Introduction
Epidemiological studies have demonstrated that
particulate matter (PM) is a serious environmental contaminant and
is responsible for multiple human diseases, including
cardiopulmonary diseases and cancers (1–5).
Toxicological studies have revealed that PM with a diameter of ≤2.5
µm (PM2.5) has a higher toxicity than larger particles (5,6).
Long-term contact with high doses of PM2.5 enhances cardiovascular
disease mortality rates (7).
However, the underlying mechanisms of PM2.5 have yet to be
elucidated.
Endothelial cell (EC) dysfunction is necessary for a
number of diseases, including cardiovascular diseases (8,9).
There have not been many studies on the effects of PM2.5 on ECs,
however, previous studies have demonstrated that PM2.5 induces
oxidative stress in human umbilical vein endothelial cells (HUVECs)
(10) and endothelial progenitor
cells (11). However, the effects
of PM2.5 on ECs and its mechanisms remain to be elucidated.
PM2.5 can be quickly released via the respiratory
tract and then affect the organs and blood vessels. PM2.5 also can
be phagocytosed by macrophages, releasing a number of
pro-inflammatory cytokines, such as tumor necrosis factor (TNF)-α
(12). PM2.5 exposure is capable
of inducing inflammation, which is regarded as the major mechanism
of PM2.5-mediated toxicity (13–15).
It has also been demonstrated that oxidative stress
is a key target of PM2.5-mediated cytotoxicity, however, these
studies mainly focused on human lung epithelial cells (16,17).
The present study focused on ECs exposed to high doses of PM2.5 and
evaluated the effect on cell viability, migration and tube
formation. The current understanding of whether PM2.5 exposure
accelerates cell damage via ROS-mediated inflammation is limited
and warrants more investigation. The present study, therefore aimed
to determine the potential mechanisms underlying PM2.5-induced
vascular toxicity.
To the best of our knowledge, the data in the
present study has demonstrated for the first time that PM2.5
effectively suppressed migration, tube formation and adhesion to
extracellular matrix (ECM) proteins in ECs. The present study also
demonstrated the mechanisms by which PM2.5 induced vascular
toxicity in ECs, at least in part, via a ROS-dependent signaling
pathway. These findings may provide a strategy for the prevention
and treatment of PM2.5-induced vascular inflammation in the
clinical setting.
Materials and methods
Materials and reagents
PM2.5 was purchased from the National Institute of
Standards and Technology (NIST; Gaithersburg, MD, USA),
2′,7′-dichlorofluorescin diacetate (DCFH-DA) and fetal bovine serum
(FBS) were purchased from Sigma-Aldrich (Merck KGaA, Darmstadt,
Germany), MTT reagents were obtained from Dojindo Molecular
Technologies, Inc. (Shanghai, China) and the Annexin V-fluorescein
isothiocyanate (FITC)/propidium iodide (PI) kit was purchased from
Nanjing KeyGen Biotech Co., Ltd. (Nanjing, China).
Cell culture
HUVECs and HMEC-1 human microvascular endothelial
cells were obtained from AllCells, LLC (H-001F; Shanghai, China).
Cells were cultured in RPMI 1640 medium with 20% FBS, 60 µg/ml of
endothelial cell growth supplement (BD Biosciences, San Jose, CA,
USA) and 100 U/ml of penicillin with 100 µg/ml of streptomycin
(Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA) in a
humidified atmosphere with 5% CO2 at 37°C.
PM2.5 was resuspended in PBS and the solutions were
stored at −20°C until use. ECs were treated with various
concentrations of PM2.5 to select the optimal concentration and
time.
Detection of cell viability
Cell viability was determined by MTT assay. The ECs
were cultured in 96-well plates (1.0×104 cells/well) in
100 µl medium for 24 h. ECs were then exposed to 0–800 µg/ml PM2.5
for 0–24 h. Following exposure, 20 µl of MTT was added to the wells
for 1 h at 37°C. Cells were then treated with 100 µl of dimethyl
sulfoxide. The absorbance was measured at 570 nm using a microplate
reader. The viability of the treated cells was calculated as a
percentage of the untreated control group, which was regarded as
100%.
Detection of cell apoptosis
ECs were cultured in 6-well plates (2×105
cells/well) for 24 h. ECs were then exposed to 0–800 µg/ml PM2.5
for 24 h. The ECs were harvested and resuspended in PBS. EC
apoptosis was evaluated by flow cytometry analysis using an Annexin
V-FITC/PI kit according to the manufacturer's protocols. The number
of apoptotic cells was calculated by FlowJo (version 9.6.2; FlowJo
LLC, Ashland, OR, USA).
Detection of cell migration
EC migration assays were performed using a modified
Boyden chamber (8 µm pore size; BD Biosciences, Oxford, UK). ECs
(5×104 cells/well) were treated with 0–800 µg/ml PM2.5 in RPMI 1640
medium for 6 h. RPMI 1640 medium supplemented with 500 µl 10 ng/ml
vascular endothelial growth factor (VEGF; 293-VE-050; R&D
Systems, Inc., Minneapolis, MN, USA) was added into the bottom
chambers and the PM2.5-treated ECs were added to the upper chambers
for 8 h. The migrating cells were stained with 1% calcein-AM and
quantified by counting under a fluorescence microscope. Experiments
were repeated at least three times, and the migrated cells were
counted in five random fields of each filter.
Detection of tube formation
The 96-well plates were pre-coated with Matrigel for
2 h. ECs were exposed to 0–800 µg/ml PM2.5 in RPMI 1640 medium for
6 h. Then 2×104 ECs/well were incubated in RPMI 1640
medium with 10 ng/ml VEGF for 24 h at 37°C. Quantification of tube
formation was performed by using IncuCyte angiogenesis version 2.0
image analysis (Essen Bioscience, Ann Arbor, MI, USA).
Detection of ECM cell adhesion
Cell adhesion of ECs was evaluated by an
Extracellular Matrix Cell Adhesion Array kit (ECM545; Chemicon; EMD
Millipore, Billerica, MA, USA). The assay was carried out according
to the manufacturer's instructions. The wells were pre-coated with
different human ECM proteins (collagen I, collagen II, collagen IV,
fibronectin, laminin, tenascin and vitronectin) and a
BSA-pre-coated well as control. The ECs were treated with 0–800
µg/ml PM2.5 for 6 h. Following washing with PBS, attached cells
were stained with CyQuant GR® Dye (ECM545; Chemicon; EMD
Millipore) according to the manufacturer's protocol and the
cell-bound stain was then extracted in extraction buffer (0.05%
trypsin in Hanks Balanced Salt Solution containing 25 mM HEPES).
