Introduction
Osteoporosis is a bone condition characterized by
low bone mass, increased fragility, decreased bone quality, and an
increased fracture risk (1). It
often becomes symptomatic when a fracture occurs accidentally. The
incidence of fractures is known to increase with age, consequently
leading to a high mortality rate and an overall functional decline
in elder patients with osteoporosis. It has been reported that the
mortality rate during the first year following a hip fracture is 36
and 21% for men and women, respectively (2). As with aging, poor glycemic control
in diabetes is a well-known risk factor for bone fracture. For
example, patients with type 1 diabetes are reported to have a
6.9-fold higher risk for hip fracture, with decreased bone mineral
density (BMD) (1). There is also
increasing evidence supporting an association between type 2
diabetes and increased fracture risk (2–4). The
Rotterdam study revealed that individuals with type 2 diabetes had
a 69% increased fracture risk compared with those without diabetes,
despite having higher BMD (5). In
addition, impaired fracture healing is also demonstrated in
diabetes (2–4,6,7).
Bone fracture healing in diabetes was reported to be prolonged by
87% with a 3.4-fold higher risk of delayed union, non-union,
redislocation, or pseudoarthrosis (3,8,9),
which are commonly observed in type 1 and type 2 diabetes. These
bone problems, such as osteoporosis and delayed fracture healing,
are thus recognized as additional complications of longstanding
diabetes.
Several mechanisms, including insulin insufficiency,
hyperglycemia, and oxidative stress, are considered to delay
fracture healing in type 1 and type 2 diabetes via the reduction of
osteoblast differentiation, increased osteoclast activity, and
induction of apoptosis in chondrocytes and osteoblasts (7,10–12).
Of these, chronic and sustained hyperglycemia is known to enhance
the glycation reaction, and ultimately results in the formation of
advanced glycation end-products (AGEs). The accumulation of AGEs
has been associated with diabetic complications, as well as various
aging-associated diseases, including Alzheimer's disease and
osteoporosis. Notably, AGEs are reported to inhibit osteoblastic
differentiation and to induce apoptosis in osteoblasts, leading to
decreased osteoblast numbers and impaired bone formation (13,14).
Methylglyoxal (MGO) is a cell-permeant molecule, highly deleterious
and the most potent glycating agent for very rapid generation of
AGEs in cellular macromolecules (15,16).
In the glycation reaction, MGO is up to 20,000-fold more reactive
compared with glucose. The most important endogenous source of MGO
is the fragmentation of the triose phosphates, glyceraldehyde
3-phosphate and dihydroxyacetone phosphate, via the glycolytic
pathway (17,18). MGO may also be produced as a
by-product of protein and fatty acid metabolism (19–21).
MGO formation is known to be increased in patients with diabetes
(22). Therefore, MGO may be
implicated in affecting bone-healing processes in diabetes;
however, direct evidence has not been provided using an in
vivo bone-healing model.
In the present study, the effect of MGO on the
differentiation, growth and cytotoxicity of osteoblastic cells
in vitro was examined, and the accumulation of MGO-derived
AGEs in the sera and femurs was measured using an in vivo
bone defect mouse model with or without streptozotocin
(STZ)-induced diabetes.
Materials and methods
Cell culture
Mouse ST2 cells (Riken Cell Bank, Tsukuba, Japan)
were cultured in α-minimum essential medium (5.5 mmol/l glucose;
Wako Pure Chemical Industries, Osaka, Japan), supplemented with 10%
fetal bovine serum, 100 U/ml penicillin, and 100 µg/ml streptomycin
in 5% CO2 at 37°C. To induce osteoblastic
differentiation in the ST2 cells, 10 mM β-glycerophosphate, 0.1 µM
dexamethasone, and 50 µM ascorbic acid were added to the cell
culture medium. For the assays, 0, 50, 200, or 400 µM MGO
(Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) was added to the
differentiation medium, and the medium was changed every other
day.
Alkaline phosphatase (ALP) activity
assay
ALP activity in the ST2 cells was determined 7 days
after the induction of osteoblastic differentiation, using a TRACP
& ALP Assay kit (Takara Bio, Inc., Otsu, Japan), according to
the manufacturer's protocol.
Mineralization assay
Calcium deposition was evaluated 28 days after
differentiation of the ST2 cells. An In Vitro Osteogenesis
Assay kit (EMD Millipore, Billerica, MA, USA) was used for alizarin
red staining and quantitative analysis, following the
manufacturer's protocols.
Cell proliferation assay
ST2 cell proliferation was assayed by the cell-based
WST-1 method using a Cell Proliferation Assay kit
(Chemicon®; EMD Millipore), at 7 and 14 days after the
induction of differentiation, according to the manufacturer's
protocol.
Cytotoxicity assay
ST2 cells were seeded on 6-well plates at a density
of 105 cells/well. After 48 h, osteoblastic
differentiation was induced by the addition of 0, 50, 200, or 400
µM MGO. After 72 h, release of the enzyme, lactate dehydrogenase
(LDH), was assayed using a Non-Radioactive Cytotoxicity assay
(Promega Corp., Madison, WI, USA), according to the manufacturer's
protocol.
Experimental animals and induction of
diabetes
Male C57BL/6J mice (weight, 21 g) at 6 weeks of age
were purchased from Charles River Japan, Inc. (Yokohama, Japan),
and acclimatized for 1 week prior to the start of the experiment.
To induce diabetes, the mice (n=37) were intraperitoneally injected
with STZ (50 mg/kg body weight) daily for 5 days, according to the
Animal Models of Diabetic Complications Consortium (AMDCC)
protocols. By contrast, sodium citrate buffer was injected into
control mice (n=28). After 3 weeks, non-fasting blood glucose
levels were measured using a glucometer (Glutest Ace, Sanwa Kagaku,
Japan), using whole blood obtained from the tail vein. Mice with
blood glucose levels >300 mg/dl were considered to be diabetic.
Hemoglobin A1c (HbA1c) was measured in tail vein blood using a DCA
Vantage analyzer (Siemens Healthineers, Erlangen, Germany). Mice
were maintained under standard cage conditions (24°C; 12-h
light/dark cycle, lights on at 8:45 a.m.) with sawdust bedding, and
access to food and water ad libitum. All animal experiments
were approved by the Committee on Animal Experimentation of
Kanazawa University, and performed in accordance with the
Fundamental Guidelines for Proper Conduct of Animal Experiment and
Related Activities in Academic Research Institutions under the
jurisdiction of the Ministry of Education, Culture, Sports,
Science, and Technology of Japan.
