Introduction
Varicella-zoster virus (VZV) is the causative agent
of chicken pox and herpes zoster, which is also known as shingles.
VZV is a member of the human herpesvirus family and has a 125 kb
double-stranded DNA genome that encodes 70 open reading frames
(ORFs), including 44 essential and 26 non-essential ORFs for viral
replication (1–7).
In 1974, chickenpox blister fluid was inoculated
into primary human embryonic lung cells, and the VZV Oka strain,
parent Oka, was isolated by Takahashi et al (8). Subsequently, a live attenuated Oka
vaccine (vOka) was successfully obtained following numerous
passages in human embryonic lung fibroblasts. While the
pathogenicity of vOka was significantly decreased, its
immunogenicity remained. The vOka strain is used in VZV vaccines
worldwide and is recommended by the World Health Organization;
however, it is still likely to cause delayed infection (9,10).
The incidence of chicken pox has decreased since the
chicken pox vaccine (vOka strain) was introduced in 1995 (11–13).
However, VZV outbreaks still occasionally occur (14,15),
and herpes zoster can cause serious harm to patient health. In
particular, VZV remains an important pathogenic factor since the
current herpes zoster vaccine only reduces the risk of infection by
50% (16).
Viral proliferation has been analyzed using highly
unstable FK506 binding protein (FKBP)12 protein mutants, which
rapidly degrade when they are expressed in mammalian cells
(17–19). Specifically, recombinant viruses
containing the destabilization domain of FKBP tagged to ORFs of
interest have been constructed using an FKBP tagged mutant method
(20). A synthetic ligand of FKBP,
Shield1, can penetrate the cell and stabilize the domain of the
FKBP fusion protein. As a result, the recombinant virus can
replicate in mammalian cells if Shield1 is added to the mammalian
cell culture medium. Conversely, if Shield1 is not added to the
mammalian cell culture medium, the FKBP-tagged fusion protein is
rapidly degraded, and the recombinant virus does not grow.
Accordingly, the FKBP-tagged fusion protein is a powerful genetic
tool for studying the role of viral genes and vaccines.
VZV ORF4 encodes transcription factors, whereas VZV
ORF48 encodes deoxyribonuclease, which is essential for the
formation of infectious virus particles (4). In the present study, recombinant VZV
containing FKBP-tagged ORF4 and 48 was constructed. The results of
the present study indicated that Shield1 can regulate replication
of the recombinant virus following its transfection into mammalian
cells.
Materials and methods
Bacterium and plasmids
The following bacterium and plasmids were used in
the present study: Escherichia coli SW102 strain,
galactokinase (galK) plasmid (pgalK), FKBP plasmid (pFKBP) and
SW102 VZV wild-type (WT) bacterial artificial chromosome
(BAC) system, which contains the whole VZVWT genome with
luciferase and chloramphenicol resistance genes. These plasmids and
bacterium were provided by Professor Hua Zhu (New Jersey Medical
School, Rutgers University, New Brunswick, NJ, USA).
Media
The following media were generated for use in the
present study: i) 1X M9 medium (500 ml):
Na2HPO4 (3 g), KH2PO4
(1.5 g), NH4Cl (0.5 g) and NaCl (0.25 g) were dissolved
in ddH2O to a final volume of 500 ml. The medium was
then autoclaved. ii) M63 minimal medium (5X M63):
(NH4)2SO4 (5 g),
KH2PO4 (34 g) and
FeSO4·7H2O (1.25 mg) were dissolved in
ddH2O to a final volume of 500 ml. The medium was then
adjusted to pH 7 with KOH and was autoclaved. Dulbecco's Modified
Eagle's medium (DMEM) with antibiotics (100 U/ml penicillin and
streptomycin) and DMEM without antibiotics were also used.
Reagents
Fetal bovine serum, Goldview II nuclear staining
dyes and lysogeny broth (LB) were provided by BioTeke Corporation
(Beijing, China). Qiagen Multiplex PCR kit, HotStarTaq DNA
polymerase, Qiagen Plasmid Mini kits and Qiaquick Gel Extraction
kit were provided by Qiagen GmbH (Hilden, Germany). Shield1 was
provided by Takara Biotechnology Co., Ltd. (Mountain View, CA,
USA).
Plate generation
The following diameter 9 cm plates were made for use
in the present study; i) M63 minimal galK plates: Initially, 7.5 g
agar was added to 400 ml H2O and was autoclaved.
Subsequently, 100 ml autoclaved 5X M63 medium was added to 0.5 ml 1
M MgSO4·7 H2O; this mixture was cooled to
50°C, after which 5 ml 20% galactose (sterile) was added as a
carbon source, 2.5 ml D-biotin (0.2 mg/ml, sterile), 2.25 ml
L-leucine (10 mg/ml, sterile) and 500 µl chloramphenicol (12.5
mg/ml, sterile) were also added and mixed. Finally, ~25 ml was
poured into each plate. ii) M63 minimal replace galK plates:
Initially, 7.5 g agar was added to 400 ml H2O and was
autoclaved. Subsequently, 100 ml autoclaved 5X M63 medium was added
to 0.5 ml 1 M MgSO4·7 H2O; this mixture was
cooled to 50°C, after which 5 ml 20% 2-deoxy-galactose (sterile),
2.5 ml D-biotin (0.2 mg/ml, sterile), 2.25 ml L-leucine (10 mg/ml,
sterile), 1.25 ml 20% glycerol (autoclaved) and 500 µl
chloramphenicol (12.5 mg/ml, sterile) were added and mixed.
Finally, ~25 ml was poured into each plate.
Cells
Human acute retinal pigment epithelial (ARPE-19)
cells were provided by Professor Hua Zhu (New Jersey Medical
School, Rutgers University, New Brunswick, NJ, USA).
Cell culture conditions
ARPE-19 cells were cultivated using 10 ml DMEM with
100 U/ml Penicillin-Streptomycin and 10% fetal bovine serum in a
diameter 10 cm petri dish, and were maintained at 37°C in a
humidified atmosphere of 5% CO2/95% air. Cell media was
changed every 2–3 days with fresh culture media.