The absorbance of the stain was determined at 450 nm. The change in
absorbance was presented as the fold of untreated control.
ROS detection
ECs (2×105 cells/well) were cultured in
35 mm dishes and exposed to PM2.5 (0–800 µg/ml) for 24 h, when the
cells were harvested for intracellular ROS detection using DCFH-DA.
First, ECs were washed with PBS and resuspended in RPMI 1640 medium
containing 10 µM of DCFH-DA at 37°C for 40 min in the dark. Then
the ECs were washed with PBS and ROS were detected using a confocal
fluorescence microscope and analyzed by flow cytometry.
ELISA
ECs (2×105 cells/well) were cultured in
35 mm dishes and exposed to PM2.5 (0–800 µg/ml) for 24 h. The ECs
were centrifuged at 10,000 × g for 10 min at 4°C, and TNF-α
(RAB1089) and interleukin (IL)-8 (RAB0595) levels were determined
by ELISA (Sigma-Aldrich; Merck KGaA) according to the
manufacturer's instructions.
In vivo matrigel plug assay
All animal care and experimental procedures complied
with the guidelines of the Animal Experimentation Ethics Committee
of Tongji University. Male C57BL/6 mice (24–25 g, 6 weeks old,
n=15) were supplied and maintained by Tongji University Laboratory
Animal Service Center at 23±2°C, 55±5% humidity on a 12 h
light/dark cycle with food and water supplied ad libitum.
Matrigel (500 µl) with heparin (10 U/ml), VEGF 100 ng/ml and PM2.5
(0–800 µg/ml) were mixed and injected into the right flanks of
mice. Negative controls were obtained by injecting mice with
Matrigel and twice-distilled water. The mice were sacrificed by
cervical dislocation after 7 days, and the Matrigel plugs were
removed and photographed. The hemoglobin content of the Matrigel
plugs was calculated using a Drabkin's reagent kit (5252;
Sigma-Aldrich; Merck KGaA).
Statistical analysis
All data are presented as mean ± standard deviation
obtained from at least three experiments. Statistical analysis of
two samples was performed with Student's t-test analysis.
Statistical analyses among multiple groups were performed using
one-way analysis of variance followed by Bonferroni's post hoc
test. P<0.05 was considered to indicate a statistically
significant difference.
Results
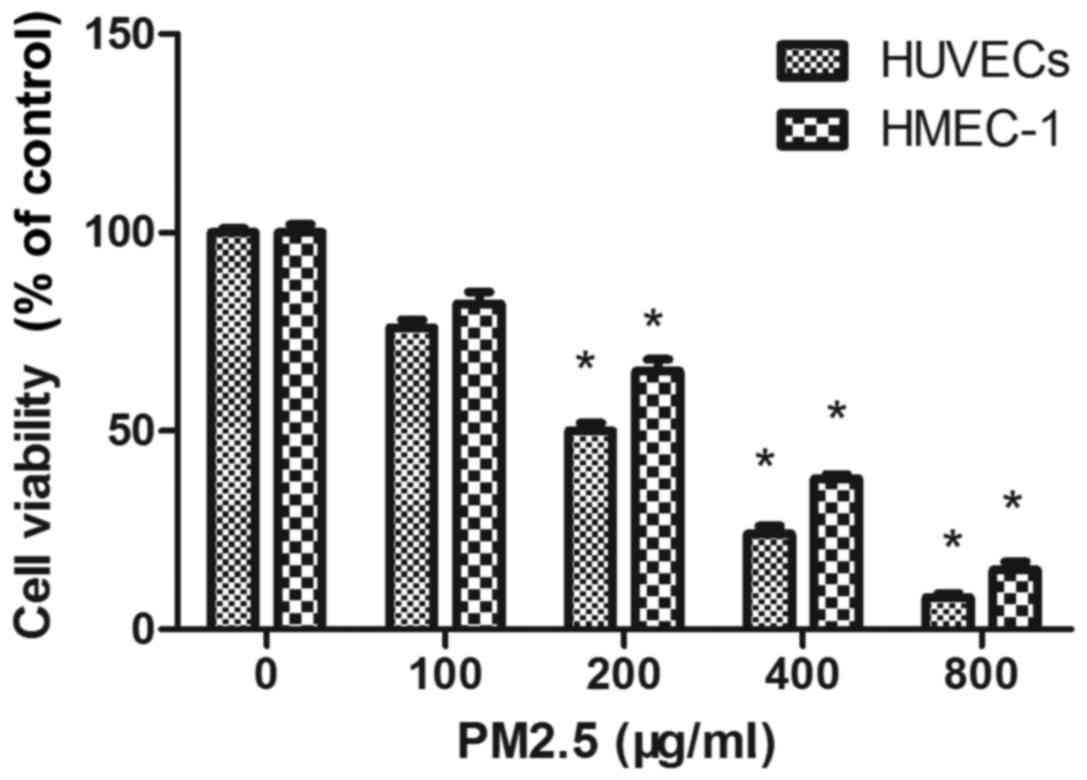
PM2.5 decreases HUVEC and HMEC-1
viability
The cytotoxicity of PM2.5 was determined by MTT
assay in HUVECs and HMEC-1 after 24 h. PM2.5 at 200–800 µg/ml
significantly reduced EC viability compared with the 0 µg/ml PM2.5
control group (Fig. 1). As
demonstrated in Fig. 1, PM2.5
significantly inhibited the viability of HUVECs and HMEC-1 in a
dose-dependent manner following 24 h treatment. The concentrations
producing 50% growth inhibition of PM2.5 on HUVEC and HMEC-1 were
~200 µg/ml and 300 µg/ml, respectively.
