Introduction
Stroke is a sudden onset cerebral circulation
disorder, and is currently the second most common cause of
mortality worldwide and the leading cause of disability in
cerebrovascular patients (1).
Since ischemic stroke is the most common type of stroke (1), the development of effective therapies
remains a medical priority. Numerous treatments currently exist for
ischemic stroke, primarily pharmacological and mechanical
therapies, including cerebral ischemic postconditioning (IPOC).
IPOC refers to the treatment of cerebral ischemia reperfusion (I/R)
injury by several circulations of short term ischemia and
reperfusion processes, which are induced before or several min
after reperfusion (2). IPOC has
been demonstrated to be an effective treatment in multiple cerebral
ischemia models, such as focal and global cerebral ischemia
reperfusion injury models (3,4) with
a high clinical value, since it can be used after the onset of
ischemia and it exerts protective effects during different stages
of ischemic stroke (2). Previous
studies have revealed that IPOC can inhibit programmed cell death,
including necrosis and apoptosis, to decrease infarct volume and
improve neurological deficit (5).
Neuronal apoptosis following cerebral I/R injury can
lead to the delayed neuronal cell death (6,7).
Autophagy is a conserved process in eukaryotic cells, which is
closely associated with apoptosis; similarly to apoptosis,
autophagy can lead to programmed cell death (8). Cerebral I/R can activate autophagy,
and since autophagy activation is involved in the process of
neuronal death, the treatment of cerebral I/R should target not
only apoptosis, but also autophagy (9,10).
IPOC can inhibit autophagy induced by cerebral ischemia (11). Since reperfusion injury is a major
component of stroke damage and the role of autophagy during I/R may
be different, the present study initially aimed to elucidate
whether IPOC can regulate autophagy during cerebral I/R. It has
been reported that the combination of light chain 3 (LC3), Beclin1,
and sequestome 1 (also known as P62) is representative of autophagy
flux (12–14), therefore these parameters were
employed to monitor the autophagy process.
High mobility group box 1 (HMGB1) is a transcription
factor expressed in the majority of eukaryotic cells, including
neurons (15). Under stress, it
translocates to the cytoplasm and then to the extracellular matrix
(16), where it induces
inflammation by interacting with receptors to activate downstream
signaling pathways (17).
Increased plasma HMGB1 levels have been observed in patients with
stroke (18) and in a mouse model
of middle cerebral artery occlusion (19), and multiple treatment methods have
been demonstrated to be effective in the treatment of cerebral I/R
by decreasing the translocation and secretion of HMGB1, including
drug treatments and hypoxic therapy (20,21).
HMGB1 is associated with autophagy (22,23).
Nuclear HMGB1 is involved in the regulation of membrane equilibrium
during autophagy and mitophagy through upregulating the level of
heat shock protein β (24). When
translocated to the cytoplasm, HMGB1 combines with the
autophagy-associated protein Beclin1 to induce autophagy (25), whereas extracellular HMGB1
activates autophagy by interaction with the advanced glycosylation
end product-specific receptor (26). However, it remains unclear whether
IPOC regulates the translocation of HMGB1, or whether IPOC can
induce autophagy through regulation of HMGB1.
The present study hypothesized that cerebral I/R
injury activates autophagy by increasing HMGB1 and Beclin1 in a
cellular modal of cerebral I/R, using oxygen and glucose
deprivation and reperfusion (OGD/R) to mimic I/R, and that IPOC
would inhibit autophagy and HMGB1 translocation.
Materials and methods
PC12 cell culture, OGD/R and IPOC
treatment
PC12 cells (American Type Culture Collection,
Manassas, VA, USA) were cultured with normal culture solution,
formed by high glucose Dulbecco's modified Eagle's medium (DMEM,
Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA)
supplemented with 10% fetal bovine serum (Gibco; Thermo Fisher
Scientific, Inc., Waltham, MA, USA) and 7.5% horse serum (Gibco;
Thermo Fisher Scientific, Inc.). Cells were incubated in a
humidified incubator at 37°C and 5% CO2. For the
induction of PC12 cells to neuronal PC12 cells, 10 nM 7S nerve
growth factor (Sigma-Aldrich, St. Louis, MO, USA) was added to the
culture medium following 3 days of cultivation.
Oxygen and glucose deprivation (OGD) experiments
were performed using PC12 cells. Oxygen deprivation was induced by
incubation of the culture dishes in a hypoxic chamber with 95%
N2/5% CO2 at 37°C for 2, 4 and 8 h. The
glucose deprivation was introduced by exposing the cells to an
ischemia-mimetic solution (140 mmol/l NaCl, 3.5 mmol/l KCl, 0.43
mmol/l KH2PO4, 1.25 mmol/l MgSO4,
1.7 mmol/l CaCl2, 5 mmol/l NaHCO3, 20 mmol/l
HEPES, pH 7.2–7.4). The buffer was exposed to 95% N2/5%
CO2 for 30 min before addition to the cells. The culture
buffer changed to normal culture solution following OGD and
incubated for 24 h to imitate the process of reperfusion. IPOC was
performed by subjecting the cells to 1–3 cycles of 10 min OGD/5 min
reperfusion at 8 h following OGD, and then cultured with normal
culture solution for 24 h. The cells of normal control group were
cultured in normal culture solution for the same period as the
experimental groups. Intralipid solution (10%; Sigma-Aldrich) at a
concentration of 50 µM was used as a vehicle control, it was used
10 min prior to and during OGD. Rapamycin (RAP; 100 nM), a specific
mTOR inhibitor, was used as an autophagy activator. Recombinant
human HMGB1 (RhHMGB1) was purchased from R&D Systems China,
Co., Ltd., (Shanghai, China; cat. no. p09429), which was diluted to
a final concentration of 0.5 µg/ml prior to the onset of IPOC.
Cell viability analysis
PC12 cell viability was determined using a Cell
Counting Kit-8 (CCK-8) cell viability assay (Dojindo Molecular
Technologies, Inc., Kumamoto, Japan). PC12 cells were incubated in
96 well plates with 10,000 cells per well for cell viability
analysis. Following the final 24 h incubation step of OGD/R or IPOC
treatments, 20 µl CCK-8 solution was added to each well containing
200 µl culture medium. After incubating the cells at 37°C for 2 h,
the absorbance at 450 nm (A450) was measured using a
Multiskan FC microplate spectrophotometer (Thermo Fisher
Scientific, Inc.). Cell viability (%) was calculated as:
(A450 of test well / A450 of control well) ×
100.
Transfection of cells with Beclin1
small interfering RNA (siRNA)
PC12 cells were transiently transfected with siRNA
against Beclin1 (Shanghai GenePharma Co., Ltd., Shanghai, China) or
non-targeting control siRNA sequences (cat. no. B01001; Shanghai
GenePharma Co., Ltd.) using Lipofectamine 2000 (Invitrogen; Thermo
Fisher Scientific, Inc.) transfection reagent. The Beclin1 siRNA
sequence was as follows:
5′-CCACCGUAAUUCACUUAGATTUCUAAGUGAAUUACGGUGGTT-3′. Cells were
cultured in 60-mm plates at a density of 4×105 cells per
plate. Following 24 h normal cultivation, the culture medium was
replaced with 1.8 ml normal culture solution mixed with 200 µl
siRNA/Lipofectamine 2000 complex (containing 10 µl siRNA, 10 µl
Lipofectamine 2000 and 180 µl high glucose DMEM solution), and
cells were incubated in the humidified incubator for 24 h.
Transfection efficiency was evaluated by western blot.
Transmission electron microscopic
(TEM) observation of autophagosomes in PC12 cells
The PC12 cells for TEM observation were cultured in
60-mm plates. Following OGD/R and IPOC treatment, cells were fixed
with 2% glutaraldehyde in 0.1 mol/l phosphate-buffered saline (PBS)
for 1 week at 4°C. The cells were post-fixed in 1% osmium tetroxide
in PBS at room temperature for 1 h. Following dehydration, the
cells were embedded in Epon 812, then sectioned with an
ultramicrotome to produce sections <100 nm in thickness, and
stained with uranyl acetate and lead citrate at room temperature
for 15 min. Finally, the sections were observed using a JEM-1200EX
transmission electron microscope (JEOL, Ltd., Tokyo, Japan). The
number of autophagosomes was determined by counting manually,
whereby 5 different fields of view per section were visualized and
3 sections for each group were analyzed.