Design and synthesis of primers
The primers for VZV ORF4 and ORF48, which were used
in the present study were designed according to the VZV gene
sequence provided by GenBank (NCBI Reference Sequence: NC_001348.1;
http://www.ncbi.nlm.nih.gov/nuccore/NC_001348). The
primers for VZV ORF4 and ORF48 galK, FKBP and Check primers (used
for verification plasmids of VZV ORF4 and ORF48 galK, FKBP) are
presented in Table I. The primers
were synthesized by Sangon Biotech Co., Ltd. (Shanghai, China).
 | Table I.Primers used in the present study. |
Table I.
Primers used in the present study.
| Primer | Sequence
(5′-3′) | Amplicon size
(bp) |
|---|
| VZV ORF4 |
|
|
| galK
F |
ggaagatacaggcaactgcaaacacgcaattgtcagatattttgcagccatgCCTGTTGACAATTAATCATCG | 1,400 |
| galK
R |
gtcttcacaaatagtagacacgtctgggtcggttggaattgaagcagaggcTCAGCACTGTCCTGCTCCTT |
|
| FKBP
F |
ggaagatacaggcaactgcaaacacgcaattgtcagatattttgcagccatgATGGGAGTGCAGGTGGAAAC |
450 |
| FKBP
R |
gtcttcacaaatagtagacacgtctgggtcggttggaattgaagcagaggcTTCCGGTTTTAGAAGCTCCAC |
|
| Check
F |
ggaagatacaggcaactgca |
|
| Check
R |
gtcttcacaaatagtagaca |
|
| VZV ORF48 |
|
|
| galK
F |
gttgttgtgtcacgataattcagaaatacgctcggatcaccctttattatgCCTGTTGACAATTAATCATCGGCA | 1,400 |
| galK
R |
ttttggctggctgggggcttatgtcgatcctatccaatcccgatcgtgcTCAGCACTGTCCTGCTCCTT |
|
| FKBP
F |
gttgttgtgtcacgataattcagaaatacgctcggatcaccctttattatgATGGGAGTGCAGGTGGAAACC |
450 |
| FKBP
R |
ttttggctggctgggggcttatgtcgatcctatccaatcccgatcgtgcTTCCGGTTTTAGAAGCTCCAC |
|
| Check
F |
gcactcaaagcttttcgaag |
|
| Check
R |
acaaaagggtgctgtagacc |
|
Preparation of SW102 VZVWT
BAC electrocompetent cells
SW102 VZVWT BAC electrocompetent cells
were prepared according to a previously described protocol
(1), and were stored at −80°C.
VZV ORF4 tagged with galK gene
Polymerase chain reaction (PCR) was used to generate
the VZV ORF4 galK cassette, as follows: The PCR reaction was
conducted in a volume of 100 µl, containing 1X PCR buffer, 3.0 mM
MgCl2, 10 µM dNTPs, 4 units HotStarTaq DNA polymerase,
10 µM VZV ORF4 galK primer sets (Table
I) and 200 ng pgalK DNA (the plasmid DNA was extracted using
the Qiagen Plasmid Mini kit (Qiagen GmbH), according to
manufacturer's protocol), using sterile water as negative control.
The PCR cycling conditions were as follows: Denaturation of the
template at 95°C for 15 min, followed by 31 cycles at 95°C for 30
sec, 55°C for 30 sec and 72°C for 1 min 30 sec, followed by a final
extension step at 72°C for 10 min and maintenance at 20°C. The PCR
products were resolved on a 1.0% agarose gel, visualized using
Goldview II nuclear staining dyes, after which the products were
cut, isolated and purified using a Qiaquick Gel Extraction kit
according to manufacturer's protocol.
The VZV ORF4 tagged with the galK gene was prepared
and verified as follows: 900 ng purified VZV ORF4 galK cassette was
electroporated into SW102-VZVWT BAC electrocompetent
cells, and the bacteria was immediately transferred to a tube
containing 1 ml LB and was incubated at 32°C for 1 h with
agitation. Following centrifugation (speed in 13,000 × g for 1 min
at room temperature), the supernatant was discarded, and the pellet
was washed twice with M9 medium. The bacteria were smeared onto M63
minimal galK plates, and colony growth was observed after
incubation at 32°C for 3 days. Two colonies were selected, and the
plasmid DNA was extracted. To verify that the SW102 colonies
contained the VZVORF4-galK-BAC, PCR was conducted as mentioned
above for VZV ORF4 galK cassette using the extracted DNA as a
template alongside the primers for ORF4 Check (Table I), and negative control using
sterile water. The correct clone was named
SW102-VZVORF4-galK-BAC.
The correct clone of SW102-VZVORF48-galK-BAC and
SW102-VZVORF4-galK-ORF48-galK-BAC was conducted in the same way as
the SW102-VZVORF4-galK-BAC protocol.
Electrocompetent cell SW102-VZVORF4-galK-BAC,
SW102-VZVORF48-galK-BAC and SW102-VZVORF4-galK-ORF48-galK-BAC were
prepared and stored at −80°C.
Counterselection to replace galK with
desired FKBP
PCR was used to generate VZVORF4-FKBP, as follows:
PCR was conducted in a 100 µl volume containing 1X PCR buffer, 3.0
mM MgCl2, 10 µM dNTPs, 4 units HotStar Taq DNA
polymerase, 10 µM VZVORF4 FKBP primer sets (Table I) and 200 ng DNA (pFKBP), using
sterile water as negative control. The PCR cycling conditions were
as follows: Denaturation of the template at 95°C for 15 min,
followed by 31 cycles at 95°C for 30 sec, 55°C for 30 sec and 72°C
for 30 sec, followed by a final extension step at 72°C for 10 min
and maintenance at 20°C. The PCR products were resolved by 1.0%
agarose gel electrophoresis, after which products were cut,
isolated and purified using a purification kit.
SW102-VZVORF4-FKBP was prepared and verified as
follows: 900 ng purified VZVORF4-FKBP PCR products were
electroporated into SW102-VZVORF4-galK-BAC electrocompetent cells.