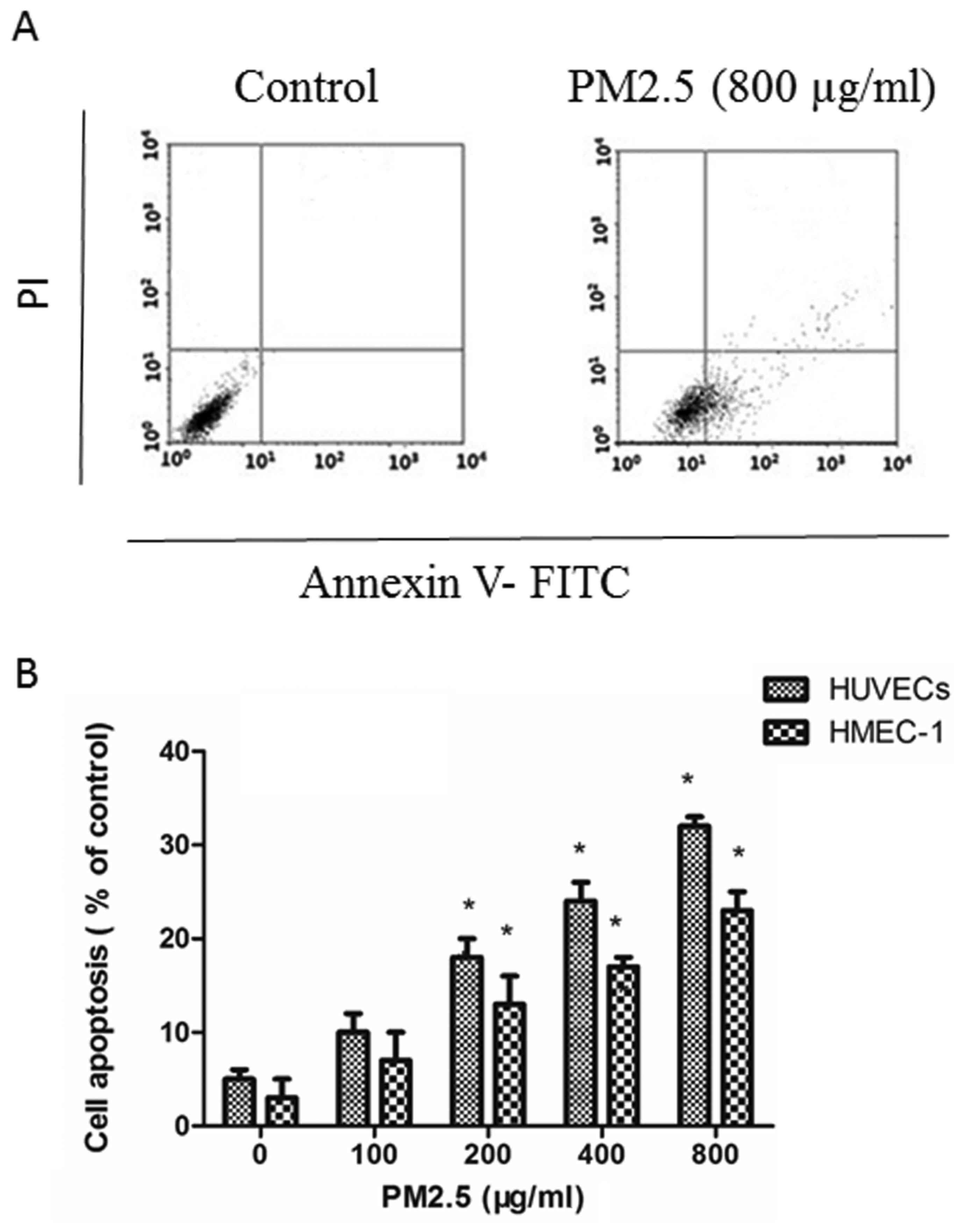
PM2.5 induces HUVECs and HMEC-1
apoptosis
Annexin V-FITC/PI assays demonstrated that PM2.5
induced apoptosis in ECs in a dose-dependent manner: PM2.5 at
200–800 µg/ml significantly increased the proportion of apoptotic
ECs after 24 h compared with the 0 µg/ml PM2.5 control group
(Fig. 2).
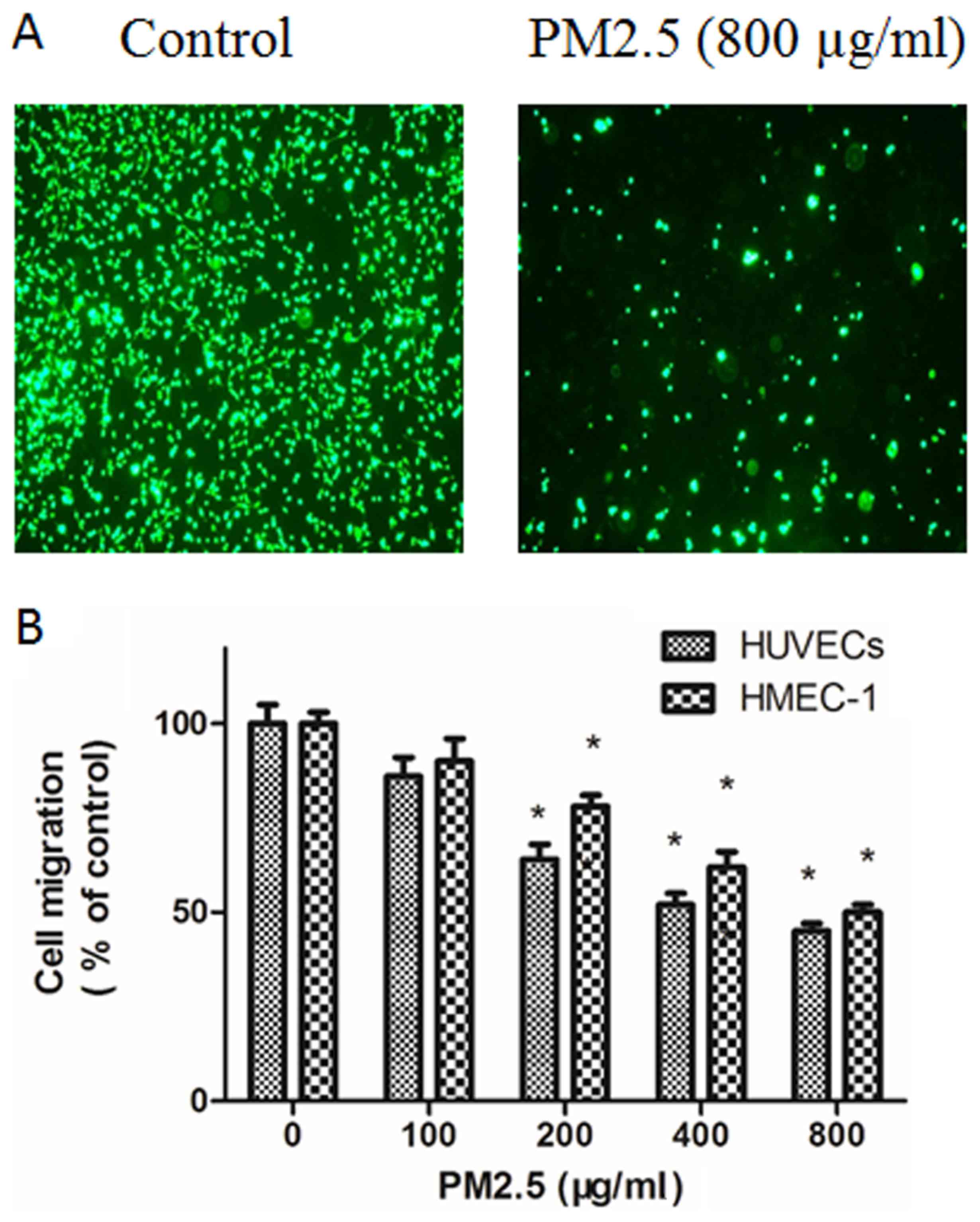
PM2.5 decreases HUVECs and HMEC-1
migration
The effect of PM2.5 on the migration of ECs was
explored using Transwell migration assays. Compared with basal
medium, RPMI 1640 medium with VEGF triggered EC migration; however,
this effect was dose-dependently inhibited by PM2.5 treatment
(Fig. 3). In the Boyden chamber
assay, 200–800 µg/ml PM2.5 treatment significantly inhibited EC
migration (Fig. 3).