Immunofluorescence detection
Immunofluorescence was used to evaluate the
distribution and expression of Beclin1, HMGB1, microtubule
associated-protein 1A/1B-LC3II and P62 in samples of PC12 cells
from normal, OGD/R and IPOC groups. Cells (~3×105) were
cultivated in 60-mm plates and were observed by confocal
fluorescence microscopy for 24 h prior to treatment. Following 3
washes in PBS, cells were fixed with 4% paraformaldehyde at room
temperature for 30 min. Plates were then washed with PBS and
blocked with 5% bovine serum albumin (Gibco; Thermo Fisher
Scientific, Inc.) at 37°C for 40 min. For staining of LC3, Beclin1,
P62 and HMGB1, the cells of the 3 groups were incubated with
antibodies against LC3 (1:800; cat. no. NB600-1384; Novus
Biologicals, LLC, Littleton, CO, USA), Beclin1 (1:100; cat. no.
ab55878; RRID: AB_879596; Abcam), P62 (1:100; cat. no. ab91526;
RRID: AB_2050336; Abcam) or HMGB1 (1:100; cat. no. ab79823; RRID:
AB_1603373; Abcam) in a humidified container at 4°C for 12 h.
Plates were washed 3 times in PBS, then incubated with
tetramethylrhodamine-conjugated anti-rabbit IgG secondary antibody
(1:100; cat. no. SA00009-1; Wuhan Sanying Biotechnology, Wuhan,
China) at room temperature for 4 h. To stain cell nuclei, plates
were washed with PBS 3 times, then incubated with 0.0001%
4,6-diamidino-2-phenylindole (DAPI; Sigma-Aldrich) for 10 min.
Finally, plates were viewed using a Digital-Eclipse C1 laser
confocal microscope (Nikon Corporation, Tokyo, Japan).
Enzyme-linked immunosorbent assay
(ELISA) examination of cell supernatant HMGB1
Cells were cultured in 60 mm dishes for ELISA
examination. Following OGD/R or IPOC treatment, cell supernatants
were collected and centrifuged at 155 × g at 4°C for 5 min
to remove the suspended cells. The concentration of HMGB1 in the
supernatant was then determined using a commercial HMGB1 ELISA kit
(cat. no. 326054329, Shino-Test Co., Tokyo, Japan) according to the
manufacturer's protocol.
Co-immunoprecipitation and western
blot analysis
PC12 cells were cultured in 100-mm plates with
3×106 cells per plate, and then collected from the
plates following treatments. Total protein was extracted from the
cells using a commercially available kit (cat. no. KGP250; Nanjing
KeyGen Biotech Co. Ltd., Nanjing, China). Cytosolic proteins were
extracted using a Nuclear and Cytoplasmic Extraction kit (Beyotime
Institute of Biotechnology, Jiangsu, China). For
co-immunoprecipitation, HMGB1 antibody (1:1,000; cat. no. ab79823;
Abcam) was added to the lysates and rotated overnight at 4°C, then
20 µl protein A agarose beads (for HMGB1 precipitation) were added
for 3 h. Co-immunoprecipitates were washed 3 times with 1X cell
lysis buffer, consisting of radioimmunoprecipitation assay buffer
(80%), phosphatase inhibitor (10%) and protease inhibitor (10%).
The supernatant was also collected as the unbound fraction and
Beclin1 antibody (1:1,000; cat. no. ab55878; Abcam) was added to
the solution and rotated overnight at 4°C. A total of 20 µl protein
A agarose beads (for Beclin1 precipitation) was then added to the
solution and incubated at 4°C for 3 h. Co-immunoprecipitates were
washed 3 times with 1X cell lysis buffer.
Whole cell lysates and immunoprecipitated proteins
were boiled in sample buffer, separated by 10–15% sodium dodecyl
sulfate-polyacrylamide gel electrophoresis, and transferred to a
polyvinylidene fluoride membrane. Membranes were blocked with 5%
nonfat milk in Tris-buffered saline +0.1% Tween-20 for 1 h at room
temperature, then incubated overnight at 4°C with anti-LC3 (1:500;
cat. no. ab62721; Abcam), anti-Beclin1 (1:1,000; cat. no. ab55878;
Abcam), anti-P62 (1:1,000; cat. no. ab91526; Abcam), anti-HMGB1
(1:1,000; cat. no. ab79823; Abcam) and glyceraldehyde 3-phosphate
dehydrogenase (GAPDH; 1:2,000; cat. no. sc365062; Santa Cruz
Biotechnology, Inc., Dallas, TX, USA), followed by an incubation
with goat anti-rabbit IgG antibody (1:3,000; cat. no. SA00002-2;
Wuhan Sanying Biotechnology). Immunoreactive bands were visualized
using an enhanced chemiluminescence kit (Santa Cruz Biotechnology,
Inc.), and protein bands were scanned using Chemi Imager 5500 V2.03
software (ProteinSimple, San Jose, CA, USA). The integrated density
value (IDV) for each band was calculated with a computer-aided
image analysis system (Fluor Chem 2.0; ProteinSimple). The IDV of
LC3II was normalized against the IDV of LC3I, while the other
proteins were normalized against the IDV of GAPDH.
Statistical analysis
All data are expressed as the mean ± standard
deviation. Data were analyzed by one-way analysis of variance
followed by the Bonferroni test for multiple comparisons. P<0.05
was considered to indicate a statistically significant
difference.
Results
IPOC inhibits autophagy in the PC12
cell OGD/R model
CCK-8 cell viability assays demonstrated that 2 h
OGD and 24 h reperfusion significantly decreased cell viability to
~84% of the level of normal control cells (P=0.017; Fig. 1A), 4 h OGD and 24 h reperfusion
decreased cell viability to ~71% of the level of normal control
cells (P=0.028; Fig. 1A), and 8 h
OGD and 24 h reperfusion decreased cell viability to ~51% of the
level of normal control cells (P=0.042; Fig. 1A). However, IPOC was observed to
increase cell viability following 8 h OGD/24 h proportionally to
the number of IPOC cycles that were performed (Fig. 1B): 1 cycle of 10 min IPOC (IPOC1)
resulted in 64% of the viability of normal control cells, 2 cycles
of 10 min IPOC (IPOC2) resulted in 71% of the viability of normal
control cells, while 3 cycles of 10 min IPOC (IPOC3) resulted in
83% of the viability of normal control cells and a significant
improvement in cell viability compared with 8 h OGD and 24 h
reperfusion (P=0.033; Fig. 1B).
Therefore, 3 cycles of 10 min OGD and 5 min reperfusion were used
as the ‘IPOC treatment’ for the remainder of the study.
Autophagosomes were examined by TEM. A small number of
autophagosomes were observed in normal control group PC12 cells
(Fig. 1C). Following OGD/R, cells
revealed some double-membrane vacuoles containing engulfed
cytoplasmic material, suggesting the formation of autophagosomes
(Fig. 1D). However, in the IPOC
group, the number of autophagosome was visibly reduced compared to
the OGD/R group (Fig. 1E).
Quantification of autophagosomes revealed a significantly higher
number of autophagosomes in the OGD/R group compared with the
normal control group (P=0.013; Fig.
1F), and significantly fewer in the IPOC group compared with
the OGD/R group (P=0.034; Fig.
1F).
