Introduction
Diabetes mellitus is a metabolic disease with an
increasing prevalence worldwide (1). Micro- and macrovascular disorders
with debilitating consequences for several organs are prevalent in
patients with diabetes (2). The
alveolar-capillary network in the lungs is a large microvascular
unit and can be affected by microangiopathy (3). Diabetes has detrimental effects on
retinal and glomerular microvasculature (4); however, pulmonary diabetic
microangiopathy has not received considerable clinical attention,
as due to the large reserve of the lung microvasculature, it is
possible that substantial pulmonary dysfunction may occur without
the appearance of dyspnea (3).
Therefore, the specific molecular mechanisms underlying the
development of metabolic defects in the lungs have yet to be
elucidated.
Previous studies have suggested that inflammation
may serve crucial roles in the pathogenesis of diabetes (5,6).
Therefore, the inhibition of inflammation has potential as a novel
therapeutic strategy for the treatment of patients with diabetes
(6). Several proinflammatory
cytokines have been implicated in the development of diabetic lung
injury (7–9). The Janus kinase (JAK)/signal
transducers and activators of transcription (STAT) intracellular
signaling pathway is activated in response to cytokine stimulation
and relays biological signals to target cells (10). The JAK/STAT pathway has been
involved in the regulation of several genes involved in cell
proliferation, inflammation and fibrosis. In addition, the JAK/STAT
pathway has been implicated in hyperglycemia-induced nephropathy in
patients with diabetes (11,12).
The JAK/STAT pathway is negatively regulated via
various mechanisms, including the suppressor of cytokine signaling
(SOCS) proteins, which have been identified as negative feedback
regulators of cytokine signaling, and have been reported to be
involved in the regulation of inflammatory responses (13–17).
The mammalian cytokine-inducible SH2-containing protein (CIS)/SOCS
family consists of 8 members, including CIS and SOCS1-7 (18). CIS/SOCS proteins are characterized
by a central SH2 domain, an amino-terminal domain of variable
length and sequence and a carboxy-terminal SOCS box (19). The presence of SOCS3 has been
demonstrated in the lungs (14).
The SH2 domain of SOCS3 does not have a high affinity for the
activation loop of JAKs; however, the kinase inhibitory region of
SOCS3 has a higher affinity for the kinase domain of JAK2 compared
with that of SOCS1 (20). Since
SOCS3 can bind to interleukin (IL)-12 receptor, which can activate
STAT3, SOCS3 can inhibit the IL-12-mediated activation of STAT3
(21).
Increasing evidence indicates that SOCS proteins may
be involved in the development of diabetes and disease-associated
complications (22). In addition,
SOCS-modulating properties have been attributed to pharmacological
agents that are currently used for the treatment of diabetes
(17). The present study aimed to
investigate the function of SOCS3 in lung cells and to explore the
implication of the JAK2/STAT3 signaling pathway in the molecular
mechanisms underlying the effects of SOCS3 in hyperglycemia-induced
lung injury.
Materials and methods
Materials
A549 human lung epithelial cells were purchased from
American Type Culture Collection (Manassas, VA, USA). Dulbecco's
modified Eagle's medium (DMEM) was purchased from Hyclone (GE
Healthcare Life Sciences, Logan, UT, USA) and fetal bovine serum
(FBS) was obtained from Gibco (Thermo Fisher Scientific, Inc.,
Waltham, MA, USA). Phosphate-buffered saline (PBS), and 0.25%
trypsin with 0.02% EDTA were obtained from Hyclone (GE Healthcare
Life Sciences). D-glucose and D-mannitol were from Sinopharm
Chemical Reagent Co., Ltd. (Shanghai, China), and tyrphostin AG490
was from Sigma-Aldrich (Merck KGaA, Darmstadt, Germany). Empty
vector plasmid pcDNA3.1 and the expression vector pcDNA3.1-SOCS3
were synthesized by Shanghai GenePharma Co., Ltd. (Shanghai,
China). Attractene transfection reagent was obtained from Qiagen,
Inc. (Valencia, CA, USA). The following antibodies were used:
Anti-JAK2 (1:1,000; catalog no. 3230), anti-STAT3 (1:1,000; catalog
no. 4904), anti-phosphorylated (p)-JAK2 (1:1,000; catalog no.
3776), anti-p-STAT3 (1:1,000; catalog no. 9145) and anti-SOCS3
polyclonal antibodies (1:1,000; catalog no. 2932) (all from Cell
Signaling Technology, Inc., Danvers, MA, USA). The β-actin antibody
(1:1,000; catalog no. 4970; Cell Signaling Technology, Inc.) served
as the loading control. The proteins were visualized using
secondary antibodies: Goat anti-rabbit polyclonal IgG (1:10,000;
catalog no. 926-32221; LI-COR Biosciences, Lincoln, NE, USA). The
following kits were used: Cell Counting Kit-8 (CCK-8; Dojindo
Molecular Technologies, Inc., Kumamoto, Japan), lactate
dehydrogenase (LDH) cytotoxicity kit, IL-6 (catalog no. H052;
Nanjing Jiancheng Bioengineering Institute, Nanjing, China) and
tumor necrosis factor (TNF)-α ELISA kits (catalog no. H007; Nanjing
Jiancheng Bioengineering Institute, Nanjing, China), and
bicinchoninic acid (BCA) protein assay kit (Beyotime Institute of
Biotechnology, Haimen, China).