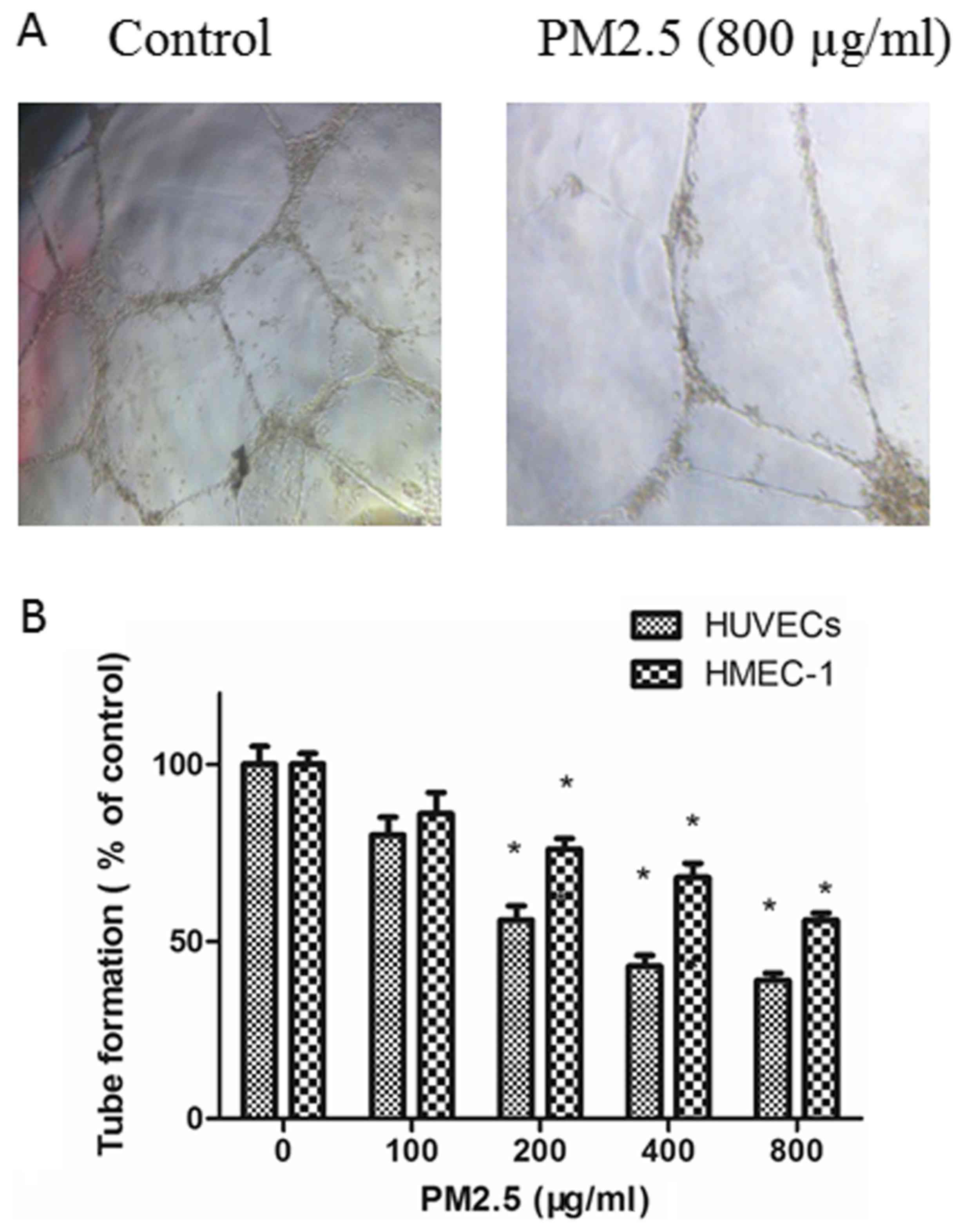
PM2.5 decreases HUVEC and HMEC-1 tube
formation
The effects of PM2.5 on the angiogenesis of ECs were
explored with a tube formation assay. ECs in RPMI 1640 medium
containing 10 ng/ml VEGF were planted on Matrigel and the amount of
tube formation was measured. As demonstrated in Fig. 4, compared with untreated ECs, tube
formation was inhibited in ECs by treatment with PM2.5 in in a
dose-dependent manner.