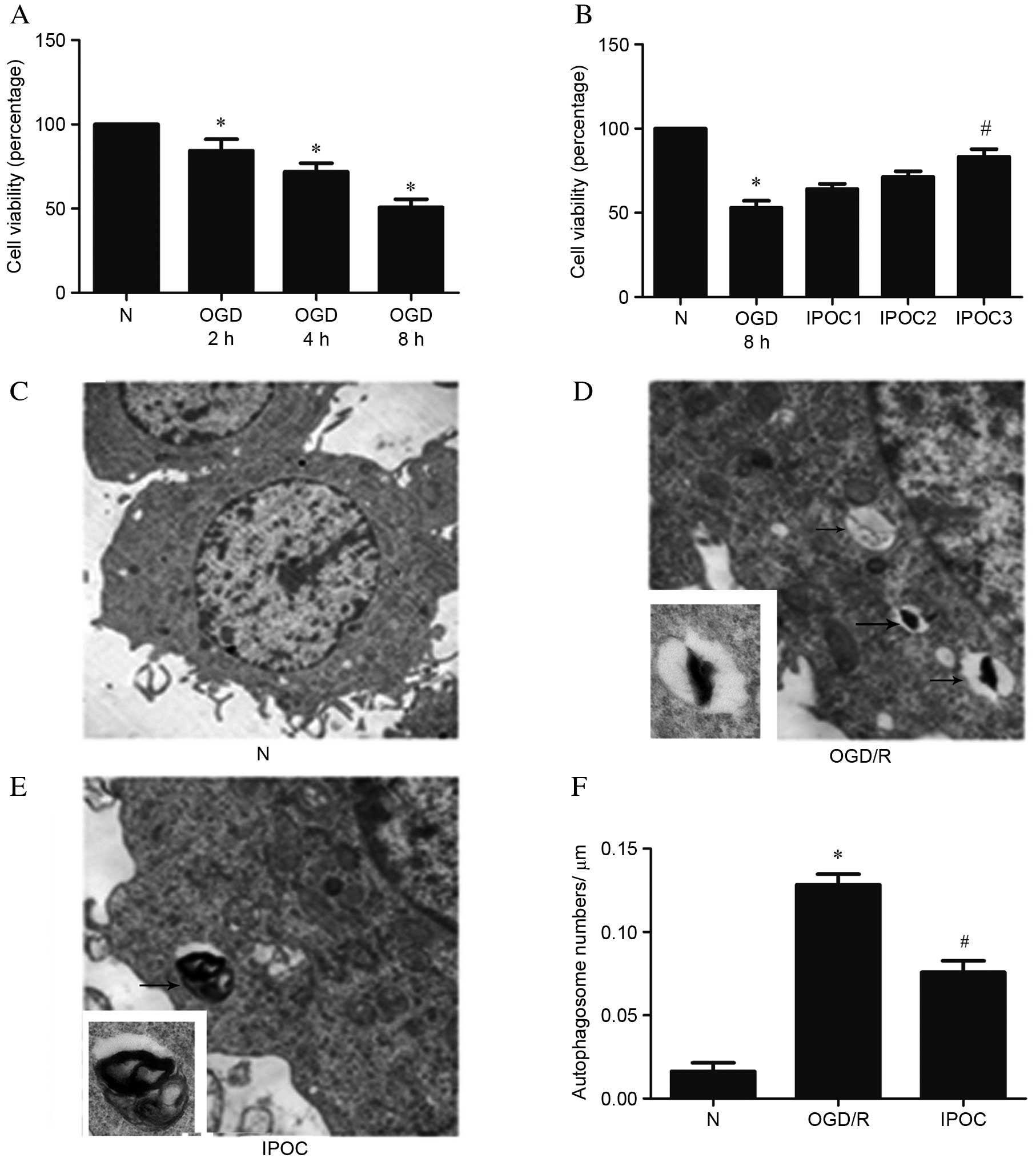 | Figure 1.Cell viability and TEM results of
cells following OGD or IPOC. (A) PC12 cell viability was evaluated
by CCK-8 assay following treatments of 2, 4 and 8 h OGD prior to
reperfusion; (B) PC12 cell viability was evaluated by CCK-8
following 8 h OGD, followed by 0, 1, 2 or 3 cycles of IPOC; (C) TEM
scanning of normal control group PC12 cells (magnification,
×3,000); (D) TEM scanning of the OGD/R group (8 h OGD followed by
24 h reperfusion; magnification, ×3,000); (E) TEM scanning of the
IPOC group (OGD/R plus 3 cycles of IPOC; magnification, ×3,000);
(F) statistical analysis of autophagosomal number following TEM.
Data are presented as the mean ± standard deviation of 3 samples
per group. *P<0.05 vs. normal control group,
#P<0.05 vs. OGD/R group. TEM, transmission electron
microscopy; CCK-8, cell counting Kit-8; N, normal control; OGD/R,
oxygen and glucose deprivation and reperfusion; IPOC, ischemic
post-conditioning. |
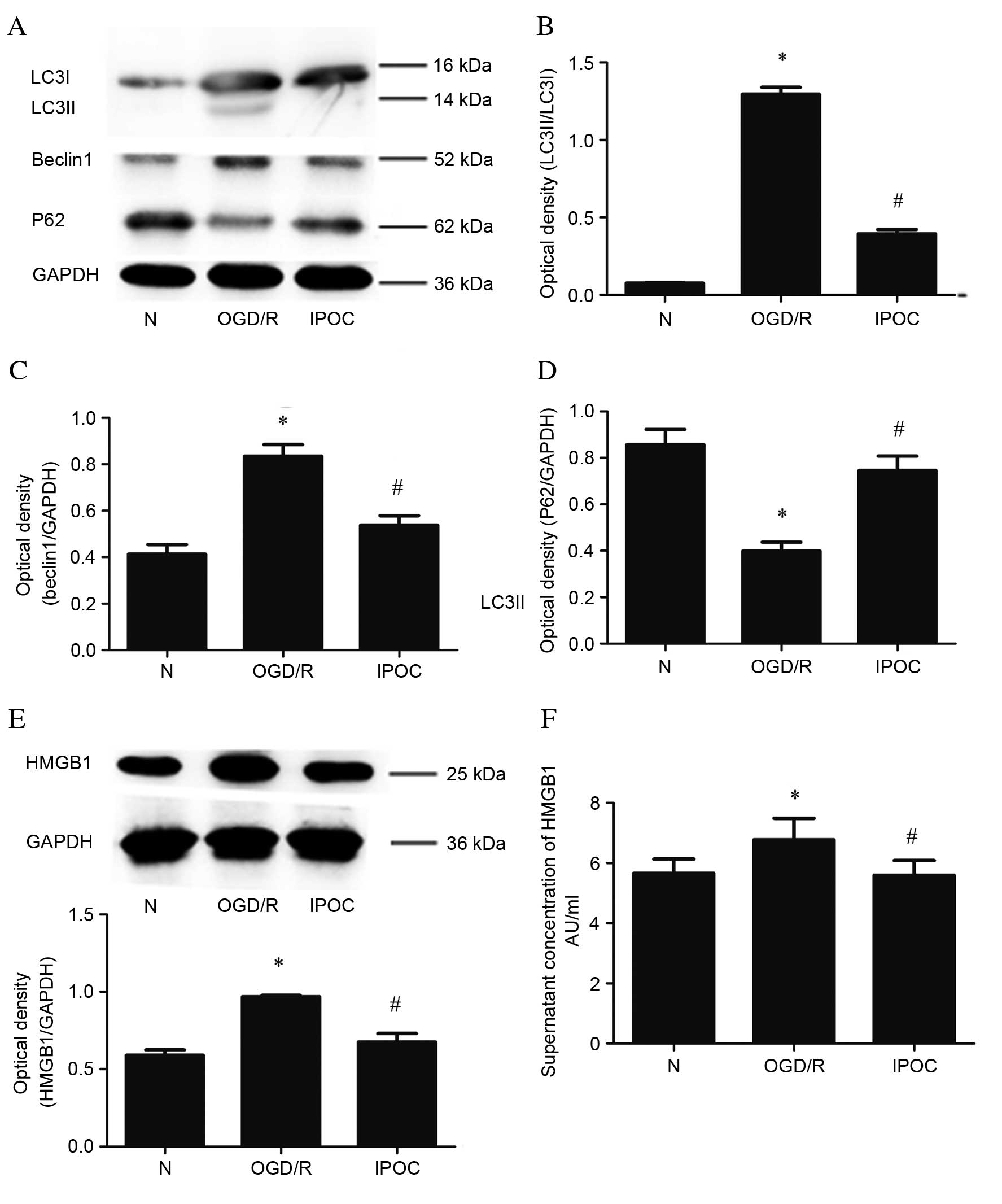
Immunofluorescence of autophagy-associated proteins
LC3II, Beclin1 and P62 revealed that OGD/R upregulated levels of
LC3II (Fig. 2A) and Beclin1
(Fig. 2B), and decreased levels of
P62 (Fig. 2C) compared with the
normal control group. IPOC abolished the OGD-mediated activation of
autophagy, which was demonstrated by the decreased expression of
LC3II and Beclin1 and increased expression of P62.
