Introduction
The oral cavity is an ideal environment for
microbial growth, with the oral microbiota comprising of a large
variety of species at high densities. It is well established that
an equilibrated microbiota contributes to a healthy status for the
host (1). Conversely, changes in
the balance between different bacterial species that inhabit the
human body contribute to the pathogenesis of several diseases
(2). An alteration in the
composition of subgingival microbiota is one of the most common
causes of periodontal disease (3).
Periodontitis is a microbial infection that manifests with
inflammation of gingival tissue (4). Bacteria, especially Gram-negative
species, invade the supporting structures of teeth, forming
subgingival oral biofilms in which pro-inflammatory molecules are
released. The affected tissues (bone, gingiva and periodontal
ligament) suffer damage, the extent of which depends on the
different immune responses of each individual (5), indicating that periodontitis may
manifest concurrently with other systemic diseases (6). Based on the severity of the
phenotype, two forms of periodontitis are identified: Chronic
periodontitis that affects up of 50% of the global population with
a slow rate of progression, and aggressive periodontitis with a
rapid loss of attachment and bone destruction (7). Although the oral microbiota is
different in each patient, several studies have demonstrated that
three principal bacteria are frequently identified in the dental
plaque of patients with periodontitis, classified as the ‘red
complex’ organisms: Tannerella forsythia, Porphyromonas
gingivalis, and Treponema denticola (8). These Gram-negative bacteria are
involved in the etiopathogenesis of periodontitis, most likely due
to their production of various virulence factors, including
collagenase, protease and endotoxin. Although subgingival
microbiota are important in the development of periodontitis, other
factors also contribute to the development and severity of the
disease, such as smoking (9). The
harmful components of smoke can alter the oral bacterial
composition, influencing the level of oxygenation and,
consequently, the growth of beneficial aerobic bacteria (10). Furthermore, smoking can modify the
host immune response making the oral cavity more susceptible to
proliferation of pathogenic bacterial species (11). Not surprisingly, the red complex
organisms associated with periodontitis are anaerobic bacteria that
can proliferate more easily in an environment worsened by smoke.
Despite previous studies demonstrating that the onset and the
progression of periodontitis may be compounded by smoking (12), analysis of the microbiota based on
culture techniques has revealed conflicting results (13,14).
Only 200 bacterial species that inhabit the oral cavity have been
cultured, however ~600 species have been identified by 16S rDNA
sequencing (15). In the present
study, a metagenomic analysis using high coverage next generation
sequencing (NGS) was performed, in order to define the microbiota
composition with high precision. The microbial composition from
gingival tissues was analyzed in order to identify both strongly
adhered and biofilm forming bacteria. The present study
demonstrated a general alteration in microbial composition in
subgingival tissues in patients with chronic periodontitis. Key
phylotypes characterized the subgingival microbiota of chronic
periodontitis patients compared with healthy ones. Finally, data
analysis revealed a potential contribution of smoking in the
alteration of microbial equilibrium in subgingival tissues, thus
worsening the severity of periodontal disease.
Materials and methods
Selection of patient study group
Twenty patients were recruited among consecutive
patients referred to the Periodontal Surgery and Osteo-integrated
Implantology Clinical Unit at Department of Neuroscience,
Reproductive Sciences and Dentistry of the University of Naples
Federico II (Naples, Italy). Patients were included in the study if
they fulfilled the following criteria: Good general health, no
previous periodontal treatment in the last 2 years, no use of
antibiotics that could affect the subgingival microbiota in the
past 6 months, no pregnancy and presence of at least 20 natural
teeth. The selected patients were then divided into three groups:
i) 8 non-smoker, periodontally healthy patients affected by
dysodontiasis of mandibular third molar [3 males and 5 females,
acting as controls (CTRLs)], ii) 6 non-smoker patients affected by
chronic moderate to severe periodontitis (1 male and 5 females;
termed here PCnoS), and iii) 6 smokers affected by chronic moderate
to severe periodontitis (2 males and 4 females; termed here PCS).
The severity of periodontitis was categorized on the basis of the
amount of clinical attachment loss, as follows: Slight, 1–2 mm;
moderate, 3–4 mm; severe, ≥5 mm (7). A patient was defined as a smoker if
he/she was currently smoking and had been smoking for at least 10
years and as a non-smoker if he/she had never smoked or quit
smoking longer than 10 years prior to recruitment for the present
study. Patients who agreed to participate in the study signed a
written informed consent. The protocol of the study conformed to
ethical regulations by the Department of Neuroscience, Reproductive
Sciences and Dentistry, University of Naples Federico II. At the
baseline appointment, the following clinical measurements were
recorded: Plaque (presence/absence), bleeding on probing, probing
pocket depth and clinical attachment loss.
