manner (P<0.05). Following treatment with BIO,
the activity of GSK-3β decreased by 15 weeks, and continued to
decline until of 18 weeks of age (P<0.05).
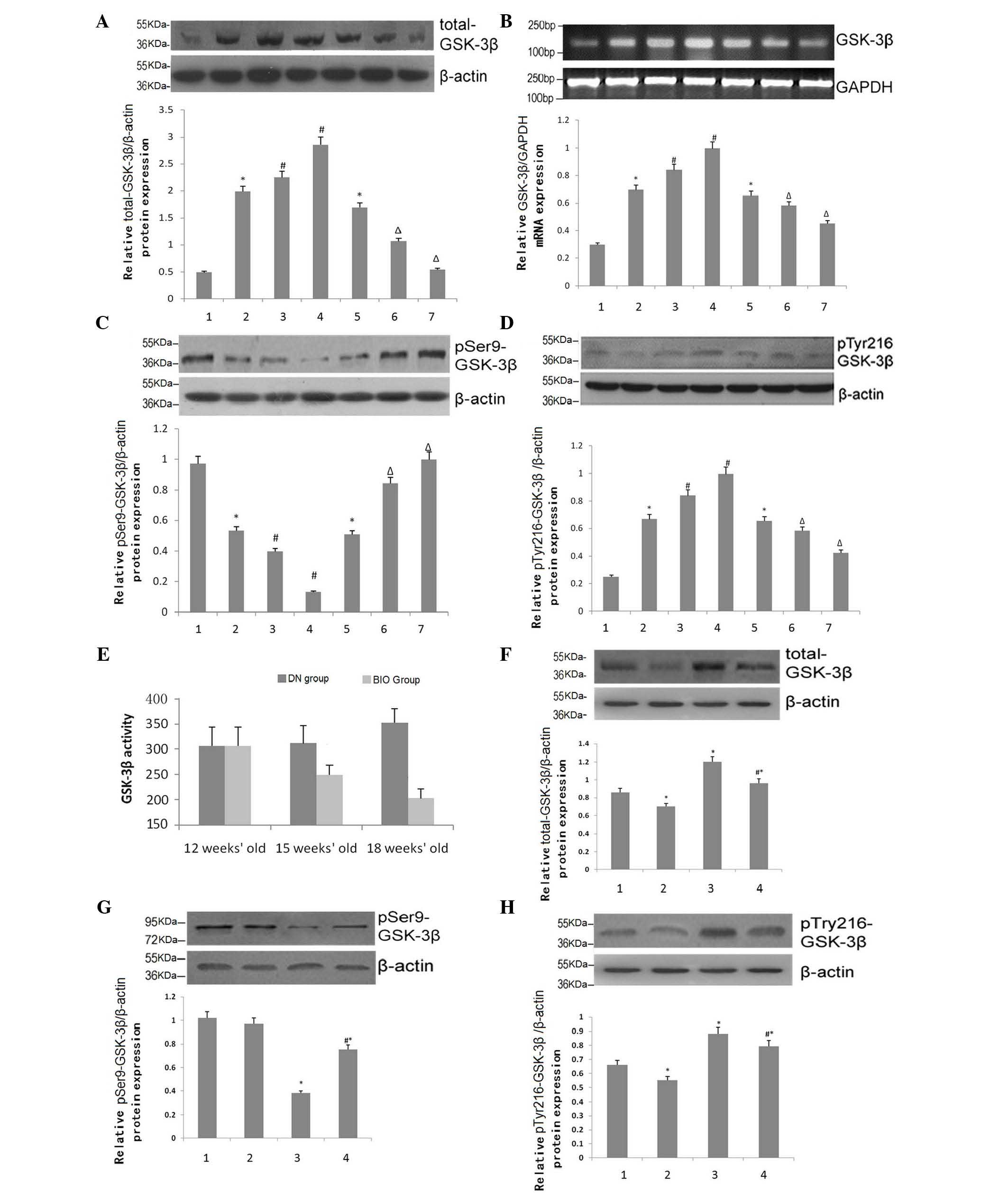 | Figure 5Effects of BIO on the expression and
activity of GSK-3β. (A) Western blotting for the protein expression
of total-GSK-3β in the kidneys of NC, DN and DN+BIO mice (n=4). (B)
Reverse transcription-quantitative polymerase chain reaction
analysis of mRNA expression of GSK-3β in the kidneys of NC, DN and
DN+BIO mice (n=6). (C) Western blotting for pSer9-GSK-3β expression
in the kidneys of NC, DN and DN+BIO mice (n=4). (D) Western
blotting for the expression of pTyr216-GSK-3β in the kidneys of NC,
DN and DN+BIO mice (n=4). 1, NC group; 2, DN group; 3, 15 week DN
group; 4, 18 week DN group; 5, 12 week BIO group; 6, 15 week BIO
group; 7, 18 week BIO group. (E) Kinase activity of GSK-3β in the
kidneys of NC, DN and DN+BIO mice. (F) Western blotting for the
protein expression of total-GSK-3β in podocytes in high or normal
glucose, with and without BIO treatment (n=5). (G) Western blotting
for the protein expression of pSer9-GSK-3β in podocytes in high or
normal glucose, with and without BIO treatment (n=5). (H) Western
blotting for the protein expression of pTyr216-GSK-3β in podocytes
in high or normal glucose, with and without BIO treatment. Each bar
represents mean ± SEM (n=5). 1–4: 1, normal glucose without BIO; 2,
normal glucose with BIO; 3, high glucose without BIO; 4, high
glucose with BIO. *P<0.05 vs. NC group or podocytes
in normal glucose group without BIO treatment;
#P<0.05, vs. 12-week-old mice in DN group or
podocytes in high glucose group without BIO treatment;
ΔP<0.05, vs. 12-week-old mice in DN+BIO group. All
data are presented as the mean ± standard error of the mean.