Immunofluorescence of HMGB1 revealed that OGD/R resulted in
translocation of HMGB1, which is usually located in the nucleus, to
the cytoplasm (Fig. 2D). However,
this translocation was also revealed to be inhibited by IPOC
(Fig. 2D).
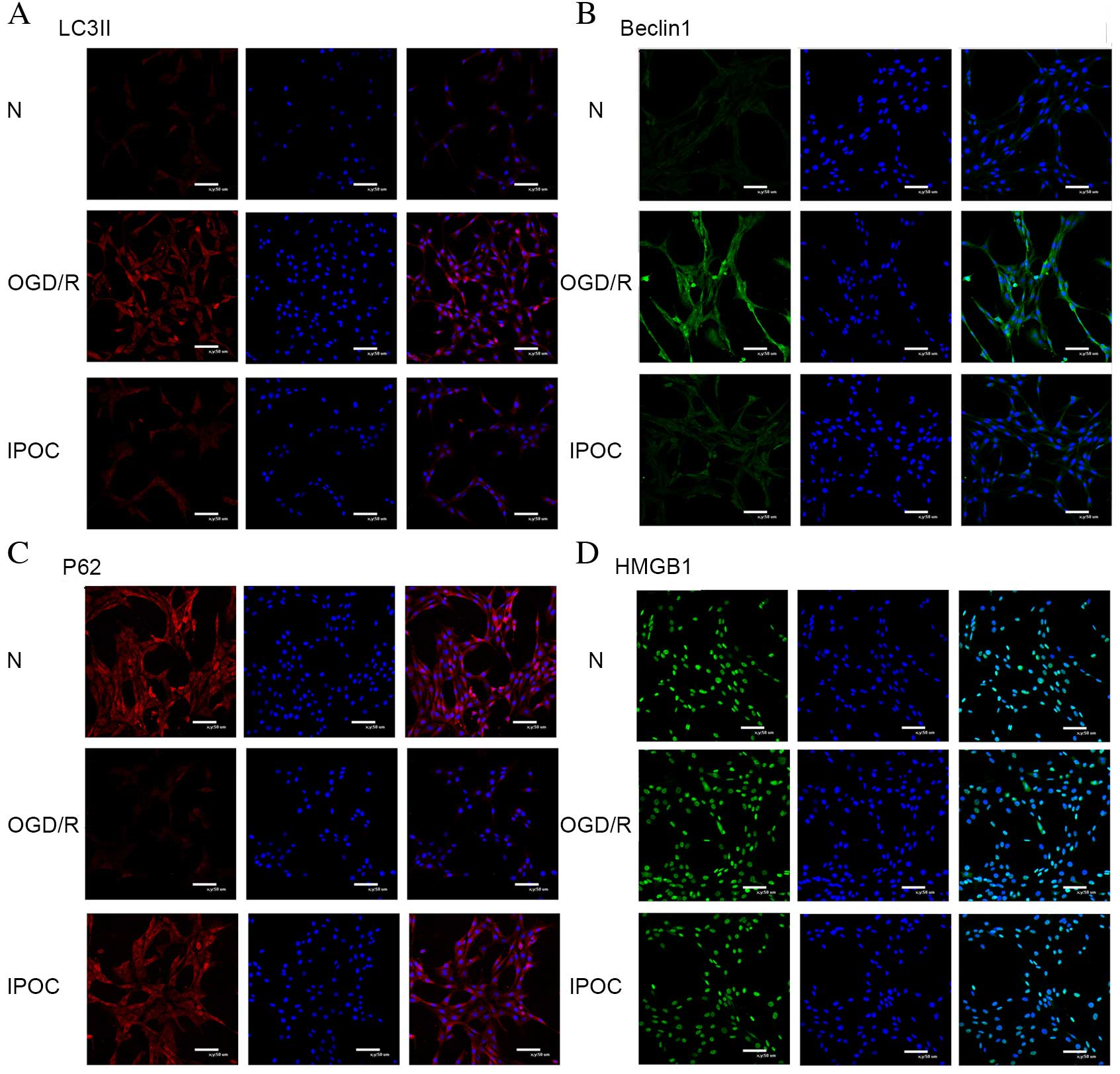 | Figure 2.Immunofluorescence images of PC12
cells to detect markers of autophagy. (A) LC3II, (B) Beclin1, (C)
P62, and (D) HMGB1 expression was demonstrated by
immunofluorescence staining in cells from the N, OGD/R (8 h OGD
followed by 24 h reperfusion) and IPOC (OGD/R plus 3 cycles of
IPOC) groups. Scale bars represent 50 µm. The left column shows
immunofluorescence staining of the specific markers, the middle
column represents staining of the cell nuclei with DAPI, and the
right column shows the left and middle images merged. N, normal
control group; OGD/R, oxygen and glucose deprivation and
reperfusion; IPOC, ischemic post-conditioning; LC3II, microtubule
associated-protein 1A/1B-light chain 3 II; P62, sequestome 1;
HMGB1, high mobility group box 1. |
Western blot analysis was also used to assess the
change in expression levels of the LC3II, Beclin1 and P62 proteins
(Fig. 3A). Similarly to
immunofluorescence, western blotting revealed that, compared with
the normal control group, OGD/R upregulated the levels of LC3II
(P=0.024; Fig. 3B), Beclin1
(P<0.05; Fig. 3C) and decreased
levels of P62 (P=0.035; Fig. 3D).
IPOC had the opposite effect, resulting in reduced levels of LC3II
(P=0.028; Fig. 3B), Beclin1
(P=0.032; Fig. 3C) and increased
levels of P62 (P=0.044; Fig. 3D)
compared with OGD/R treatment. Quantitative western blot analysis
also revealed that levels of cytoplasmic HMGB1 were increased by
OGD/R compared with the normal control group (P=0.031; Fig. 3E), and decreased by IPOC compared
with OGD/R (P=0.018; Fig. 3E). In
addition, ELISA was used to evaluate altered HMGB1 levels in the
cell supernatant; the level of extracellular HMGB1 was
significantly higher in the OGD/R group compared with the normal
control group (P=0.037; Fig. 3F),
but this effect was attenuated by IPOC (P=0.029 OGD/R vs. IPOC;
Fig. 3F).
IPOC inhibits autophagy to reduce the
secretion of HMGB1
An autophagy activator, RAP, was used prior to IPOC
to examine the effect on autophagy inhibition. CCK-8 cell viability
assays demonstrated that RAP significantly eliminated the
protective effect of IPOC (P=0.023, IPOC vs. IPOC + RAP; Fig. 4A), reducing cell viability to 54%
of the normal control group. Western blot analysis demonstrated
that the reduction in LC3II and Beclin1 expression as a result of
IPOC was significantly abolished by pre-treatment with RAP (P=0.032
and P=0.041, IPOC vs. IPOC + RAP; Fig.
4C and D, respectively), while the reverse effect on P62
expression was observed (P=0.021, IPOC vs. IPOC + RAP; Fig. 4E). The quantitative analysis of
cytoplasmic (Fig. 4F and G) and
extracellular HMGB1 (Fig. 4H)
revealed that expression and secretion of HMGB1 was reactivated by
the use of RAP (P=0.017 and P=0.037, IPOC vs. IPOC + RAP; Fig. 4G and H, respectively).