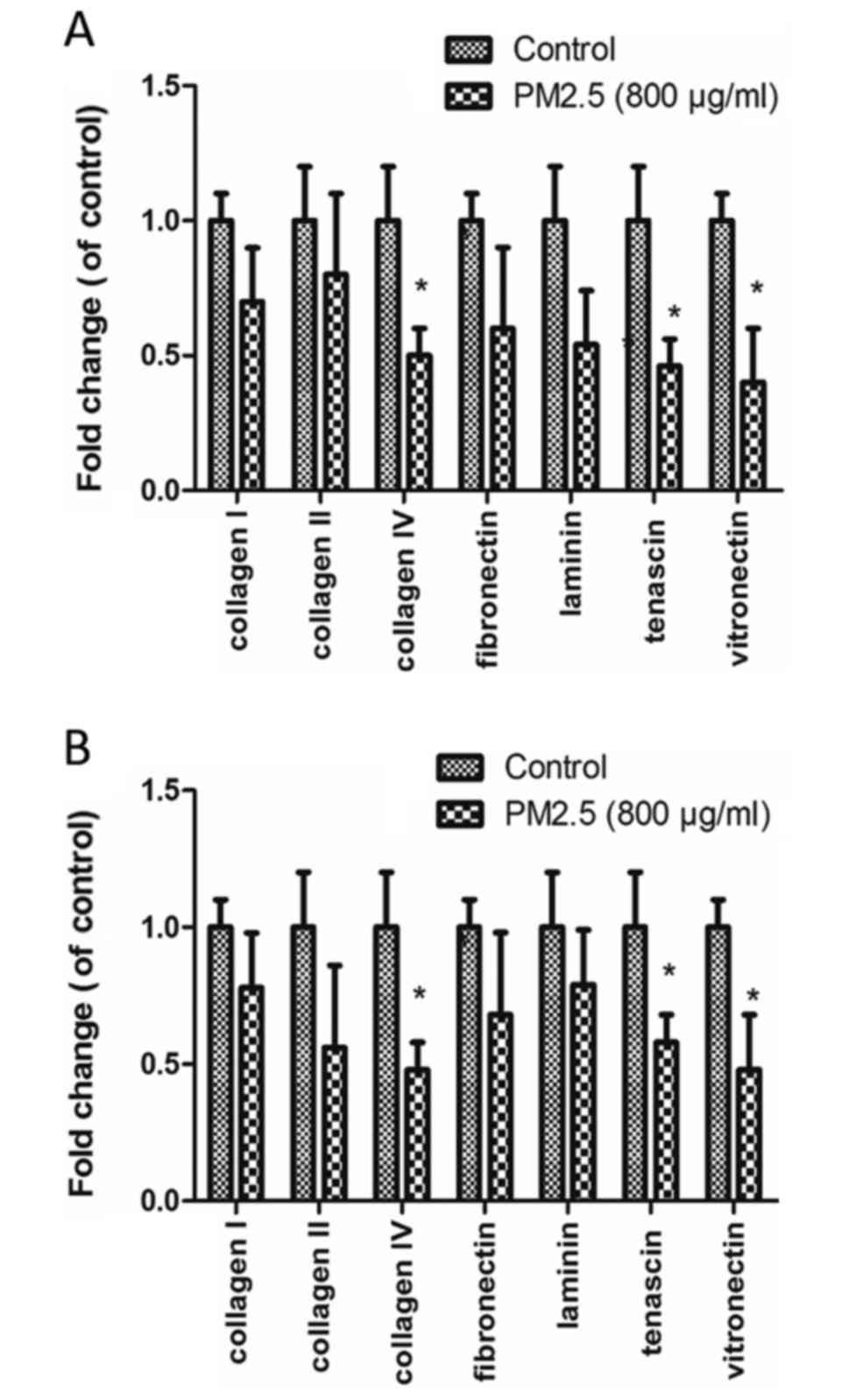
PM2.5 decreases EC adhesion to ECM
proteins
EC adhesion to various ECM proteins (collagen I,
collagen II, collagen IV, fibronectin, laminin, tenascin and
vitronectin) was measured in cells were treated with 0 or 800 µg/ml
PM2.5. The decreased adhesions were observed in the two cell lines
(P<0.05). Collagen IV, tenascin and vitronectin adhesion was
significantly reduced in PM2.5-treated HUVECs and HMEC-1s, compared
with control (P<0.05; Fig.
5).
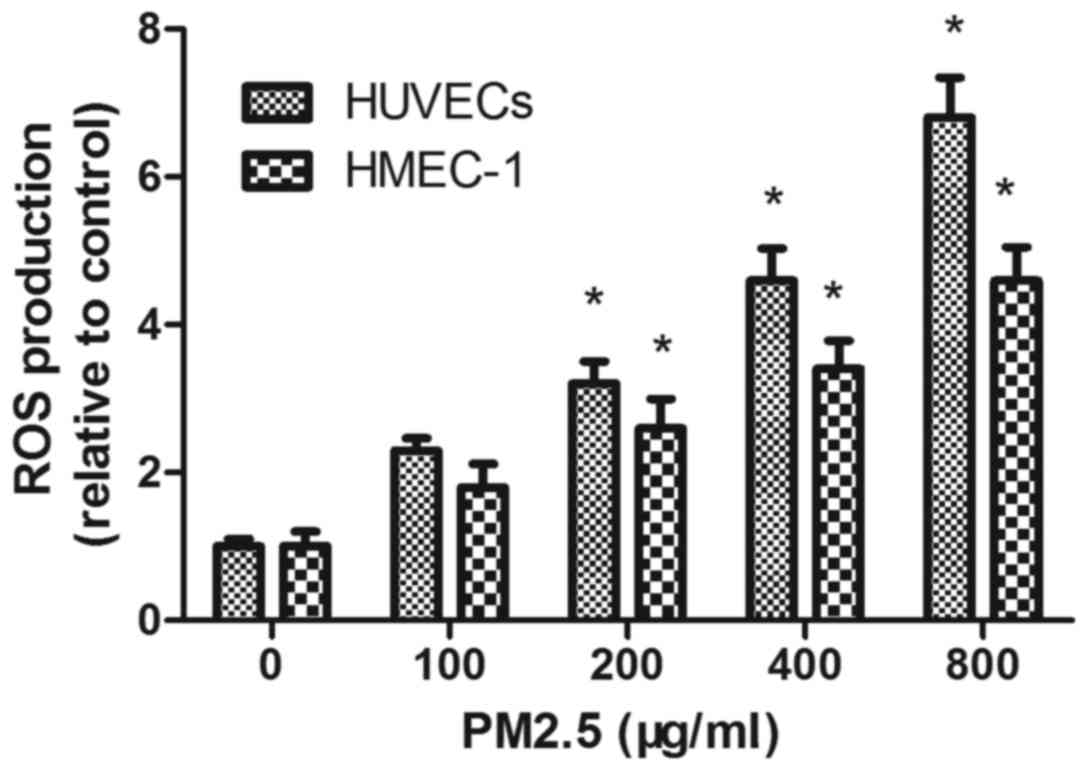
PM2.5 increases intracellular HUVEC
and HMEC-1 ROS generation
DCFH-DA staining demonstrated that PM2.5 treatment
increased ROS accumulation in a dose-dependent manner; flow
cytometry demonstrated that the mean fluorescence intensity of ECs
incubated with PM2.5 at 200, 400 and 800 µg/ml for 24 h was
significantly increased compared with the 0 µg/ml PM2.5 control
group (P<0.05; Fig. 6).