GSK-3β, glycogen synthase kinase 3β; NC, normal control group
(db/+); DN, diabetic neuropathy group (db/db); BIO, (2′Z,
3′E)-6-bromoindirubin-3′-oxime. |
The expression of total-GSK-3β in podocytes was also
assessed, using RT-qPCR with β-actin as an internal reference
(Fig. 5F). Under conditions of
high glucose, the expression of total-GSK-3β increased
significantly, compared with normal glucose (P<0.05). Treatment
with BIO partially alleviated the high glucose-mediated increases
in total-GSK-3β (P<0.05). Similar patterns of expression were
observed for pTyr216-GSK-3β in the podocytes, whereas, consistent
with previous observations, the protein expression patterns of
pSer9-GSK-3β were reversed (P<0.05; Figs. 5G and H).
Effects of BIO on the expression levels
of β-catenin, Snail and VDR
The expression levels of β-catenin in the mouse
kidney tissues were compared, with β-actin as a loading control,
using RT-qPCR (Fig. 6A). As DN
progressed, the expression of β-catenin in the DN group increased
significantly, in a time-dependent matter (Fig. 6A). However, the levels were
substantially attenuated by BIO treatment at 15 and 18 weeks of
age, compared with the DN group. RT-qPCR analysis of β-catenin in
the kidney tissues of the 15-week-old mice confirmed these
observations (Fig. 6B). Similar
expression patterns for Snail were observed (Fig. 6C). However, the opposite patterns
of expression were observed for VDR (P<0.05; Fig. 5D).
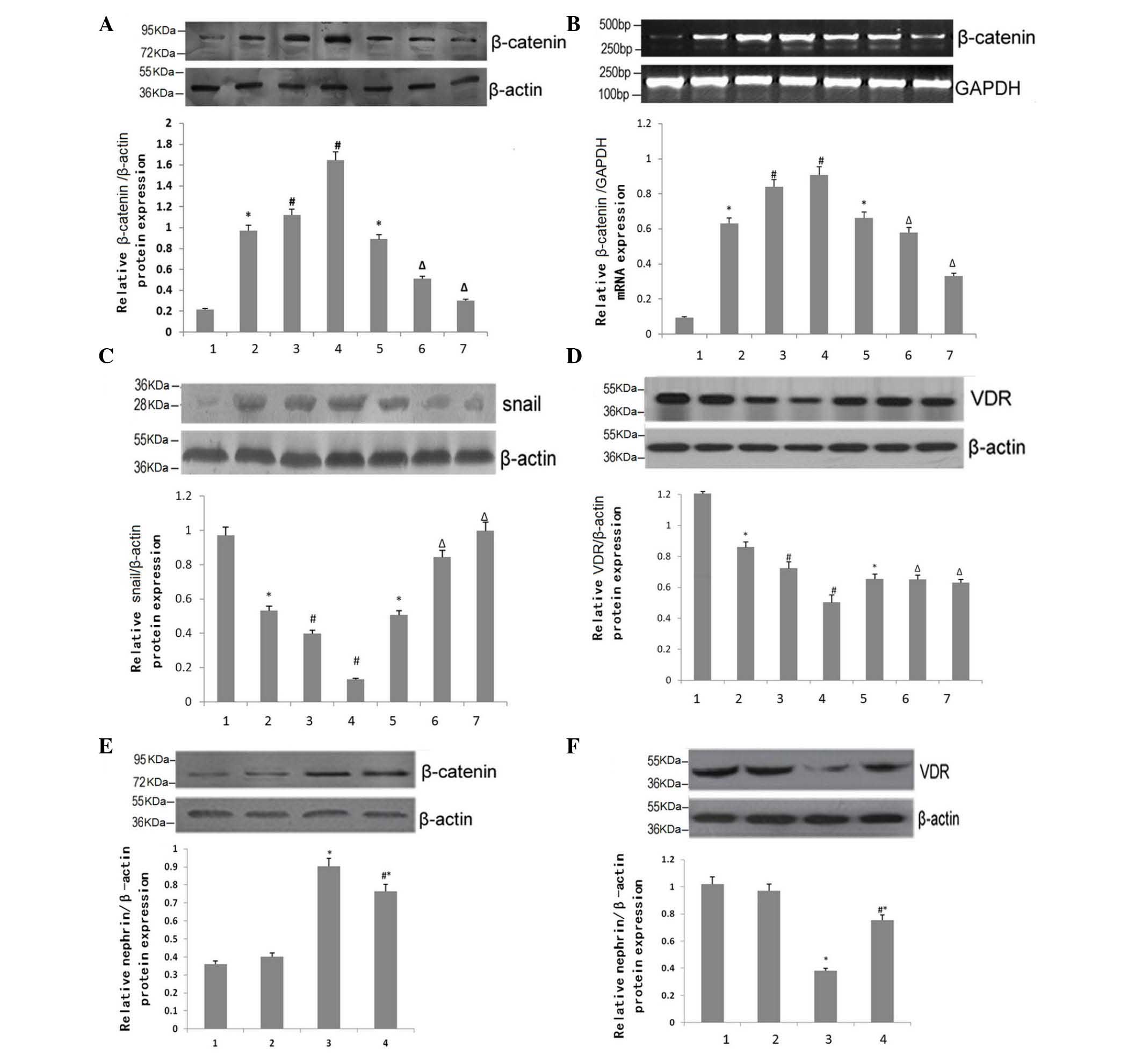 | Figure 6Effects of BIO on the expression
levels of β-catenin, Snail and VDR. (A) Western blotting for the
protein expression of β-catenin in the kidneys of NC, DN and DN+BIO
mice. Data are presented as the mean ± standard error of the mean
(n=6). (B) Reverse transcription-quantitative polymerase chain
reaction analysis of the mRNA expression of β-catenin in the
kidneys of NC, DN and DN+BIO mice. Data are presented as the mean ±
standard error of the mean (n=6). (C) Western blotting for the
protein expression of Snail expression in the kidneys of NC, DN and
DN+BIO mice. Data are presented as the mean ± standard error of the
mean (n=4). (D) Western blotting for the protein expression of VDR
in the kidneys of NC, DN and DN+BIO mice. Data are presented as the
mean ± standard error of the mean (n=6). 1, NC group; 2, DN group;
3, 15 week DN group; 4, 18 week DN group; 5, 12 week BIO group; 6,
15 week BIO group; 7, 18 week BIO group. (E) Western blotting for
the protein expression of β-catenin in podocytes in high or normal
glucose, with and without BIO treatment. Data are presented as the
mean ± standard error of the mean (n=5). (F) Western blotting for
the protein expression of VDR in podocytes in high or normal
glucose, with and without BIO treatment. 1–4: 1, normal glucose
without BIO; 2, normal glucose with BIO; 3, high glucose without
BIO; 4, high glucose with BIO. *P<0.05, vs. mice in
NC group or podocytes in normal glucose group without BIO
treatment; #P<0.05, vs. 12-week-old mice in DN group,
or podocytes in high glucose group without BIO treatment;
ΔP<0.05, vs.12-week-old mice in DN+BIO group. VDR,
vitamin D receptor; NC, normal control group (db/+); DN, diabetic
neuropathy group (db/db); BIO, (2′Z,
3′E)-6-bromoindirubin-3′-oxime. |
The expression of β-catenin in podocytes was also
assessed and compared with β-actin, as a loading control, using
RT-qPCR (Fig. 6E). Under normal
glucose conditions, BIO treatment had no effect (P>0.05).