Induction of drill-hole injury in the
femur
Under anesthesia with a mixture of medetomidine (0.3
mg/kg), midazolam (4.0 mg/kg) and butorphanol (5.0 mg/kg), a
straight, longitudinal skin incision (5 mm) was made in the distal
femur of the mice, and the periosteal membrane was subsequently
stripped away, following muscle splitting, to expose the bone
surface. A drill-hole injury was then introduced by inserting a
drill bit with a diameter of 0.9 mm fixed to a finger handle at the
medial portion of the diaphysis of the left femur, 5 mm above the
knee joint. The hole was drilled through the collateral cortical
bone and bone marrow, and thus a round defect measuring ~1.0 mm in
diameter was made. The hole was irrigated with saline solution to
prevent thermal necrosis of the margins. The incised skin was
subsequently sutured in a sterile manner, and anesthesia was
discontinued. During the surgery, the body temperature was
maintained at 37°C using a heating pad.
Computed tomography (CT) scanning
Mice were anesthetized using the aforementioned
procedure, and femurs were scanned using an X-ray CT system
(Latheta LCT-200; Hitachi Aloka Medical, Tokyo, Japan) at 3, 7, 10,
and 14 days after bone injury. The healing process in the bone
defect area was evaluated using suitable analysis software (AzeWin;
AZE, Ltd., Tokyo, Japan). Images were processed in a multiplanar
reconstruction, according to oblique coronal planes, maintaining
working axes parallel to the center line of the bone defect. CT
values at the area of bone defect were calculated at each
phase.
Measurement of MGO-derived AGEs
Collected serum was stored at −80°C prior to
analysis. Serum levels of the MGO-derived AGEs,
Nε-(carboxymethyl)-lysine (CML) and
Nδ-(5-hydro-5-methyl-4-imidazolone-2-yl)-ornithine
(MG-H1), were measured using liquid chromatography-tandem mass
spectrometry (LC-MS/MS), as previously described (23). Briefly, low-molecular-weight
fractions (<3,000 kDa) in the serum (50 µl) were separated using
a Vivaspin® centrifugal concentrator
(Vivaspin® 500; Sartorius Stedim Biotech, Goettingen,
Germany) according to the manufacturer's protocol, and then reduced
with sodium borohydride (NaBH4) (Sigma-Aldrich; Merck
KGaA). Standard [2H2]CML (0.01 nmol) and
[2H2]MG-H1 (0.01 nmol) (PolyPeptide
Laboratories, Strasbourg, France), and
[13C6]lysine (5 nmol) (Cambridge Isotope
Laboratories, Inc., Tewksbury, MA, USA), were added to the reduced
samples. The prepared samples were subsequently passed through a
Strata-X-C column (Phenomenex, Torrance, CA, USA) and assayed by
LC-MS/MS using a TSQ Vantage triple-stage quadrupole mass
spectrometer (Thermo Fisher Scientific, Waltham, MA, USA). AGE
structures, lysine, and the standards were detected using
electrospray positive ionization-mass spectrometric multiple
reaction monitoring.
The femur samples were cleaned of periosteal
membrane and bone marrow, and subsequently frozen and pulverized in
liquid nitrogen, as previously described (24). The levels of CML and MG-H1 in the
specimens were also quantified by LC-MS/MS serum sample measurement
(24). Briefly, bone powder was
demineralized with 0.5 M EDTA in 50 mM Tris buffer (pH 7.4) for 96
h at 4°C. Demineralized bone residues were reduced at room
temperature with NaBH4. Specimens were hydrolyzed in 6 M
HCl at 100°C for 18 h. The prepared samples were then assayed using
[2H3]hydroxyproline (C/D/N Isotopes Inc.,
Quebec, Canada).
Statistical analysis
Data are expressed as the mean ± standard error of
the mean. P-values were calculated using a two-tailed Student's
t-test for pairwise comparisons, and one-way analysis of variance
followed by Bonferroni's test for multiple comparisons
(Ekuseru-Toukei 2012 software; SSRI, Tokyo, Japan), unless
otherwise stated. P<0.05 was considered to indicate a
statistically significant difference.
Results
Bone defect repair in STZ-induced
diabetic mice
Compared with non-diabetic control mice, the
diabetic mice exhibited marked decreases in body weight (27±0.3 vs.
23±0.3 g) and significantly increased levels of non-fasting blood
glucose (140±3.8 vs. 493±14.1 mg/dl) and HbA1c (4.0±0.05% vs.