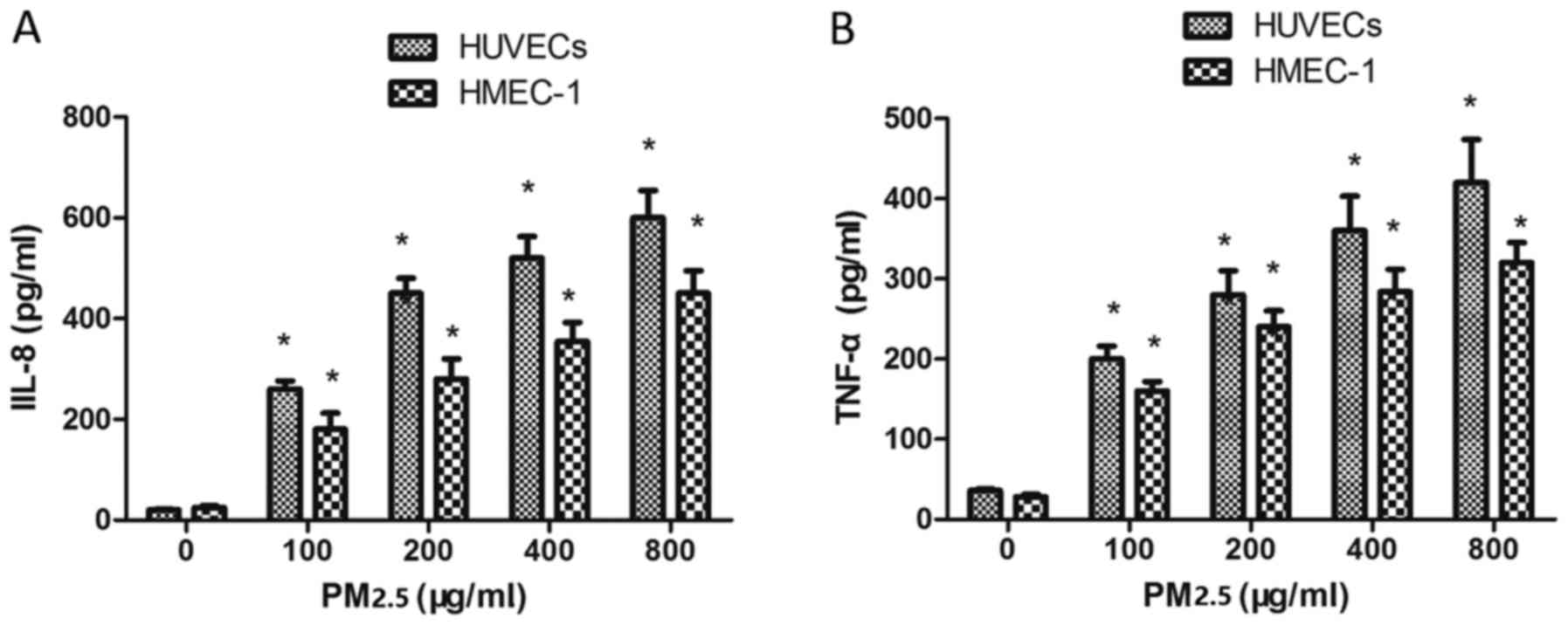
Effect of PM2.5 on IL-8 and TNF-α
expression in HUVECs and HMEC-1
To investigate whether PM2.5 treatment affected IL-8
and TNF-α expression, ECs were incubated with PM2.5 for 24 h. PM2.5
(100–800 µg/ml) induced IL-8 (Fig.
7A) and TNF-α (Fig. 7B)
expression in ECs compared with the 0 µg/ml PM2.5 control group,
indicating that PM2.5 induced vascular inflammation via IL-8 and
TNF-α overexpression.