Subgingival samples collection
Eight periodontally healthy patients aged 21–63
years who required removal of impacted mandibular third molars were
included in this study. No acute infection of the soft tissues
covering the impacted tooth was observed. Following administration
of locoregional anesthesia, a full-thickness mucoperiosteal flap
was raised for the extraction of the third molar. The flap incision
extended from the vestibular side of the retromolar trigon to the
second molar, corresponding to its distolingual cuspid. The
incision continued vestibularly around the intrasulcular surface of
the second molar, then an additional cut was made distally to the
papilla between the first and second molars, on a 45° angle, which
extended vestibularly for 2 to 3 cm, as described by Sammartino
et al (16). A small
quantity of soft tissue was picked up from the surgical site and
stored in a 1.5 ml microcentrifuge tube and frozen at −80°C for
further processing. Subsequently, an osteotomy was performed using
a Lindemann bur under constant irrigation, followed by an
odontotomy using a diamond bur. Following the extraction of the
included third molar, the root surface of the second molar was
scaled. The flap was sutured by the use of 2–0 silk sutures. Twelve
patients aged 42–68 for the PCnoS group and aged 39–55 for the PCS
group, affected by severe to moderate chronic periodontitis were
recruited for surgery. Modified Widman Flap was performed (17). Following administration of
locoregional anesthesia, intrasulcular and interdental incisions
were made. A flap was raised and reflected by the use of an
elevator. Granulation tissue was isolated, removed and stored in a
1.5 ml microcentrifuge tube and frozen at −80°C for further
processing. Scaling and root planning of the exposed root surfaces
were accurately made and finally the flap was repositioned and
sutured using 4–0 silk sutures.
DNA extraction and 16S metagenomic
sequencing
Bacterial genomic DNA was extracted from liquid
nitrogen-pulverized tissue samples using the DNeasy Blood &
Tissue kit (Qiagen Gmbh, Hilden, Germany) according to
manufacturer's instructions. The quantity and quality of DNA was
determined by spectrophotometric measurements (NanoDrop; Thermo
Fisher Scientific Inc., Waltham, MA, USA), and all DNA samples were
stored at −20°C until further processing for sequencing.
The V3-V4 region of the 16S ribosomal RNA (rRNA)
gene from each DNA sample was amplified and prepared for sequencing
according to the protocol 16S Metagenomic Sequencing Library
Preparation for Illumina Miseq System (18). This region of the rRNA gene has
previously been described as an optimal marker for bacterial
taxonomic classification (19).
Barcoded amplicons were mixed in equal amounts based
on concentrations determined by Qubit Fluorometer (Invitrogen;
Thermo Fisher Scientific, Inc.) and library sizes were assessed
using a Bioanalyzer DNA 1000 chip (Agilent Technologies Gmbh,
Waldbronn, Germany). Normalized libraries (4 nM) were pooled,
denatured with NaOH (0.2 N) for 5 min at room temperature, diluted
to 10 pM with HT1 buffer (provided by the Illumina MiSeq v3 Reagent
kit; Illumina, Inc., San Diego, CA, USA) and combined with 25%
(v/v) denatured 10 pM PhiX (Illumina, Inc.), according to Illumina
guidelines (20). Sequencing run
was performed on the Illumina MiSeq system using v3 reagents for
2×281 cycles (Illumina, Inc.).
Metagenomic data analysis
V3-V4 16S rDNA FASTQ paired-end reads were
pre-processed with PEAR (21) in
order to assemble reads with an overlap of at least 40 nucleotides,
and to remove low quality sequences (PHRED score ≤33) that were
shorter than 400 bp and longer than 500 bp. Passing filter
sequences were then processed with PRINSEQ (22) in order to obtain FASTA and quality
files for further analyses. Pick of operational taxonomic units
(OTUs), taxonomic assignment and diversity analyses were conducted
using Quantitative Insights Into Microbial Ecology (QIIME, version
1.8.0) (23). High-quality
sequences were used to pick OTUs at 97% sequence similarity from
Greengenes 16S gene database (GG; May 2013 version) (24) with a closed reference-based OTU
picking method. The GG database was used to taxonomically classify
the identified OTUs and to compute their distribution across
different taxonomic levels. To avoid sample size biases in
subsequent analyses, samples were normalized to 34,559
sequences/sample using a sequence rarefaction procedure. Species
heterogeneity in each sample was assessed employing two Alpha
diversity metrics (the number Observed species and the Shannon
entropy) and compared using one-way analysis of variance (ANOVA)
followed by Tukey's multiple comparison post-hoc test. Unweighted
Unifrac distances were calculated to analyze OTUs diversity among
sample communities (beta diversity). Nonparametric multivariate
analysis of changes in community structure was performed using the
analysis of similarities (ANOSIM) method (25) with 999 permutations, to test the
force of microbial communities grouping. Statistical differences in
OTU frequencies among groups across different taxonomic levels were
assessed with linear discriminative analysis (LDA) effect size
analysis (LEfSe, logarithmic LDA score >2 and P<0.05 for
Kruskal-Wallis test) (26). The
normalized OTU table, corrected for multiple 16S rRNA gene copy
number, was used for metagenome prediction in PICRUSt (27). Kyoto encyclopedia of genes and
genomes (KEGG) (28) ortholog
abundances were categorized by function at hierarchical pathway
level 3 and compared among groups with LEfSe (LDA score >2 and
P<0.05).