8.0±0.16%; control vs. diabetic mice, respectively) at 3 weeks
after the STZ injection (P<0.05; Table I). After confirming the presence of
overt diabetic conditions in the mice, a drill-hole injury (1.0 mm
in diameter) was introduced at the medial portion of the diaphysis
of the left femur (5 mm above the knee joint) to check the recovery
rate from bone injury. The healing process in the bone defect area
was subsequently evaluated using CT scanning at days 3, 7, 10 and
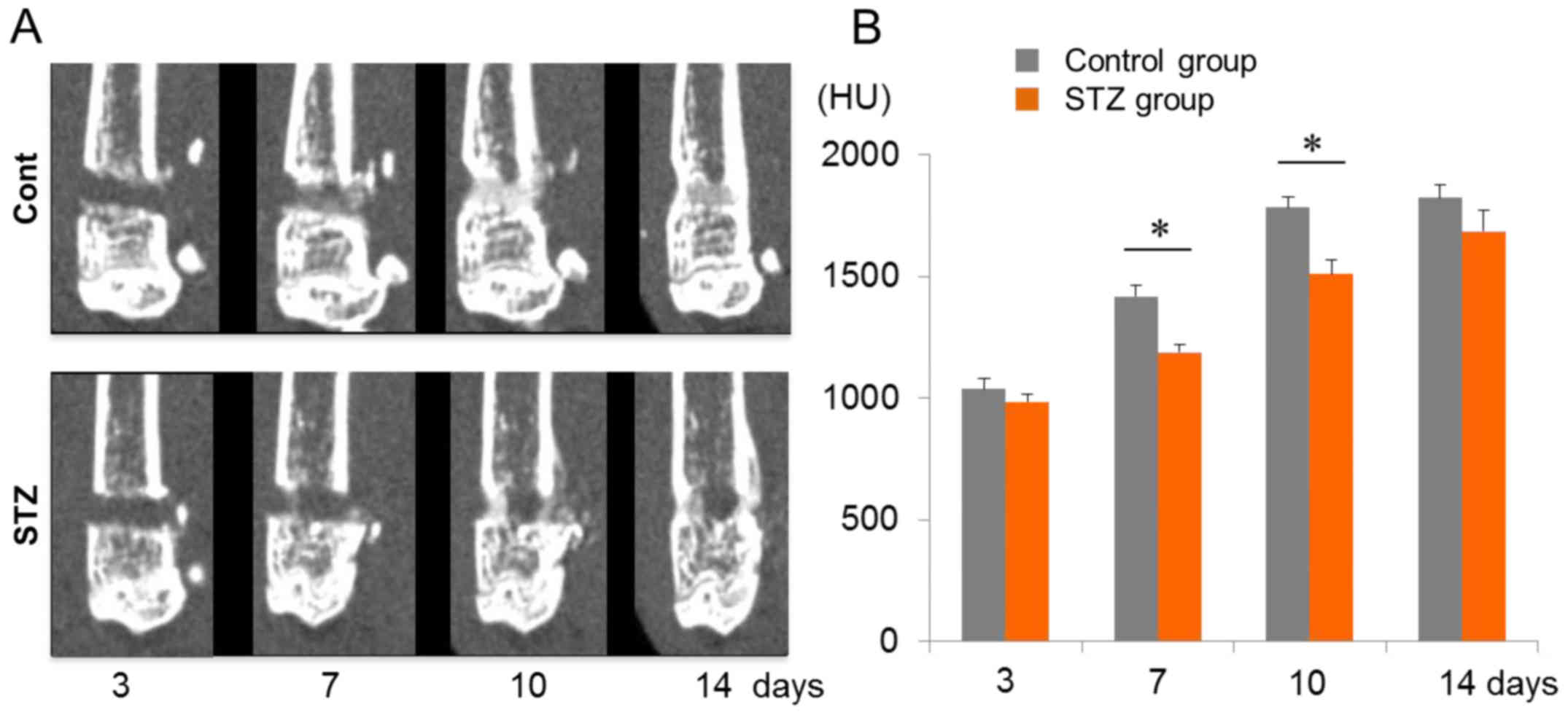
14 after injury (Fig. 1A). CT
imaging of the bone defect lesions revealed no significant
differences in ossification between the non-diabetic and the
diabetes-induced mice at the early healing stage on day 3 (Fig. 1A and B). However, repair of the
damaged site on the femur was significantly delayed at days 7 and
10 in diabetic mice, corresponding to intramembranous bone
formation, chondrogenesis and early endochondral bone phases. In
non-diabetic controls, recovery peaked at day 10, and reached a
plateau thereafter (Fig. 1A and
B).
 | Table I.Diabetic status of STZ-induced
mice. |
Table I.
Diabetic status of STZ-induced
mice.
| Measurement | Control (n=28) | STZ-induced
(n=37) |
|---|
| Body weight
(g) | 27±0.3 | 23±0.3a |
| Blood glucose
(mg/dl) | 140±3.8 |
493±14.1a |
| HbA1c (NGSP,
%) | 4.0±0.05 |
8.0±0.16a |
Diabetes altered bone quality through
increased accumulations of CML and MG-H1
Subsequently, the accumulation of MGO-derived AGEs,
CML, and MG-H1 in sera and femurs under diabetic conditions was
investigated. In the sera from diabetic mice, the levels of CML and