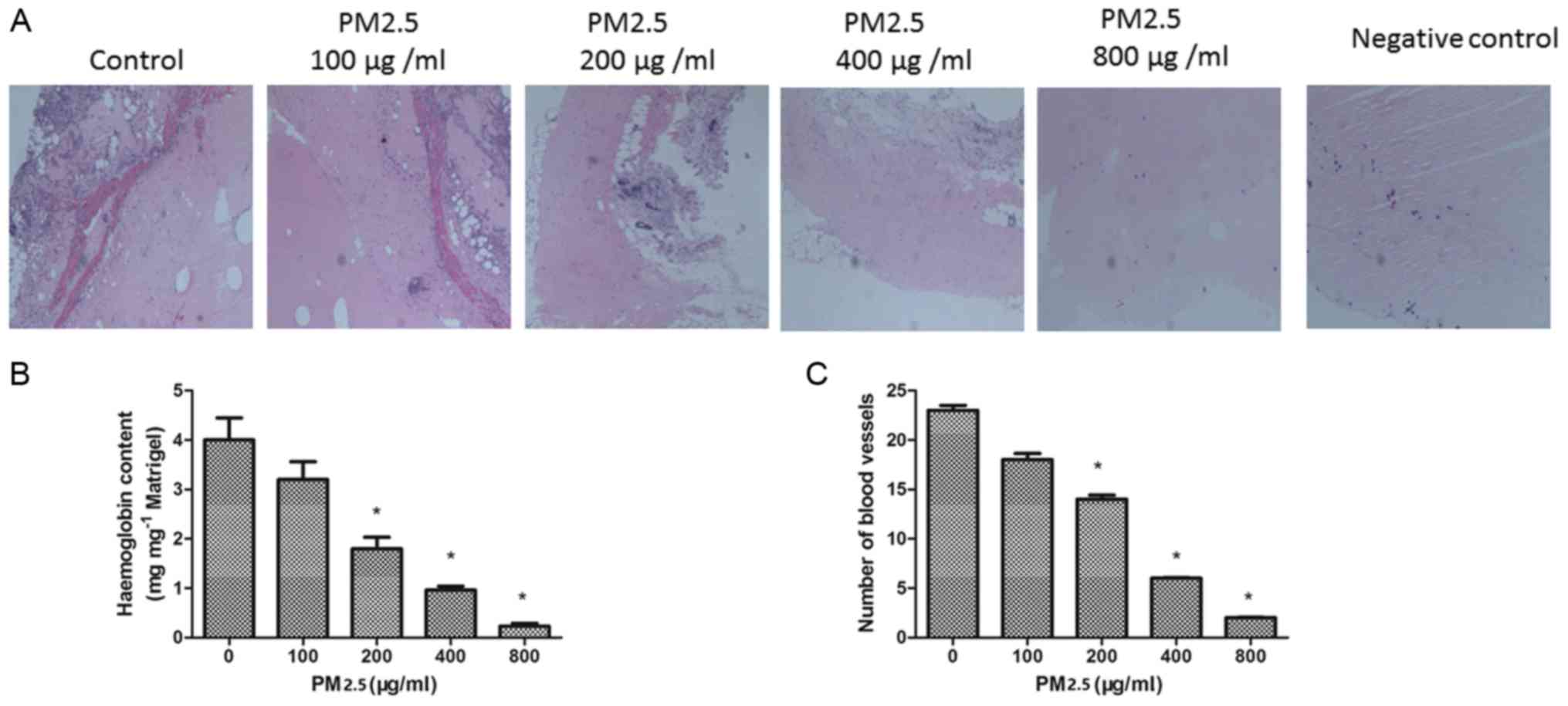
PM2.5 inhibits angiogenesis in
vivo
To confirm the effect of PM2.5 on angiogenesis in
vivo, a Matrigel plug assay was performed. The plugs containing
VEGF demonstrated greater levels of angiogenesis compared with the
negative control diluent plugs. In the presence of PM2.5 treatment,
the number of blood vessels in the plugs was decreased. The
hemoglobin concentration was quantified in the plugs; the
hemoglobin content was significantly decreased in the 200–800 µg/ml
PM2.5 treated groups compared with the 0 µg/ml PM2.5 group
(P<0.05; Fig. 8B). This result
is consistent with the inhibition of angiogenesis by PM2.5 in
vivo (Fig. 8C).
Discussion
PM2.5 has previously been demonstrated to exercise
adverse effects on ECs (18–20);
however, its effect on angiogenesis in ECs remains unclear. In the
present study, the vascular inflammation activities of PM2.5 were
demonstrated for the first time in HUVEC and HMEC-1. Although the
molecular mechanisms of PM2.5 in ECs have not been verified, the
data from the present study demonstrated that high doses of PM2.5
decreased EC viability, migration and tube formation. High doses of
PM2.5 also increased ROS production and IL-8 and TNF-α expression.
Based on the results of the ECs lines, high doses (>200 µg/ml)
of PM2.5 are able to significantly inhibit angiogenesis. Therefore,
it is possible that high-doses of PM2.5 can induce vascular
inflammation.
Angiogenesis requires a number of steps, including
cell proliferation, migration, tube formation and remodeling
(21). The results of the present
study demonstrated that the inhibitory effect of PM2.5 on viability
were stronger in HUVECs than HMEC-1s. PM2.5 treatment may also
inhibit migration and tube formation in the two EC lines, implying
that PM2.5 can inhibit angiogenesis. The endothelial recovery
involved endothelial proliferation and migration to the injury
site; the findings of the present study add a previously
unrecognized role of PM2.5 exposure in the regulation of ECs
biological function following vascular injury.
To further investigate mechanisms through which
PM2.5 induces inhibitory effects on migration of ECs, the effects
of PM2.5 on the ECs ability to adhere to ECM proteins were
investigated. PM2.5 decreased the ability to adhere to collagen
type IV, tenascin and vitronectin. Collagen IV serves an important
role in cell adhesion and motility (22) and tenascin has an important role in
promoting cell survival and migration (23,24).
Vitronectin is a high molecular weight glycoprotein known to
promote cell adhesion and affect cell migration (25). Therefore, treatment with PM2.5
resulted in decreased adhesion of endothelial cells to these matrix
proteins. It is suggested that the consequent effect was to disrupt
endothelial cell-cell junctions leading to an increase in vascular
permeability to the environment affected by local injury to blood
vessels.
The effect of PM2.5 on ROS production in ECs was
also evaluated. ROS have key roles in EC apoptosis. The data from
the present study suggested that PM2.5-induced ECs apoptosis may
act through the ROS overproduction. PM2.5 also caused the release
of the pro-inflammatory cytokines IL-8 and TNF-α. A previous study
demonstrated that intracellular ROS contributed to the release of
pro-inflammatory cytokines (26).
In the present study, TNF-α and IL-8 were secreted from the ECs
following 24 h exposure to PM2.5, which may have induced
endothelial permeability. Promotion of inflammation is considered a
key step in the adverse health effects associated with PM2.5
exposure.
The effects of PM2.5 were studied using an in
vivo Matrigel plug model. PM2.5 decreased the hemoglobin
content in plugs in comparison with the 0 µg/ml VEGF group
(Fig. 8B). The data are consistent
with the inhibition of angiogenesis by PM2.5 in vitro.
In conclusion, the anti-angiogenic effects by
high-dose PM2.5 exposure on ECs may be exerted through increased
ROS production and IL-8 and TNF-α expression, leading to reduced EC
proliferation and migration. These findings suggest that high-dose
PM2.5 exposure may be an important mediator of vascular
inflammation in ECs.
References
|
1
|
Aaron CP, Chervona Y, Kawut SM, Roux AV
Diez, Shen M, Bluemke DA, Van Hee VC, Kaufman JD and Barr RG:
Particulate matter exposure and cardiopulmonary differences in the
multi-ethnic study of atherosclerosis. Environ Health Perspect.
124:1166–1173. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Thurston G and Lippmann M: Ambient
particulate matter air pollution and cardiopulmonary diseases.