Following electroporation, the bacteria were allowed to recover in
1 ml LB medium at 32°C for 4–5 h with agitation. This long recovery
period enriched for bacteria that only contained the desired
recombined BAC, and resulted in the loss of any BAC that still
contained the galK markers. The bacterial suspension was then
centrifuged (speed in 13,000 × g for 1 min at room temperature),
and the pellet was washed twice with M9 buffer. After the second
wash, the pellet was resuspended in 1 ml M9 medium, and 100 µl
bacteria (1:100 dilution) were plated onto M63 minimal plates
containing 2-deoxygalactose. The plates were incubated at 32°C for
3 days, after which colonies were observed. Two clones were
selected, and the plasmid DNA was extracted. To verify that the
SW102 colonies contained the VZVORF4-FKBP-BAC, PCR was conducted as
described above for VZVORF4-FKBP using the extracted DNA as a
template and the primers for ORF4 Check, negative control using
VZVWT DNA as a PCR template. The correct clone was named
SW102-VZVORF4-FKBP-BAC. SW102-VZVORF4-FKBP-BAC
electroporation-competent cells were then prepared and stored at
−80°C.
SW102-VZVORF48-FKBP-BAC and
SW102-VZVORF4-FKBP-ORF48-FKBP-BAC were obtained in the same manner
as described for SW102-VZVORF4-FKBP-BAC.
Maxiprep
DNA was extracted from VZVORF4-FKBP-BAC,
VZVORF48-FKBP-BAC and VZVORF4-FKBP-ORF48-FKBP-BAC according to a
previously published protocol (1).
The BAC DNA concentration was measured, and the DNA was stored at
4°C.
Transfection of BAC DNA and regulation
of Shield1
ARPE-19 were incubated in a 6-well-plate at 37°C in
a humidified atmosphere containing 5% CO2. Transfection
was performed using VZVWT−BAC, VZVORF4-FKBP-BAC,
VZVORF48-FKBP-BAC and VZVORF4-FKBP-ORF48-FKBP-BAC once the ARPE-19
cells reached 60% confluence.
The BAC DNA was transfected into ARPE-19 cells using
the FuGene 6 transfection kit (Roche Diagnostics, Indianapolis, IN,
USA), as follows: 6.0 µg BAC DNA was diluted to 200 µl using DMEM
(without antibiotics). Concurrently, 24 µl transfection reagent was
diluted to 400 µl with serum-free medium. The two solutions were
then mixed and incubated for 20 min at room temperature.
Subsequently, BAC DNA and transfection reagent mixtures for
VZVWT−BAC (335 ng/µl, 18 µl), VZVORF4-FKBP-BAC (338
ng/µl, 18 µl), VZVORF48-FKBP-BAC (365 ng/µl, 16.5 µl) and
VZVORF4-FKBP-ORF48-FKBP-BAC (355 ng/µl, 17 µl) were added to
ARPE-19 cells. Each transfection mixture was added to four wells of
cells, of which Shield1 (200 µM, 2 µl) was added to two wells, but
not to the remaining two wells. The medium was replaced 24 h
post-transfection. Subsequently, the medium was changed every 2–3
days. To observe viral growth, the cells were checked every day
with a fluorescence microscope.
Detection of VZVORF4-FKBP,
VZVORF48-FKBP and VZVORF4-FKBP-ORF48-FKBP in transfected cells
The transfected cells were cultured using DMEM with
100 U/ml Penicillin-Streptomycin, 10% fetal bovine serum, treated
with or without Shield1, and maintained at 37°C in humidified
atmosphere of 5% CO2/95% air. The media were changed
every 2–3 days. Then, the cells were collected after 20 days of
cultivation, and total RNA was isolated using the Micro-to-Midi
Total RNA Purification system kit (BioTeke Corporation) according
to the manufacturer's protocol. Reverse transcription (RT) of
isolated mRNA was then performed to produce double-stranded DNA.
The cDNA of transfected cells was verified by PCR (same as the
VZVORF4-FKBP PCR protocol) using ORF4 and ORF48 Check primers.
VZVWT cDNA was used as a control.
Titering and viral growth curve
analysis
Recombinant VZVORF4-FKBP, VZVORF48-FKBP and
VZVORF4-FKBP-ORF48-FKBP viruses were titered by infectious focus
assay. Briefly, ARPE-19 cells (2×105 cell/ml) were
suspended in DMEM with 100 U/ml penicillin-streptomycin, and 10%
fetal bovine serum, and treated with or without Shield1. Then, they
were inoculated in 12-well plates and were mixed with serial
dilutions of virus-infected ARPE-19 cells. Plaques were counted by
fluorescence microscopy 3 days postinfection. Growth curve analyses
were conducted based on live-cell bioluminescence. Briefly, ARPE-19
cells (2×105 cell/ml) were suspended in DMEM (with 100
U/ml penicillin-streptomycin) and 10% fetal bovine serum, and
treated with or without Shield1 were infected with the same amount
(1×105 PFU/ml) of VZVWT, VZVORF4-FKBP,
VZVORF48-FKBP and VZVORF4-FKBP-ORF48-FKBP viruses in 12-well
plates. Each virus was used to inoculate cells in 3 wells per
12-well plate. After 2 h at 37°C, 150 g/ml D-luciferin was added to
the cell culture media, and bioluminescent signals were quantified
and recorded using an in vivo imaging system (Biocompare,
South San Francisco, CA, USA) following incubation for 10 min at
37°C. Finally, cell culture media was replaced with new cell
culture media for further incubation. Measurements were repeated
every 24 h, for 7 days. Viral growth curves for each sample were
thus generated.
Results
Preparation of VZV ORFs tagged with
galK and FKBP
PCR was used to generate the VZV ORF4 and ORF48 galK
and FKBP cassettes. Successful PCR amplification of the VZV ORF4
and ORF48 galK cassettes was confirmed by the detection of 1,400 bp
PCR products on a 1.0% agarose gel (Fig. 1A). Consistent with the expected
results, ~500 bp PCR products, representing the VZV ORF4 and ORF48
FKBP cassettes (Fig. 1B), were
observed on a 1.0% agarose gel.