MG-H1, as quantified by LC-MS/MS, were significantly increased
compared with those in non-diabetic controls (Fig. 2A). Similarly, higher levels of
femur CML and MG-H1 were observed in diabetic mice compared with
control mice (Fig. 2B). These
findings suggested that MGO generated under diabetic conditions is
able to affect bone defect healing in our animal model.
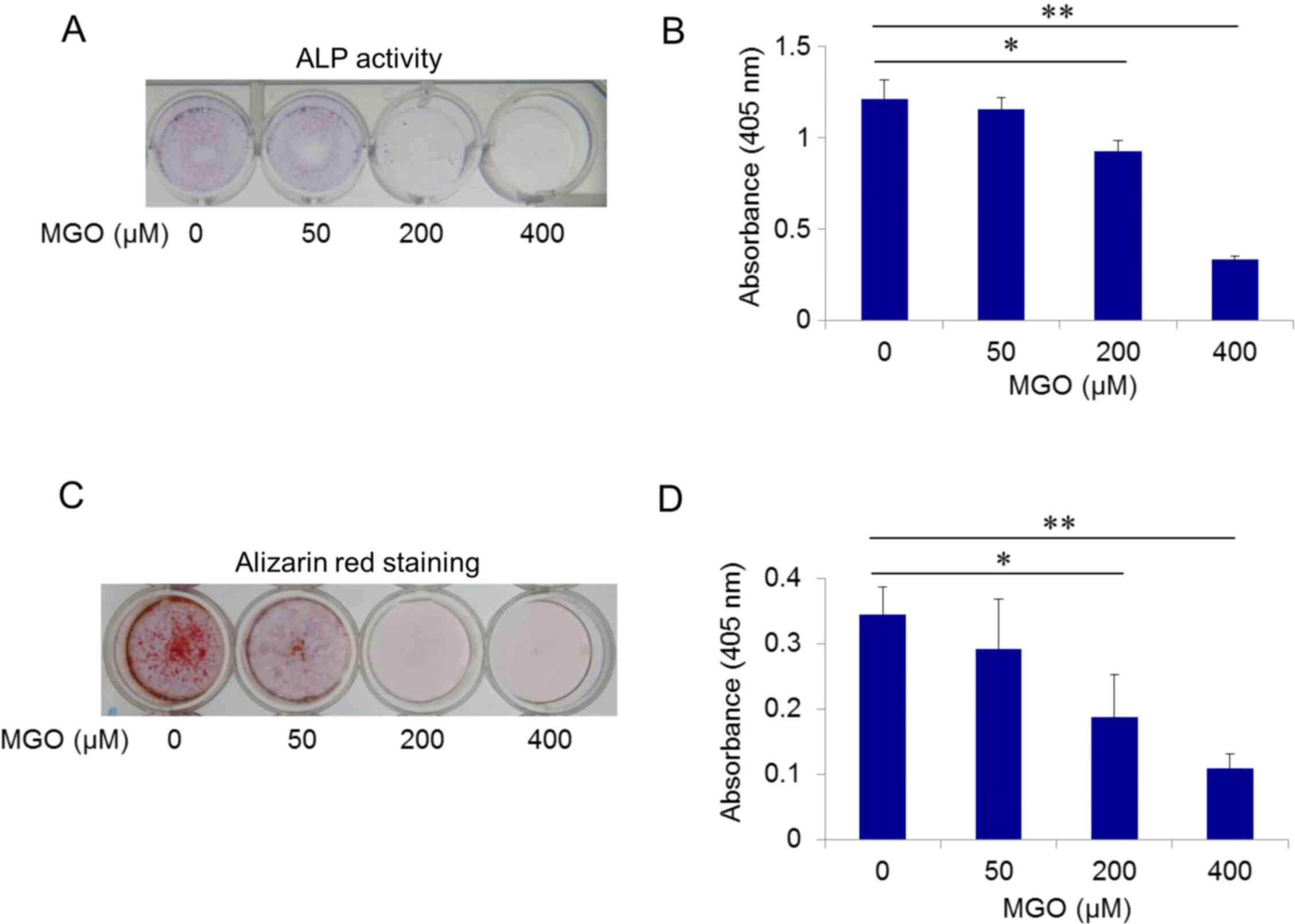
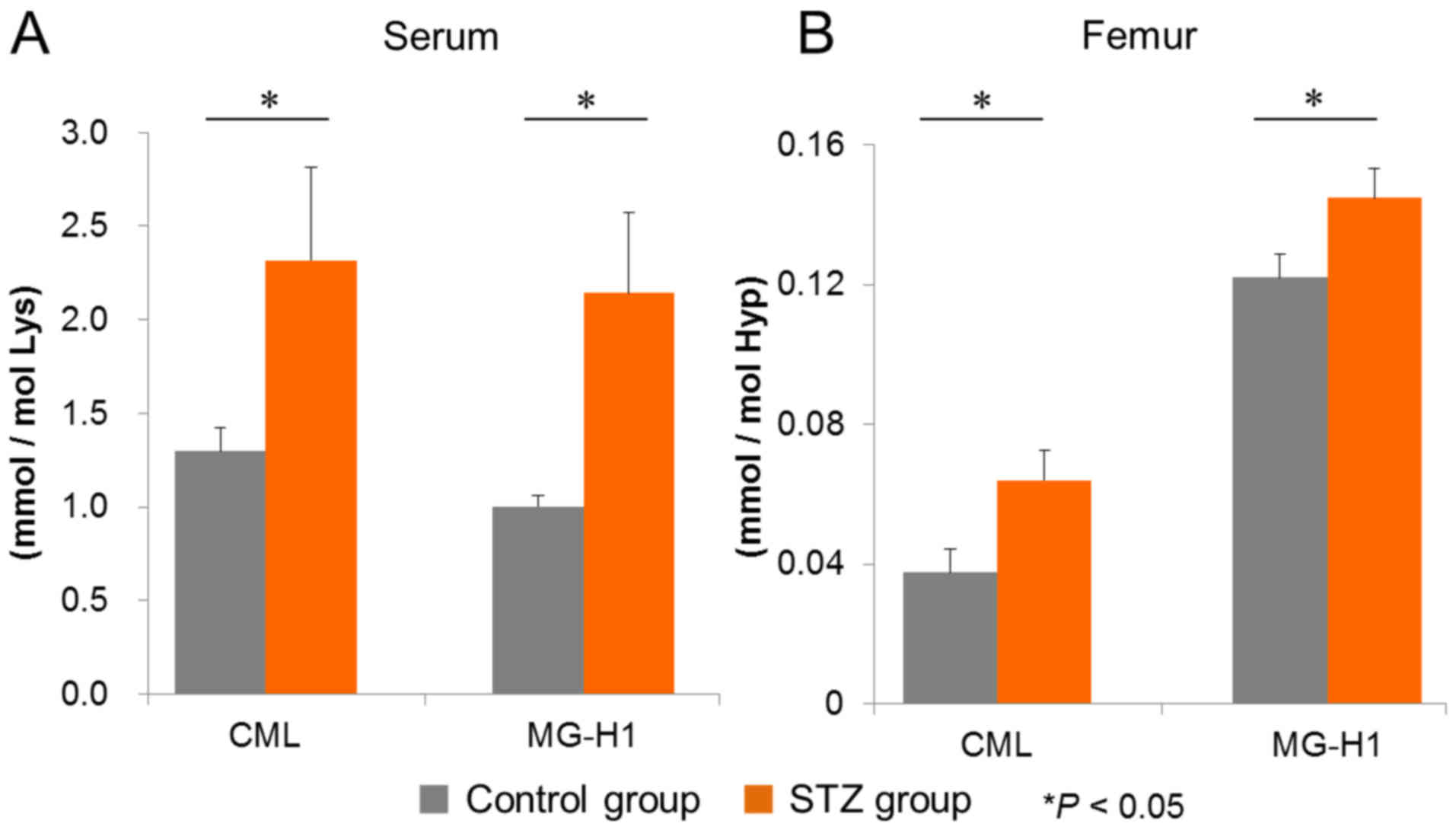 | Figure 2.Quantitative evaluation of AGEs in
sera and femurs by LC-MS/MS. (A) Serum and (B) femur levels of the
MGO-derived AGEs, CML and MG-H1, were measured by LC-MS/MS, as
described in the Materials and methods section. The control and the
STZ diabetes-induced groups both contained n=6 mice. Values are
presented as the mean ± standard error of the mean. *P<0.05 vs.
the control. MGO, methylglyoxal; AGEs, advanced glycation
end-products; LC-MS/MS, liquid chromatography-tandem mass
spectrometry; CML, Nε-(carboxymethyl)-lysine; MG-H1,
Nδ-(5-hydro-5-methyl-4-imidazolone-2-yl)-ornithine; STZ,
streptozotocin; Lys, lysine; Hyp, hydroxyproline. |
Effect of MGO on in vitro osteoblastic
differentiation of ST2 cells
Impairment of osteoblastic differentiation is an
important factor in the pathogenesis of detrimental bone repair. To
address the effect of MGO on osteoblastic differentiation, the
mouse stromal ST2 cell line was used, which is an established cell
culture model characterized by its potency to differentiate into
cells with the typical characteristics of osteoblasts, and the
formation of mineralized nodules (25,26).