Cell culture
A549 cells were cultured in DMEM containing 5.5 mM
glucose, supplemented with 10% FBS. Following 24 h, cells were
subcultured using 0.25% trypsin with 0.02% EDTA following washing
with PBS twice, when they reached 70–80% confluence. The following
treatment groups were used: Normal glucose (NG) group, fresh 5.5 mM
glucose was added to the medium; high glucose (HG) group, 25 mM
glucose was added to the medium; and high osmosis (OG) group, 5.5
mM glucose and 19.5 mM D-mannitol were added to the medium. All
cells were incubated at 37°C in a5% CO2 atmosphere for
48 h (23).
When the A549 cells reached 70–80% confluence they
were cultured in DMEM containing 5.5 mM glucose without FBS for 24
h. Subsequently, they were treated with 10 µmol/l tyrphostin AG490
in serum-free DMEM containing 25 mM glucose (HG+AG490 group) and
incubated at 37°C in a 5% CO2 atmosphere for 24 h. Cells
were then collected for subsequent experiments.
Plasmid transfection
A549 cells were seeded at a density of
2×105 cells/ml in DMEM supplemented with FBS 24 h prior
to transfection. Cells were 40–80% confluent on the day of
transfection and were treated according to the manufacturer's
protocol. Cells were transfected, using Attractene, with empty
vector control (HG+SOCS3− group) or with pcDNA3.1-SOCS3
expression vector (HG+SOCS3+group) and incubated in
serum-free medium for 6 h. Subsequently, the medium was replaced
with fresh medium containing 5.5 mM glucose and cells were cultured
for 24 h. Then, the medium was replaced with 25 mM
glucose-containing medium and cells were incubated for an
additional 24 h.
Cell viability assay
A total of 100 µl adherent A549 cells from each
experimental group were cultured in 96-well plates
(1×104cells/well), and the supernatant was collected for
the LDH toxicity assay. The assay was performed according to the
manufacturer's protocol and the absorbance of each sample was
measured at 450 nm using a microplate reader. For the CCK-8 assay,
the medium was removed and cells were washed twice with PBS. Fresh
medium and 10 µl CCK-8 solution were added to each well and cells
were incubated for 2 h at 37°C in 5% CO2. Subsequently,
the absorbance of each sample was measured of 450 nm using a
microplate reader. The mean optical density (OD) values of six
randomly selected wells from each treatment group were used as an
index of cell viability.
Proinflammatory cytokine ELISA
A549 cells were seeded in a 6-well plate at a
density of 2×105cells/ml and stimulated with different
conditions. Following treatment, cells were washed three times with
PBS and their IL-6 and TNF-α contents were measured using ELISA
kits, according to the manufacturer's protocol.
Western blot analysis
Cells were rinsed twice with PBS and dissolved in
SDS sample loading buffer. Total proteins were extracted using a
radioimmunoprepitation assay lysis buffer (Beyotime Institute of
Biotechnology). Protein concentrations were determined using a BCA
assay. Equal amounts of extracted protein samples (30 µg) were
separated by 10% SDS-PAGE and transferred onto a
polyvinylidenedifluoride membrane. The membrane was blocked with 5%
bovine serum albumin (BSA; Beyotime Institute of Biotechnology) at
room temperature for 2 h under agitation to prevent non-specific
antibody binding. The membranes were then incubated with the
following primary antibodies at 4°C for 12 h: Anti-STAT3,
anti-SOCS3, anti-JAK2, anti-p-JAK2 and anti-p-STAT3, and
anti-β-actin. Subsequently, the membrane was rinsed with PBS and
incubated with goat anti-rabbit horseradish peroxidase-conjugated
secondary antibody for 1 h at room temperature. The protein bands
were visualized by enhanced chemiluminescence using an Odyssey
Infrared Imaging system (LI-COR Biosciences), and blots were
semi-quantified by densitometry using Quantity One software version
4.6.2 (Bio-Rad Laboratories, Inc., Hercules, CA, USA).
Statistical analysis
Data are presented as the mean ± standard error of
the mean. Statistical analysis was performed using GraphPad Prism
software version 5.0 (GraphPad Software, Inc., La Jolla, CA, USA).
The statistical significance of the differences between groups was
evaluated using an unpaired Student's t-test for pair-wise
comparisons, or a one-way analysis of variance followed by the
Tukey post hoc test for multiple comparisons. P<0.05 was
considered to indicate a statistically significant difference.