Data Deposition
The sequences reported in this study are deposited
on the ‘European Nucleotide Archive’ server (www.ebi.ac.uk/ena) under the accession number
PRJEB17957.
Results
Subgingival microbial diversity
associated with chronic periodontitis
The subgingival microbiota of patients with chronic
periodontitis and healthy controls (CTRLs) was analyzed. Among
periodontitis patients, the samples were classified into 2 groups:
Samples from smoker patients with chronic periodontitis (PCS) and
samples from non-smoker patients with chronic periodontitis
(PCnoS). The different subgingival microbiota compositions in 8
CTRLs, 6 PCS and 6 PCnoS patients were studied by high-throughput
sequencing of the 16S rDNA V3-V4 hypervariable region on an
Illumina MiSeq platform. This region is commonly amplified in 16S
rRNA gene-based metagenomic studies, as it has been determined to
provide optimal bacterial taxonomic coverage and taxonomic
resolution (19,29). High quality reads (>2,000,000),
with an average of 110,169 sequences/sample, were assigned into
3,891 OTUs; following the rarefaction procedure, 34,559 rarefied
sequences/sample were considered, representing 2,937 OTUs, in order
to describe the differences in subgingival bacterial communities
between samples. The alpha and beta diversity among the three
groups were measured in order to evaluate the bacterial diversity
in terms of species richness and community diversity. Alpha
diversity results revealed that the subgingival bacterial
population of PCS and PCnoS groups exhibited a significant decrease
of species richness, demonstrated by number of OTUs (P<0.05;
Fig. 1A) and of microbial
diversity (Shannon index; P<0.05; Fig. 1B), compared with CTRLs. Beta
diversity, representing the distances among samples and groups in
terms of bacterial community composition, was then measured. Using
the ANOSIM R statistic, an index based on rank dissimilarity,
dissimilarity between groups based on Unweighted Unifract distances
was measured. ANOSIM analysis revealed significant differences in
subgingival bacterial assortment between PCS and CTRL groups
(ANOSIM R=0.21, P=0.03), while no significant differences in
bacterial assortment were identified between PCnoS and the other
groups. In fact, a Principal Coordinate Analysis (PCoA) plot
displayed clustering of smoker patients away from healthy controls,
while non-smoker samples clustered closely to healthy controls
(Fig. 1C).
Description of subgingival microbiota
in PCS and PCnoS patients compared with CTRL: identification of key
phylotypes
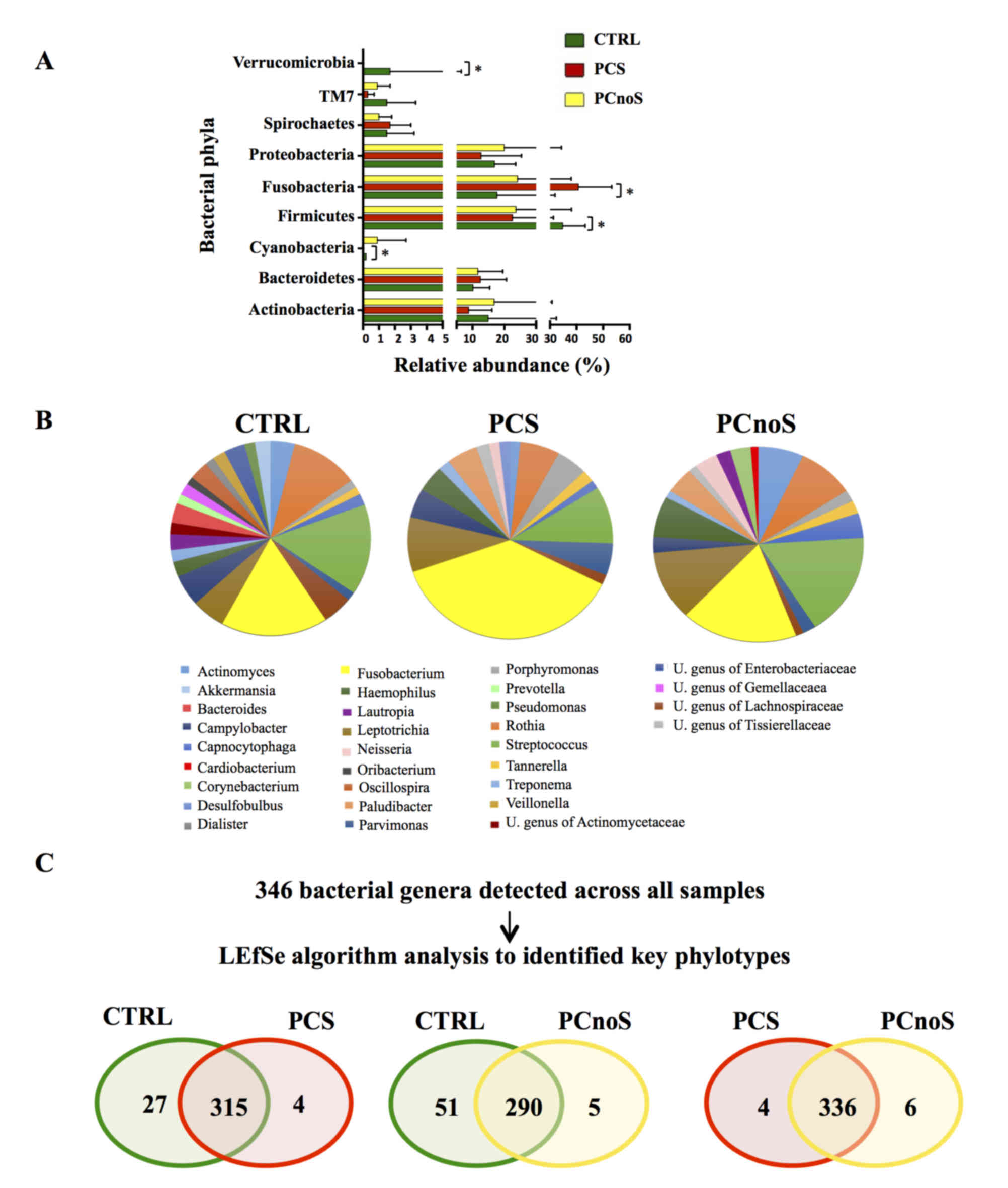
Sequencing analysis revealed that Actinobacteria,
Bacteroidetes, Firmicutes, Fusobacteria and Proteobacteria were the
dominant phyla of subgingival microbiota in all groups (Fig. 2A). Amongst the 22 bacterial phyla
identified, comparison of relative abundances by LEfSe analysis