Using ALP activity as an osteoblastic differentiation marker, ST2
cells were cultured in induction media with MGO at a final
concentration of 0, 50, 200, or 400 µM. MGO exposure for 7 days
decreased ALP activity in a dose-dependent manner, and
significantly inhibited osteoblastic differentiation of ST2 cells
at 200 and 400 µM MGO (P<0.05 and P<0.01, respectively;
Fig. 3A and B). Matrix
mineralization, the final step of osteoblast differentiation,
serves a critical role in maintaining the mechanical integrity of
bone tissues.
Subsequently, the effects of MGO on mineralization
were examined using alizarin red staining after 28 days of
differentiation. In the presence of MGO, dose-dependent decreases
in calcium deposition in ST2 cells were observed, which were
statistically significant at 200 and 400 µM MGO (P<0.05 and
P<0.01, respectively; Fig. 3C and
D). These results indicated that exposure to MGO led to
disrupted osteoblastic differentiation.
Effect of MGO on cell viability and
cytotoxicity in ST2 cells
Subsequently, it was examined whether MGO may affect
cell viability and proliferation, and induce cytotoxicity in ST2
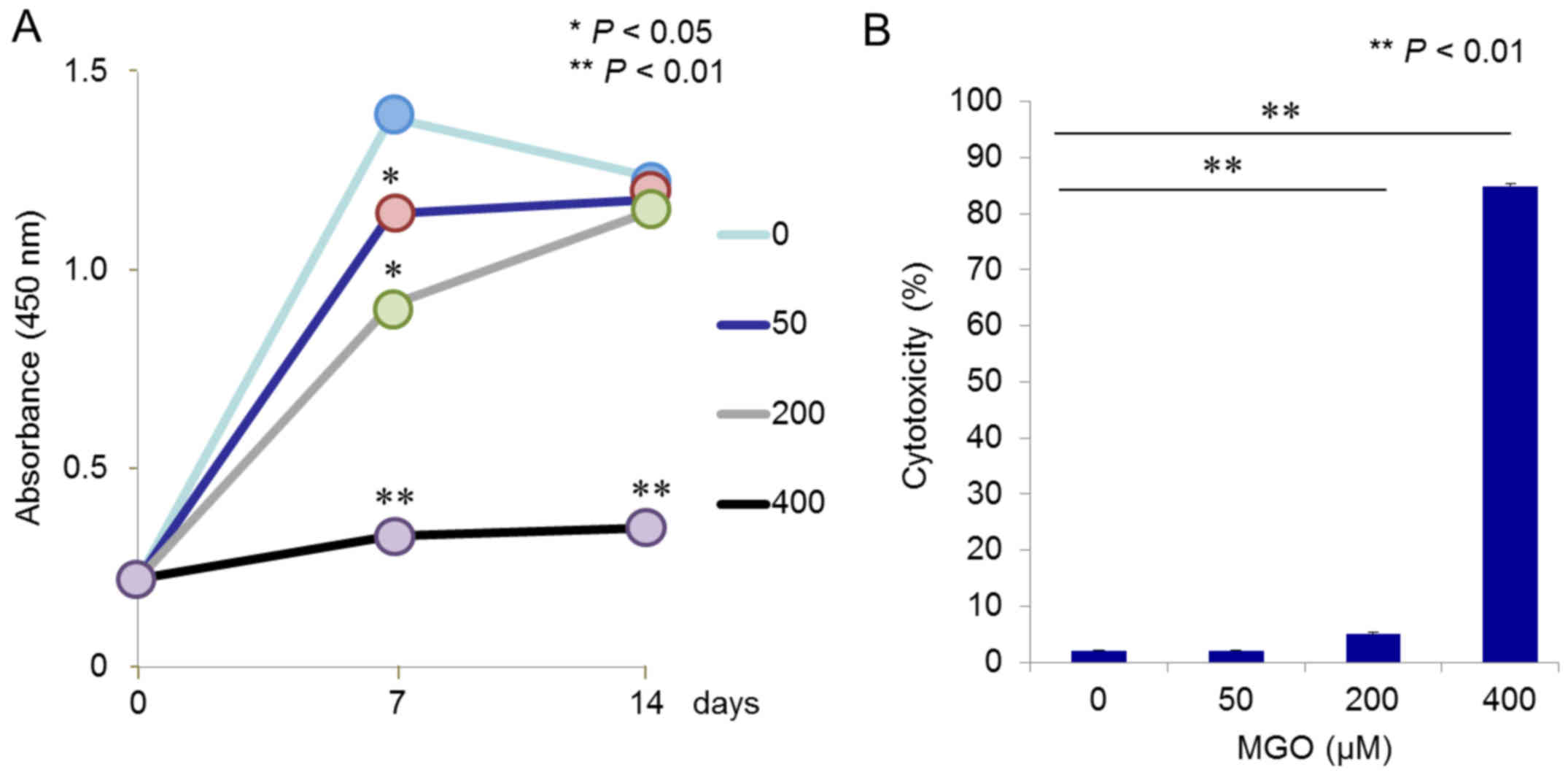
cells. Cell proliferation assays clearly demonstrated that 400 µM
MGO severely inhibited ST2 cell growth during the observation
period, and lower concentrations of MGO (50 and 200 µM) could
significantly, although to a much lesser extent, inhibit
proliferation at day 7 of cultivation (Fig. 4A). However, no significant
difference was observed among cells exposed to 0, 50, and 200 µM
MGO at day 14, which had already achieved confluency (Fig. 4A). Finally, ST2 cell damage was
examined using an LDH assay, which monitored release of the
cytoplasmic enzyme into the culture media. As expected, a
significant (P<0.01) and dramatic increase in LDH leakage was
seen with 400 µM MGO (Fig. 4B),
whereas 50 µM MGO elicited no effect, and 200 µM slightly induced
cytotoxicity in cultured ST2 cells (Fig. 4B).
Discussion
In the present study, it has been demonstrated that
MGO may exert a role in inhibiting osteoblastic differentiation,
mineralization, and proliferation of mouse stromal ST2 cells
(Figs. 3 and 4). MGO was also found to increase cell
damage at higher concentrations (≥ 200 µM) (Fig. 4B). Concentrations of MGO in the
range 50–400 µM were used in these experiments, as described
previously (27). The
concentration of MGO that occurs in vivo is still under
debate. It has been reported that plasma MGO levels are estimated
to be ~0.5 µM in healthy subjects, and to increase to several µM in
patients with diabetes (22,28).