Results
SOCS3 is overexpressed in A549 cells
following transfection
To assess the effects of SOCS3 activation in
hyperglycemia-induced lung injury, human A549 lung epithelial cells
were transfected with pcDNA3.1-SCOS3 expression vector expressing
green fluorescence or empty vector control expressing no
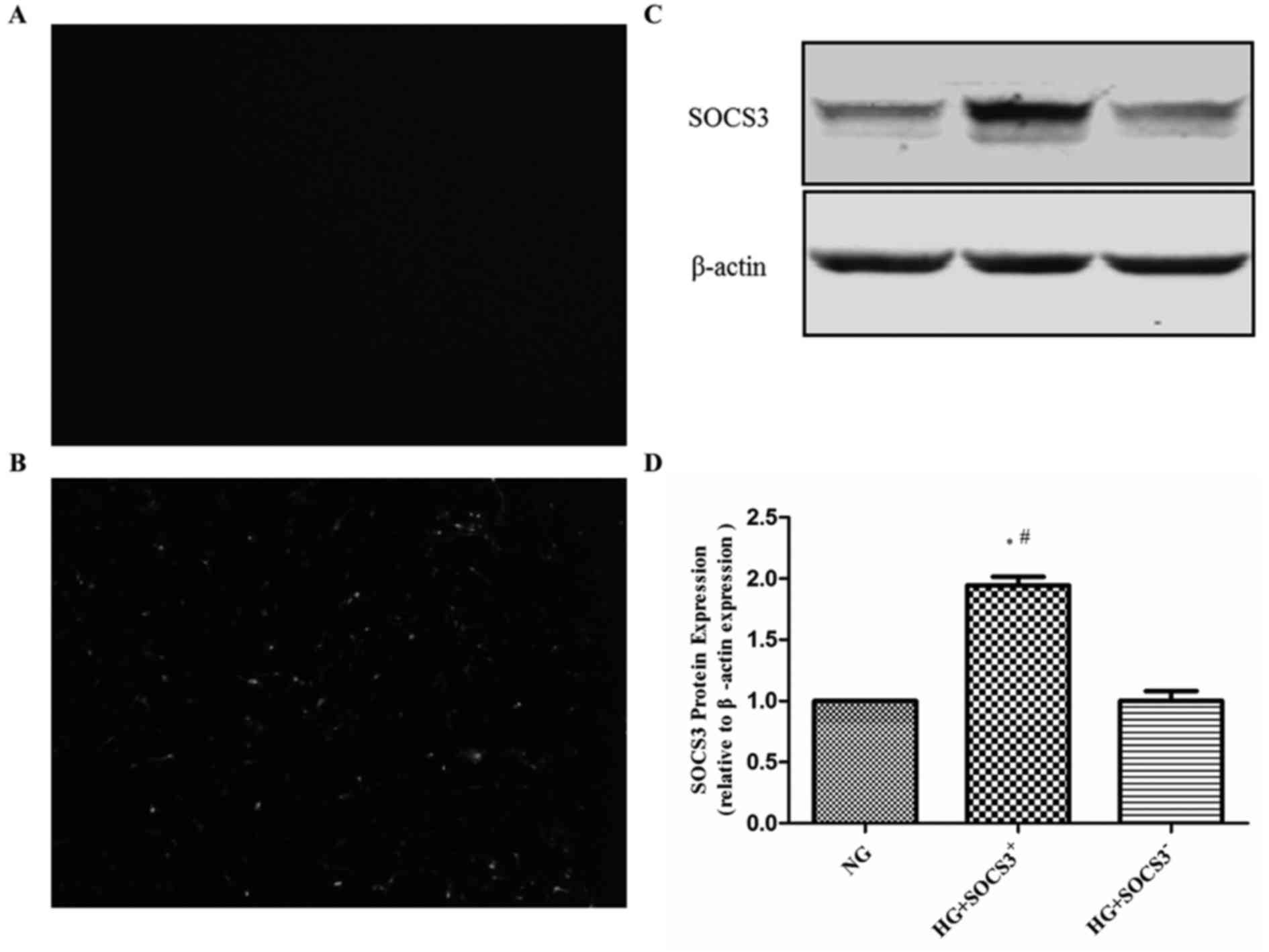
fluorescence. As presented in Fig. 1A
and B, intense green fluorescence was observed in
pcDNA3.1-SCOS3-transfected cells compared with control
vector-transfected cells. Furthermore, western blot analysis
demonstrated that the protein expression of SOCS3 was significantly
increased by transfection with the SOCS3 overexpression plasmid
compared with the empty vector-transfected group (Fig. 1C and D).
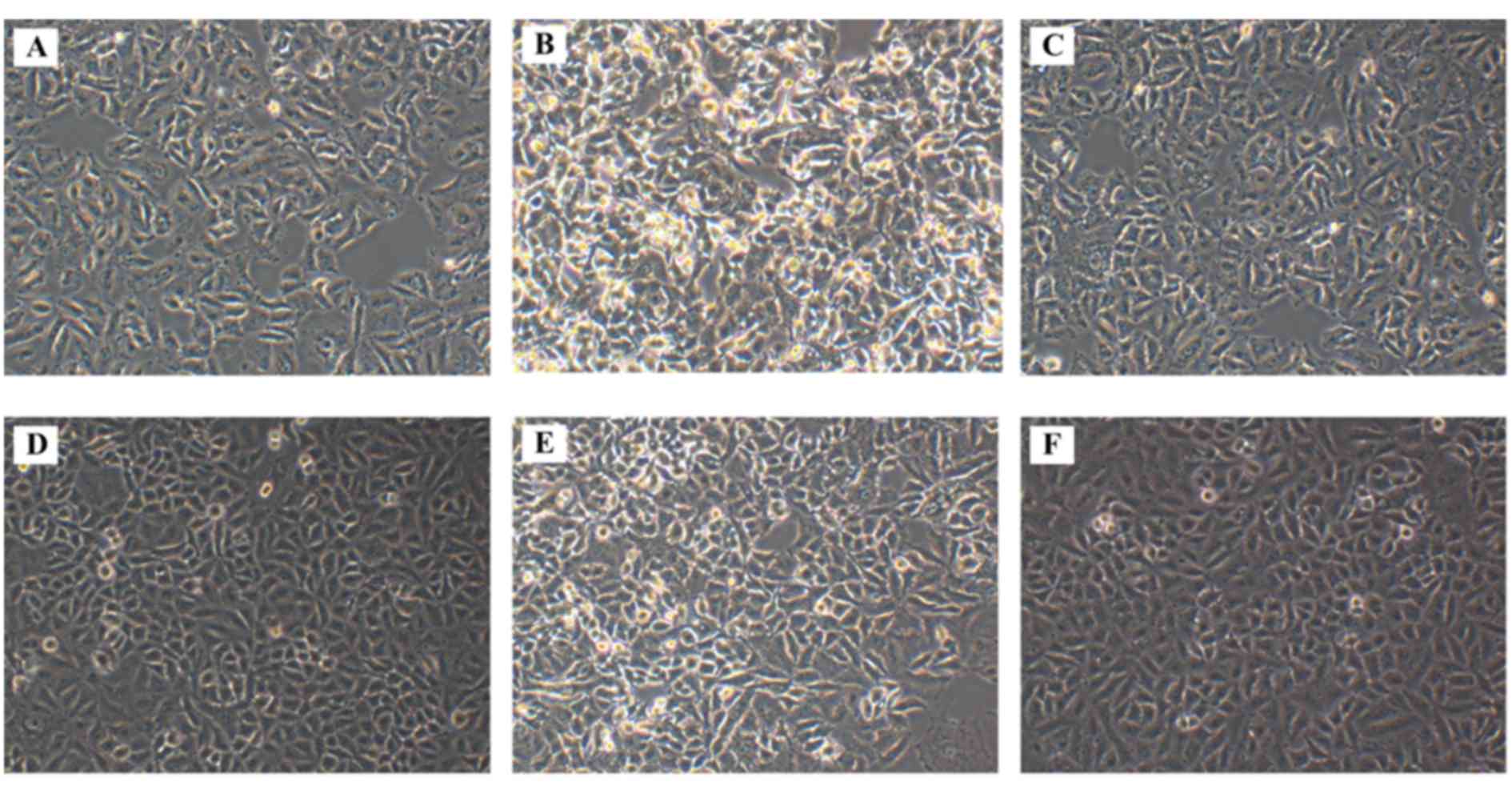
Morphological alterations following
SOCS3 overexpression in HG-treated A549 cells
The morphology of A549 cells from the various
treatment groups was observed under an inverted microscope. A549
cells cultured in normal control medium (NG group) exhibited
shuttle-like shapes, and long and fine cell bodies (Fig. 2A). However, exposure to HG markedly
altered the morphology of the cells in the HG group, which
exhibited shorter and less extended cell bodies, and the cells were
dead and lysed with more cell fragments (Fig. 2B), thus suggesting that HG exposure
may induce apoptotic morphological characteristics in A549 cells.
When exposed to high osmotic pressure, the cells exhibited fusiform
shapes with failed outgrowth of processes, whereas cell loss was
also observed (Fig. 2C). Treatment