revealed no significant differences between PCnoS and CTRL
(Fig. 2A). By contrast,
significantly higher levels of Fusobacteria and lower levels of
Cyanobacteria, Firmicutes, SR1 (also termed Absconditabacteria)
(30) and Verrucomicrobia phyla
distinguished PCS and CTRL bacterial communities (P<0.05;
Fig. 2A). At the genus level,
periodontitis patients were characterized by alterations in
relative abundance of several bacterial genera compared to the CTRL
group. According to alpha diversity results, the microbial
communities of CTRL and PCnoS groups were not dominated by a single
genus, while the PCS group showed a predominance in the
Fusobacterium genus (Fig.
2B). In fact, Fusobacterium was more abundant in the PCS
group (32.84% of total microbiota) compared with CTRL and PCnoS
groups (13.78 and 14.96%, respectively; Fig. 2B), even though these effects were
not statistically significant.
Key phylotypes were identified from direct
comparisons between the patient groups (PCS/CTRL, PCnoS/CTRL and
PCS/PCnoS) using LEfSe algorithm analysis. Among the 346 genera
detected across all samples, 56 genera differentiated between the
CTRL and PCS groups. A distinct and smaller set of bacterial genera
(31 genera) differentiated the PCnoS and CTRLs groups, while only
10 different genera were evident between the PCS and PCnoS groups
(Fig. 2C). Table I describes the relative abundances
of key phylotypes identified by LEfSe analysis as significantly
different between groups. The dysbiotic profile of PCS subgingival
microbiota compared with CTRL was characterized by significant
increase of Desulfobulbus, Sphaerochaeta, and unclassified
genera of Mogibacteriaceaea and Tissierellaceae, and significant
decrease of Pedobacter, Granulicatella, Paracoccus,
Gemellaceae, Clostridiaceae, Ruminococcaceae, Sphingomonadaceae,
Pseudomonadaceae, Xanthomondaceae, and an unclassified genus of
S24-7 (Table I). Notably,
Pseudoramibacter was significantly more abundant in both PCS
and PCnoS compared with CTRL (Table
I). Finally, Bacteroides, Prevotella, Veillonella,
Pseudomonas, and unclassified genera of Enterobacteriaceae,
Rikenellaceae, Lachnospiraceae and Clostridiales were significantly
less abundant or even absent in the PCS and PCnoS groups compared
with the CTRL group (Table I).
 | Table I.Key genera among PCS, PCnoS and CTRL
groups. |
Table I.
Key genera among PCS, PCnoS and CTRL
groups.
| Phyla | Genera | % in CTRL | % in PCS | % in PCnoS |
|---|
| Actinobacteria |
Atopobium | 0.19±0.05 |
0.57±0.37c | 0.57±0.37 |
|
|
Bifidobacterium | 0.25±0.10 | 0.12±0.07 |
0.02±0.02b |
|
| U.
Actinomycetales | 0.06±0.06 | 0c | 0.01±0.00 |
| Bacteriodetes |
Bacteroides | 2.52±2.21 |
0.01±0.01a |
0.03±0.02b |
|
|
Pedobacter | 0.003±0.00 | 0a | 0.001±0.00 |
|
|
Prevotella | 1.46±0.27 |
0.40±0.14a |
0.26±0.09b |
|
| U.