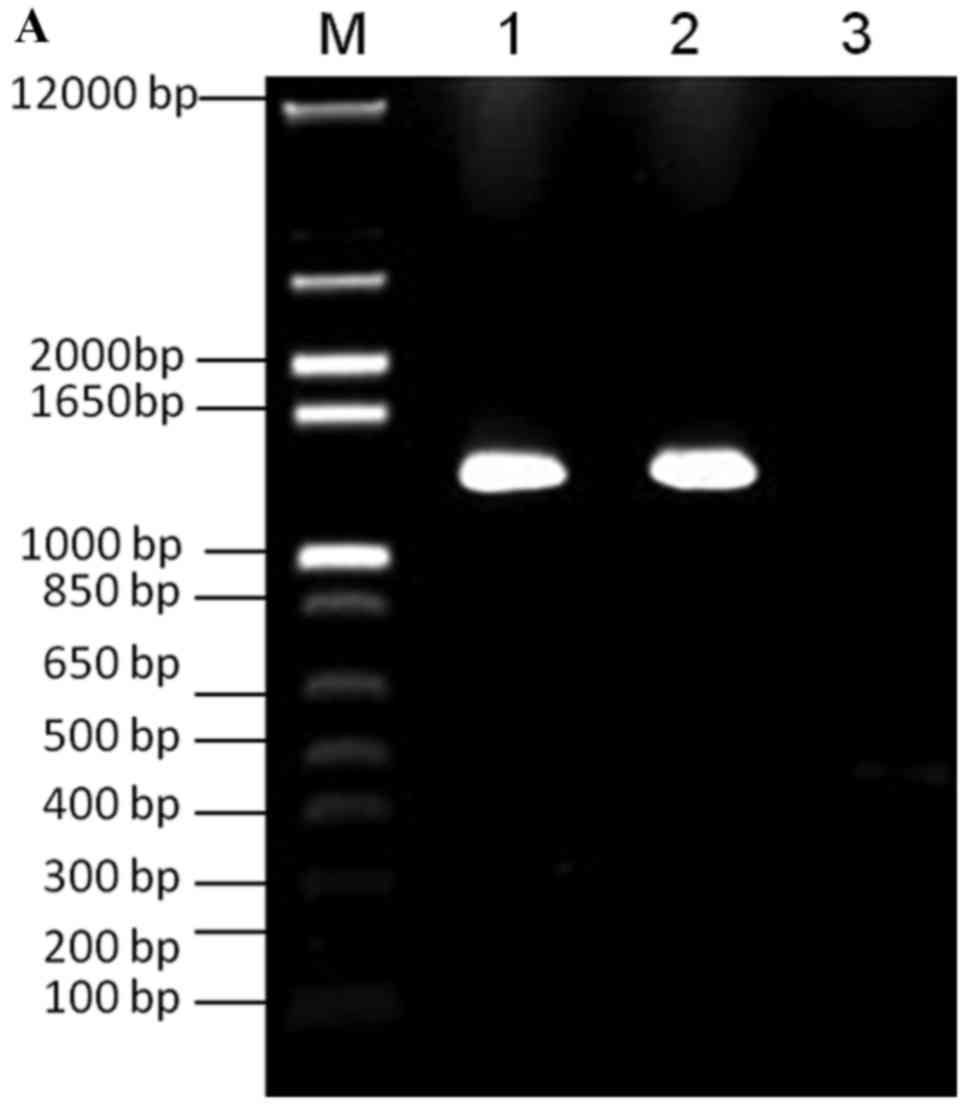 | Figure 1.PCR amplification of galK and FKBP.
(A) Lane M, 100 bp DNA ladder; lane 1, PCR amplification of VZV
ORF4 galK cassette; lane 2, PCR amplification of VZV ORF48 galK
cassette; lane 3, negative control. (B) Lane M, 100 bp DNA ladder;
lane 1, PCR amplification of VZVORF4 FKBP cassette; lane 2, PCR
amplification of VZV ORF48 FKBP cassette; lane 3, negative control.
FKBP, FK506 binding protein; galK, galactokinase; ORF, open reading
frame; PCR, polymerase chain reaction; VZV, varicella-zoster
virus. |
Verification of VZV ORF4, VZV ORF48
and VZV ORF4-ORF48 tagged with galK and FKBP
Two colonies were selected from the M63 minimal galK
plates following transformation of ORF4 and ORF48 galK cassettes
into SW102-VZVWTBAC electrocompetent cells. Plasmid DNA
was extracted and was used as a template for PCR verification of
the presence of galK. As expected, 1,400 bp PCR products were
detected on a 1.0% agarose gel (Fig.
2).
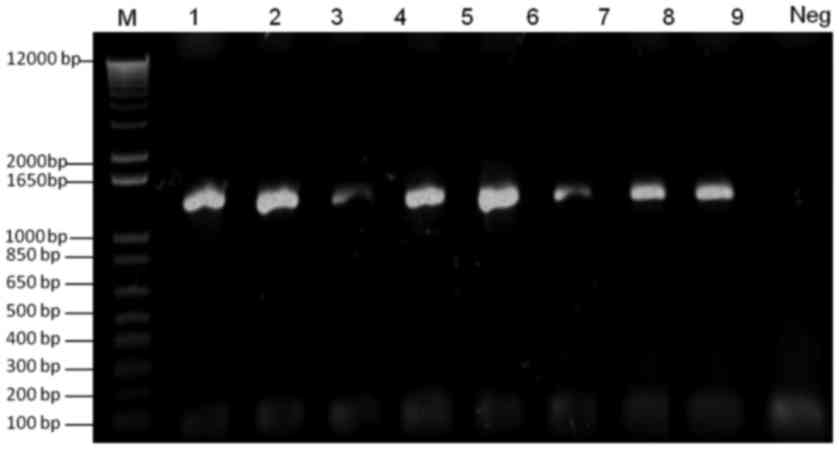 | Figure 2.Verification of galK-tagged VZV ORF4
and ORF48. Lane M, 100 bp DNA ladder; lanes 1 and 2, verification
of galK-tagged ORF4 from two clones of SW102-VZVORF4-galK-BAC;
lanes 3 and 4, verification of galK-tagged ORF48 from two clones of
SW102-VZVORF48-galK-BAC; lanes 5–8, verification of galK-tagged
ORF4 and ORF48 from four clones of
SW102-VZVORF4-galK-ORF48-galK-BAC; Neg, negative control. BAC,
bacterial artificial chromosome; galK, galactokinase; ORF, open
reading frame; PCR, polymerase chain reaction; VZV,
varicella-zoster virus. |
Counterselection was conducted to replace galK with
FKBP. Two colonies were selected from the M63 minimal plates
containing 2-deoxygalactose and chloramphenicol, following
electroporation to generate SW102-VZVORF4-galK-BAC,
SW102-VZVORF48-galK-BAC and SW102-VZVORF4-galK-ORF48-galK-BAC
electrocompetent cells. Plasmid DNA was extracted from the two
selected colonies, and used as a template for PCR verification.