with tyrphostin AG490 appeared to attenuate the morphological
changes in HG-treated cells, and to increase their density
(Fig. 2D). Cell morphology
appeared to be similar between cells in the HG and
HG+SOCS3− groups (Fig.
2E); whereas SOCS3 overexpression appeared to produce similar
effects in HG-exposed cells (Fig.
2F) appeared to attenuate the morphological changes caused by
HG.
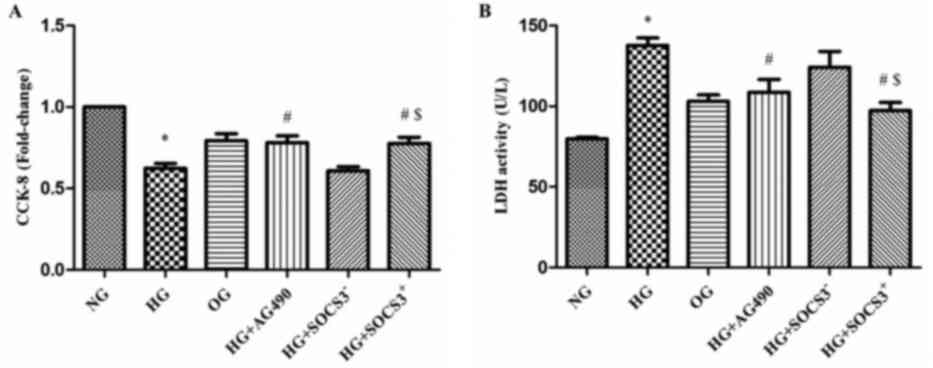
HG exposure suppresses the viability
of A549 cells
A CCK-8 assay was used to evaluate the viability of
A549 cells from the various treatment groups in vitro. As
presented in Fig. 3A, the
viability of cells exposed to HG was significantly decreased
compared with the NG group (P<0.05). Notably, the viability of
cells in the HG+AG490 and HG+SOCS3+ groups was
significantly enhanced compared with cells in the HG group
(P<0.05; Fig. 3A). In addition,
cells in the HG+SOCS3+ group exhibited increased
viability compared with cells in the HG+SOCS3− group
(P<0.05; Fig. 3A).
An LDH cytotoxicity assay was also used to evaluate
cell viability. The present results demonstrated that LDH activity
was significantly increased in HG-treated cells compared with the
NG group (P<0.05; Fig. 3B).
Conversely, LDH activity in cells from the HG+AG490 and
HG+SOCS3+ groups was significantly suppressed compared
with the HG group, indicating an increase in cell viability
(P<0.05; Fig. 3B). Furthermore,
the viability of HG+SOCS3+ cells was significantly
enhanced compared with HG+SOCS3− cells (P<0.05;
Fig. 3B). Notably, hyperosmolarity
did not appear to exert an effect on A549 cell viability (OG group;
Fig. 3A and B).