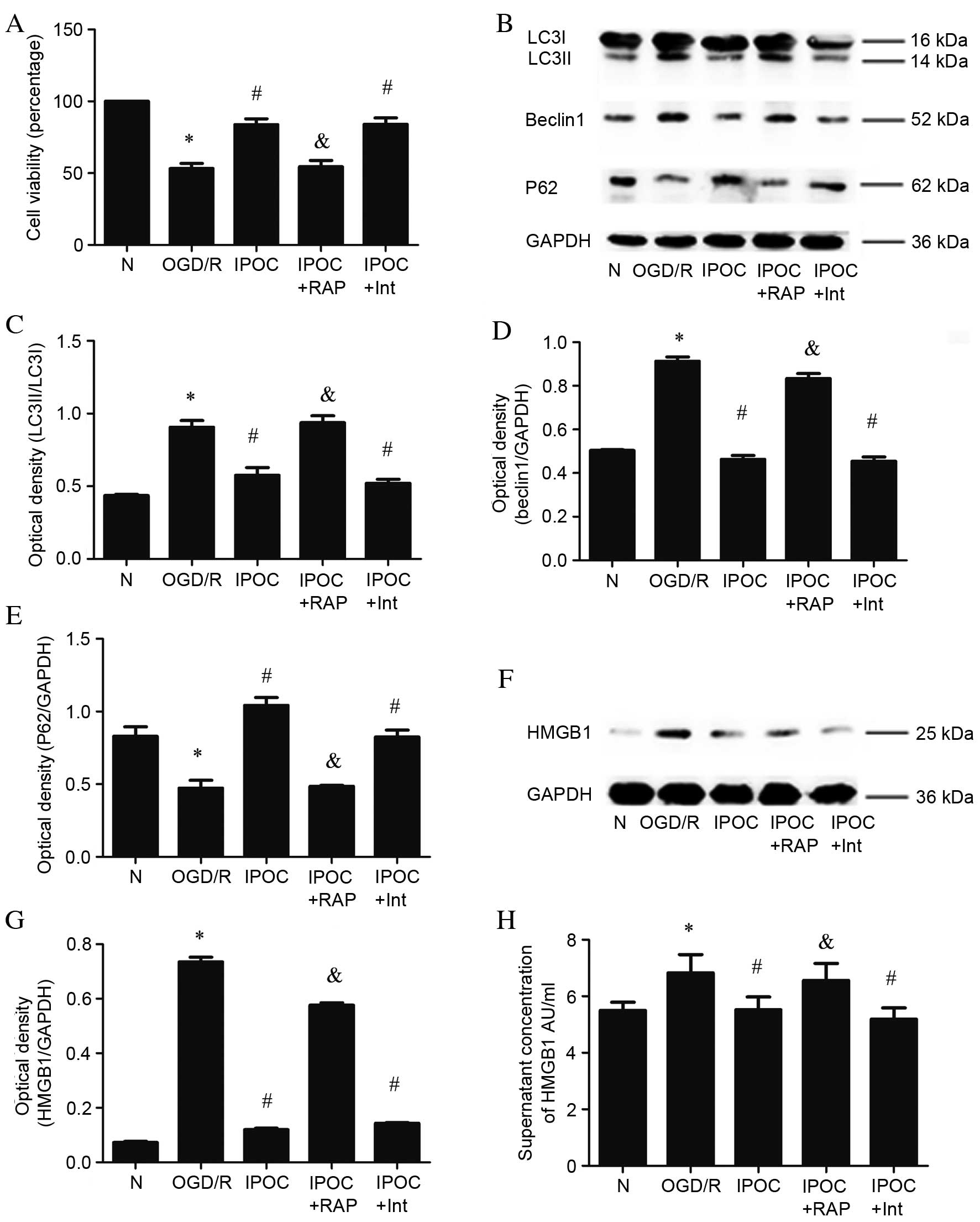 | Figure 4.The effect of the autophagy
activator, RAP, on IPOC (OGD/R plus 3 cycles of IPOC). (A) Cell
viability evaluated using a CCK-8 assay; (B) expression levels of
autophagy related proteins were detected by western blot; (C)
quantitative analysis of LC3II protein relative to LC3I; (D)
quantitative analysis of Beclin1 protein relative to GAPDH; (E)
quantitative analysis of P62 protein relative to GAPDH; (F)
cytoplasmic HMGB1 detected by western blot; (G) quantitative
analysis of cytoplasmic HMGB1 protein relative to GAPDH; (H)
quantitative analysis of cell supernatant HMGB1 protein detected by
enzyme-linked immunosorbent assay. Data are presented as the mean ±
standard deviation of 3 samples per group. *P<0.05 vs. normal
control group, #P<0.05 vs. OGD/R group,
&P<0.05 vs. IPOC group. N, normal control group;
OGD/R (8 h OGD followed by 24 h reperfusion), oxygen and glucose
deprivation and reperfusion; IPOC, ischemic post-conditioning; RAP,
rapamycin; Int, Intralipid vehicle control; LC3, microtubule
associated-protein 1A/1B-light chain 3; P62, sequestome 1; GAPDH,
glyceraldehyde 3-phosphate dehydrogenase; HMGB1, high mobility
group box 1. |
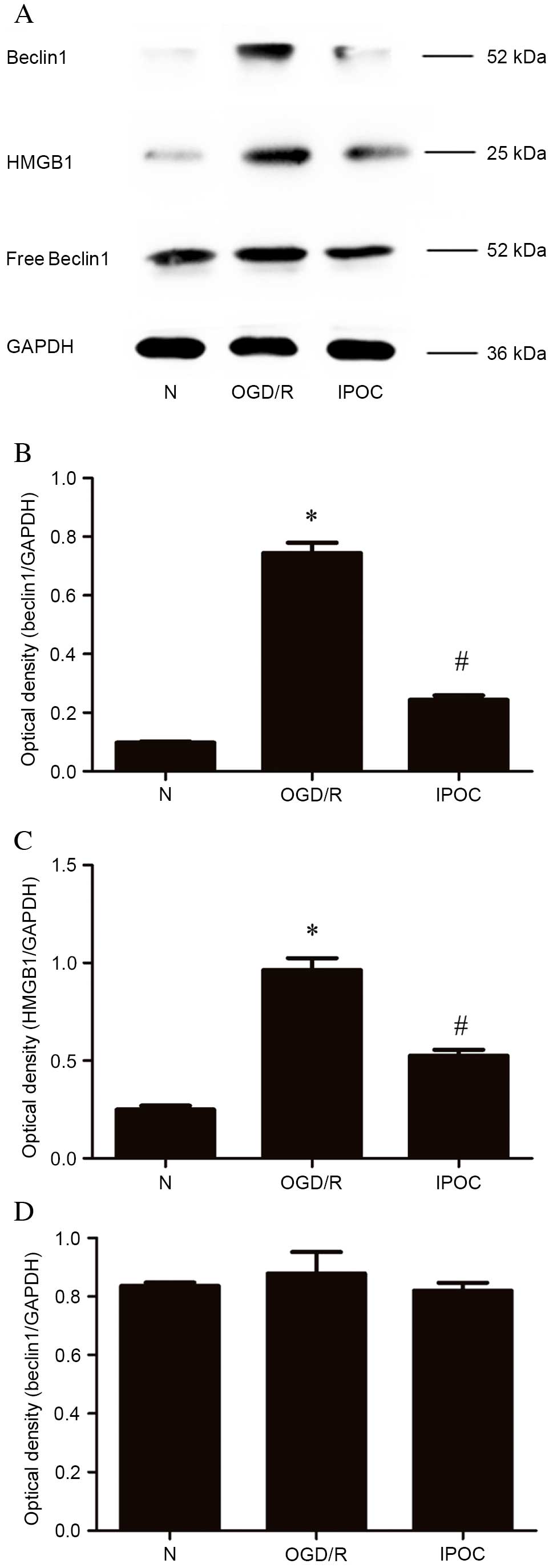
IPOC attenuates the interaction
between Beclin1 and HMGB1 to inhibit autophagy
To examine whether OGD/R and IPOC affected the
interaction between Beclin1 and HMGB1, co-immunoprecipitation was
performed. Anti-HMGB1 antibody was used to immunoprecipitate the
intracellular protein complex, revealing that OGD/R increased the
quantity of Beclin1 bound to HMGB1 compared with the normal control
group (P=0.022; Fig 5A and B),
while IPOC reversed this effect (P=0.012, OGD/R vs. IPOC; Fig 5A and B). The effect on HMGB1 was
same as that of Beclin1: OGD/R increased the quantity of HMGB1
detected compared with the normal control group (P=0.042; Fig 5A and C), while IPOC again reversed
this effect (P=0.035, OGD/R vs. IPOC; Fig 5A and C). The proportion of free
Beclin1 isolated from the cell supernatant remained unchanged
(Fig. 5A and D). These results
indicate that OGD may increase the interaction of HMGB1 with
Beclin1, whereas IPOC abolishes their interaction.