Rikenellaceae | 0.80±0.50 |
0.01±0.00a |
0.06±0.06b |
|
| U. S24–7 | 0.30±0.22 |
0.03±0.02a | 0.04±0.04 |
| Firmicutes |
Faecalibacterium | 0.14±0.09 | 0 | 0b |
|
|
Granulicatella | 0.78±0.32 |
0.11±0.07a | 0.22±0.02 |
|
|
Oribacterium | 1.01±0.36 | 0.92±0.52 |
0.19±0.00b |
|
|
Oscillospira | 2.33±1.41 | 0.02±0.01 |
0.12±0.02b |
|
|
Pseudoramibacter | 0.01±0.01 | 0.02±0.01 |
0.24±0.01b |
|
|
Ruminococcus | 0.19±0.11 | 0.001±0.00 | 0b |
|
|
Veillonella | 1.68±0.39 |
0.27±0.12a |
0.23±0.04b |
|
| U.
Clostridiaceae | 0.67±0.50 |
0.04±0.04a | 0.09±0.07 |
|
| U.
Clostridiales | 0.07±0.03 |
0.17±0.05a |
0.50±0.07b |
|
| U. Gemellaceae | 1.53±0.41 |
0.07±0.04a | 0.43±0.04 |
|
| U.
Lachnospiraceae | 4.21±0.79 | 1.42±0.83 |
1.02±1.07b |
|
| U.
Mogibacteriaceae | 0.68±0.29 |
1.83±0.30a | 0.79±1.05 |
|
| U.
Ruminococcaceae | 1.02±0.64 |
0.02±0.01a | 0.07±0.06 |
|
| U.
Tissierellaceae | 0.08±0.07 |
1.78±1.27a | 1.07±1.08 |
| Fusobacteria | U.
Leptotrichiaceae | 0.34±0.34 |
0.33±0.21c | 0 |
| Proteobacteria |
Cardiobacterium | 0.48±0.17 |
0.18±0.12c | 1±0.01 |
|
|
Desulfobulbus | 0.05±0.02 |
1.49±0.64a,c | 0.14±1.02 |
|
|
Lautropia | 1.85±1.55 |
0.61±0.51c | 1.85±0.07 |
|
|
Ochrobactrum | 0.01±0.01 | 0c | 0.01±0.00 |
|
|
Paracoccus | 0.28±0.24 |
0.003±0.00a | 0.10±0.09 |
|
|
Pseudomonas | 1.51±0.95 |
0.02±0.01a |
0.12±0.05b |
|
| U.
Enterobacteriaceae | 2.40±2.05 |
0.01±0.01a |
0.02±0.01b |
|
| U.
Pseudomonadaceae | 0.73±0.34 |
0.01±0.00a | 0.16±0.01 |
|
| U.
Sphingomonadaceae | 0.01±0.00 |
0.001±0.00a | 0.01±0.01 |
|
| U.
Xanthomonadaceae | 0.70±0.27 |
0.01±0.01a | 0.10±0.04 |
| Spirochaetes |
Sphaerochaeta | 0 |
0.01±0.01a | 0 |
| Tenericutes | ML615J_28 | 0.004±0.00 |
0.01±0.00c | 0 |
|
Verrucomicrobia |
Akkermansia | 1.72±1.71 | 0 | 0b |
Predictive subgingival microbiota
functions in chronic periodontitis patients
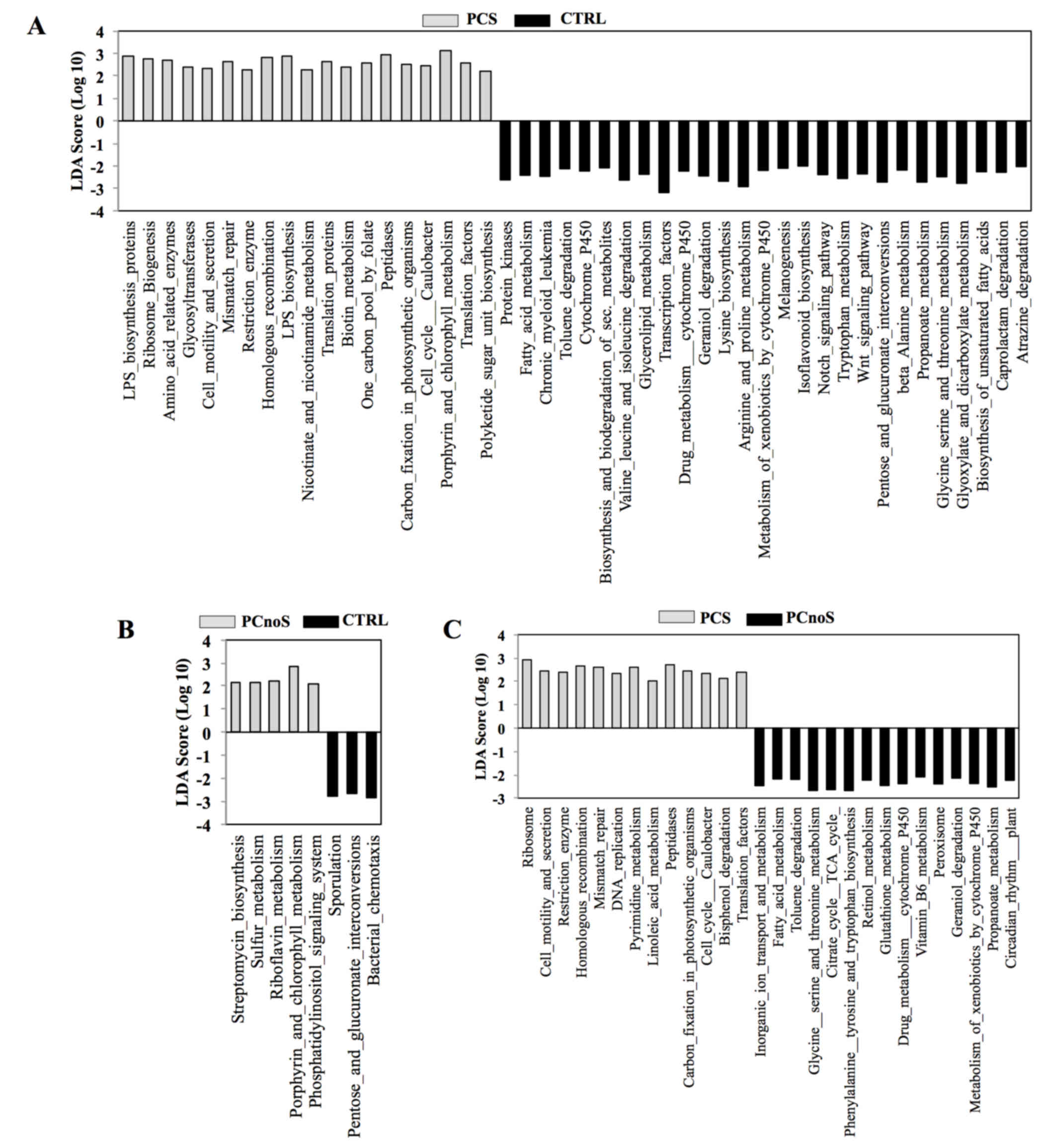
In order to evaluate putative biological functions
of PCS, PCnoS and CTRL subgingival microbial communities, a PICRUSt
analysis was performed. Based on bacterial sequences, the
comparison of microbiota-related gene functions between
periodontitis and healthy samples demonstrated alterations in 46
metabolic pathways in PCS and in 8 pathways in PCnoS subgingival
microbiota compared with CTRL (Fig
3). Among the altered metabolic functions in the PCS group
compared with CTRL, an enrichment of genes encoding peptidases,
enzymes for lipopolysaccharide biosynthesis, and proteins involved
in cell motility and secretions was evident (Fig. 3). Conversely, enzymes for
biosynthesis and biodegradation of secondary metabolites, fatty
