Introduction
Inflammasomes are cytosolic multiprotein complexes
which induce the production of proinflammatory cytokines, primarily
interleukin (IL)-1β and IL-18, in response to pathogens or danger
signals (1,2). Inflammasome complexes are composed
primarily of the apoptosis-associated speck-like protein containing
a caspase activation and recruitment domain, a sensor protein and
caspase-1. Distinct inflammasome complexes specifically respond to
various signals via a diverse set of associated sensor proteins.
The nucleotide binding domain and leucine-rich repeat pyrin 3
domain (NLRP3) is a well-characterized sensor protein that forms
the NLRP3 inflammasome, which is activated by exogenous pathogens
or endogenous danger signals, including mitochondrial dysfunction
and the accumulation of reactive oxygen species (ROS) (3–5).
Activation of the NLRP3 inflammasome leads to the auto-activation
of procaspase-1; procaspase-1 subsequently catalyzes pro-IL-1β
maturation to IL-1β, which is then secreted (1–5).
Rheumatoid arthritis (RA) is a chronic inflammatory
disease, which is induced by genetic and/or environmental factors.
Of these, pro-inflammatory cytokines, such as tumor necrosis
factor-α and IL-1β, are well-characterized inducers (6–8).
Notably, the NLRP3 inflammasome is reportedly associated with RA
susceptibility and severity in certain animal models and human
studies (9–11). A previous study involving a
spontaneous arthritis mouse model (A20mye-KO mice)
demonstrated that the pathology of arthritis may be associated with
the NLRP3 inflammasome/IL-1β axis (12).
Cellular metabolism has been increasingly recognized
as an endogenous controller of NLRP3 inflammasome activation
(3,5). Saturated free fatty acids are potent
simulators of the NLRP3 inflammasome and IL-1β secretion (13). In addition, mitochondrial
dysfunction and ROS accumulation may induce NLRP3 expression and
inflammasome activation (4,5).
Modulation of cellular metabolism has been demonstrated to regulate
NLRP3 inflammasome activity and associated disease pathologies,
such as Alzheimer's disease and liver fibrosis, in a number of
previous studies (1,14,15).
MicroRNAs (miRs) form one of the largest groups of
post-transcriptional regulatory factors (16,17).
miRs possess 2–8 bases at the 5′-terminal that bind to the
3′-untranslated region (3′-UTR) of a target mRNA, thereby
repressing gene translation and decreasing protein expression
(17). By regulating the
expression of key enzymes or regulatory factors, miRs serve a role
in regulating cellular metabolism (18). miR-33 has been previously
demonstrated to regulate cellular lipid metabolism and to repress
the expression of genes associated with cholesterol efflux
(19), high-density lipoprotein
biogenesis (20) and fatty acid
oxidation (21). A recent study
reported that a macrophage-specific miR-33 deletion increases
oxidative respiration, enhances spare respiratory capacity and
induces the expression of an M2 macrophage polarization-associated
gene profile (22). However, the
mechanism associating miR-33 with macrophage activation remains to
be completely elucidated.
In the present study, the authors hypothesized that
miR-33 may regulate the mitochondrial function-dependent activity
of the NLRP3 inflammasome in macrophages. Through gain and loss of
function studies, the role of miR-33 in regulating the NLRP3
inflammasome/IL-1β axis in primary mouse macrophages was
investigated. In addition, the expression of miR-33 and NLRP3
inflammasome-associated genes was measured in peripheral blood
monocytes obtained from patients with RA and compared with those of
healthy donors.
Materials and methods
Isolation of peritoneal
macrophages
A total of 130 C57BL/6 mice, provided by Model
Animal Resource Information Platform of Nanjing University
(Nanjing, China), were used the present study. Mice, housed five
per cage, were supplied with sterile water and food ad libitum in a
pathogen-free facility with a 12-h light/dark cycle. The
temperature was maintained at 25°C and the humidity at 55%. Each
mouse (male; age, 8 weeks; weight, 20–24 g) was injected
intraperitoneally with 2 ml 3% thioglycollate (Difco; BD
Biosciences, Franklin Lakes, NJ, USA) on day 1, and was sacrificed
with CO2 or isoflurane on day 3. Following an
intraperitoneal injection of 5 ml Dulbecco's modified Eagle's
medium (cat. no. 11965118; Gibco; Thermo Fisher Scientific, Inc.,
Waltham, MA, USA) containing 10% fetal bovine serum (cat. no.
10100147; Thermo Fisher Scientific, Inc.) and 1% penicillin and
streptomycin (cat. no. 15070063; Gibco; Thermo Fisher Scientific,
Inc.), the peritoneal cells were collected and transferred to cell
culture dishes maintained at 37°C in a humidified 5% CO2
atmosphere. Following 2 h, the floating cells were removed by
washing the cells with phosphate-buffered saline (PBS). The
adherent cells were considered to be peritoneal macrophages and
employed for subsequent experiments. The use of animals in the
present study was approved by the Ethics Committee of Chengdu
Military General Hospital (Chengdu, China).
Treatment of the peritoneal
macrophages
All treated macrophages were maintained at 37°C in a
humidified 5% CO2 atmosphere.
For measurement of mRNA and protein expression,
peritoneal macrophages were transfected with PBS (control), a
scramble miR (100 nM, cat. no. AM17010), 100 nM miR-33 mimic (cat.
no. MC12410) or 100 nM anti-miR-33 (cat. no. AM12410) (all from
Thermo Fisher Scientific, Inc.) for 5 h and then treated with PBS
or lipopolysaccharide (LPS; 50 ng/ml, cat. no. L2630;
Sigma-Aldrich) for 24 h before cells were harvested. For
measurement of ELISA, peritoneal macrophages were treated as
aforementioned before collecting the supernatants.
For measurement of ROS levels, peritoneal
macrophages were treated as aforementioned. Alternatively,
peritoneal macrophages were transfected with a scramble miR (100
nM) or an miR-33 mimic (100 nM) for 5 h and subsequently treated
with PBS (control), 2 µg/ml oligomycin, 1 µg/ml carbonyl cyanide
p-(trifluoromethoxy) phenylhydrazone (FCCP), 1 µg/ml rotenone + 1
µg/ml antimycin A (all the mitochondrial inhibitors were from the
Agilent Seahorse XF Cell Mito Stress test kit; cat. no. 103015-100;
Agilent Technologies, Inc., Santa Clara, CA, USA), 20 mM
N-acetylcysteine (NAC; cat. no. S0077; Beyotime Institute of
Biotechnology, Shanghai, China) or 10 mM glutathione (GSH; cat. no.