To further demonstrate the association between
Beclin1 and HMGB1 interactions and autophagy activation, Beclin1
siRNA and rhHMGB1 were, respectively, used to inhibit the
expression of Beclin1 and simulate overexpression of HMGB1.
Expression levels of Beclin1 were significantly reduced following
transfection with Beclin1 siRNA compared with the normal control
group (P=0.032; Fig. 6A). Beclin1
expression was similarly decreased in cells transfected with
Beclin1 siRNA and treated with OGD/R compared with the OGD/R
condition (P=0.031; Fig. 6B).
Compared with IPOC alone, the use of rhHMGB1 in addition to IPOC
significantly increased the level of expression of LC3II (P=0.021;
Fig. 6B and C), Beclin1 (P=0.024;
Fig. 6B and D), attenuated P62
expression (P=0.033; Fig. 6B and
E) and increased the level of HMGB1 (P=0.012; Fig. 6B and F). The simultaneous
application of IPOC, Beclin1 siRNA and rhHMGB1 induced a similar
effect to IPOC alone, resulting in significantly reduced levels of
LC3II (P=0.024; Fig. 6B and C),
Beclin1 (P=0.015; Fig. 6C and D),
increased levels of P62 (P=0.014; Fig.
6C and E) and decrease the expression of HMGB1 (P=0.041;
Fig. 6B and F) compared with OGD/R
treatment. In conclusion, the results demonstrate that the
autophagy inhibition effect of IPOC may be due to its role in
decreasing interactions between HMGB1 and Beclin1.
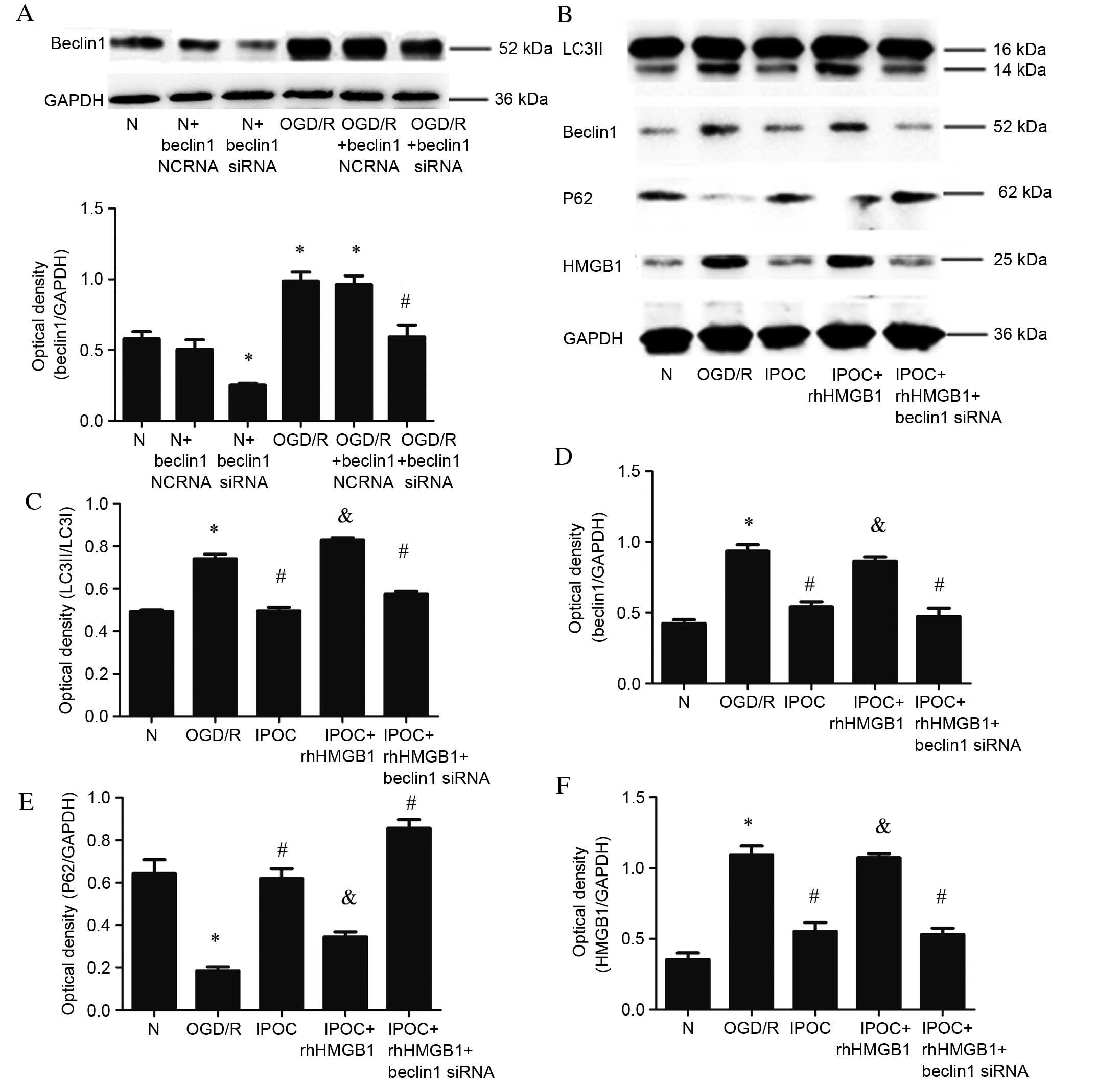 | Figure 6.Effect of rhHMGB1 and Beclin1 siRNA.
(A) Beclin1 protein levels detected by western blot following
Beclin1 siRNA treatment in normal and OGD/R (8 h OGD followed by 24
h reperfusion) groups and quantitative analysis of Beclin1 was
performed following Beclin1 siRNA treatment relative to GAPDH; (B)
autophagy related proteins detected by western blot; (C)
quantitative analysis of LC3II protein relative to LC3I; (D)
quantitative analysis of Beclin1 protein relative to GAPDH; (E)
quantitative analysis of P62 protein relative to GAPDH; (F)
quantitative analysis of cytoplasmic HMGB1 protein relative to
GAPDH. Data are presented as the mean ± standard deviation of 3
samples per group. *P<0.05 vs. normal control group,
#P<0.05 vs. OGD/R group, &P<0.05
vs. IPOC group. N, normal control group; NCRNA, negative control
siRNA; siRNA, small interfering RNA; OGD/R, oxygen and glucose
deprivation and reperfusion; LC3, microtubule associated-protein
1A/1B-light chain 3; P62, sequestome 1; HMGB1, recombinant human
high mobility group box 1; GAPDH, glyceraldehyde 3-phosphate
dehydrogenase; IPOC (OGD/R plus 3 cycles of IPOC), ischemic
post-conditioning; rhHMGB1, recombinant human HMGB1. |
Discussion
IPOC is an effective treatment for cerebral I/R
injury, while autophagy has been considered as a target for the
treatment of ischemic stroke (2).
The elevation of plasma HMGB1 in cerebral ischemic model and
patients with stroke is associated with worsened injury, and there
is a close association between HMGB1 secretion and autophagy
activation (19,23). Thus, it will be important to
examine the mutual interaction between HMGB1 secretion and
autophagy activation following IPOC. The current study demonstrated
that IPOC inhibits autophagy activation and HMGB1 release in the
OGD/R model. Additionally, it was revealed that IPOC inhibited
autophagy to reduce the translocation of HMGB1 from the nucleus to
the cytoplasm, and the autophagy inhibition effect of IPOC may be
caused by attenuation of the interaction between HMGB1 and
Beclin1.
Cellular models are important in mechanistic
studies, therefore, the present study developed and applied a novel
and convenient PC12 cell model of OGD/R and IPOC, which may be
useful in the future studies of IPOC. In the present study, PC12
cells were subjected to 8 h OGD plus 24 h reperfusion to mimic I/R,
followed by 1–3 cycles of 10 min OGD and 5 min reperfusion to mimic
IPOC in a cellular model. CCK-8 viability assays demonstrated that
3 cycles of IPOC (10 min OGD and 5 min reperfusion) had the most
potent effect on cell viability in the OGD/R model, increasing cell
viability from 51 to 83%. This was, therefore, the IPOC model used
for the remainder of the study.