However, under high-glucose conditions, the expression of β-catenin
increased significantly, compared with the normal glucose group
(P<0.05). Treatment with BIO partially attenuated the high
glucose-induced increase in the expression of β-catenin
(P<0.05). Consistent with the data from the mouse kidney
tissues, the protein expression patterns of VDR in the podocytes
were the reverse (Fig. 6F).
Discussion
Selection and establishment of animal
models
db/db mice are one of the most commonly used animal
models in studies investigating type II diabetes (25). The mouse contains a G>T point
mutation in the leptin receptor (LepRdb/db), leading to its
aberrant splicing derived by fat cells, and the formation of
defective receptors (26,27). As a result, mice overeat, causing
symptoms including obesity, hyperinsulinemia and insulin
resistance. In addition, db/db mice develop progressive nephrotic
disease, making the model suitable for long-term
investigations.
The LepRdb/db mutation has been verified in
C57BLKS/J (28), C57BL/6J
(29) and FVB/NJ strains. The
blood glucose levels of male C57BLKS/J db/db mice gradually
increases from ~4 weeks of age, and hyperglycemia appears at ~8
weeks (>16 mmol/l). Microalbuminuria occurs at ~10–12 weeks
(30–32), with morphological changes,
including glomerular hypertrophy, basement membrane thickening,
mesangial widening and foot process fusion (33) observed from 12–14 weeks of age.
Finally, blood glucose levels peak at ~16 weeks (34–37).
Therefore, the present study used male db/db C57BLKS/J mice to
establish a model of DN.
The characteristics of the DN model used in the
present study were as follows: i) at 12 weeks-of-age, mice
exhibited significant polyuria, polydipsia and polyphagia, whereas
body weight was 29.91±4.71 g and kidney weight was 180.00±25.66 mg,
all of which were significantly different, compared with the NC
group (P<0.05). ii) By 12 weeks of age, the blood glucose level
in the DN mice was 27.5000±7.78 mmol/l and protein was detected in
the urine. These observations were significantly different,
compared with the NC group (P<0.05). iii) UAE over a 24 h period
in the DN model mice was 239.24±37.80 µg at 12 weeks of age,
which was also significantly different from that in the NC group
(P<0.05). Finally, histopathological examination of the kidneys
from the DN mice at 12 weeks of age revealed glomerular
hypertrophy, basement membrane thickening and mesangial area
widening, all of which increased in severity with disease
progression. By 18 weeks of age, certain glomeruli exhibited
lobulated sclerosis, a characteristic of DN mesangial cells, and
capillary endothelial cells. Podocytes, which are glomerular
epithelial cells attached to the outside of the GBM, constitute the
glomerular filtration barrier between the GBM and capillary
endothelium. Podocyte processes protrude from podocyte cell bodies,
covering the outer surface of the GBM, and interacting with the GBM
via proteoglycan adhesion molecules (38,39).
The slit diaphragm, a zipper-like structure situated between two
adjacent podocytes, is the final barrier of plasma protein
filtration (35). Podocyte injury
is involved in the progression of glomerulonephritis, DN, renal
failure and other kidney diseases (40). Specifically, reductions in the
number and density of podocytes have been linked to the
pathological changes, which occur during DN (41). It has been suggested that podocyte
phenotypic transformation, the process of its transdifferentiation
into mesenchymal cells during cell damage, leads to the loss of
specific podocyte protein markers, disrupted cell function and
proteinuria (42).
Podocytes are highly differentiated epithelial
cells. Several mature protein markers, including nephrin (43), podocin (44) and synaptopodin (45), are found on the surface and slit
diaphragms of podocytes, and constitute a charge barrier of the
GBM. They are also involved in cell-cell connections and signal
transduction, and can maintain the integrity of podocyte foot
processes and the normal function of the membrane hole (38,39).
Phenotypic markers of mesenchymal cells include α-SMA and
fibronectin, which are important for cellular activation and
transdifferentiation.
A previous study has revealed that elevated blood
glucose can cause mice to produce increased levels of fibronectin
and laminin β1 in the extracellular matrix (46). In addition, Kang et al
revealed that glucose levels can be raised following the induction
of the expression of ILK, inducing podocyte transdifferentiation
and injury (47).
Selection and application of treatments
for DN
Clinically, DN treatment includes regulating blood
glucose, controlling blood lipids and diet adjustment (48). Frequently used medications include
angiotensin-converting enzyme inhibitors and angiotensin receptor
blockers (49). However, these
therapies only delay the development of ESRD, and do not
effectively prevent the development of DN (50–52).
GSK-3β is a serine/threonine kinase, which is expressed in all
eukaryotic cells. The key amino acids for regulating GSK-3β
activity are Tyr-216 for activation and Ser-9 for inhibition.
Studies have shown that GSK-3β not only phosphorylates glycogen
synthase to regulate the activity of glycolytic enzymes, but is
also involved in signaling pathways, including insulin, Hedgehog,
Notch and Wnt/β-catenin, to affect cellular differentiation,
metabolism and apoptosis (53).
During insulin signaling, the insulin receptor phosphorylates
GSK-3β, leading to decreased GSK-3β activity in liver and muscle.