Consistent with the expected results, 500 bp PCR products were
detected on a 1.0% agarose gel (Fig.
3). The clones were named SW102-VZVORF4-FKBP-BAC,
SW102-VZVORF48-FKBP-BAC and SW102-VZVORF4-FKBP-ORF48-FKBP-BAC.
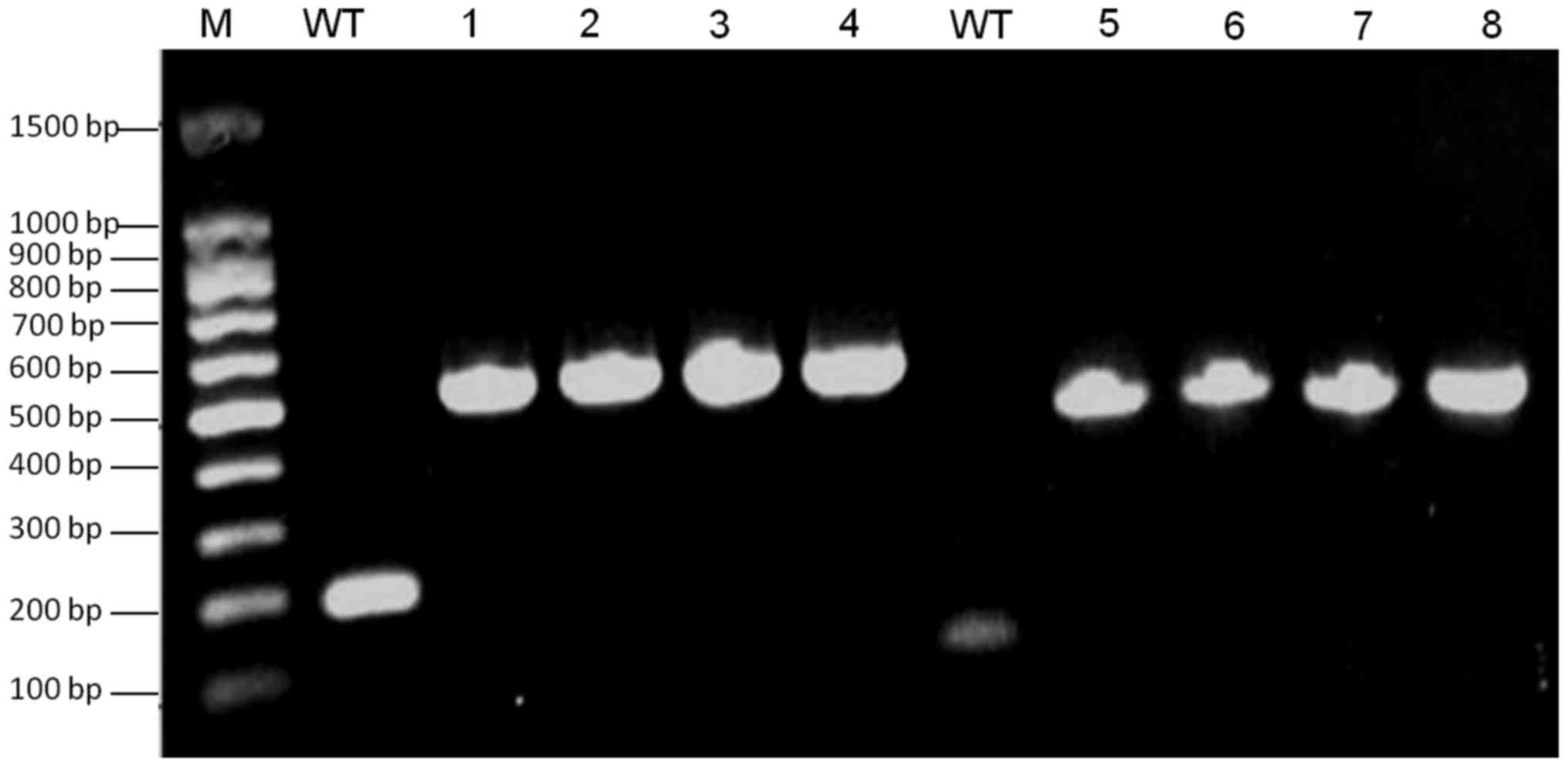 | Figure 3.Verification of FKBP-tagged VZV ORF4
and ORF48. Lane M, 100 bp DNA ladder; WT, negative control using
VZVWT DNA as a PCR template; lanes 1 and 2, detection of
ORF48-FKBP from SW102-VZVORF48-FKBP-BAC clones; lanes 3 and 4,
detection of ORF48-FKBP from SW102-VZVORF4-FKBP-ORF48-FKBP-BAC
clones; lanes 5 and 6 detection of ORF4-FKBP from
SW102-VZVORF4-FKBP-BAC clones; lanes 7 and 8, detection ORF4-FKBP
from SW102-VZVORF4-FKBP-ORF48-FKBP-BAC clones. BAC, bacterial
artificial chromosome; FKBP, FK506 binding protein; ORF, open
reading frame; PCR, polymerase chain reaction; VZV,
varicella-zoster virus; WT, wild type. |
Transfection
Plasmid DNA from SW102-VZVORF4-FKBP-BAC,
SW102-VZVORF48-FKBP-BAC and SW102-VZVORF4-FKBP-ORF48-FKBP-BAC was
transfected into ARPE-19 cells (Fig.