S0073; Beyotime Institute of Biotechnology) for 24 h. For
measurement of mitochondrial oxygen consumption rate (OCR),
peritoneal macrophages were transfected with a scrambled miR (100
nM) or an miR-33 mimic (100 nM) for 24 h, followed by the treatment
of oligomycin (2 µg/ml), FCCP (1 µg/ml) and rotenone (1 µg/ml) plus
antimycin A (1 µg/ml) one after another in the XFe96 Extracellular
Flux analyzer (Seahorse Bioscience; Agilent Technologies, Inc.).
For measurement of caspase-1 activity, peritoneal macrophages were
transfected with a scrambled miR (100 nM) or miR-33 mimic (100 nM)
for 5 h, and then treated with LPS (50 ng/ml) or PBS as a control,
plus DMSO, NAC (20 mM), GSH (10 mM) or FCCP (1 µg/ml) for 24 h.
Collection of human samples
All experiments involving human subjects were
approved by the Ethics Committee of the Chengdu Military General
Hospital, and informed consent was provided by all subjects. Blood
samples were obtained from 10 male patients with RA (age, 54.3±3.2
years) and from 10 healthy male donors (age, 52.5±3.6 years).
Peripheral blood monocytes were isolated for the subsequent
analysis of mRNA or protein expression. The plasma was subjected to
enzyme-linked immunosorbent assay (ELISA) analysis to detect IL-1β
expression. Patients enrolled to the present study were originally
admitted to the Chengdu Military General Hospital (Chengdu,
Sichuan, China) from January to June in 2015.
Gain or loss of function studies
Primary peritoneal macrophages were isolated and
seeded into a 6-well plate at 80–90% density, and maintained at
37°C in a humidified 5% CO2 atmosphere 10 h before
transfection. Overexpression of miR-33 was achieved by transfecting
the mouse peritoneal macrophages with a miR-33 mimic. Inhibition of
miR-33 was achieved by transfecting the macrophages with an
anti-miR-33. A scrambled miR was used as a control. NLRP3
expression in macrophages was silenced using a pre-designed small
interfering (si)RNA sequence targeting NLRP3 (cat. no. 4390771;
Thermo Fisher Scientific, Inc.). The negative siRNA control was
also provided commercially (cat. no. 4390843; Thermo Fisher
Scientific, Inc.). For macrophage transfection, the working
concentration of the miRs was 100 nM, and for the siRNAs it was 20
nmol/ml. The transfection reagent was Lipofectamine 2000 (cat. no.
11668500; Thermo Fisher Scientific, Inc.). The cells were incubated
with miR-33, anti-miR-33, si-NLRP3 or the scramble for 5 h, and
then stimulated with PBS (1,000X) or LPS (50 ng/ml) for 24 h prior
to further analysis.
Protein extraction and immunoblotting
assays
Mouse peritoneal macrophages were lysed with
radioimmunoprecipitation Lysis and Extraction Buffer (cat. no.
89900; Thermo Fisher Scientific, Inc.) supplemented with 1%
protease cocktails 1, 2 and 3, and 1 mM phosphatase inhibitors (all
Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) and shaken for 30
min prior to centrifugation at 12,000 × g for 30 min at 4°C. The
supernatant was then collected and quantified using a bicinchoninic
acid assay kit (Roche Applied Science, Penzberg, Germany). The
extracted proteins (50 mg/well) were separated via SDS-PAGE on a
10% gel, and transferred to a polyvinylidene difluoride membrane.
The membrane was blocked with 5% non-fat milk at 4°C overnight, and
then incubated with anti-NLRP3 (cat. no. 15101, dilution 1:1,000;
Cell Signaling Technology, Inc., Danvers, MA, USA), anti-caspase-1
p20 (cat. no. 1780, dilution 1:1,000; Santa Cruz Biotechnology,
Inc., Dallas, TX, USA) or anti-β-actin (cat. no. 8457, dilution
1:1,000; Cell Signaling Technology, Inc.) for 10 h at 4°C. The
membrane was rinsed 3 times with PBS containing 0.1% Tween-20, and
incubated for 1 h with the appropriate horseradish
peroxidase-conjugated secondary antibodies (cat. nos. A0181 and
A0208, dilution 1:1,000; Beyotime Institute of Biotechnology) at
37°C. The membrane was then washed with PBS containing 0.1%
Tween-20 and incubated with enhanced chemiluminescence substrate
(cat. no. NEL105001EA; PerkinElmer, Inc., Waltham, MA, USA) for 1
min at 4°C. The signals were captured using a ChemiDoc MP System
(cat. no. 170-8280; Bio-Rad Laboratories, Inc., Hercules, CA,
USA).
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA from the mouse peritoneal macrophages and
human blood monocytes was isolated using TRIzol reagent
(Invitrogen; Thermo Fisher Scientific, Inc.). RNA (1 mg) was
reverse transcribed into cDNA using the Omniscript RT kit (Qiagen
GmbH, Hilden, Germany), according to the manufacturer's protocol.
qPCR was performed using a 7900HT Fast Real-Time PCR system
(Applied Biosystems; Thermo Fisher Scientific, Inc.). The mRNA
expression levels were normalized to β-actin. Reactions were
performed in duplicate using Applied Biosystems TaqMan Gene
Expression assays and Universal PCR Master Mix (Applied Biosystems;
Thermo Fisher Scientific, Inc.) The probes (human NLRP3, cat. no.
Hs00366465_m1; human IL-1β, cat. no. Hs01555410_m1; human β-actin,
cat. no. Hs99999903_m1; mouse NLRP3, cat. no. Mm04210227_g1; mouse
IL-1β, cat. no. Mm00434228_m1; mouse β-actin, cat. no.
Mm99999915_g1) were provided commercially by Applied Biosystems;
Thermo Fisher Scientific, Inc. The thermocycling conditions were as
follows: Denaturation at 95°C, followed by 40 cycles of
denaturation at 95°C for 15 sec and annealing at 60°C for 1 min.