However, others reported that plasma MGO levels were in the range
of ~400 µM in patients with poorly controlled diabetes (29). Additionally, it has been
demonstrated that cells are able to produce large amounts of MGO
(30), and therefore intracellular
levels are probably much higher compared with plasma levels
(31). Our hypothesis was
therefore that the MGO concentrations used in the present study
were neither in a non- nor a ‘super’-physiological range. Chan
et al (32) revealed that
MGO treatment (~20 µM) triggered apoptotic biochemical changes in
human osteoblasts. In addition, Suh et al (27) demonstrated that MGO toxicity (~400
µM) in MC3T3-E1 cells, an osteoblast-like cell line, was due to
oxidative stress-mediated mitochondrial dysfunction (27). This oxidative stress was also able
to impair MGO detoxification processes, with a subsequent increase
in endogenous MGO, which, in turn, was able to potentiate
MGO-mediated protein glycation and AGE formation, with the further
overproduction of reactive oxygen species (27). The findings of the present study,
together with those of previous studies, suggest that MGO that
accumulates under diabetic conditions may act on cells of the
osteoblast lineage, thereby damaging their viability and functional
differentiation.
In the present study, it was also demonstrated that
bone repair was delayed in STZ-induced diabetic mice compared with
non-diabetic control mice, using CT image analysis (Fig. 1). It is commonly known that
fracture repair involves hematoma formation following injury, and
the subsequent production of cytokines and growth factors, leading
to inflammatory responses and the recruitment of mesenchymal stem
cells (33,34). These cells proliferate and
differentiate into chondrocytes, which subsequently form new
cartilage in the endochondral phase. The cells along the periosteum
differentiate into osteoblasts to produce new bone. The cartilage
mineralizes and mechanically stabilizes the fracture site, and the
mineralized cartilage is subsequently removed by osteoclasts. The
transition phase of bone formation from cartilage is involved in
angiogenesis (33). Remodeling is
the final step, in which osteoclasts and osteoblasts reshape the
bone lesion. Previous studies have demonstrated that diabetes may
impair fracture healing in almost all the above-described phases of
fracture repair (2–4,6,7). In
patients with diabetes, impaired recruitment of bone marrow-derived
mesenchymal stem cells to the site of injury has been reported
(35). Decreased and delayed bone
formation was observed with the reduction of growth factor levels
in diabetic animals (36,37). Dysregulation of the transition from
cartilage to bone has also been observed due to chondrocyte
apoptosis, premature removal of cartilage, reduced osteoblast
differentiation, and insufficient vascularization (7,10–12,38,39).
Supernormal osteoclast activity was reported to disturb remodeling
of the osseous callus (7). The
drill-hole injury and recovery data featured in the present study
were compatible with those of previous reports, clearly indicating
that diabetes may delay and damage bone healing.
In addition, representative MGO-derived AGE levels
of CML and MG-H1 in the sera and femurs in STZ-induced diabetic
mice, as well as in non-diabetic control mice, were measured. AGE
levels were significantly increased in the sera and femurs of
diabetic mice compared with those of the non-diabetic controls
(Fig. 2). Notably, higher levels
of MG-H1 accumulated in the femur compared with those of CML,
whereas similar levels of the two AGEs were observed in sera
(Fig. 2). These data suggest that
the production and accumulation of MGO may be upregulated, and
subsequently the concentrations of MGO-derived AGEs may build up,
thereby affecting biological functions in diabetes. Okazaki et
al (40) reported that AGEs,
and not high glucose exposure, were able to inhibit osteoblastic
differentiation and mineralization of ST2 cells by decreasing the
expression of osterix, and partly by increasing the expression of
receptor for AGEs (RAGE). AGEs were also shown to inhibit cell
growth and increase apoptosis in ST2 cells (40). CML and MG-H1 are well-known ligands
for RAGE, and their interactions may lead to diabetic complications
(41). Therefore, MGO in itself,
and its derivatives, such as AGE, all account for detrimental bone
healing in diabetes.
Consequently, anti-glycating and quenching agents
against reactive MGO may prove to be useful for the prevention and
treatment of bone problems, including delayed fracture healing, in
diabetes. The natural vitamin B6 analog, pyridoxamine, is a drug
that may potentially be effective in diabetic nephropathy and
retinopathy (42–51). Metformin is also a putative
candidate (52–54). Further studies are required to
investigate the effectiveness and application of anti-glycating
remedies to bone problems in diabetes.
In conclusion, the present study has revealed that
MGO was able to inhibit the differentiation and proliferation of
osteoblasts, possibly resulting in delayed bone healing. Therefore,
the detoxification of MGO, or of MGO scavengers, may improve bone
repair in patients with diabetes.
Acknowledgements
We would like to thank Ms. Yoko Kasai for her
assistance, and acknowledge financial support from Grants-in-Aid
for Scientific Research (nos. 16K19031, 24590375, 25461335,
26450152 and 26861176) from the Japan Society for the Promotion of
Science.
References
|
1
|
Vestergaard P: Discrepancies in bone
mineral density and fracture risk in patients with type 1 and type
2 diabetes-a meta- analysis. Osteoporosis Int. 18:427–444. 2007.
View Article : Google Scholar
|
|
2
|
Kayal RA, Alblowi J, McKenzie E,
Krothapalli N, Silkman L, Gerstenfeld L, Einhorn TA and Graves DT:
Diabetes causes the accelerated loss of cartilage during fracture
repair which is reversed by insulin treatment. Bone. 44:357–363.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Retzepi M and Donos N: The effect of
diabetes mellitus on osseous healing. Clin Oral Implants Res.
21:673–681. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Simpson CM, Calori GM and Giannoudis PV:
Diabetes and fracture healing: The skeletal effects of diabetic
drugs. Expert Opin Drug Saf. 11:215–220. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
de Liefde II, van der Klift M, de Laet CE,
van Daele PL, Hofman A and Pols HA: Bone mineral density and
fracture risk in type-2 diabetes mellitus: The Rotterdam study.