acid metabolism and amino acid biosynthesis were highly represented
in healthy CTRL subgingival microbiotacompared with the PCS group
(Fig. 3). In addition, enzymes
involved in cell motility and secretion, homologous recombination,
peptidase and DNA replication were more heavily represented in the
subgingival microbiota of PCS compared with PCnoS (Fig. 3).
Discussion
The bacterial composition of healthy and periodontal
gingival sulcus has largely been described (31,32).
The present study expanded on previous published information about
periodontal subgingival microbiota, by next generation sequencing
on an Illumina Miseq platform. This approach allows identification
of a larger range of bacterial sequences, increasing the
sensitivity of the analysis. In order to avoid loss of bacterial
taxa that are able to adhere to subgingival oral biofilms and
aggravate periodontitis pathology, bacterial DNA was extracted
directly from gingival tissue. Furthermore, the involvement of
smoking as an environmental factor and its role in altering the
microbiota composition of periodontitis patients was assessed. The
present study demonstrated that alpha diversity decreased in both
smoker (PCS) and non-smoker (PCnoS) periodontitis patients compared
with healthy controls. The lower bacterial richness of
periodontitis samples was confirmed by taxonomic classification of
the majority (41%) of total reads in Gram-negative and anaerobic
Fusobacteria phylum, specifically in the PCS group. In addition,
beta diversity analysis demonstrated a different composition in
subgingival microbiota between PCS and CTRLs. The PCoA plot
revealed a separate clustering of the two groups, indicating a
diversity in their bacterial communities.
In the present study, the potential pathogenic role
of bacteria identified in high levels in subgingival microbiota of
PCS and PCnoS groups was also explored. Genera including
Parvimonans, Desulfubulbus, Paludibacter, Haemophilus, and
Sphaerochaeta were discovered to be associated with
periodontitis. Although Parvimonans normally inhabits the
oral cavity, it has been isolated in high numbers in periodontitis
patients and is associated with destruction of periodontal tissue
(33). Desulfubulbus and
Paludibacter have also previously been associated with
periodontitis (34) and high
concentrations have been linked to worse periodontal therapeutic
response (35). In the present
study, identification of Desulfobulbus and
Paludibacter was increased in PCS samples compared with
PCnoS and CTRLs, corroborating the hypothesis that smoking
interferes with microbiota composition during periodontal disease.