Relative target gene expression was calculated using the
2−ΔΔCq method (23).
ROS assays
miR-33 mimic or anti-miR-33-transfected peritoneal
macrophages were seeded in 96-well plates (80–90% fusion) and
treated with 50 µM
5-(and-6)-chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate
acetyl ester (DCFDA; cat. no. D6883; Sigma-Aldrich; Merck KGaA) for
6 h at 37°C. ROS production was determined by measuring the level
of DCFDA hydrolysis to fluorescent 2′,7′-dichlorofluorescein, which
is promoted by a number of reactive radical species, and allows for
the assessment of general oxidative stress. DCFDA conversion was
kinetically measured at 0, 2, 4 and 6 h using a microplate reader
(BioTek Instruments, Inc., Winooski, VT, USA) at 488 nm excitation
and 535 nm emission wavelengths.
Caspase-1 activity assay
Caspase-1 activity was measured using the
Caspase-1/ICE Colorimetric Assay kit (cat. no. BF15100; R&D
Systems, Inc., Minneapolis, MN, USA), according to the
manufacturer's instructions. The macrophages and human blood
monocytes were lysed in the kit lysis buffer and diluted to a final
protein concentration of 2–4 mg/ml. The enzymatic reaction was
performed in a 96-well flat-bottom microplate. Each reaction
contained 50 µl cell lysate and 50 µl 2X reaction buffer, to which
5 µl caspase-1 colorimetric substrate was added and incubated at
37°C for 1.5 h. The plate was read using a microplate reader at a
wavelength of 405 nm. The activity of caspase-1 was displayed as
optical density (OD)405/mg protein.
Mitochondrial biogenesis and function
assays
Mitochondrial oxygen consumption rates (OCRs) of the
scramble miR or miR-33 mimic-transfected macrophages were measured
following different treatments in 96-well plates (10,000
cells/well) using a Seahorse XF Cell Mito Stress Test kit (cat. no.
101706-100; Seahorse Bioscience; Agilent Technologies, Inc.) on the
XFe96 Extracellular Flux Analyzer (Seahorse Bioscience; Agilent
Technologies, Inc.) according to the manufacturer's instructions.
Briefly, basal cellular OCRs were recorded in the absence of
metabolic inhibitors or uncouplers. ATP synthase was inhibited with
2 µg/ml oligomycin, followed by uncoupling of the respiratory chain
from oxidative phosphorylation by stepwise titration with 1 µg/ml
FCCP to achieve maximal OCRs. Subsequently, rotenone (Mito