Autophagy has been viewed as a double-edged sword
during the process of cardiac I/R (27); autophagosomes are formed to
encapsulate damaged organelles and other large molecular structures
to promote the reuse of materials and energy (28), however, the formation of too many
autophagosomes results in depletion of necessary substances for
cell maintenance, resulting in the death of the cell (8). Autophagy is protective during the
process of cardiac ischemia but causes destruction during
reperfusion (27). Its role in
cerebral I/R injury has, thus far, not been fully elucidated.
Numerous studies have demonstrated a protective role of autophagy
(29,30), while others have confirmed a
destructive role in cerebral ischemia (31). Apoptosis has been considered as a
key target for the treatment of cerebral I/R, since it is a form of
programmed cell death, which is involved in the process of delayed
neuronal death (6,7). Recent studies have confirmed that,
like apoptosis, autophagy can result in the death of neurons
(8,31). The processes of apoptosis and
autophagy are linked; the anti-apoptotic protein B cell lymphoma 2
apoptosis regulator (Bcl-2) is also involved in autophagic pathways
(32). The autophagic marker
Beclin1 possesses a Bcl-2-homology-3 (BH3)-only domain, which
facilitates its binding with the BH3 binding groove of multi-domain
proteins, thus, Bcl-2 interacts with Beclin1 to target
Beclin1-dependent autophagic pathway (33). Numerous methods have been developed
to target autophagy in the treatment of cerebral I/R injury
(10,34). IPOC can inhibit autophagy
activation induced by cerebral ischemia (11), however, the involvement of IPOC in
the process of autophagy in cerebral I/R has, thus far, not been
thoroughly investigated. The present study demonstrated that the
activation of autophagy reduced cell viability in an OGD/R cellular
model of I/R, and that IPOC inhibited autophagy activation and
HMGB1 release. In addition, the use of an autophagy activator, RAP,
reversed the protective effect of IPOC. IPOC was also demonstrated
to reduce the translocation of HMGB1 from the nucleus to the
cytoplasm, and that the autophagy inhibition effect of IPOC may be
caused by reduced interaction between HMGB1 and Beclin1. These
results are consistent with the findings of our previous study
(35), which confirmed that IPOC
decreased the translocation of HMGB1 from the nucleus to the
cytoplasm to exert its autophagy inhibition effect in an in
vivo model of focal cerebral ischemia and IPOC. The current
study adds further evidence for the mechanism of HMGB1-induced
autophagy in ischemic stroke and IPOC.
Autophagy activation has been demonstrated to be
induced by the secretion of HMGB1 (23), which interacts with the
autophagy-associated protein, Beclin1, and displaces Bcl-2
(25). HMGB1 exerts different
functions depending on its cellular locations. Nuclear HMGB1 is
considered to be a non-histone nuclear DNA-binding protein, which
helps to maintain the homeostasis of the nucleus and can facilitate
the bending of DNA to regulate gene expression (36). However, extracellular HMGB1 acts as
an inflammatory factor by binding with its receptor to stimulate
downstream pathways (37). HMGB1
is released following ischemic stroke and is regarded as a marker
of the severity of ischemic stroke in clinical studies (18). In a rat cerebral I/R model, plasma
HMGB1 levels were rapidly elevated following injury (38,39),
while in clinical studies, elevated HMGB1 levels can be observed in
patient serum for 7 days following subarachnoid hemorrhage
(40). Methods have, therefore,
been developed to decrease the level of plasma HMGB1 (19,20).
The present study revealed that IPOC inhibits the secretion of
HMGB1, and confirmed this by demonstrating a decrease of
cytoplasmic and extracellular HMGB1 in cells in the IPOC group
compared with the OGD/R group. Furthermore, the decreased secretion
of HMGB1 in the IPOC group was a result of autophagy inhibition,
since the use of RAP reversed the downregulation of HMGB1 induced
by IPOC. HMGB1 has been demonstrated to associate with Beclin1 to
activate autophagy in the cytoplasm (25); the present study used
co-immunoprecipitation to study the interaction between HMGB1 and
Beclin1. While the level of combined Beclin1 and HMGB1 was low in
normal cells, OGD/R increased the incidence of combined Beclin1 and
HMGB1, while IPOC reversed this effect. However, as the HMGB1
levels were also increased by OGD/R and decreased by IPOC it is
unclear whether OGD/R and IPOC affected the level of interaction
between the two proteins, or whether the level of expression of
HMGB1 was altered without affecting its interaction with Beclin1.
The levels of total Beclin1 protein and free Beclin1 in the
supernatant were then assessed following HMGB1 immunoprecipitation
in all treatment groups. The level of free Beclin1 was revealed to
be unchanged between treatment groups, while the level of total
Beclin1 was increased by OGD/R and decreased by IPOC. These
results, therefore, indicated that the combination of HMGB1 and
Beclin1 is increased by OGD/R and decreased by IPOC. To further
elucidate the association between HMGB1, Beclin1 combination and
autophagy activation, recombinant human HMGB1 and Beclin1 siRNA
were used prior to IPOC to upregulate HMGB1 and decrease Beclin1,
respectively. Overexpression of HMGB1 was revealed to reverse the
autophagy inhibition effect of IPOC, but did not induce the
activation of autophagy under the conditions of Beclin1 inhibition.
Therefore, IPOC was demonstrated to attenuate the interaction
between HMGB1 and Beclin1 to induce autophagy inhibition. HMGB1
inhibition may also result in other effects besides autophagy
inhibition, which will be examined in future studies.
In conclusion, IPOC simultaneously inhibits
autophagy and HMGB1 secretion, and mutual regulation exists between
these two processes; autophagy inhibition leads to decreased HMGB1
secretion, while the inhibition of HMGB1 release causes the
attenuation of cytoplasmic HMGB1, decreasing interactions between
HMGB1 and Beclin1 to further inhibit the process of autophagy.
Therefore, a positive feedback mechanism exists between IPOC
inhibition of autophagy process and decreased HMGB1 secretion. The
present study has further elucidated the mechanisms of IPOC in a
cellular model of cerebral I/R injury. As a method, which
simultaneously affects two key therapeutic targets of cerebral I/R
injury (autophagy and HMGB1), IPOC has huge potential for clinical
application.
Acknowledgements
This study was partially supported by a grant from
the Liaoning Province Science and Technology Project-Animal
Scientific Research and Clinical Application for Major Disease of
Liaoning Province (grant no. 2012225021) and Science and Technology
Projects of Liaoning Province (grant no. 2009225010-2) to Dr
Feng.
References
|
1
|
Writing Group Members, ; Mozaffarian D,
Benjamin EJ, Go AS, Arnett DK, Blaha MJ, Cushman M, Das SR, de
Ferranti S, Després JP, et al: Executive summary: Heart disease and
stroke statistics-2016 update: A report from the American heart
association. Circulation. 133:447–454. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Zhao H: Ischemic postconditioning as a
novel avenue to protect against brain injury after stroke. J Cereb
Blood Flow Metab. 29:873–885. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Duanmu WS, Cao L, Chen JY, Ge HF, Hu R and
Feng H: Ischemic postconditioning protects against ischemic brain
injury by up-regulation of acid-sensing ion channel 2a. Neural
Regen Res. 11:641–645. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Han D, Zhang S, Fan B, Wen LL, Sun M,
Zhang H and Feng J: Ischemic postconditioning protects the
neurovascular unit after focal cerebral ischemia/reperfusion
injury. J Mol Neurosci. 53:50–58. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Yuan Y, Guo Q, Ye Z, Pingping X, Wang N
and Song Z: Ischemic postconditioning protects brain from
ischemia/reperfusion injury by attenuating endoplasmic reticulum
stress-induced apoptosis through PI3K-Akt pathway. Brain Res.