Sphaerochaeta has been previously linked with a large number
of pathologies, such as syphilis and Lyme disease (36), but no association to periodontitis
had been previously reported. Similarly, the presence of
Haemophilus in subgingival microbiota of periodontal
patients had not been demonstrated to date. Because
Haemophilus grows well in blood-enriched culture media
(37), its presence may be
explained by the bleeding gums typically featured in periodontal
disease. Periodontitis-associated dysbiosis was also characterized
by low abundance or absence of specific bacteria normally present
in healthy subgingival microbiota. Low levels of Pedobacter,
Granulicatella, Paracoccus, Gemellaceae, Clostridiaceae,
Ruminococcaceae, Sphingomonadaceae, Pseudomonadaceae and of
Xanthomondaceae and an unclassified genus of S24-7 were identified
in PCS samples compared with CTRL, and low levels of Atopobium,
Bifidobacterium, Coprococcus, Oridobacterium, Peptococcus,
Oscillospira, Akkermansia and an unclassified genus of
Moraxellaceae in PCnoS samples. These bacteria may, therefore, be
relevant to the onset of periodontitis. To assess how an alteration
of subgingival microbiota could impact periodontal disease,
metagenomic function prediction was performed by PICRUSt analysis.
Notably, genes associated with tissue damage were increased in PCS
patients, while genes associated with amino acid metabolism and
nutrient biosynthesis were reduced in PCS patients compared with
CTRLs.
Although the PCS and PCnoS groups both exhibited a
shift in subgingival bacterial composition compared with healthy
subjects, several results highlight a major alteration in PCS
smoker patients. In particular, the PCS subgingival microbiota was
dominated by the Fusobacteria phylum, consistent with a lower
bacterial richness. In addition, PCS samples displayed significant
alterations in the relative abundance of several phyla compared to
PCnoS, and a specific bacterial signature discriminated PCS from
CTRLs at the genus level. According to these results, predictive
functional analysis designated PCS subgingival microbiota as
potentially able to promote tissue damage, more so than the PCnoS
microbiota.
Although the present study reports a detailed
description of periodontitis-associated subgingival microbiota,
further studies, involving larger cohorts and considering potential
gender differences, would enhance our knowledge about the role of
subgingival dysbiosis and smoking in periodontal disease. The
present study indicated that chronic periodontitis results in a
global perturbation of oral microbial communities, and elucidated
the role of key bacteria in the complex ecology of subgingival
microbiota. Finally, the present study confirmed that smoking
aggravates the polymicrobial synergy of pathogenic bacteria and the
subgingival dysbiosis that characterize chronic periodontal
disease.
Acknowledgements
This work was partially funded by Regione Campania
(2007 legge 5, to LoCh).
References
|
1
|
Avila M, Ojcius DM and Yilmaz O: The oral
microbiota: Living with a permanent guest. DNA Cell Biol.
28:405–411. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Cho I and Blaser MJ: The human microbiome:
At the interface of health and disease. Nat Rev Genet. 13:260–270.
2012.PubMed/NCBI
|
|
3
|
Haffajee AD, Teles RP and Socransky SS:
The effect of periodontal therapy on the composition of the
subgingival microbiota. Periodontol 2000. 42:219–258. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Dentino A, Lee S, Mailhot J and Hefti AF:
Principles of periodontology. Periodontol 2000. 61:16–53. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Al Harthi LS, Cullinan MP, Leichter JW and
Thomson WM: The impact of periodontitis on oral health-related
quality of life: A review of the evidence from observational
studies. Aust Dent J. 58:274–277; quiz 384. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Seymour GJ, Ford PJ, Cullinan MP, Leishman
S and Yamazaki K: Relationship between periodontal infections and
systemic disease. Clin Microbiol Infect. 13 Suppl 4:S3–S10. 2007.
View Article : Google Scholar
|
|
7
|
Armitage CG: Comparison of the
microbiological features of chronic and aggressive periodontitis.
Periodontol 2000. 53:70–88. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Socransky SS, Haffajee AD, Cugini MA,
Smith C and Kent RL Jr: Microbial complexes in subgingival plaque.
J Clin Periodontol. 25:134–44. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Page RC and Kornman KS: The pathogenesis
of human periodontitis: An introduction. Periodontol 2000. 14:9–11.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lee J, Taneja V and Vassallo R: Cigarette
smoking and inflammation: Cellular and molecular mechanisms. J Dent
Res. 91:142–149. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Labriola A, Needleman I and Moles DR:
Systematic review of the effect of smoking on nonsurgical
periodontal therapy. Periodontol 2000. 37:124–137. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Haffajee AD and Socransky SS: Relationship
of cigarette smoking to the subgingival microbiota. J Clin
Periodontol. 28:377–88. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Preber H, Bergström J and Linder LE:
Occurrence of periopathogens in smoker and non-smoker patients. J
Clin Periodontol. 19:667–671. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Van der Velden U, Varoufaki A, Hutter JW,
Xu L, Timmerman MF, Van Winkelhoff AJ and Loos BG: Effect of
smoking and periodontal treatment on the subgingival microflora. J
Clin Periodontol. 30:603–610. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Dewhirst FE, Chen T, Izard J, Paster BJ,
Tanner AC, Yu WH, Lakshmanan A and Wade WG: The human oral
microbiome. J Bacteriol. 192:5002–5017. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Sammartino G, Tia M, Gentile E, Marenzi G
and Claudio PP: Platelet-rich plasma and resorbable membrane for
prevention of periodontal defects after deeply impacted lower third
molar extraction. J Oral Maxillofac Surg. 67:2369–2373. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ramfjord SP: Present status of the
modified Widman flap procedure. J Periodontol. 48:558–565. 1977.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Illumina. 16S Metagenomic Sequencing
Library Preparation. 2013.http://support.illumina.com/downloads/16s_metagenomic_sequencing_library_preparation.html
|
|
19
|
Mizrahi-Man O, Davenport ER and Gilad Y:
Taxonomic classification of bacterial 16S rRNA genes using short
sequencing reads: Evaluation of effective study designs. PLoS One.