Inhibitor B), a complex I inhibitor (1 µg/ml), and antimycin A
(Mito Inhibitor A), a complex III inhibitor (1 µg/ml), were
combined to completely inhibit the mitochondrial respiratory chain.
The results are presented as the OCR (pmol/min).
ELISA analysis
IL-1β levels in the supernatants of cultured
macrophages were measured using a IL-1β ELISA kit (cat. no. MLB00C;
R&D Systems, Inc.), and IL-1β levels in blood plasma were
measured using another ELISA kit (cat. no. DLB50; R&D Systems,
Inc.), according to the manufacturer's instructions. The final
cytokine concentration in the supernatants of cultured cells was
normalized to the total number of cells. The mouse or human IL-1β
standard was included in the corresponding kit. These tests were
performed using a microplate reader (BioTek Instruments, Inc.) set
to 450 nm.
Statistical analysis
Statistical analysis was performed using GraphPad
Prism version 5.01 (GraphPad Software, Inc., La Jolla, CA, USA).
The results are expressed as the mean ± standard error of the mean
and were analyzed using one-way analysis of variance followed by
the Tukey post hoc test for multiple comparisons, and a two-tailed
unpaired Student's t-test. P<0.05 was considered to indicate a
statistically significant difference.
Results
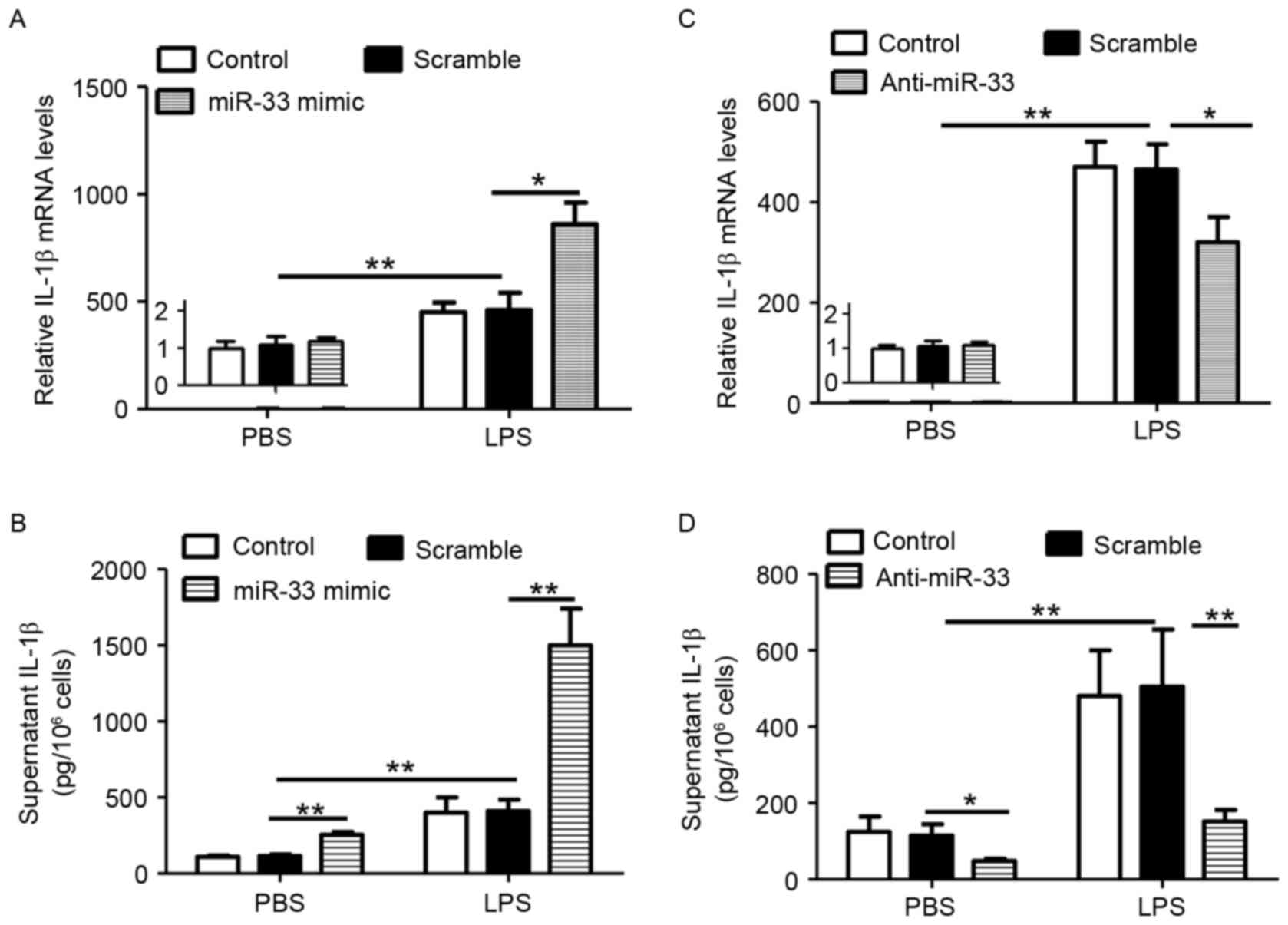
miR-33 potentiates the secretion of
IL-1β in primary macrophages
In order to determine whether miR-33 regulates the
inflammasome pathway, the expression of IL-1β in miR-33 deficient
or proficient macrophages was measured. To increase inflammation
signaling, LPS was also employed. As presented in Fig. 1A and B, treatment with a miR-33
mimic significantly increased LPS-stimulated IL-1β expression at
the mRNA and protein levels when compared with scramble
miR-transfected controls. By contrast, miR-33 did not stimulate
IL-1β mRNA levels in the absence of LPS treatment (Fig. 1A), however, it significantly
induced IL-1β secretion (Fig. 1B).
Consistent with these observations, treatment of macrophages with
anti-miR-33 and/or LPS resulted in the inverse effects on IL-1β
expression and secretion (Fig. 1C and
D). Therefore, the results indicated that miR-33 may increase
IL-1β secretion by stimulating the inflammasome pathway.
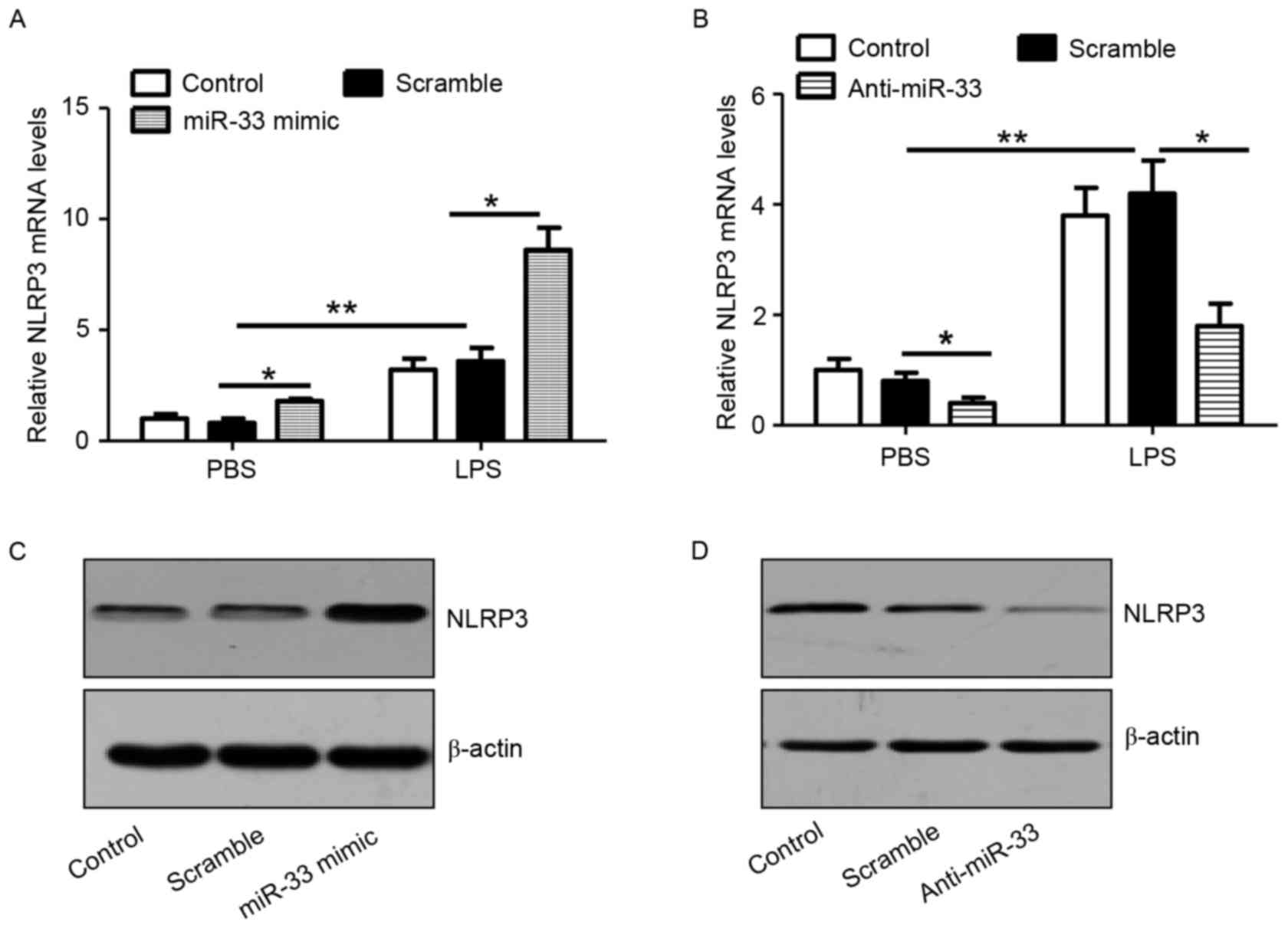
miR-33 increases the expression of
NLRP3
As shown in Fig.