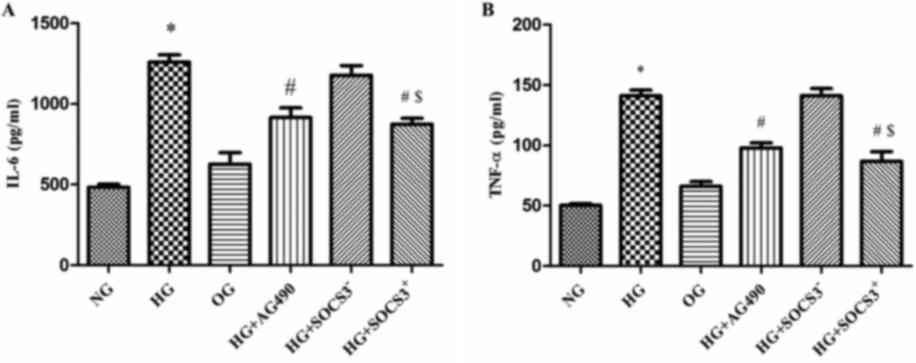
HG induces the expression of IL-6 and
TNF-α in A549 cells
As presented in Fig.
4, IL-6 levels were increased by >2-fold in HG-treated cells
compared with in NG cells (P<0.05; Fig. 4A). In addition, TNF-α levels were
increased by~3-fold following HG exposure (P<0.05; Fig. 4B). Notably, IL-6 and TNF-α levels
in cells from the HG+AG490 and HG+SOCS3+ groups were
significantly decreased compared with in cells from the HG group
(P<0.05). Furthermore, IL-6 and TNF-α levels were significantly
increased in the HG+SOCS3− group compared with in
HG+SOCS3+ cells (P<0.05; Fig. 4A and B).
Effects of SOCS3 overexpression on the
expression of JAK2/STAT3 pathway proteins
As demonstrated in Fig.
5, p-JAK2 and p-STAT3 densitometric units were normalized to
that of total JAK2 and STAT3 respectively. The protein expression
levels of p-JAK2 and p-STAT3 were significantly upregulated in HG
cells compared with in cells from the NG group (P<0.05).
Following treatment of HG-exposed cells with the JAK2/STAT3
signaling pathway inhibitor tyrphostin AG490, the protein levels
of, p-JAK2 and p-STAT3 were significantly downregulated compared
with the HG group (P<0.05; Fig.
5). In addition, SOCS3 protein expression was enhanced in A549
cells following exposure to HG compared with the NG group
(P<0.05; Fig. 5). To
investigate the putative regulatory effects of SOCS3 on the
JAK2/STAT3 pathway, SOCS3 was overexpressed in A549 cells by
plasmid transfection. The results revealed that SOCS3
overexpression significantly inhibited HG-induced upregulation of
both phosphorylated JAK2 and STAT3 proteins compared with the
control vector-transfected groups (P<0.05; Fig. 5). Notably, the hyperosmolarity
control did not exert an effect on the expression of JAK2/STAT3
pathway proteins in A549 cells (Fig.
5).
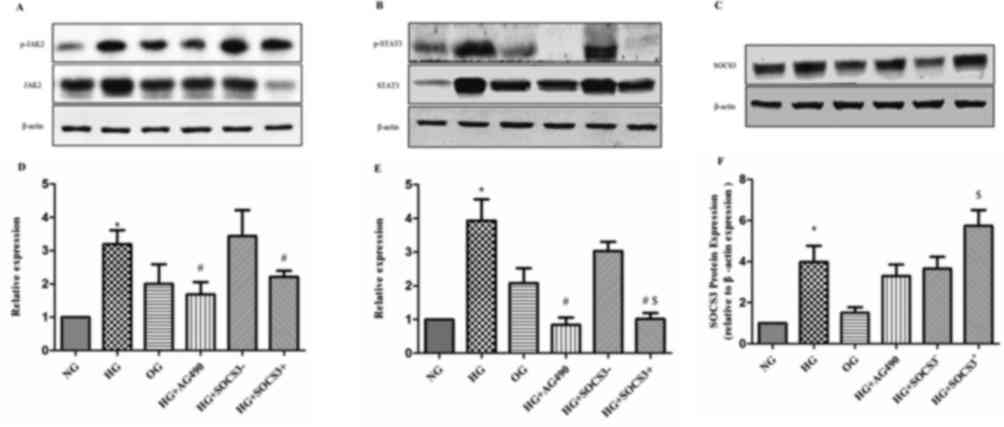 | Figure 5.Protein expression levels of p-JAK2,
p-STAT3 and SOCS3 in human A549 pulmonary epithelial following
various treatments. Protein expression levels of (A) p-JAK2 and
total JAK2, (B) p-STAT3 and total STAT3 and (C) SOCS3, were
detected using western blot analysis. β-actin was used as the
loading control. Blots were semi-quantified using densitometry.
Densitometric analysis of (D) p-JAK2, (E) p-STAT3 and (F) SOCS3.
Cells were cultured under NG, HG and OG conditions, with HG +
tyrphostin AG490, and with HG + pcDNA3.1-SOCS3 expression vector.