This leads to reduced blood glucose levels and an increase in
glycogen synthesis. In the Wnt pathway, GSK-3β, β-catenin,
adenomatosis polyposis coli and Axin form a complex in the
cytoplasm, leading to the degradation of β-catenin by the
proteasome and the inhibition of Wnt gene transcription (54). Snail is a zinc finger transcription
factor, which can transform epithelial cells into mesenchymal
cells. GSK-3β can regulate the expression and activity of Snail,
thereby regulating EMT (55,56).
Therefore, the present study used BIO, an inhibitor of GSK-3β, to
assess the effects of the expression and activity of GSK-3β on the
progression of DN.
Effects of BIO on the development of
diabetic nephropathy in db/db mice
In the present study, the db/db mice, as a model of
DN, were treated with the GSK-3β inhibitor, BIO, and the effects on
DN progression were assessed. At 18 weeks of age, the symptoms of
the mice in BIO group, including polyuria, polydipsia and
polyphagia, were significantly reduced. In addition, body weight
was 32.49±4.77 g and kidney weight was 159.90±22.53 mg, which were
significantly different, compared with those in the DN group
(P<0.05). By 18 weeks of age, the blood glucose level in the BIO
group was 28.7625±2.71 mmol/l, which was comparable to that in the
DN group, suggesting that BIO had no effect on blood glucose
(P>0.05). The UAE of mice in BIO group was 135.65±18.96
µg over 24 h, which was decreased significantly from the DN
group (P<0.05). At ~18 weeks of age, renal pathological lesions,
including mesangial cell proliferation and matrix, increased
significantly in the DN group, but were alleviated by treatment
with BIO. These data suggested that the GSK-3β inhibitor reduced
proteinuria and slowed the progression of early DN. Of note, the
mechanism underlying this protection of the kidney may involve
adjusting EMT, but not lowering blood glucose.
Effects of BIO on GSK-3β in the db/db
mouse kidney
Normal kidney tissue expresses small quantities of
total-GSK-3β, however protein and mRNA expression levels were
observed to increase with the progression of DN, in a
time-dependent manner (P<0.05). Following treatment with BIO,
the upregulation of total-GSK-3β was lower, compared with the
control group (P<0.05). Similar expression patterns were
observed for Tyr216-phosphorylated GSK-3β (pTyr216-GSK-3β) were
observed. By contrast, Ser-9 phosphorylated GSK-3β (pSer9-GSK-3β)
was expressed at a high level in normal renal tissues, but
decreased with the development of DN, in a time-dependent manner
(P<0.05). Following treatment with BIO, the levels of
pSer9-GSK-3β were reduced, compared with the DN group (P<0.05).
With the progression of DN, GSK-3β kinase activity in the DN group
increased in a time-dependent matter, whereas treatment with BIO
for 3 weeks led to a partial decrease in GSK-3β kinase activity,
compared with the DN group. Following 6 weeks of BIO therapy, the
activity of GSK-3β was further reduced (P<0.05). These results
suggested that BIO, an inhibitor of GSK-3β, effectively affected
the activity of GSK-3β, and may be useful for the treatment of
DN.
Effects of BIO on EMT in the db/db mouse
kidney
In the present study, as EMT developed in the renal
cortex of mice in the DN group, the expression levels of total
GSK-3β and pTyr216-GSK-3β, and the activity of GSK-3β all
increased, whereas the levels of pSer9-GSK-3β declined. Therefore,
it was hypothesized that the activation of GSK-3β is important in
EMT, and that inhibiting the activity of GSK-3β may delay the
development of EMT.
BIO was used to inhibit the activation of GSK-3β in
the DN mice. When the mice were treated with BIO, the protein and
mRNA expression levels of epithelial phenotype markers, including
podocin, podocin and synaptopodin, increased in a time-dependent
manner, which was significantly different, compared with the DN
group (P<0.05). In addition, the expression levels of
mesenchymal markers, including fibronectin and α-SMA, were
decreased significantly, compared with those in the DN group
(P<0.05). These results suggested that BIO, effectively reduced
EMT in the db/db mice with DN.
During Wnt signaling, the upregulation of GSK-3β
activity can lead to degradation of β-catenin (54) and decreased expression of Snail
(57,58). Previous studies have revealed that
using small interfering (si)RNA against GSK-3β can cause
transdifferentiation of mammary gland tissue and skin epithelial
cells (59). In addition, the
activity of GSK-3β may maintain the structure of epithelial cells
and their surface role in the normal podocyte phenotype (60,61).
In contrast with these data, the present study revealed that normal
kidney tissue expressed low levels of β-catenin and Snail. During
the progression of DN, the expression levels of β-catenin and Snail
increased, and the activity of GSK-3β was also upregulated in a
time-dependent manner (P<0.05). Following treatment with BIO,
the upregulation in the levels of β-catenin and Snail were
inhibited significantly, compared with those in the DN group
(P<0.05). These observations contradict previous observations in
traditional Wnt signaling (62).
It has been suggested that the activation of GSK-3β may directly
stimulate transdifferentiation by triggering the transcription of
relevant genes, and inhibiting the expression of epithelial cell
markers (63). In the resting
state, β-catenin is bound to fibronectin, which anchors in the cell
membrane. This causes transdifferentiation of epithelial cells,
following which β-catenin dissociates and migrates to the
cytoplasm. Intracellular β-catenin then tends to migrate towards
the nucleus (64), further
promoting the transcription of transdifferentiation-associated
genes.
Abnormalities in the expression of vitamin D are
frequently observed in patients with chronic kidney disease
(65). VDR, a receptor specific
for vitamin D, has a protective effect in the kidneys, and its
expression in the renal tissues of patients with DN is decreased
(66). Once activated, VDR
specifically combines with the DNA structural domain of vitamin D,
and undergoes conformational change allowing it to combine with
specific DNA regions. This leads to novel gene expression,
resulting in a series of biological effects. In a previous study on
human bone marrow stromal cells, the 1, 25 (OH)-2-vitamin D3 system
activated VDR, which then combined with β-catenin in the nucleus,
leading to the transport of β-catenin out of the nucleus (67). When activated by its ligand, VDR
competitively combines with T cell transcription factor-4, which is
normally bound to β-catenin, thus inhibiting the activity of
β-catenin in colon cancer (68).