2A, transfection of macrophages with the miR-33 mimic
significantly stimulated NLRP3 mRNA expression in cells with or
without LPS treatment when compared with scramble miR-transfected
controls. Consistently, anti-miR-33 significantly attenuated the
expression levels of NLRP3 mRNA in PBS or LPS-treated macrophages
when compared with scramble miR-transfected controls (Fig. 2B). In addition, the results
confirmed that treatment with a miR-33 mimic or anti-miR-33 induced
or attenuated NLRP3 protein expression in peritoneal macrophages,
respectively (Fig. 2C and D).
miR-33 increases the activity of
caspase-1
The NLRP3 inflammasome activates caspase-1, and
caspase-1 cleaves pro-IL-1β protein to produce mature IL-1β, which
is subsequently secreted (2,5).
Therefore, the present study investigated the role of miR-33 in
regulating caspase-1 activity. Treatment of PBS or LPS-stimulated
peritoneal macrophages with the miR-33 mimic significantly induced
the activity of caspase-1 when compared with scramble
miR-transfected controls (Fig.
3A). Consistent with this result, the expression of the active
form of caspase-1, p20, was increased by miR-33 transfection when
compared with the controls (Fig.
3B). By contrast, transfection with anti-miR-33 attenuated
caspase-1 activity in PBS and LPS-stimulated cells (Fig. 3C). These results indicated that
miR-33 may regulate the inflammasome pathway in macrophages. As
demonstrated in Fig. 3D,
transfection of macrophages with si-NLRP3 effectively silenced
NLRP3 protein expression. In addition, miR-33 was observed to
induce caspase-1 activity and IL-1β secretion in an NLRP3-dependent
manner (Fig. 3E and F).
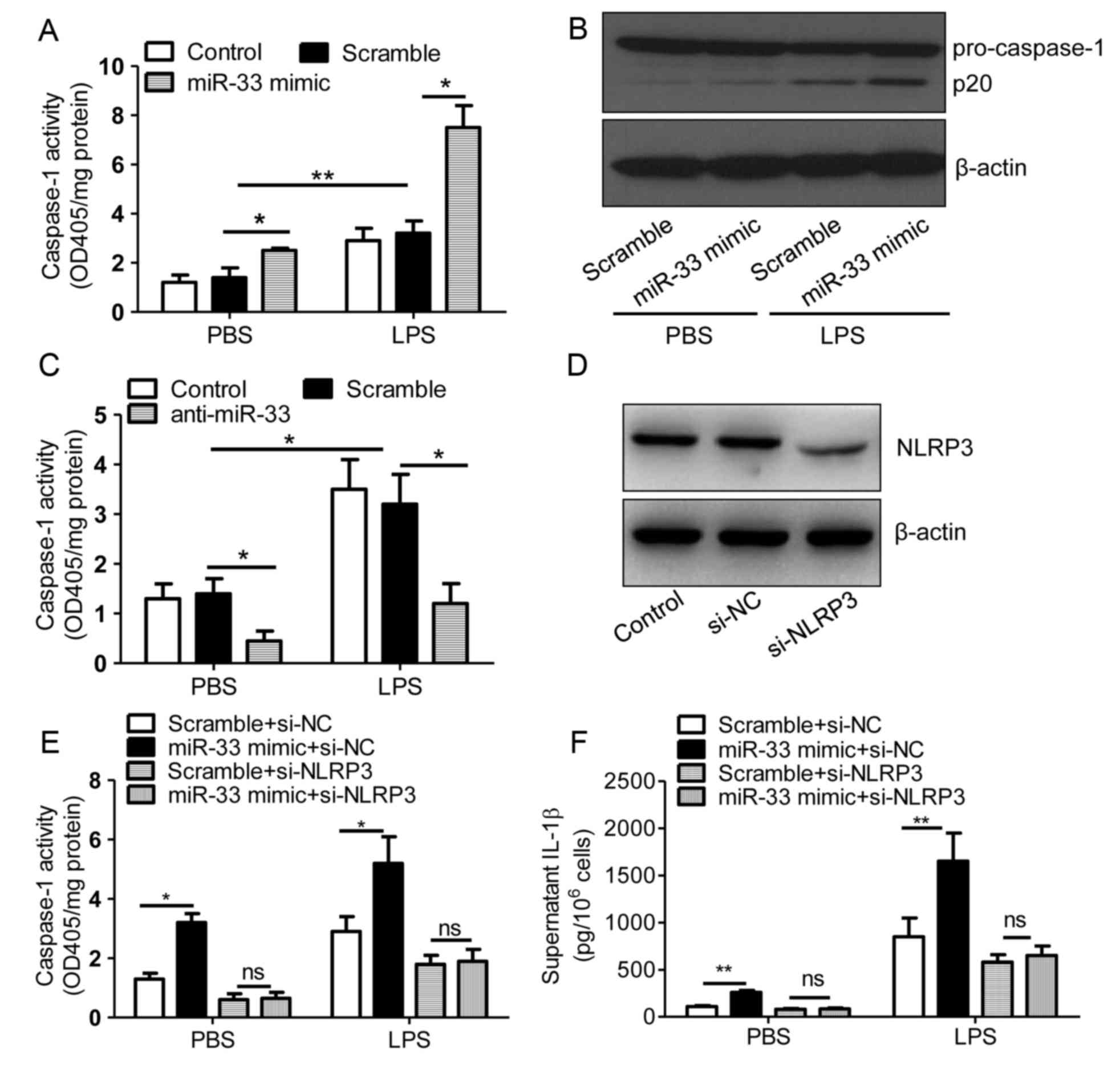 | Figure 3.miR-33 increases the activity of
caspase-1. (A) The activity of caspase-1 in untransfected control
mouse peritoneal macrophages or those transfected with a scramble
miR (100 nM) or an miR-33 mimic (100 nM). (B) Immunoblotting assays
for pro-caspase-1 expression in mouse peritoneal macrophages
transfected with a scrambled miR (100 nM) or an miR-33 mimic (100
nM). (C) Activity of caspase-1 in untransfected control mouse
peritoneal macrophages or those transfected with a scramble miR
(100 nM) or an anti-miR-33 (100 nM). At 5 h following transfection
with miR-33 mimics, anti-miR-33 or scramble controls, cells were
treated with LPS (50 ng/ml) or PBS. (D) Immunoblotting assays for
NLRP3 expression in mouse peritoneal macrophages transfected with
si-NC (20 nmol/ml) or si-NLRP3 (20 nmol/ml) for 24 h. (E) The
activity of caspase-1 and (F) the supernatant IL-1β levels in mouse
peritoneal macrophages transfected with a scrambled miR (100 nM) or
an miR-33 mimic (100 nM) in addition to si-NC (20 nmol/ml) or
si-NLRP3 (20 nmol/ml) for 5 h, followed by treatment with LPS (50
ng/ml) or PBS for 24 h. The results are presented as the mean ±
standard error of the mean (n=4). *P<0.05 and **P<0.01, as
indicated. miR, microRNA; LPS, lipopolysaccharide; PBS,
phosphate-buffered saline; NLRP3, nucleotide binding domain and
leucine-rich repeat pyrin 3 domain; siRNA, short interfering RNA;
si-NC, negative control siRNA; IL-1β, interleukin-1β; ns, not
significant. |
miR-33 attenuates mitochondrial oxygen
consumption and increases the production of cellular ROS
According to previous studies, ROS stimulate NLRP3
expression (3,5). Therefore, the present study
investigated whether miR-33 regulates ROS production in
macrophages. As presented in Fig. 4A
and B, treatment with an miR-33 mimic significantly induced
cellular ROS production in peritoneal macrophages with or without
LPS stimulation when compared with scramble miR controls.