4). A total of 3 days post-transfection, green fluorescence,
indicating viral plaques, was observed in cells with and without
Shield1. After a longer incubation period, more fluorescence
plaques were observed in cells transfected with
SW102-VZVORF4-FKBP-ORF48-FKBP-BAC and treated with Shield1
(Fig. 4A), and in cells
transfected with VZVWT−BAC and treated with or without
Sheild1 (Fig. 4C and D), and in
cells transfected with SW102-VZVORF4-FKBP-BAC and
SW102-VZVORF48-FKBP-BAC and treated with or without Shield1 (data
not shown). Conversely, weak fluorescence was observed in cells
transfected with SW102-VZVORF4-FKBP-ORF48-FKBP-BAC DNA without
Shield1 (Fig. 4B).
Verification of FKBP from transfected
cells by RT-PCR
Transfected cells were collected and underwent RNA
extraction, after which RT-PCR was conducted. A ~500 bp product was
observed in cells transfected with SW102-VZVORF4-FKBP-BAC and
SW102-VZVORF48-FKBP-BAC, with or without Shield1 treatment, and in
cells transfected with SW102-VZVORF4-FKBP-ORF48-FKBP-BAC and
treated with Shield1 (Fig. 5). The
results of Fig. 5 demonstrated
that VZVWT, VZVORF4-FKBP and VZVORF48-FKBP may grow in
infected ARPE-19 cells if the cell media is treated with or without
Shield1, respectively; however, VZVORF4-FKBP-ORF48-FKBP did not
grow if the cell media was treated without Shield1, and
VZVORF4-FKBP-ORF48-FKBP grew if the ARPE-19 cells were treated with
Shield1.
 | Figure 5.Verification of FKBP from transfected
cells by reverse transcription polymerase chain reaction. Lanes 1
and 5, verification of VZVORF4-FKBP and VZVORF48-FKBP from ARPE-19
cells transfected with VZVORF4-FKBP-ORF48-FKBP-BAC without Shield1
treatment, respectively; lanes 2–4, verification of VZVORF4-FKBP
from ARPE-19 cells transfected with VZVORF4-FKBP-ORF48-FKBP-BAC and
treated with Shield1, and from ARPE-19 cells transfected with
VZVORF4-FKBP-BAC treated with or without Shield1, respectively;
lanes 6–8, verification of VZVORF48-FKBP from ARPE-19 cells
transfected with VZVORF4-FKBP-ORF48-FKBP-BAC and treated with
Shield1, and from ARPE-19 cells transfected with VZVORF48-FKBP-BAC
treated with or without Shield1, respectively; lanes 9 and 10,
negative control: ARPE-19 cells transfected with
VZVWT−BAC treated with or without Shield1, respectively.
BAC, bacterial artificial chromosome; FKBP, FK506 binding protein;
ORF, open reading frame; VZV, varicella-zoster virus; WT, wild
type. |
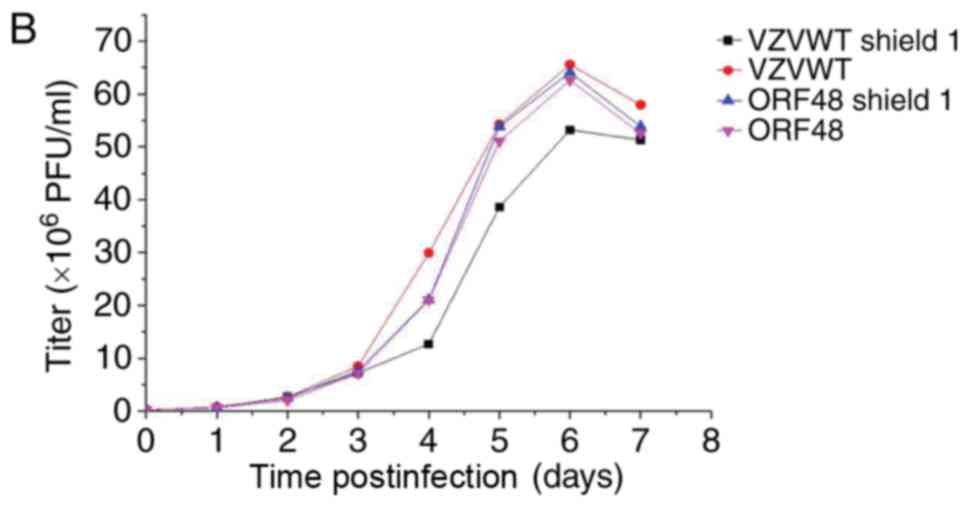
Titering and growth properties of
recombinant viruses in the presence and absence of Shield1
Each sample was tittered using an infectious focus
assay, and data were used to generate a final curve (data not
shown). For the bioluminescence assay, 3 wells from 12-well plates
were inoculated with the recombinant viruses, in order to measure
their growth properties in the presence and absence of Shield1 2 h
postinfection. Measurements were repeated every 24 h for 7 days
following addition of the reaction substrate D-luciferin to cells.