In a mouse model of unilateral ureteral obstruction, it was
observed that VDR was associated with the extent and development of
renal fibrosis. Loss of the expression of VDR caused epithelial
cell transdifferentiation, which may be mediated by β-catenin.
Specifically, when the cells were treated with VDR siRNA, the
expression of β-catenin was increased and transported to the
nucleus, whereas the over expression of VDR inhibited the
expression of β-catenin (69). The
data presented in the present study demonstrated that the
expression of VDR was decreased in the DN group, and the levels of
β-catenin and Snail were increased. As expected, these changes were
ameliorated following treatment with BIO, suggesting that GSK-3β
may modulate the expression levels of β-catenin and Snail by
regulating the expression of VDR. Therefore, the present study
hypothesized that GSK-3β uses pathways in addition to the classical
Wnt pathway to regulate and control EMT. This may explain why the
activation of GSK-3β increased, and treatment with the GSK-3β
inhibitor decreased, the expression levels of β-catenin and Snail,
which were observations contradictory to the classical Wnt
pathway.
Advantages and disadvantages of the
clinical application of GSK-3β inhibitors
GSK-3β is a multifunctional protein, which is
involved in several biological processes, including the
transdifferentiation, proliferation, apoptosis and migration of
several different cell types. During the transdifferentiation of
renal tubular epithelial cells induced by high glucose, reports on
GSK-3β activity are conflicting. It has been suggested that GSK-3β
activity is reduced during the transdifferentiation of renal
tubular epithelial cells induced by high glucose (70), whereas other have suggested that
the activity of GSK-3β increases during high-glucose-mediated
apoptosis in mesangial cells (71). In addition, studies have reported
that the expression of GSK-3β increases in patients with type 2
diabetes and in rodent models of obesity and insulin resistance
(72,73).
Further investigations are required to clarify the
role of GSK-3β in high-glucose-stimulated DN, and specifically to
clarify the association between GSK-3β, EMT and DN proteinuria. The
data in the present study suggested that BIO, as an inhibitor of
GSK-3β, can cause transdifferentiation of podocytes. Additional
data suggested that LiCl, another inhibitor of GSK-3β has a similar
role in EMT (data not shown) (74). Furthermore, the effects of GSK-3β
in podocyte EMT were verified using GSK-3β RNA interference in
podocytes, the results of which supported the conclusions of the
present study. However, additional investigations are required to
define the mechanisms underlying the effects of GSK-3β inhibitors
in the treatment of DN, and the downstream signal transduction
pathways. In addition, investigations on the long-term treatment
with BIO is essential prior to its consideration as a therapeutic
agent for the treatment of DN.
In conclusion, the present study demonstrated that,
during the development of diabetic nephropathy, the expression
levels of glomerular epithelial cell markers decreased, and the
levels of mesenchymal-like markers increased in a time-dependent
manner, suggesting the occurrence of EMT. In addition, the
expression and activity of total-GSK-3β increased, the expression
of pSer9-GSK-3β (the inhibitory site of GSK-3β) decreased and the
expression of pTyr216-GSK-3β (the active site of GSK-3β) increased.
This suggested that the activity of GSK-3β is closely associated
with the development of DN in db/db mice. Following treatment with
BIO, the expression of kidney epithelial cell markers in mice with
DN increased, whereas the expression of mesenchymal-like markers
decreased, compared with the untreated DN mice, suggesting that the
inhibitor of GSK-3β partially reversed EMT. The present study also
demonstrated that treatment with BIO reduced proteinuria leakage,
delayed the deterioration of renal function and prevented
structural changes in the kidneys of the mice with DN. This
suggested that GSK-3β may regulate podocyte EMT, and that the
GSK-3β inhibitor, BIO, may protect the filtration barrier of the
glomerulus, reversing EMT and delaying the development of DN.
The present study also observed that the activation
of GSK-3β increased the expression levels of β-catenin and Snail.
Following GSK-3β inhibition by BIO, the expression levels of
β-catenin and snail decreased. These effects may be due to the fact
that EMT is directly associated with GSK-3β activity, which may
inhibit the gene transcription of epithelial cell marker proteins.
When EMT occurs, β-catenin dissociates from the cytomembrane and
translocates to the cytoplasm, increased concentrations of
intracellular β-catenin migrate and accumulate in the nucleus,
accelerating the transcription of transdifferentiation-associated
genes. In addition, it is possible that GSK-3β regulated the
expression of VDR, and was consequently involved in regulating the
expression of β-catenin and Snail. Therefore, the present study
hypothesized that, rather than relying on the classical Wnt pathway
to control EMT, GSK-3β can also regulate podocytes by modifying the
levels of VDR, or by stimulating the nuclear accumulation of
β-catenin.
Acknowledgments
The authors would like to thank Dr Li-Bin Zhang
(Basic Medical College, Xinxiang Medical University) for
discussion, and Dr Xin-Lai Qian (The Third Affiliated Hospital of
Xinxiang Medical University, Xinxiang, China) and Dr Song-Xia Quan
(The First Affiliated Hospital of Zhengzhou University) for their
invaluable assistance in histological analyses. The authors would
also like to thank Dr Ying Zhao (Department of Foreign Language,
Henan Institute of Science and Technology, Zhengzhou, China) and Dr
Peng Wan (Henan Time Culture Media Company, Zhengzhou, China) for
their editorial assistance. This study was supported by grants from
the National Basic Research Program of China 973 Program (grant
nos, 2012CB517600 and 2012CB517606) and the National Natural
Science Foundation of China (grant nos. 81070574 and 81270807).
References
|
1
|
US Renal Data System: 2009 USRDS Annual
Data Report: Epidemiology of kidney disease in the United States.