8:e536082013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Illumina. MiSeq System Denature and Dilute
Libraries Guide. 2016.http://support.illumina.com/downloads/prepare_libraries_for_sequencing_miseq_15039740.html
|
|
21
|
Zhang J, Kobert K, Flouri T and Stamatakis
A: PEAR: A fast and accurate Illumina Paired-End reAd mergeR.
Bioinformatics. 30:614–620. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Schmieder R and Edwards R: Quality control
and preprocessing of metagenomic datasets. Bioinformatics.
27:863–864. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Caporaso JG, Kuczynski J, Stombaugh J,
Bittinger K, Bushman FD, Costello EK, Fierer N, Peña AG, Goodrich
JK, Gordon JI, et al: QIIME allows analysis of high-throughput
community sequencing data. Nat Methods. 7:335–336. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
DeSantis TZ, Hugenholtz P, Larsen N, Rojas
M, Brodie EL, Keller K, Huber T, Dalevi D, Hu P and Andersen GL:
Greengenes, a chimera-checked 16S rRNA gene database and workbench
compatible with ARB. Appl Environ Microbiol. 72:5069–5072. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Clarke KR: Non-parametric multivariate
analyses of changes in community structure. Aust J Ecol.
18:117–143. 1993. View Article : Google Scholar
|
|
26
|
Segata N, Izard J, Waldron L, Gevers D,
Miropolsky L, Garrett WS and Huttenhower C: Metagenomic biomarker
discovery and explanation. Genome Biol. 12:R602011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Langille MG, Zaneveld J, Caporaso JG,
McDonald D, Knights D, Reyes J, Clemente JC, Burkepile DE, Thurber
RL Vega, Knight R, et al: Predictive functional profiling of
microbial communities using 16S rRNA marker gene sequences. Nat
Biotechnol. 31:814–821. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kanehisa M and Goto S: KEGG: kyoto
encyclopedia of genes and genomes. Nucleic Acids Res. 28:27–30.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kim M, Morrison M and Yu Z: Evaluation of
different partial 16S rRNA gene sequence regions for phylogenetic
analysis of microbiomes. J Microbiol Methods. 84:81–87. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Hug LA, Baker BJ, Anantharaman K, Brown
CT, Probst AJ, Castelle CJ, Butterfield CN, Hernsdorf AW, Amano Y,
Ise K, et al: A new view of the tree of life. Nat Microbiol.
1:160482016. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wang J, Qi J, Zhao H, He S, Zhang Y, Wei S
and Zhao F: Metagenomic sequencing reveals microbiota and its
functional potential associated with periodontal disease. Sci Rep.
3:18432013.PubMed/NCBI
|
|
32
|
Kirst ME, Li EC, Alfant B, Chi YY, Walker
C, Magnusson I and Wang GP: Dysbiosis and alterations in predicted
functions of the subgingival microbiome in chronic periodontitis.
Appl Environ Microbiol. 81:783–793. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Arora N, Mishra A and Chugh S: Microbial
role in periodontitis: Have we reached the top? Some unsung
bacteria other than red complex. J Indian Soc Periodontol. 18:9–13.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Bizzarro S, Loos BG, Laine ML, Crielaard W
and Zaura E: Subgingival microbiome in smokers and non-smokers in
periodontitis: An exploratory study using traditional targeted
techniques and a next-generation sequencing. J Clin Periodontol.
40:483–492. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Colombo AP, Boches SK, Cotton SL, Goodson
JM, Kent R, Haffajee AD, Socransky SS, Hasturk H, Van Dyke TE,
Dewhirst F and Paster BJ: Comparisons of subgingival microbial
profiles of refractory periodontitis, severe periodontitis, and
periodontal health using the human oral microbe identification
microarray. J Periodontol. 80:1421–1432. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Gupta RS, Mahmood S and Adeolu M: Erratum:
A phylogenomic and molecular signature based approach for
characterization of the Phylum Spirochaetes and its major clades:
Proposal for a taxonomic revision of the phylum. Front Microbiol.
4:3222013. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Bergeron MG, Simard P and Provencher P:
Influence of growth medium and supplement on growth of Haemophilus
influenzae and on antibacterial activity of several antibiotics. J
Clin Microbiol. 25:650–655. 1987.PubMed/NCBI
|