Bioluminescence data were collected and analyzed to generate viral
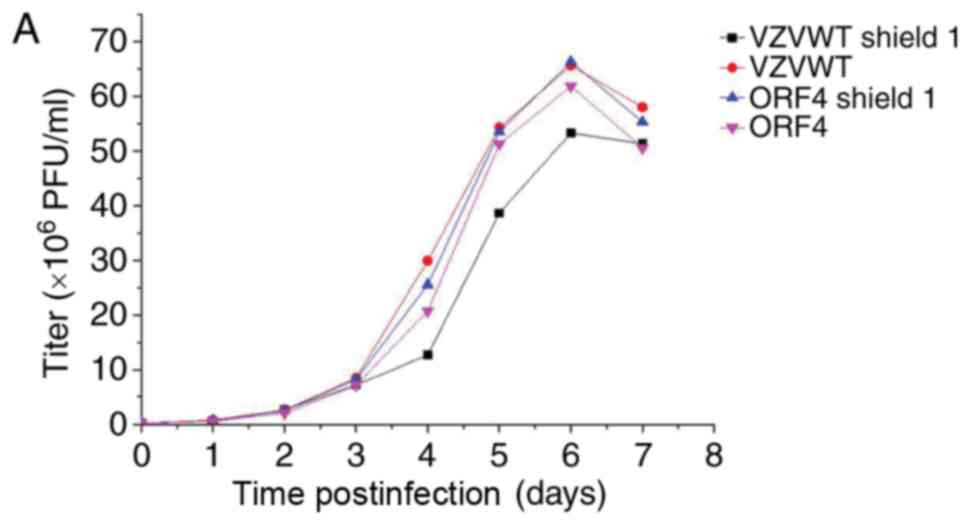
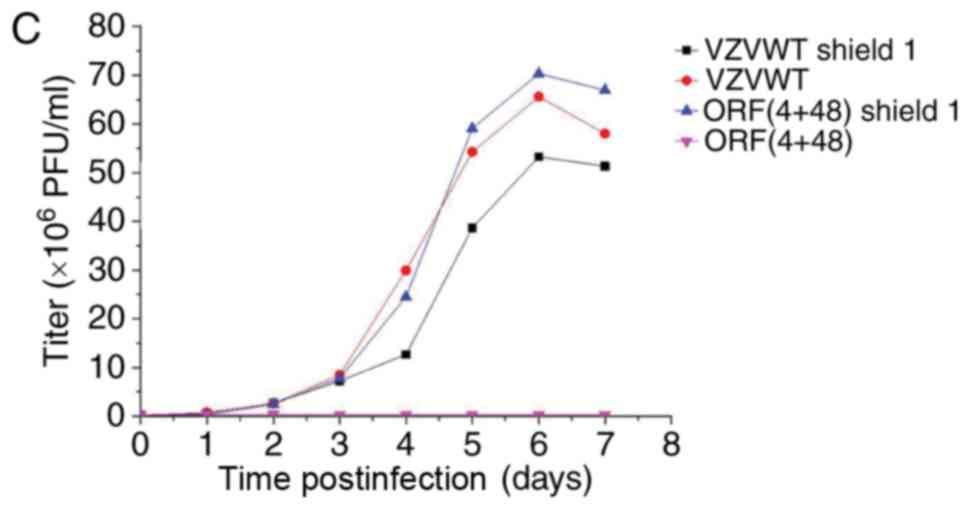
growth curves (Fig. 6A-C). The
results of Fig. 6 demonstrated
that the titer of the FKPB-tagged virus may be regulated by Shield1
in infected ARPE-19 cells.
Discussion
The development of BAC has been a major breakthrough
in virology, as BAC clones enable large fragments of viral genomes
to be cloned. BAC clones of viruses remain stable and can spread in
bacterial cells; therefore, the viral genome can easily operate in
bacterial cells. In addition, any required mutants can be easily
and quickly generated in bacterial cells (21–23).
The ability to rapidly regulate the functions of
specific proteins in living cells is a desirable tool for
biological research. Banaszynski et al (20) developed a general technique to
regulate the stability of the 12-kDa FKBP protein that can be fused
at either the N- or C-terminus when expressed in mammalian cells
(24). The synthetic ligand
Shield1 can bind to the destabilization domains and shield them
from degradation, thus allowing fused proteins to perform their
cellular functions.
Maximum stabilization typically was observed using
Shield1 with achieved maximum protein levels depending on FKBP
protein in live animals (25).
However, FKBP protein is degraded to background levels within 2–4 h
if Shield1 is removed. In addition, Shield1 stabilization resulted
in a >50-fold increase in mean fluorescence intensity of yellow
fluorescent protein in vitro (26). Ma et al (27) previously constructed a modified
pTREX vector, the N-terminal of which expressed
3flag-destabilization domain FKBP. The vector was subsequently
transfected into epimastigotes. The results demonstrated that the
fusion protein was gradually degraded in the absence of Shield1;
however, following addition of Shield1 to the parasites, the fusion
protein once again became detectable, indicating that fusion
protein expression and efficient function were regulated by Shield1
in various Trypanosoma cruzi life cycle stages.
The present study used a BAC system to clone long
fragments of viral DNA, and VZVORF4-FKBP-BAC, VZVORF48-FKBP-BAC and
VZVORF4-FKBP-ORF48-FKBP-BAC were constructed using the homologous
recombination method. A previously described method was used for
FKBP tagging (20), and the
ability of Shield1 to regulate protein degradation in FKBP-tagged
VZV ORF4-, ORF48- and ORF4-48-transfected cells during VZV
replication was studied. Human ARPE-19 cells were transfected with
the recombinant virus, and the FKPB-tagged viral proteins were
rapidly degraded by proteases. However, degradation of the
FKPB-tagged viral proteins could be prevented by the addition of
Shield1, thereby allowing viral replication in these epithelial
cells. The results demonstrated that only FKBP-tagged VZV ORF4-48
proteins were regulated by Shield1 in transfected ARPE-19 cells.
Conversely, FKBP-tagged VZV ORF4 and ORF48 proteins were not
regulated by Shield1 during VZV replication in transfected ARPE-19
cells. These results indicated that Shield1 may regulate
replication of VZVORF4-FKBP-ORF48-FKBP in infected ARPE-19 cells.
In conclusion, the present study is one of few studies that aimed
to determine the viral gene functions of VZV ORFs using FKBP tags,
and the recombinant VZVORF4-FKBP-ORF48-FKBP has the potential of
developing into a VZV vaccine, as Shield1 may regulate its
replication, and lay the foundation for preventing clinical
associated diseases. In the future, the authors of the present
study will analyze the immunological effects of Shield1 regulating
the replication of VZVORF4-FKBP-ORF48-FKBP in infected animals.
Acknowledgements
The present study was supported by the Project of
Science and Technology for Overseas Scholars in Hebei Province
(grant no. C201400559) and the Project of Hebei Education
Department (grant no. ZD2016003).