Osteoporosis Int. 16:1713–1720. 2005. View Article : Google Scholar
|
|
6
|
Kayal RA, Tsatsas D, Bauer MA, Allen B,
Al-Sebaei MO, Kakar S, Leone CW, Morgan EF, Gerstenfeld LC, Einhorn
TA and Graves DT: Diminished bone formation during diabetic
fracture healing is related to the premature resorption of
cartilage associated with increased osteoclast activity. J Bone
Miner Res. 22:560–568. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kayal RA, Siqueira M, Alblowi J, McLean J,
Krothapalli N, Faibish D, Einhorn TA, Gerstenfeld LC and Graves DT:
TNF-alpha mediates diabetes-enhanced chondrocyte apoptosis during
fracture healing and stimulates chondrocyte apoptosis through
FOXO1. J Bone Miner Res. 25:1604–1615. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Loder RT: The influence of diabetes
mellitus on the healing of closed fractures. Clin Orthop Relat Res.
210–216. 1988.PubMed/NCBI
|
|
9
|
Folk JW, Starr AJ and Early JS: Early
wound complications of operative treatment of calcaneus fractures:
Analysis of 190 fractures. J Orthop Trauma. 13:369–372. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Botushanov NP and Orbetzova MM: Bone
mineral density and fracture risk in patients with type 1 and type
2 diabetes mellitus. Folia Med (Plovdiv). 51:12–17. 2009.PubMed/NCBI
|
|
11
|
Stolzing A, Sellers D, Llewelyn O and
Scutt A: Diabetes induced changes in rat mesenchymal stem cells.
Cells Tissues Organs. 191:453–465. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Sheweita SA and Khoshhal KI: Calcium
metabolism and oxidative stress in bone fractures: Role of
antioxidants. Curr Drug Metab. 8:519–525. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Sanguineti R, Storace D, Monacelli F,
Federici A and Odetti P: Pentosidine effects on human osteoblasts
in vitro. Ann N Y Acad Sci. 1126:166–172. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Alikhani M, Alikhani Z, Boyd C, MacLellan
CM, Raptis M, Liu R, Pischon N, Trackman PC, Gerstenfeld L and
Graves DT: Advanced glycation end products stimulate osteoblast
apoptosis via the MAP kinase and cytosolic apoptotic pathways.
Bone. 40:345–353. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Brownlee M: Biochemistry and molecular
cell biology of diabetic complications. Nature. 414:813–820. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Westwood ME and Thornalley PJ: Molecular
characteristics of methylglyoxal-modified bovine and human serum
albumins. Comparison with glucose-derived advanced glycation end
product-modified serum albumins. J Protein Chem. 14:359–372. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Phillips SA and Thornalley PJ: The
formation of methylglyoxal from triose phosphates. Investigation
using a specific assay for methylglyoxal. Eur J Biochem.
212:101–105. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Silva Sousa M, Gomes RA, Ferreira AE,
Ponces Freire A and Cordeiro C: The glyoxalase pathway: The first
hundred years… and beyond. Biochem J. 453:1–15. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Thornalley PJ: Pharmacology of
methylglyoxal: Formation, modification of proteins and nucleic
acids, and enzymatic detoxification-a role in pathogenesis and
antiproliferative chemotherapy. Gen Pharmacol. 27:565–573. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kalapos MP: Methylglyoxal in living
organisms: Chemistry, biochemistry, toxicology and biological
implications. Toxicol Lett. 110:145–175. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Vander Jagt DL and Hunsaker LA:
Methylglyoxal metabolism and diabetic complications: Roles of
aldose reductase, glyoxalase-I, betaine aldehyde dehydrogenase and
2-oxoaldehyde dehydrogenase. Chem Biol Interact. 143–144:341–351.
2003. View Article : Google Scholar
|
|
22
|
Scheijen JL and Schalkwijk CG:
Quantification of glyoxal, methylglyoxal and 3-deoxyglucosone in
blood and plasma by ultra performance liquid chromatography tandem
mass spectrometry: Evaluation of blood specimen. Clin Chem Lab Med.
52:85–91. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Yamanaka M, Matsumura T, Ohno R, Fujiwara
Y, Shinagawa M, Sugawa H, Hatano K, Shirakawa J, Kinoshita H, Ito
K, et al: Non-invasive measurement of skin autofluorescence to
evaluate diabetic complications. J Clin Biochem Nutr. 58:135–140.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Shirakawa J, Arakawa S, Tagawa T, Gotoh K,
Oikawa N, Ohno R, Shinagawa M, Hatano K, Sugawa H, Ichimaru K, et
al: Salacia chinensis L. extract ameliorates abnormal glucose
metabolism and improves the bone strength and accumulation of AGEs
in type 1 diabetic rats. Food Funct. 7:2508–2515. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Otsuka E, Yamaguchi A, Hirose S and
Hagiwara H: Characterization of osteoblastic differentiation of
stromal cell line ST2 that is induced by ascorbic acid. Am J
Physiol. 277:C132–C138. 1999.PubMed/NCBI
|
|
26
|
Yamaguchi A, Ishizuya T, Kintou N, Wada Y,
Katagiri T, Wozney JM, Rosen V and Yoshiki S: Effects of BMP-2,
BMP-4 and BMP-6 on osteoblastic differentiation of bone
marrow-derived stromal cell lines, ST2 and MC3T3-G2/PA6. Biochem
Biophys Res Commun. 220:366–371. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Suh KS, Choi EM, Rhee SY and Kim YS:
Methylglyoxal induces oxidative stress and mitochondrial
dysfunction in osteoblastic MC3T3-E1 cells. Free Radic Res.
48:206–217. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
McLellan AC, Thornalley PJ, Benn J and
Sonksen PH: Glyoxalase system in clinical diabetes mellitus and
correlation with diabetic complications. Clin Sci (Lond). 87:21–29.