National Institutes of Health, National Institute of Diabetes and
Digestive and Kidney Diseases; Bethesda, MD: 2009
|
|
2
|
de Boer IH, Rue TC, Hall YN, Heagerty PJ,
Weiss NS and Himmelfarb J: Temporal trends in the prevalence of
diabetic kidney disease in the United States. JAMA. 305:2532–2539.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Panchapakesan U, Pegg K, Gross S, Komala
MG, Mudaliar H, Forbes J, Pollock C and Mather A: Effects of SGLT2
inhibition in human kidney proximal tubular cells-renoprotection in
diabetic nephropathy? PLoS One. 8:e544422013. View Article : Google Scholar
|
|
4
|
Dronavalli S, Duka I and Bakris GL: The
pathogenesis of diabetic nephropathy. Nat Clin Pract Endocrinol
Metab. 4:444–452. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kanwar YS, Wada J, Sun L, Xie P, Wallner
EI, Chen S, Chugh S and Danesh FR: Diabetic nephropathy: Mechanisms
of renal disease progression. Exp Biol Med (Maywood). 233:4–11.
2008. View Article : Google Scholar
|
|
6
|
Wolf G and Ziyadeh FN: Molecular
mechanisms of diabetic renal hypertrophy. Kidney Int. 56:393–405.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wolf G and Ziyadeh FN: Cellular and
molecular mechanisms of proteinuria in diabetic nephropathy.
Nephron Physiol. 106:p26–p31. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lomelí C, Rosas-Peralta M, Lorenzo A and
Saucedo N; Grupo de investigadores participantes en México para el
estudio I-Search: Microalbuminuria and associated cardiovascular
risk factors in patients with arterial systemic hypertension. A
subanalysis of the I-Search study. Arch Cardiol Mex. 82:93–104.
2012.In Spanish.
|
|
9
|
Monhart V: Microalbuminuria. From diabetes
to cardiovascular risk. Vnitr Lek. 57:293–298. 2011.In Czech.
PubMed/NCBI
|
|
10
|
Ozyol A, Yucel O, Ege MR, Zorlu A and
Yilmaz MB: Microalbuminuria is associated with the severity of
coronary artery disease independently of other cardiovascular risk
factors. Angiology. 63:457–460. 2012. View Article : Google Scholar
|
|
11
|
Udenze IC, Azinge EC, Ebuehi OA, Awolola
NA, Adekola OO, Menkiti I and Irurhe NK: The relationship between
microalbuminuria, cardiovascular risk factors and disease
management in type 2 diabetes. Nig Q J Hosp Med. 22:34–38.
2012.PubMed/NCBI
|
|
12
|
Harris RC: Podocyte ACE2 protects against
diabetic nephropathy. Kidney Int. 82:255–256. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wiggins RC: The spectrum of
podocytopathies: A unifying view of glomerular diseases. Kidney
Int. 71:1205–1214. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Reidy K and Susztak K:
Epithelial-mesenchymal transition and podocyte loss in diabetic
kidney disease. Am J Kidney Dis. 54:590–593. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Steffes MW, Schmidt D, McCrery R and
Basgen JM; International Diabetic Nephropathy Study Group:
Glomerular cell number in normal subjects and in type 1 diabetic
patients. Kidney Int. 59:2104–2113. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Susztak K, Raff AC, Schiffer M and
Böttinger EP: Glucose-induced reactive oxygen species cause
apoptosis of podocytes and podocyte depletion at the onset of
diabetic nephropathy. Diabetes. 55:225–233. 2006. View Article : Google Scholar
|
|
17
|
Thibodeau JF, Holterman CE, Burger D, Read
NC, Reudelhuber TL and Kennedy CR: A novel mouse model of advanced
diabetic kidney disease. PLoS One. 9:e1134592014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hu P, Wang G, Shen M, Zhang P, Zhang J, Du
J and Liu Q: Intratumoral polymorphonuclear granulocyte is
associated with poor prognosis in squamous esophageal cancer by
promoting epithelial-mesenchymal transition. Future Oncol.
11:771–783. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Dai HY, Zheng M, Lv LL, Tang RN, Ma KL,
Liu D, Wu M and Liu BC: The roles of connective tissue growth
factor and integrin-linked kinase in high glucose-induced
phenotypic alterations of podocytes. J Cell Biochem. 113:293–301.
2012. View Article : Google Scholar
|
|
20
|
Liang Y, Jing Z, Deng H, Li Z, Zhuang Z,
Wang S and Wang Y: Soluble epoxide hydrolase inhibition ameliorates
proteinuria-induced epithelial-mesenchymal transition by regulating
the PI3K-Akt-GSK-3β signaling pathway. Biochem Biophys Res Commun.
463:70–75. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Obligado SH, Ibraghimov-Beskrovnaya O, Zuk
A, Meijer L and Nelson PJ: CDK/GSK-3 inhibitors as therapeutic
agents for parenchymal renal diseases. Kidney Int. 73:684–690.
2008. View Article : Google Scholar
|
|
22
|
Mundel P, Reiser J, Zúñiga Mejía Borja A,
Pavenstädt H, Davidson GR, Kriz W and Zeller R: Rearrangements of
the cytoskeleton and cell contacts induce process formation during
differentiation of conditionally immortalized mouse podocyte cell
lines. Exp Cell Res. 236:248–258. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Dai C, Stolz DB, Kiss LP, Monga SP,
Holzman LB and Liu Y: Wnt/beta-catenin signaling promotes podocyte
dysfunction and albuminuria. J Am Soc Nephrol. 20:1997–2008. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Jonathan Ryves W, Dalton EC, Harwood AJ
and Williams RS: GSK-3 activity in neocortical cells is inhibited
by lithium but not carbamazepine or valproic acid. Bipolar Disord.
7:260–265. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Hummel KP, Dickie MM and Coleman DL:
Diabetes, a new mutation in the mouse. Science. 153:1127–1128.
1966. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Chen H, Charlat O, Tartaglia LA, Woolf EA,
Weng X, Ellis SJ, Lakey ND, Culpepper J, Moore KJ, Breitbart RE, et
al: Evidence that the diabetes gene encodes the leptin receptor:
Identification of a mutation in the leptin receptor gene in db/db
mice. Cell. 84:491–495. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Lee GH, Proenca R, Montez JM, Carroll KM,
Darvishzadeh JG, Lee JI and Friedman JM: Abnormal splicing of the
leptin receptor in diabetic mice. Nature. 379:632–635. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Hummel KP, Coleman DL and Lane PW: The
influence of genetic background on expression of mutations at the
diabetes locus in the mouse. I. C57BL-KsJ and C57BL-6J strains.