References
|
1
|
Visalli MA, House BL, Selariu A, Zhu H and
Visalli RJ: The varicella-zoster virus portal protein is essential
for cleavage and packaging of viral DNA. J Virol. 88:7973–7986.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Khalil MI, Arvin A, Jones J and Ruyechan
WT: A sequence within the varicella-zoster virus (VZV) OriS is a
negative regulator of DNA replication and is bound by a protein
complex containing the VZV ORF29 protein. J Virol. 85:12188–12200.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Berarducci B, Rajamani J, Zerboni L, Che
X, Sommer M and Arvin AM: Functions of the unique N-terminal region
of glycoprotein E in the pathogenesis of varicella-zoster virus
infection. Proc Natl Acad Sci USA. 107:282–287. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Zhang Z, Selariu A, Warden C, Huang G,
Huang Y, Zaccheus O, Cheng T, Xia N and Zhu H: Genome-wide
mutagenesis reveals that ORF7 is a novel VZV skin-tropic factor.
PLoS Pathog. 6:e10009712010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Zhang Z, Huang Y and Zhu H: An efficient
protocol for VZV BAC-based mutagenesis. Methods Mol Biol.
634:75–86. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zhang Z, Huang Y and Zhu H: A highly
efficient protocol of generating and analyzing VZV ORF deletion
mutants based on a newly developed luciferase VZV BAC system. J
Virol Methods. 148:197–204. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zhang Z, Rowe J, Wang W, Sommer M, Arvin
A, Moffat J and Zhu H: Genetic analysis of varicella-zoster virus
ORF0 to ORF4 by use of a novel luciferase bacterial artificial
chromosome system. J Virol. 81:9024–9033. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Takahashi M, Asano Y, Kamiya H, Baba K,
Ozaki T, Otsuka T and Yamanishi K: Development of varicella
vaccine. J Infect Dis. 197 Suppl 2:S41–S44. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Arvin AM: Varicella-zoster virus. Clin
Microbiol Rev. 9:361–381. 1996.PubMed/NCBI
|
|
10
|
Cohen JI: Varicella-zoster vaccine virus:
Evolution in action. Proc Natl Acad Sci USA. 104:7–8. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Centers for Disease Control and Prevention
(CDC): Decline in annual incidence of varicella-selected states,
1990–2001. MMWR Morb Mortal Wkly Rep. 52:884–885. 2003.PubMed/NCBI
|
|
12
|
Agopian A, Lopez A, Wilson D, Peralta V,
El Amin AN and Bialek S: Varicella hospitalizations in Los Angeles
during the varicella vaccination era, 2003–2011: Are they
preventable? Vaccine. 32:5353–5356. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Baxter R, Tran TN, Ray P, Lewis E, Fireman
B, Black S, Shinefield HR, Coplan PM and Saddier P: Impact of
vaccination on the epidemiology of varicella: 1995–2009.
Pediatrics. 134:24–30. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Galil K, Lee B, Strine T, Carraher C,
Baughman AL, Eaton M, Montero J and Seward J: Outbreak of varicella
at a day-care center despite vaccination. N Engl J Med.
347:1909–1915. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Gan L, Wang M, Yang S, Gershon AA and Chen
JJ: Transmission of varicella vaccine virus to a non-family member
in China. Vaccine. 29:2015–2017. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Oxman MN, Levin MJ, Johnson GR, Schmader
KE, Straus SE, Gelb LD, Arbeit RD, Simberkoff MS, Gershon AA, Davis
LE, et al: A vaccine to prevent herpes zoster and postherpetic
neuralgia in older adults. N Engl J Med. 352:2271–2284. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Das S, Ortiz DA, Gurczynski SJ, Khan F and
Pellett PE: Identification of human cytomegalovirus genes important
for biogenesis of the cytoplasmic virion assembly complex. J Virol.
88:9086–9099. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Maetzig T, Kuehle J, Schwarzer A, Turan S,
Rothe M, Chaturvedi A, Morgan M, Ha TC, Heuser M, Hammerschmidt W,
et al: All-in-One inducible lentiviral vector systems based on drug
controlled FLP recombinase. Biomaterials. 35:4345–4356. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Perng YC, Qian Z, Fehr AR, Xuan B and Yu
D: The human cytomegalovirus gene UL79 is required for the
accumulation of late viral transcripts. J Virol. 85:4841–4852.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Banaszynski LA, Chen LC, Maynard-Smith LA,
Ooi AG and Wandless TJ: A rapid, reversible, and tunable method to
regulate protein function in living cells using synthetic small
molecules. Cell. 126:995–1004. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Murphy E, Yu D, Grimwood J, Schmutz J,
Dickson M, Jarvis MA, Hahn G, Nelson JA, Myers RM and Shenk TE:
Coding potential of laboratory and clinical strains of human
cytomegalovirus. Proc Natl Acad Sci USA. 100:14976–14981. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Warden C, Tang Q and Zhu H: Herpesvirus
BACs: Past, present, and future. J Biomed Biotechnol.
2011:1245952011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Yu D, Silva MC and Shenk T: Functional map
of human cytomegalovirus AD169 defined by global mutational
analysis. Proc Natl Acad Sci USA. 100:12396–12401. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Bonger KM, Chen LC, Liu CW and Wandless
TJ: Small molecule displacement of a cryptic degron causes
conditional protein degradation. Nat Chem Biol. 7:531–537. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Banaszynski LA, Sellmyer MA, Contag CH,
Wandless TJ and Thorne SH: Chemical control of protein stability
and function in living mice. Nat Med. 14:1123–1127. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Sellmyer MA, Thorne SH, Banaszynski LA,
Contag CH and Wandless TJ: A general method for conditional
regulation of protein stability in living animals. Cold Spring Harb
Protoc. 2009:pdb.prot51732009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Ma YF, Weiss LM and Huang H: A method for
rapid regulation of protein expression in Trypanosoma cruzi.
Int J Parasitol. 42:33–37. 2012. View Article : Google Scholar : PubMed/NCBI
|