1367:85–93. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zhu C, Xu F, Wang X, Shibata M, Uchiyama
Y, Blomgren K and Hagberg H: Different apoptotic mechanisms are
activated in male and female brains after neonatal
hypoxia-ischaemia. J Neurochem. 96:1016–1027. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Li J, Han B, Ma X and Qi S: The effects of
propofol on hippocampal caspase-3 and Bcl-2 expression following
forebrain ischemia-reperfusion in rats. Brain Res. 1356:11–23.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Bowen ID, Mullarkey K and Margen SM:
Programmed cell death during meta-morphosis in the blow-fly
Calliphora vormitoria. Microsc Res Tech. 34:202–217. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wen YD, Sheng R, Zhang LS, Han R, Zhang X,
Zhang XD, Han F, Fukunaga K and Qin ZH: Neuronal injury in rat
model of permanent focal cerebral ischemia is associated with
activation of autophagic and lysosomal pathways. Autophagy.
4:762–769. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Koike M, Shibata M, Tadakoshi M, Gotoh K,
Komatsu M, Waguri S, Kawahara N, Kuida K, Nagata S, Kominami E, et
al: Inhibition of autophagy prevents hippocampal pyramidal neuron
death after hypoxic-ischemic injury. Am J Pathol. 172:454–469.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Gao L, Jiang T, Guo J, Liu Y, Cui G, Gu L,
Su L and Zhang Y: Inhibition of autophagy contributes to ischemic
postconditioning-induced neuroprotection against focal cerebral
ischemia in rats. PLoS One. 7:e460922012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Chakrabarti L, Eng J, Ivanov N, Garden GA
and La Spada AR: Autophagy activation and enhanced mitophagy
characterize the Purkinje cells of pcd mice prior to neuronal
death. Mol Brain. 2:242009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Erlich S, Shohami E and Pinkas-Kramarski
R: Neurodegeneration induces upregulation of Beclin 1. Autophagy.
2:49–51. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kabeya Y, Mizushima N, Ueno T, Yamamoto A,
Kirisako T, Noda T, Kominami E, Ohsumi Y and Yoshimori T: LC3, a
mammalian homologue of yeast Apg8p, is localized in autophagosome
membranes after processing. EMBO J. 19:5720–5728. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Goodwin GH, Sanders C and Johns EW: A new
group of chromatin associated proteins with a high content of
acidic and basic amino acids. Eur J Biochem. 38:14–19. 1973.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wang H, Bloom O, Zhang M, Vishnubhakat JM,
Ombrellino M, Che J, Frazier A, Yang H, Ivanova S, Borovikova L, et
al: HMGB-1 as a late mediator of endotoxin lethality in mice.
Science. 285:248–251. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Yang QW, Wang JZ, Li JC, Zhou Y, Zhong Q,
Lu FL and Xiang J: High-mobility group protein box-1 and its
relevance to cerebral ischemia. J Cereb Blood Flow Metab.
30:243–254. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Huang JM, Hu J, Chen N and Hu ML:
Relationship between plasma high-mobility group box-1 levels and
clinical outcomes of ischemic stroke. J Crit Care. 28:792–797.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Rickenbacher A, Jang JH, Limani P,
Ungethüm U, Lehmann K, Oberkofler CE, Weber A, Graf R, Humar B and
Clavien PA: Fasting protects liver from ischemic injury through
sirt1-mediated downregulation of circulating HMGB1 in mice. J
Hepatol. 61:301–308. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Nakamura T, Yamada S and Yoshioka T: Brain
hypothermic therapy dramatically decreases elevated blood
concentrations of high mobility group box 1 in neonates with
hypoxic-ischemic encephalopathy. Dis Markers. 35:327–330. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wang Q, Wang F, Li X, Yang Q, Li X, Xu N,
Huang Y, Zhang Q, Gou X, Chen S and Xiong L: Electroacupuncture
pretreatment attenuates cerebral ischemic injury through α7
nicotinic acetylcholine receptor-mediated inhibition of
high-mobility group box 1 release in rats. J Neuroinflammation.
9:242012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kang R, Zeh HJ, Lotze MT and Tang D: The
Beclin 1 network regulates autophagy and apoptosis. Cell Death
Differ. 18:571–580. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Thorburn J, Frankel AE and Thorburn A:
Regulation of HMGB1 release by autophagy. Autophagy. 5:247–249.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Tang D, Kang R, Livesey KM, Kroemer G,
Billiar TR, Van Houten B, Zeh HJ III and Lotze MT: High-mobility
group box 1 is essential for mitochondrial quality control. Cell
Metab. 13:701–711. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Tang D, Kang R, Livesey KM, Cheh CW,
Farkas A, Loughran P, Hoppe G, Bianchi ME, Tracey KJ, Zeh HJ III
and Lotze MT: Endogenous HMGB1 regulates autophagy. J Cell Biol.
190:881–892. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Tang D, Kang R, Cheh CW, Livesey KM, Liang
X, Schapiro NE, Benschop R, Sparvero LJ, Amoscato AA, Tracey KJ, et
al: HMGB1 release and redox regulates autophagy and apoptosis in
cancer cells. Oncogene. 29:5299–5310. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Juhaszova M, Zorov DB, Yaniv Y, Nuss HB,
Wang S and Sollott SJ: Role of glycogen synthase kinase-3beta in
cardioprotection. Circ Res. 104:1240–1252. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Cuervo AM: Autophagy: In sickness and in
health. Trends Cell Biol. 14:70–77. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Sheng R, Zhang LS, Han R, Liu XQ, Gao B
and Qin ZH: Autophagy activation is associated with neuroprotection
in a rat model of focal cerebral ischemic preconditioning.
Autophagy. 6:482–494. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Su J, Zhang T, Wang K, Zhu T and Li X:
Autophagy activation contributes to the neuroprotection of remote
ischemic perconditioning against focal cerebral ischemia in rats.
Neurochem Res. 39:2068–2077. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wang JY, Xia Q, Chu KT, Pan J, Sun LN,
Zeng B, Zhu YJ, Wang Q, Wang K and Luo BY: Severe global cerebral
ischemia-induced programmed necrosis of hippocampal CA1 neurons in
rat is prevented by 3-methyladenine: A widely used inhibitor of
autophagy. J Neuropathol Exp Neurol. 70:314–322. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Pattingre S, Tassa A, Qu XP, Garuti R,
Liang XH, Mizushima N, Packer M, Schneider MD and Levine B: Bcl-2
antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell.
122:927–939. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Funderburk SF, Wang QJ and Yue Z: The
Beclin 1-VPS34 complex-at the crossroads of autophagy and beyond.
Trends Cell Biol. 20:355–362. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Cui D, Wang L, Qi A, Zhou Q, Zhang X and
Jiang W: Propofol prevents autophagic cell death following oxygen
and glucose deprivation in PC12 cells and cerebral
ischemia-reperfusion injury in rats. PLoS One. 7:e353242012.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Wang J, Han D, Sun M and Feng J: A
combination of remote ischemic perconditioning and cerebral
ischemic postconditioning inhibits autophagy to attenuate plasma
HMGB1 and induce neuroprotection against stroke in rat. J Mol
Neurosci. 58:424–431. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Zhang CC, Krieg S and Shapiro DJ: HMG-1
stimulates estrogen response element binding by estrogen receptor
from stably transfected HeLa cells. Mol Endocrinol. 13:632–643.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Wang H, Bloom O, Zhang M, Vishnubhakat JM,
Ombrellino M, Che J, Frazier A, Yang H, Ivanova S, Borovikova L, et
al: HMG1 as a late mediator of endotoxin lethality in mice.
Science. 285:248–251. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Hayakawa K, Mishima K, Nozako M, Hazekawa
M, Mishima S, Fujioka M, Orito K, Egashira N, Iwasaki K and
Fujiwara M: Delayed treatment with minocycline ameliorates
neurologic impairment through activated microglia expressing a
high-mobility group box 1-inhibiting mechanism. Stroke. 39:951–958.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Qiu J, Nishimura M, Wang Y, Sims JR, Qiu
S, Savitz SI, Salomone S and Moskowitz MA: Early release of HMGB-1
from neurons after the onset of brain ischemia. J Cereb Blood Flow
Metab. 28:927–938. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Nakahara T, Tsuruta R, Kaneko T, Yamashita
S, Fujita M, Kasaoka S, Hashiguchi T, Suzuki M, Maruyama I and
Maekawa T: High-mobility group box 1 protein in CSF of patients
with subarachnoid hemorrhage. Neurocrit Care. 11:362–368. 2009.
View Article : Google Scholar : PubMed/NCBI
|