Biochem Genet. 7:1–13. 1972. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Chua S Jr, Liu SM, Li Q, Yang L,
Thassanapaff VT and Fisher P: Differential beta cell responses to
hyperglycaemia and insulin resistance in two novel congenic strains
of diabetes (FVB-Lepr (db)) and obese (DBA-Lep (ob)) mice.
Diabetologia. 45:976–990. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Cohen MP, Clements RS, Cohen JA and
Shearman CW: Prevention of decline in renal function in the
diabetic db/db mouse. Diabetologia. 39:270–274. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Lim AK, Ma FY, Nikolic-Paterson DJ, Thomas
MC, Hurst LA and Tesch GH: Antibody blockade of c-fms suppresses
the progression of inflammation and injury in early diabetic
nephropathy in obese db/db mice. Diabetologia. 52:1669–1679. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Chow FY, Nikolic-Paterson DJ, Ozols E,
Atkins RC and Tesch GH: Intercellular adhesion molecule-1
deficiency is protective against nephropathy in type 2 diabetic
db/db mice. J Am Soc Nephrol. 16:1711–1722. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Chodavarapu H, Grobe N, Somineni HK, Salem
ES, Madhu M and Elased KM: Rosiglitazone treatment of type 2
diabetic db/db mice attenuates urinary albumin and angiotensin
converting enzyme 2 excretion. PLoS One. 8:e628332013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Vivanco I and Sawyers CL: The
phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat Rev
Cancer. 2:489–501. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
35
|
Ward SG and Finan P: Isoform-specific
phosphoinositide 3-kinase inhibitors as therapeutic agents. Curr
Opin Pharmacol. 3:426–434. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Franke TF, Hornik CP, Segev L, Shostak GA
and Sugimoto C: PI3 K/Akt and apoptosis: Size matters. Oncogene.
22:8983–8998. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Chua S Jr, Li Y, Liu SM, Liu R, Chan KT,
Martino J, Zheng Z, Susztak K, D'Agati VD and Gharavi AG: A
susceptibility gene for kidney disease in an obese mouse model of
type II diabetes maps to chromosome 8. Kidney Int. 78:453–462.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Taneda S, Honda K, Ohno M, Uchida K, Nitta
K and Oda H: Podocyte and endothelial injury in focal segmental
glomerulosclerosis: An ultrastructural analysis. Virchows Arch.
467:449–458. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Lenoir O, Jasiek M, Hénique C, Guyonnet L,
Hartleben B, Bork T, Chipont A, Flosseau K, Bensaada I, Schmitt A,
et al: Endothelial cell and podocyte autophagy synergistically
protect from diabetes-induced glomerulosclerosis. Autophagy.
11:1130–1145. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Dai C, Stolz DB, Bastacky SI, St-Arnaud R,
Wu C, Dedhar S and Liu Y: Essential role of integrin-linked kinase
in podocyte biology: Bridging the integrin and slit diaphragm
signaling. J Am Soc Nephrol. 17:2164–2175. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Xu W, Chen J, Lin J, Liu D, Mo L, Pan W,
Feng J, Wu W and Zheng D: Exogenous H2S protects H9c2 cardiac cells
against high glucose-induced injury and inflammation by inhibiting
the activation of the NF-κB and IL-1β pathways. Int J Mol Med.
35:177–186. 2015.
|
|
42
|
El-Aouni C, Herbach N, Blattner SM, Henger
A, Rastaldi MP, Jarad G, Miner JH, Moeller MJ, St-Arnaud R, Dedhar
S, et al: Podocyte-specific deletion of integrin-linked kinase
results in severe glomerular basement membrane alterations and
progressive glomerulosclerosis. J Am Soc Nephrol. 17:1334–1344.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Langham RG, Kelly DJ, Cox AJ, Thomson NM,
Holthöfer H, Zaoui P, Pinel N, Cordonnier DJ and Gilbert RE:
Proteinuria and the expression of the podocyte slit diaphragm
protein, nephrin, in diabetic nephropathy: Effects of angiotensin
converting enzyme inhibition. Diabetologia. 45:1572–1576. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Nakatsue T, Koike H, Han GD, Suzuki K,
Miyauchi N, Yuan H, Salant DJ, Gejyo F, Shimizu F and Kawachi H:
Nephrin and podocin dissociate at the onset of proteinuria in
experimental membranous nephropathy. Kidney Int. 67:2239–2253.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Mundel P, Heid HW, Mundel TM, Krüger M,
Reiser J and Kriz W: Synaptopodin: An actin-associated protein in
telencephalic dendrites and renal podocytes. J Cell Biol.
139:193–204. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Liu W, Hu M, Wang Y, Sun B, Guo Y, Xu Z,
Li J and Han B: Overexpression of interleukin-18 protein reduces
viability and induces apoptosis of tongue squamous cell carcinoma
cells by activation of glycogen synthase kinase-3β signaling. Oncol
Rep. 33:1049–1056. 2015.PubMed/NCBI
|
|
47
|
Kang YS, Li Y, Dai C, Kiss LP, Wu C and
Liu Y: Inhibition of integrin linked kinase blocks podocyte
epithelial mesenchymal transition and ameliorates proteinuria.
Kidney Int. 78:363–373. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Cai X, Han X, Luo Y and Ji L: Analysis of
insulin doses of Chinese type 2 diabetic patients with intensive
insulin treatment. PLoS One. 7:e389622012. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Tang L, Yi R, Yang B, Li H, Chen H and Liu
Z: Valsartan inhibited HIF-1α pathway and attenuated renal
interstitial fibrosis in streptozotocin-diabetic rats. Diabetes Res
Clin Pract. 97:125–131. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Berwick DC, Hers I, Heesom KJ, Moule SK
and Tavare JM: The identification of ATP-citrate lyase as a protein
kinase B (Akt) substrate in primary adipocytes. J Biol Chem.
277:33895–33900. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo
P, Hu LS, Anderson MJ, Arden KC, Blenis J and Greenberg ME: Akt
promotes cell survival by phosphorylating and inhibiting a Forkhead
transcription factor. Cell. 96:857–868. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Figueroa C, Tarras S, Taylor J and Vojtek
AB: Akt2 negatively regulates assembly of the POSH-MLK-JNK
signaling complex. J Biol Chem. 278:47922–47927. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Kim WY and Snider WD: Functions of GSK-3
Signaling in development of the nervous system. Front Mol Neurosci.