Semin Respir Crit Care Med. 36:422–432. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Amatullah H, North ML, Akhtar US, Rastogi
N, Urch B, Silverman FS, Chow CW, Evans GJ and Scott JA:
Comparative cardiopulmonary effects of size-fractionated airborne
particulate matter. Inhal Toxicol. 24:161–171. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Cui P, Huang Y, Han J, Song F and Chen K:
Ambient particulate matter and lung cancer incidence and mortality:
A meta-analysis of prospective studies. Eur J Public Health.
25:324–329. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Fu J, Jiang D, Lin G, Liu K and Wang Q: An
ecological analysis of PM2.5 concentrations and lung cancer
mortality rates in China. BMJ Open. 5:e0094522015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Yue H, Yun Y, Gao R, Li G and Sang N:
Winter polycyclic aromatic hydrocarbon-bound particulate matter
from peri-urban North China promotes lung cancer cell metastasis.
Environ Sci Technol. 49:14484–14493. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Hu H, Dailey AB, Kan H and Xu X: The
effect of atmospheric particulate matter on survival of breast
cancer among US females. Breast Cancer Res Treat. 139:217–226.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lin CP, Lin FY, Huang PH, Chen YL, Chen
WC, Chen HY, Huang YC, Liao WL, Huang HC, Liu PL, et al:
Endothelial progenitor cell dysfunction in cardiovascular diseases:
Role of reactive oxygen species and inflammation. Biomed Res Int.
2013:8450372013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Gimbrone MA Jr and García-Cardeña G:
Endothelial cell dysfunction and the pathobiology of
atherosclerosis. Circ Res. 118:620–636. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yang GZ, Wang ZJ, Bai F, Qin XJ, Cao J, Lv
JY and Zhang MS: Epigallocatechin-3-gallate protects HUVECs from
PM2.5-induced oxidative stress injury by activating critical
antioxidant pathways. Molecules. 20:6626–6639. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Cui Y, Xie X, Jia F, He J, Li Z, Fu M, Hao
H, Liu Y, Liu JZ, Cowan PJ, et al: Ambient fine particulate matter
induces apoptosis of endothelial progenitor cells through reactive
oxygen species formation. Cell Physiol Biochem. 35:353–363. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Tong GQ, Zhang ZH, Zhao Y, Liu JJ and Han
JB: Traffic-related PM2.5 induces cytosolic [Ca2+]
increase regulated by Orai1, alters the CaN-NFAT signaling pathway,
and affects IL-2 and TNF-α cytoplasmic levels in Jurkat T-cells.
Arch Environ Contam Toxicol. 68:31–37. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Li R, Kou X, Xie L, Cheng F and Geng H:
Effects of ambient PM2.5 on pathological injury, inflammation,
oxidative stress, metabolic enzyme activity, and expression of
c-fos and c-jun in lungs of rats. Environ Sci Pollut Res Int.
22:20167–20176. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ostro B, Malig B, Broadwin R, Basu R, Gold
EB, Bromberger JT, Derby C, Feinstein S, Greendale GA, Jackson EA,
et al: Chronic PM2.5 exposure and inflammation: Determining
sensitive subgroups in mid-life women. Environ Res. 132:168–175.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Potera C: Toxicity beyond the lung:
Connecting PM2.5, inflammation, and diabetes. Environ Health
Perspect. 122:A292014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wang Y, Lin Z, Huang H, He H, Yang L, Chen
T, Yang T, Ren N, Jiang Y, Xu W, et al: AMPK is required for
PM2.5-induced autophagy in human lung epithelial A549 cells. Int J
Clin Exp Med. 8:58–72. 2015.PubMed/NCBI
|
|
17
|
Deng X, Zhang F, Rui W, Long F, Wang L,
Feng Z, Chen D and Ding W: PM2.5-induced oxidative stress triggers
autophagy in human lung epithelial A549 cells. Toxicol In Vitro.
27:1762–1770. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wang FF, Geng CM, Hao WD, Zhao YD, Li Q,
Wang HM and Qian Y: The cellular toxicity of PM2.5 emitted from
coal combustion in human umbilical vein endothelial cells. Biomed
Environ Sci. 29:107–116. 2016.PubMed/NCBI
|
|
19
|
Montiel-Dávalos A, Alfaro-Moreno E and
López-Marure R: PM2.5 and PM10 induce the expression of adhesion
molecules and the adhesion of monocytic cells to human umbilical
vein endothelial cells. Inhal Toxicol. 19:(Suppl 1). 91–98. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Rui W, Guan L, Zhang F, Zhang W and Ding
W: PM2.5-induced oxidative stress increases adhesion molecules
expression in human endothelial cells through the
ERK/AKT/NF-κB-dependent pathway. J Appl Toxicol. 36:48–59. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Abdelrahim M, Konduri S, Basha R, Philip
PA and Baker CH: Angiogenesis: An update and potential drug
approaches (Review). Int J Oncol. 36:5–18. 2010.PubMed/NCBI
|
|
22
|
Favreau AJ, Vary CP, Brooks PC and
Sathyanarayana P: Cryptic collagen IV promotes cell migration and
adhesion in myeloid leukemia. Cancer Med. 3:265–272. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Hessel M, Steendijk P, den Adel B, Schutte
C and van der Laarse A: Pressure overload-induced right ventricular
failure is associated with re-expression of myocardial tenascin-C
and elevated plasma tenascin-C levels. Cell Physiol Biochem.
24:201–210. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ide M, Saito K, Tsutsumi S, Tsuboi K,
Yamaguchi S, Asao T, Kuwano H and Nakajima T: Over-expression of
14-3-3σ in budding colorectal cancer cells modulates cell migration
in the presence of tenascin-C. Oncol Rep. 18:1451–1456.
2007.PubMed/NCBI
|
|
25
|
Madsen CD, Ferraris GM, Andolfo A,
Cunningham O and Sidenius N: uPAR-induced cell adhesion and
migration: Vitronectin provides the key. J Cell Biol. 177:927–939.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zhu G, Huang Q, Zheng W, Huang Y, Hua J,
Yang S, Zhuang J, Wang J, Chang J, Xu J and Ye J: LPS upregulated
VEGFR-3 expression promote migration and invasion in colorectal
cancer via a mechanism of increased NF-κB binding to the promoter
of VEGFR-3. Cell Physiol Biochem. 39:1665–1678. 2016. View Article : Google Scholar : PubMed/NCBI
|