Consistent with these observations, treatment with anti-miR-33
significantly decreased ROS production when compared with controls
(Fig. 4C and D). Mitochondrial
function was analyzed in the present study, as mitochondria are
known to be a major source of ROS. It was demonstrated that
treatment with the miR-33 mimic significantly decreased the basal
OCR in mitochondria when compared with scramble miR-transfected
controls (Fig. 4E). ROS in
macrophages were significantly increased in response to treatment
with the miR-33 mimic at basal levels (Fig. 4F). Treatment with the NAC or GSH
antioxidants efficiently reduced cellular ROS (Fig. 4F). The results of the present study
indicate that miR-33-induced mitochondrial dysfunction may account
for the increased production of cellular ROS in macrophages.
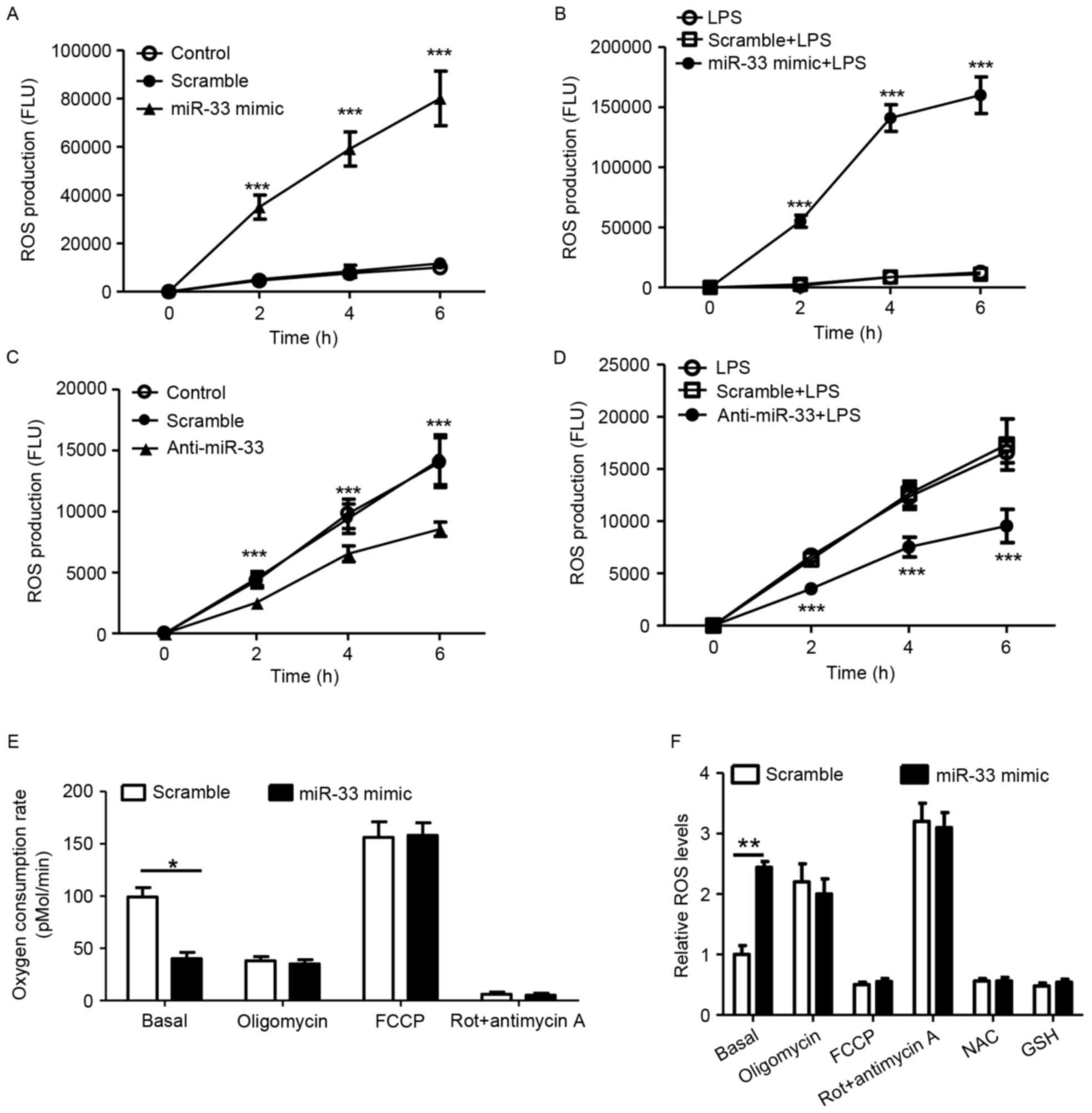 | Figure 4.miR-33 attenuates mitochondrial
oxygen consumption and increases the production of cellular ROS.