Data areexpressed as the mean ± standard error of the mean of 10
independent experiments. *P<0.05 vs. NG group;
#P<0.05 vs. HG group; $P<0.05 vs.HG +
SOCS3− group. JAK, Janus kinase; NG, normal glucose; HG,
high glucose; OG, hyperosmotic group; SOCS3, suppressor of cytokine
signaling 3; p-, phosphorylated; STAT, signal transducers and
activators of transcription. |
Discussion
The present study demonstrated that the
SOCS3/JAK2/STAT3 pathway was activated in HG-treated A549 lung
epithelial cells. Previous studies reported that HG exposure
enhanced the phosphorylation of proteins of the JAK/STAT pathway in
human renal tubular epithelial and glomerular mesangial cells
(24–26). Similarly, in the present study,
treatment of pulmonary epithelial cells with HG resulted in the
significant increase of p-JAK2 and p-STAT3 protein levels. In
addition, HG exposure was revealed to upregulate the protein
expression of SOCS3 in A549 cells. Notably, SOCS3 overexpression
was demonstrated to attenuate the HG-induced increases in p-JAK2
and p-STAT3, showing that SOCS3, which are negative regulators of
the JAK2/STAT3 signaling pathway, are involved in
hyperglycemia-induced cell responses during the development of
diabetic lung injury. It has also been suggested that modulation of
this pathway may prevent lung complications of diabetes.
The rapid worldwide increase in the prevalence of
type 2 diabetes and diabetic lung injury has become a serious
public health concern (1). Dyspnea
upon exertion in patients with diabetes is often mistakenly
associated with cardiovascular diseases and/or physical unfitness
(3). Accumulating evidence
suggests that inflammation may serve a crucial role during the
pathogenesis of type 2 diabetes, thus linking diabetes to various
common comorbidities that also involve inflammatory mechanisms
(7,9). The SOCS, JAK and Src families of
kinases have been implicated in systemic responses to hyperglycemia
(10,27–29);
however, their roles in inflammatory responses in the lungs have
yet to be elucidated.
Previous studies have reported that SOCS3 expression
is induced by various stimuli, including cytokines, Toll-like
receptor ligands, bacteria and immune complexes (30,31).
In addition, SOCS3 was demonstrated to be activated by
lipopolysaccharide in neutrophils and macrophages (32). The functions of SOCS3 in the lung
have previously been investigated. However, the role of the
SOCS3/JAK2/STAT3 signaling pathway in diabetes-induced lung injury
has not yet to be elucidated. The present study demonstrated that
SOCS3 may be involved in the regulation of JAK2/STAT3-mediated
signaling in HG-exposed A549 cells. To the best of our knowledge,
this is the first time that SOCS3 has been associated with the
modulation of JAK2/STAT3 signalingin an in vitro model of
diabetic lung injury.
During the pathogenesis of diabetic lung injury,
hyperglycemia triggers several intracellular processes in lung
cells, including the generation of reactive oxygen species, the
activation of protein kinase C and of various proinflammatory
cytokines (8). TNF-α and IL-6 are
proinflammatory cytokines that have been revealed to be upregulated
following exposure to HG (33). In
addition, the prolonged increase in IL-6 production during
inflammatory-induced lung injury has been associated with increased
mortality (34). Notably, SOCS3
has been implicated in the regulation of signaling by the IL-6
family of cytokines, through the inhibition of STAT3 activation
(35). In the present study, HG
exposure was revealed to potentiate TNF-α and IL-6 levels in A549
cells, whereas treatment with the JAK2/STAT3 inhibitor tyrphostin
AG490 attenuated the HG-induced increases in cytokine production.
Furthermore, the present findings demonstrated that SOCS3
overexpression similarly prevented the HG-induced upregulation of
TNF-α and IL-6 levels. In addition, the viability of A549 cells was
significantly decreased following exposure to HG, indicating the
development of HG-induced lung cell injury. These results suggested
that SOCS3 may inhibit the HG-induced upregulation of JAK2/STAT3
proteins, adhesion molecules and cytokines in the lungs, thus
suggesting a critical role for the SOCS3/JAK2/STAT3 pathway during
the inflammatory responses to hyperglycemia.
SOCS proteins are activated by several stimuli and
inhibit JAK/STAT signaling in a negative feedback loop involving
various mechanisms (36). In
agreement with previous data, HG increased the tyrosine
phosphorylation of JAK/STAT members in human MCs and HK2 cells
(37). HG may induce the
transcriptional activation of STAT3. Along with STAT activation, HG
transiently induced SOCS expression (21). In the present study, western blot
analysis demonstrated that HG exposure potentiated the expression
of SOCS3, JAK2 and STAT3. In addition, HG induced the
phosphorylation of JAK2 and STAT3 proteins compared with the
control groups, indicating that HG is a potent inducer of both JAK2
and STAT3 tyrosine phosphorylation. The JAK2 specific inhibitor
AG490 inhibited the HG-induced p-STAT3 protein expression. The
results also suggested that JAK2 serves an important role in HG
activation of STAT3. Similarly, SOCS3 overexpression significantly
inhibited HG-induced tyrosine phosphorylation JAK2 and STAT3. A549
cells were treated with D-mannitol to confirm that the effects of
HG treatment were not a result of hyperosmolarity.
Previous studies have suggested that the inhibition
of JAK/STAT signaling through various mechanisms, including JAK2
inhibition, STAT3 knockdown and pharmacological intervention, may
counteract HG-induced JAK/STAT activation and prevent the
development of HG-associated injury (25,38).
In the present study, SOCS3 overexpression was revealed to prevent
tyrosine phosphorylation of JAK2 and STAT3 induced by HG in A549
cells, thus suggesting that SOCS3 may protect against HG-induced
lung injury through the inhibition of the JAK2/STAT3 pathway to the
progression of chronic inflammatory diseases, as previous studies
have demonstrated (39–42). Therefore, it may be hypothesized
that strategies aiming to upregulate the expression of SOCS
proteins in the lungs have potential for the treatment of patients
with diabetic lung injury.