1994. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Lapolla A, Reitano R, Seraglia R, Sartore
G, Ragazzi E and Traldi P: Evaluation of advanced glycation end
products and carbonyl compounds in patients with different
conditions of oxidative stress. Mol Nutr Food Res. 49:685–690.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Chaplen FW, Fahl WE and Cameron DC:
Evidence of high levels of methylglyoxal in cultured Chinese
hamster ovary cells. Proc Natl Acad Sci USA. 95:5533–5538. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Webb-Robertson BJ, Lowry DF, Jarman KH,
Harbo SJ, Meng QR, Fuciarelli AF, Pounds JG and Lee KM: A study of
spectral integration and normalization in NMR-based metabonomic
analyses. J Pharm Biomed Anal. 39:830–836. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Chan WH, Wu HJ and Shiao NH: Apoptotic
signaling in methylglyoxal-treated human osteoblasts involves
oxidative stress, c-Jun N-terminal kinase, caspase-3, and
p21-activated kinase 2. J Cell Biochem. 100:1056–1069. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Ai-Aql ZS, Alagl AS, Graves DT,
Gerstenfeld LC and Einhorn TA: Molecular mechanisms controlling
bone formation during fracture healing and distraction
osteogenesis. J Dent Res. 87:107–118. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Gerstenfeld LC, Wronski TJ, Hollinger JO
and Einhorn TA: Application of histomorphometric methods to the
study of bone repair. J Bone Miner Res. 20:1715–1722. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Shin L and Peterson DA: Impaired
therapeutic capacity of autologous stem cells in a model of type 2
diabetes. Stem Cells Transl Med. 1:125–135. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Follak N, Kloting I and Merk H: Influence
of diabetic metabolic state on fracture healing in spontaneously
diabetic rats. Diabetes Metab Res Rev. 21:288–296. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Kawaguchi H, Kurokawa T, Hanada K, Hiyama
Y, Tamura M, Ogata E and Matsumoto T: Stimulation of fracture
repair by recombination human basic fibroblast growth factor in
normal and streptozotocin-diabetic rats. Endocrinology.
135:774–781. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Bahney CS, Hu DP, Miclau T III and
Marcucio RS: The multifaceted role of the vasculature in
endochondral fracture repair. Front Endocrinol (Lausanne).
6:42015.PubMed/NCBI
|
|
39
|
Liao YF, Chen LL, Zeng TS, Li YM, Fan Y,
Hu LJ and Ling Yue: Number of circulating endothelial progenitor
cells as a marker of vascular endothelial function for type 2
diabetes. Vasc Med. 15:279–285. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Okazaki K, Yamaguchi T, Tanaka K, Notsu M,
Ogawa N, Yano S and Sugimoto T: Advanced glycation end products
(AGEs), but not high glucose, inhibit the osteoblastic
differentiation of mouse stromal ST2 cells through the suppression
of osterix expression, and inhibit cell growth and increasing cell
apoptosis. Calcif Tissu Int. 91:286–296. 2012. View Article : Google Scholar
|
|
41
|
Xue J, Ray R, Singer D, Böhme D, Burz DS,
Rai V, Hoffmann R and Shekhtman A: The receptor for advanced
glycation end products (RAGE) specifically recognizes
methylglyoxal-derived AGEs. Biochemistry. 53:3327–3335. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Voziyan PA and Hudson BG: Pyridoxamine as
a multifunctional pharmaceutical: Targeting pathogenic glycation
and oxidative damage. Cell Mol Life Sci. 62:1671–1681. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Degenhardt TP, Alderson NL, Arrington DD,
Beattie RJ, Basgen JM, Steffes MW, Thorpe SR and Baynes JW:
Pyridoxamine inhibits early renal disease and dyslipidemia in the
streptozotocin-diabetic rat. Kidney Int. 61:939–950. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Zhu P, Lin H, Sun C, Lin F, Yu H, Zhuo X,
Zhou C and Deng Z: Synergistic effects of telmisartan and
pyridoxamine on early renal damage in spontaneously hypertensive
rats. Mol Med Rep. 5:655–662. 2012.PubMed/NCBI
|
|
45
|
Waanders F, van den Berg E, Nagai R, van
Veen I, Navis G and van Goor H: Renoprotective effects of the
AGE-inhibitor pyridoxamine in experimental chronic allograft
nephropathy in rats. Nephrol Dial Transplant. 23:518–524. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Alderson NL, Chachich ME, Youssef NN,
Beattie RJ, Nachtigal M, Thorpe SR and Baynes JW: The AGE inhibitor
pyridoxamine inhibits lipemia and development of renal and vascular
disease in Zucker obese rats. Kidney Int. 63:2123–2133. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Tanimoto M, Gohda T, Kaneko S, Hagiwara S,
Murakoshi M, Aoki T, Yamada K, Ito T, Matsumoto M, Horikoshi S and
Tomino Y: Effect of pyridoxamine (K-163), an inhibitor of advanced
glycation end products, on type 2 diabetic nephropathy in
KK-A(y)/Ta mice. Metabolism. 56:160–167. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Williams ME, Bolton WK, Khalifah RG,
Degenhardt TP, Schotzinger RJ and McGill JB: Effects of
pyridoxamine in combined phase 2 studies of patients with type 1
and type 2 diabetes and overt nephropathy. Am J Nephrol.
27:605–614. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Lewis EJ, Greene T, Spitalewiz S,
Blumenthal S, Berl T, Hunsicker LG, Pohl MA, Rohde RD, Raz I,
Yerushalmy Y, et al: Pyridorin in type 2 diabetic nephropathy. J Am
Soc Nephrol. 23:131–136. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Garg S, Syngle A and Vohra K: Efficacy and
tolerability of advanced glycation end-products inhibitor in
osteoarthritis: A randomized, double-blind, placebo-controlled
study. Clin J Pain. 29:717–724. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Nagaraj RH, Sarkar P, Mally A, Biemel KM,
Lederer MO and Padayatti PS: Effect of pyridoxamine on chemical
modification of proteins by carbonyls in diabetic rats:
Characterization of a major product from the reaction of
pyridoxamine and methylglyoxal. Arch Biochem Biophys. 402:110–119.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Ruggiero-Lopez D, Lecomte M, Moinet G,
Patereau G, Lagarde M and Wiernsperger N: Reaction of metformin
with dicarbonyl compounds. Possible implication in the inhibition
of advanced glycation end product formation. Biochem Pharmacol.
58:1765–1773. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Effect of intensive blood-glucose control
with metformin on complications in overweight patients with type 2
diabetes (UKPDS 34). UK Prospective Diabetes Study (UKPDS) Group.
Lancet. 352:854–865. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Kender Z, Fleming T, Kopf S, Torzsa P,
Grolmusz V, Herzig S, Schleicher E, Rácz K, Reismann P and Nawroth
PP: Effect of metformin on methylglyoxal metabolism in patients
with type 2 diabetes. Exp Clin Endocrinol Diabetes. 122:316–319.
2014. View Article : Google Scholar : PubMed/NCBI
|