(A) Cellular ROS production of peritoneal macrophages treated with
a scramble miR (100 nM) or an miR-33 mimic (100 nM). ***P<0.005
vs. scramble. (B) Cellular ROS production of peritoneal macrophages
that were treated with a scramble miR (100 nM) or an miR-33 mimic
(100 nM) for 5 h, followed by treatment with LPS (50 ng/ml) for 24
h. ***P<0.005 vs. scramble + LPS. (C) Cellular ROS production of
peritoneal macrophages treated with a scramble miR (100 nM) or
anti-miR-33 (100 nM). ***P<0.005, anti-miR-33 vs. scramble. (D)
Cellular ROS production of peritoneal macrophages that were treated
with a scramble miR (100 nM) or anti-miR-33 (100 nM) for 5 h,
followed by treatment with LPS (50 ng/ml) for 24 h. ***P<0.005
vs. scramble + LPS. (E) The oxygen consumption rate of peritoneal
macrophages treated with a scrambled miR (100 nM) or an miR-33
mimic (100 nM) for 24 h, followed by mitochondrial function assays.
(F) Relative ROS production in peritoneal macrophages that were
transfected with a scramble miR (100 nM) or an miR-33 mimic (100
nM) and subsequently treated with various regulators of
mitochondrial function or antioxidants for 24 h. The results are
presented as the mean ± standard error of the mean (n=5).
*P<0.05 and **P<0.01, as indicated. miR, microRNA; ROS,
reactive oxygen species; LPS, lipopolysaccharide; Rot, rotenone;
FCCP, carbonyl cyanide p-(trifluoromethoxy) phenylhydrazone; NAC,
N-acetylcysteine; GSH, glutathione. |
miR-33 stimulates the inflammasome
pathway by impairing mitochondrial function and inducing ROS
production in peritoneal macrophages
In order to further verify the role of
miR-33-mediated mitochondrial function and ROS production in
inflammasome activation, NLRP3 expression, caspase-1 activity and
IL-1β secretion were analyzed in peritoneal macrophages treated
with a miR-33 mimic, antioxidants (NAC and GSH) or the
mitochondrial stimulator FCCP. The results demonstrated that
miR-33-induced NLRP3 expression was prevented by NAC treatment