In conclusion, the present study demonstrated that
HG exposure increased SOCS3 expression, induced the activation of
the JAK2/STAT3 pathway and potentiated the production of
proinflammatory cytokines in A549 cells. Furthermore, SOCS3
overexpression and JAK2/STAT3 inhibition attenuated the HG-induced
morphological alterations in lung cells, enhanced their viability
and suppressed cytokine production. These findings suggested the
involvement of the JAK/STAT/SOCS pathway in hyperglycemia-induced
cell responses during the development of diabetic lung injury.
Further studies are required to elucidate the molecular mechanisms
underlying the roles of SOCS3 in the regulation of JAK2/STAT3
signaling and the modulation of inflammatory responses in the
lungs.
Acknowledgements
The present study was supported by the Renmin
Hospital Central Laboratory of Wuhan University (Wuhan, China), and
the National Natural Science Foundation of China (grant nos.
81471844 and 81501648).
References
|
1
|
Klein OL, Krishnan JA, Glick S and Smith
LJ: Systematic review of the association between lung function and
type 2 diabetes mellitus. Diabet Med. 27:977–987. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Wang HJ, Huang YL, Shih YY, Wu HY, Peng CT
and Lo WY: MicroRNA-146a decreases high glucose/thrombin-induced
endothelial inflammation by inhibiting NAPDH oxidase 4 expression.
Mediators Inflamm. 2014:3795372014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Hsia CC and Raskin P: Lung involvement in
diabetes: Does it matter? Diabetes Care. 31:828–829. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Orasanu G and Plutzky J: The pathologic
continuum of diabetic vascular disease. J Am CollCardiol. 53:(5
Suppl). S35–S42. 2009. View Article : Google Scholar
|
|
5
|
Pradhan AD, Manson JE, Rifai N, Buring JE
and Ridker PM: C-reactive protein, interleukin 6, and risk of
developing type 2 diabetes mellitus. JAMA. 286:327–334. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lee YS and Jun HS: Anti-inflammatory
effects of GLP-1-based therapies beyond glucose control. Mediators
Inflamm. 2016:30946422016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wang X, Bao W, Liu J, Ouyang YY, Wang D,
Rong S, Xiao X, Shan ZL, Zhang Y, Yao P and Liu LG: Inflammatory
markers and risk of type 2 diabetes: A systematic review and
meta-analysis. Diabetes Care. 36:166–175. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Williams MD and Nadler JL: Inflammatory
mechanisms of diabetic complications. Curr Diab Rep. 7:242–248.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Stuart MJ and Baune BT: Depression and
type 2 diabetes: Inflammatory mechanisms of a psychoneuroendocrine
co-morbidity. Neurosci Biobehav Rev. 36:658–676. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Liu Q, Xing L, Wang L, Yao F, Liu S, Hao
J, Liu W and Duan H: Therapeutic effects of suppressors of cytokine
signaling in diabetic nephropathy. J Histochem Cytochem.
62:119–128. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Hu C, Sun L, Xiao L, Han Y, Fu X, Xiong X,
Xu X, Liu Y, Yang S, Liu F and Kanwar YS: Insights into the
mechanisms involved in the expression and regulation of
extracellular matrix proteins in diabetic nephropathy. Curr Med
Chem. 22:2858–2870. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Marrero MB, Banes-Berceli AK, Stern DM and
Eaton DC: Role of the JAK/STAT signaling pathway in diabetic
nephropathy. Am J Physiol Renal Physiol. 290:F762–F768. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Dalpke A, Heeg K, Bartz H and Baetz A:
Regulation of innate immunity by suppressor of cytokine signaling
(SOCS) proteins. Immunobiology. 213:225–235. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Inagaki-Ohara K, Kondo T, Ito M and
Yoshimura A: SOCS, inflammation, and cancer. JAKSTAT.
2:e240532013.PubMed/NCBI
|
|
15
|
Krebs DL and Hilton DJ: SOCS proteins:
Negative regulators of cytokine signaling. Stem Cells. 19:378–387.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yasukawa H, Sasaki A and Yoshimura A:
Negative regulation of cytokine signaling pathways. Annu Rev
Immunol. 18:143–164. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Suchy D, Łabuzek K, Machnik G, Kozłowski M
and Okopień B: SOCS and diabetes-ups and downs of a turbulent
relationship. Cell Biochem Funct. 31:181–195. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Piessevaux J, Lavens D, Peelman F and
Tavernier J: The many faces of the SOCS box. Cytokine Growth Factor
Rev. 19:371–381. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Yoshimura A, Suzuki M, Sakaguchi R, Hanada
T and Yasukawa H: SOCS, inflammation, and autoimmunity. Front
Immunol. 3:202012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Sasaki A, Yasukawa H, Suzuki A, Kamizono
S, Syoda T, Kinjyo I, Sasaki M, Johnston JA and Yoshimura A:
Cytokine-inducible SH2 protein-3 (CIS3/SOCS3) inhibits Janus
tyrosine kinase by binding through the N-terminal kinase inhibitory
region as well as SH2 domain. Genes Cells. 4:339–351. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Carow B and Rottenberg ME: SOCS3, a major
regulator of infection and inflammation. Front Immunol. 5:582014.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Feng X, Tang H, Leng J and Jiang Q:
Suppressors of cytokine signaling (SOCS) and type 2 diabetes. Mol
Biol Rep. 41:2265–2274. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Alisson-Silva F, Freire-de-Lima L, Donadio