4:442011. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Lee J and Kim MS: The role of GSK3 in
glucose homeostasis and the development of insulin resistance.
Diabetes Res Clin Pract. 77(Suppl 1): S49–S57. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Yoshino J, Monkawa T, Tsuji M, Inukai M,
Itoh H and Hayashi M: Snail1 is involved in the renal
epithelial-mesenchymal transition. Biochem Biophys Res Commun.
362:63–68. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Bachelder RE, Yoon SO, Franci C, de
Herreros AG and Mercurio AM: Glycogen synthase kinase-3 is an
endogenous inhibitor of Snail transcription: Implications for the
epithelial-mesenchymal transition. J Cell Biol. 168:29–33. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Jiang J and Griffin JD: Wnt/β-catenin
pathway modulates the sensitivity of the mutant flt3 receptor
kinase inhibitors in a GSK-3β dependent manner. Genes Cancer.
1:164–176. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Zhou BP, Deng J, Xia W, Xu J, Li YM,
Gunduz M and Hung MC: Dual regulation of Snail by
GSK-3beta-mediated phosphorylation in control of
epithelial-mesenchymal transition. Nat Cell Biol. 6:931–940. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Wang Y, Feng W, Xue W, Tan Y, Hein DW, Li
XK and Cai L: Inactivation of GSK-3beta by metallothionein prevents
diabetes-related changes in cardiac energy metabolism,
inflammation, nitrosative damage and remodeling. Diabetes.
58:1391–1402. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Shi WR, Liu Y, Xie JD, Zhuo S, Tu CX and
Xie ZF: Changes in Wnt pathway inhibiting factors in
nitrosamine-induced esophageal precancerosis lesions and effect of
gexia zhuyu decoction. Zhongguo Zhong Yao Za Zhi. 39:3131–3135.
2014.In Chinese. PubMed/NCBI
|
|
61
|
Mariappan MM, Shetty M, Sataranatarajan K,
Choudhury GG and Kasinath BS: Glycogen synthase kinase 3beta is a
novel regulator of high glucose- and high insulin-induced
extracellular matrix protein synthesis in renal proximal tubular
epithelial cells. J Biol Chem. 283:30566–30575. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Yao CJ, Lai GM, Yeh CT, Lai MT, Shih PH,
Chao WJ, Whang-Peng J, Chuang SE and Lai TY: Honokiol eliminates
human oral cancer stem-like cells accompanied with suppression of
Wnt/β-catenin signaling and apoptosis induction. Evid Based
Complement Alternat Med. 2013:1461362013. View Article : Google Scholar
|
|
63
|
Li Y, Kang YS, Dai C, Kiss LP, Wen X and
Liu Y: Epithelial-to-mesenchymal transition is a potential pathway
leading to podocyte dysfunction and proteinuria. Am J Pathol.
172:299–308. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Solanas G, Porta-de-la-Riva M, Agustí C,
Casagolda D, Sánchez-Aguilera F, Larriba MJ, Pons F, Peiró S,
Escrivà M, Muñoz A, et al: E-cadherin controls beta-catenin and
NF-kappaB transcriptional activity in mesenchymal gene expression.
J Cell Sci. 121:2224–2234. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Cozzolino M, Brenna I, Volpi E, Ciceri P,
Mehmeti F and Cusi D: Restoring the physiology of vitamin D
receptor activation and the concept of selectivity. Contrib
Nephrol. 171:151–156. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Zhang Y, Deb DK, Kong J, Ning G, Wang Y,
Li G, Chen Y, Zhang Z, Strugnell S, Sabbagh Y, et al: Long-term
therapeutic effect of vitamin D analog doxercalciferol on diabetic
nephropathy: Strong synergism with AT1 receptor antagonist. Am J
Physiol Renal Physiol. 297:F791–F801. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Fretz JA, Zella LA, Kim S, Shevde NK and
Pike JW: 1, 25-Dihydroxyvitamin D3 regulates the expression of
low-density lipoprotein receptor-related protein 5 via
deoxyribonucleic acid sequence elements located downstream of the
start site of transcription. Mol Endocrinol. 20:2215–2230. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Rincon-Choles H, Vasylyeva TL, Pergola PE,
Bhandari B, Bhandari K, Zhang JH, Wang W, Gorin Y, Barnes JL and
Abboud HE: ZO-1 expression and phosphorylation in diabetic
nephropathy. Diabetes. 55:894–900. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Xiong M, Gong J, Liu Y, Xiang R and Tan X:
Loss of vitamin D receptor in chronic kidney disease: A potential
mechanism linking inflammation to epithelial-to-mesenchymal
transition. Am J Physiol Renal Physiol. 303:F1107–F1115. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Song H, Deng B, Zou C, Huai W, Zhao R and
Zhao W: GSK3β negatively regulates LPS-induced osteopontin
expression via inhibiting its transcription. Scand J Immunol.
81:186–191. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Liu Y: New insights into
epithelial-mesenchymal transition in kidney fibrosis. J Am Soc
Nephrol. 21:212–222. 2010. View Article : Google Scholar
|
|
72
|
Jope RS, Yuskaitis CJ and Beurel E:
Glycogen synthase kinase-3 (GSK3): Inflammation, diseases and
therapeutics. Neurochem Res. 32:577–595. 2007. View Article : Google Scholar
|
|
73
|
Lin CL, Wang JY, Huang YT, Kuo YH,
Surendran K and Wang FS: Wnt/beta-catenin signaling modulates
survival of high glucose-stressed mesangial cells. J Am Soc
Nephrol. 17:2812–2820. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Costabile V, Duraturo F, Delrio P, Rega D,
Pace U, Liccardo R, Rossi GB, Genesio R, Nitsch L, Izzo P and de
Rosa M: Lithium chloride induces mesenchymal-to-epithelial
reverting transition in primary colon cancer cell cultures. Int J
Oncol. 46:1913–1923. 2015.PubMed/NCBI
|