JL, Lucena MC, Penha L, Sá-Diniz JN, Dias WB and Todeschini AR:
Increase of O-glycosylated oncofetal fibronectin in high
glucose-induced epithelial-mesenchymal transition of cultured human
epithelial cells. PLoS One. 8:e604712013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Huang JS, Chuang LY, Guh JY, Huang YJ and
Hsu MS: Antioxidants attenuate high glucose-induced hypertrophic
growth in renal tubular epithelial cells. Am J Physiol Renal
Physiol. 293:F1072–F1082. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wang X, Shaw S, Amiri F, Eaton DC and
Marrero MB: Inhibition of the Jak/STAT signaling pathway prevents
the high glucose-induced increase in tgf-beta and fibronectin
synthesis in mesangial cells. Diabetes. 51:3505–3509. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Shi Y, Zhang Y, Wang C, Du C, Zhao S, Qi
Z, Zhang Q and Duan H: Suppressor of cytokine signaling-1 reduces
high glucose-induced TGF-beta1 and fibronectin synthesis in human
mesangial cells. FEBS Lett. 582:3484–3488. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Nakashima T, Yokoyama A, Onari Y, Shoda H,
Haruta Y, Hattori N, Naka T and Kohno N: Suppressor of cytokine
signaling 1 inhibits pulmonary inflammation and fibrosis. J Allergy
Clin Immunol. 121:1269–1276. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Severgnini M, Takahashi S, Tu P, Perides
G, Homer RJ, Jhung JW, Bhavsar D, Cochran BH and Simon AR:
Inhibition of the Src and Jak kinases protects against
lipopolysaccharide-induced acute lung injury. Am J RespirCrit Care
Med. 171:858–867. 2005. View Article : Google Scholar
|
|
29
|
Ortiz-Muñoz G, Lopez-Parra V, Lopez-Franco
O, Fernandez-Vizarra P, Mallavia B, Flores C, Sanz A, Blanco J,
Mezzano S, Ortiz A, et al: Suppressors of cytokine signaling
abrogate diabetic nephropathy. J Am Soc Nephrol. 21:763–772. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Gao H, Hoesel LM, Guo RF, Rancilio NJ,
Sarma JV and Ward PA: Adenoviral-mediated overexpression of SOCS3
enhances IgG immune complex-induced acute lung injury. J Immunol.
177:612–620. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Yan C, Ward PA, Wang X and Gao H: Myeloid
depletion of SOCS3 enhances LPS-induced acute lung injury through
CCAAT/enhancer binding protein δ pathway. FASEB J. 27:2967–2976.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Qin H, Roberts KL, Niyongere SA, Cong Y,
Elson CO and Benveniste EN: Molecular mechanism of
lipopolysaccharide-induced SOCS-3 gene expression in macrophages
and microglia. J Immunol. 179:5966–5976. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Venieratos PD, Drossopoulou GI,
Kapodistria KD, Tsilibary EC and Kitsiou PV: High glucose induces
suppression of insulin signalling and apoptosis via upregulation of
endogenous IL-1beta and suppressor of cytokine signalling-1 in
mouse pancreatic beta cells. Cell Signal. 22:791–800. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Goodman RB, Pugin J, Lee JS and Matthay
MA: Cytokine-mediated inflammation in acute lung injury. Cytokine
Growth Factor Rev. 14:523–535. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Croker BA, Krebs DL, Zhang JG, Wormald S,
Willson TA, Stanley EG, Robb L, Greenhalgh CJ, Förster I, Clausen
BE, et al: SOCS3 negatively regulates IL-6 signaling in vivo. Nat
Immunol. 4:540–545. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
36
|
Alexander WS and Hilton DJ: The role of
suppressors of cytokine signaling (SOCS) proteins in regulation of
the immune response. Annu Rev Immunol. 22:503–529. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Shi YH, Zhao S, Wang C, Li Y and Duan HJ:
Fluvastatin inhibits activation of JAK and STAT proteins in
diabetic rat glomeruli and mesangial cells under high glucose
conditions. Acta Pharmacol Sin. 28:1938–1946. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Manea SA, Manea A and Heltianu C:
Inhibition of JAK/STAT signaling pathway prevents
high-glucose-induced increase in endothelin-1 synthesis in human
endothelial cells. Cell Tissue Res. 340:71–79. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Hernández-Vargas P, López-Franco O,
Sanjuán G, Rupérez M, Ortiz-Muñoz G, Suzuki Y, Aguado-Roncero P,
Pérez-Tejerizo G, Blanco J, Egido J, et al: Suppressors of cytokine
signaling regulate angiotensin II-activated Janus kinase-signal
transducers and activators of transcription pathway in renal cells.
J Am SocNephrol. 16:1673–1683. 2005.
|
|
40
|
Gómez-Guerrero C, López-Franco O, Sanjuán
G, Hernández-Vargas P, Suzuki Y, Ortiz-Muñoz G, Blanco J and Egido
J: Suppressors of cytokine signaling regulate Fc receptor signaling
and cell activation during immune renal injury. J Immunol.
172:6969–6977. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Suzuki A, Hanada T, Mitsuyama K, Yoshida
T, Kamizono S, Hoshino T, Kubo M, Yamashita A, Okabe M, Takeda K,
et al: CIS3/SOCS3/SSI3 plays a negative regulatory role in STAT3
activation and intestinal inflammation. J Exp Med. 193:471–481.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Wong PK, Egan PJ, Croker BA, O'Donnell K,
Sims NA, Drake S, Kiu H, McManus EJ, Alexander WS, Roberts AW and
Wicks IP: SOCS-3 negatively regulates innate and adaptive immune
mechanisms in acute IL-1-dependent inflammatory arthritis. J Clin
Invest. 116:1571–1581. 2006. View Article : Google Scholar : PubMed/NCBI
|