(Fig. 5A). In addition,
transfection with the miR-33 mimic induced caspase-1 activity,
whereas additional treatment with antioxidants or FCCP abolished
this effect (Fig. 5B).
Furthermore, miR-33 mimic-induced IL-1β secretion was prevented by
treatment with NAC, GSH or FCCP (Fig.
5C).
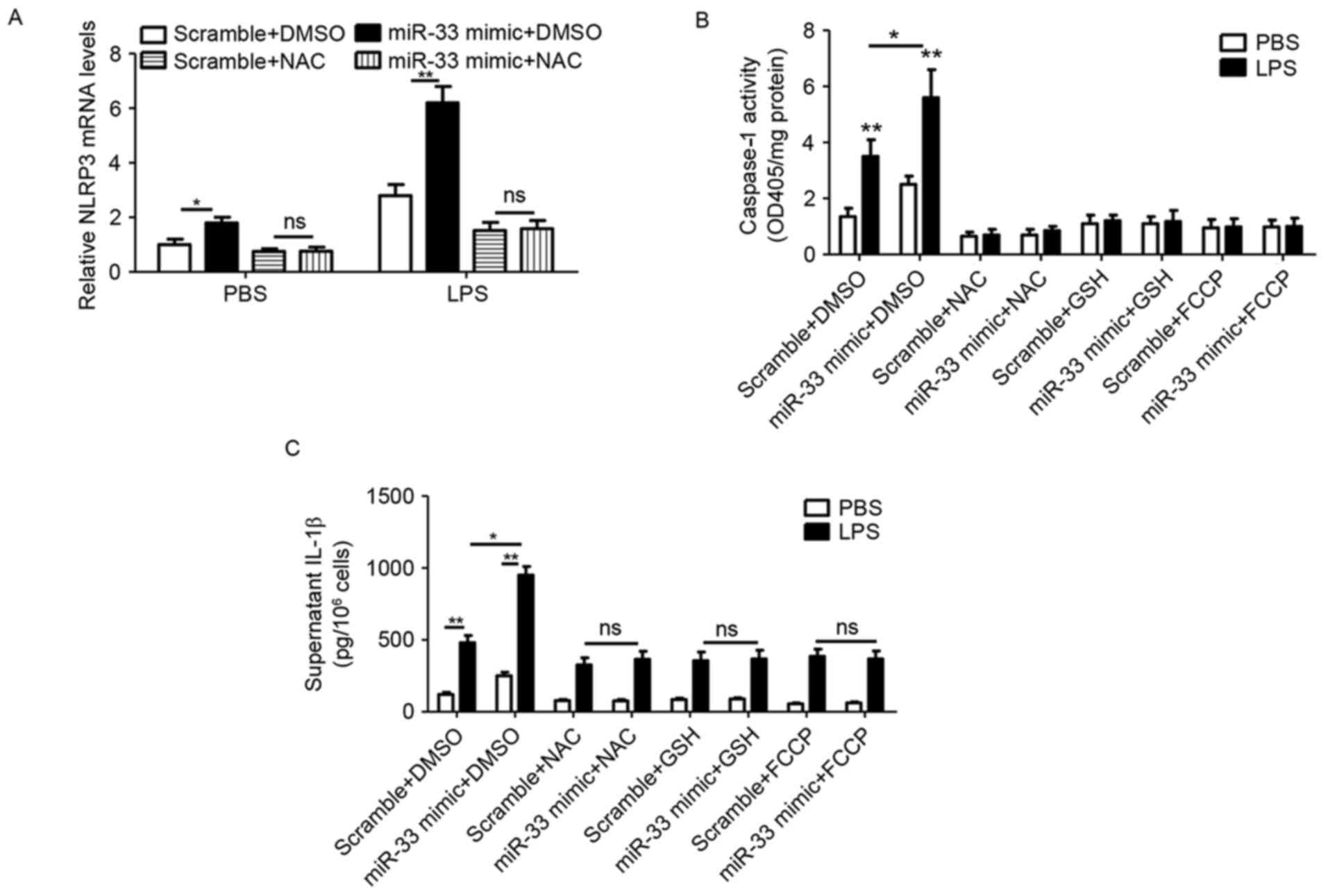 | Figure 5.miR-33 stimulates the inflammasome
signaling pathway by impairing mitochondrial function and inducing
ROS production in peritoneal macrophages. (A) The mRNA levels of
NLRP3 in mouse peritoneal macrophages that were transfected with a
scramble miR (100 nM) or an miR-33 mimic (100 nM) for 5 h, followed
by treatment with LPS (50 ng/ml) or PBS as a control, plus NAC (20
mM) or DMSO for 24 h. (B) The activity of caspase-1 and (C) the
level of IL-1β in the supernatant of mouse peritoneal macrophages
that were transfected with a scramble miR (100 nM) or miR-33 mimic
(100 nM) for 5 h, followed by treatment with LPS (50 ng/ml) or PBS
as a control, plus NAC (20 mM), GSH (10 mM) or FCCP (1 µg/ml) for
24 h. The results are presented as the mean ± standard error of the
mean (n=4). *P<0.05 and **P<0.01, as indicated. miR,
microRNA; NLRP3, nucleotide binding domain and leucine-rich repeat
pyrin 3 domain; LPS, lipopolysaccharide; PBS, phosphate-buffered
saline; NAC, N-acetylcysteine; DMSO, dimethyl sulfoxide; IL-1β,
interleukin-1β; GSH, glutathione; FCCP, carbonyl cyanide
p-(trifluoromethoxy)phenylhydrazone; OD, optical density; ns, not
significant. |
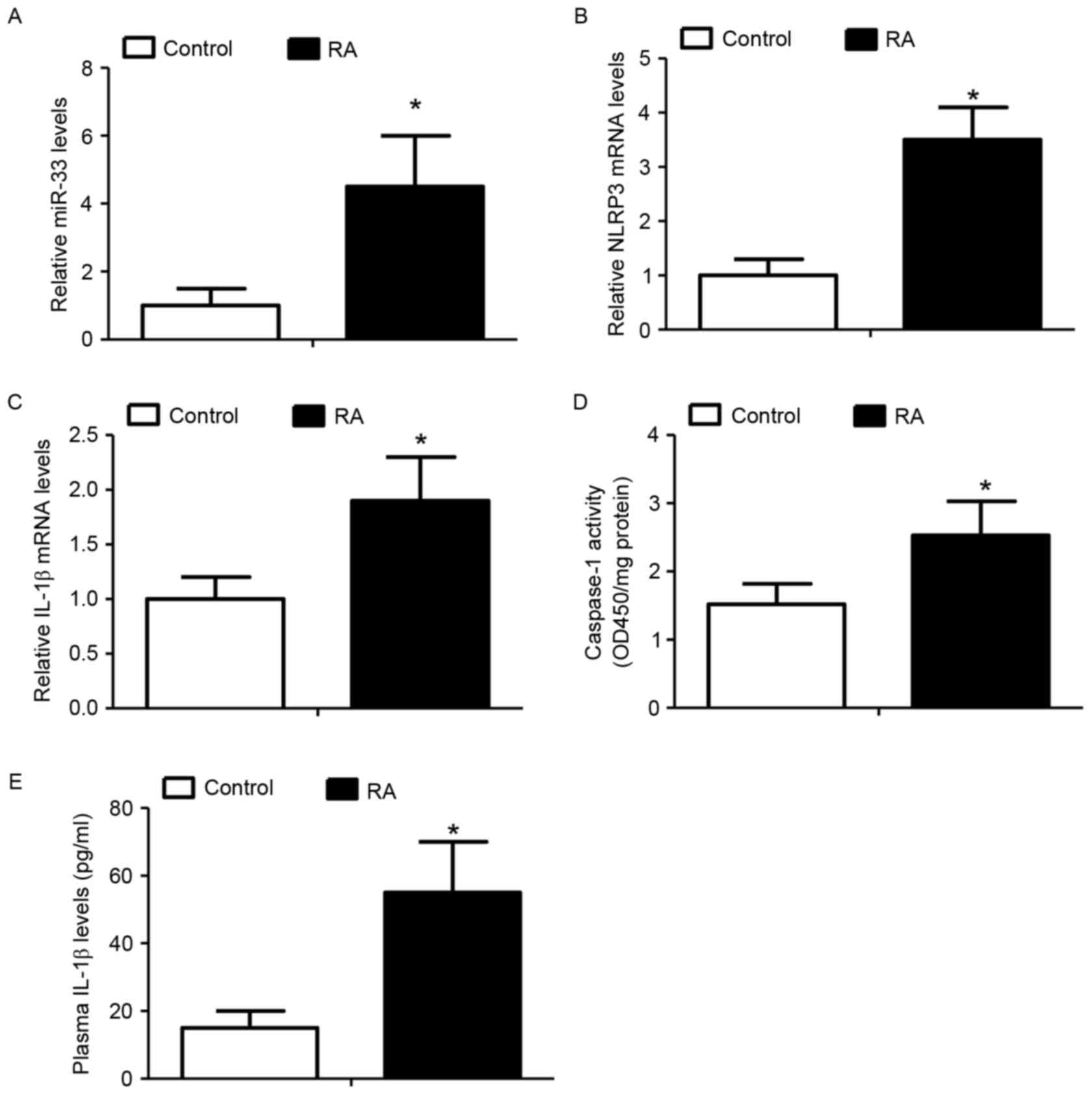
Elevated miR-33 levels are associated
with activation of the NLRP3 inflammasome pathway in RA-associated
monocytes
The expression of miR-33 and activation of the NLRP3
inflammasome signaling pathway was analyzed in peripheral blood
monocytes derived from patients with RA and compared with those
from healthy donors, as IL-1β is a well-documented inducer of RA
(24,25). It was demonstrated that
RA-associated monocytes expressed significantly increased miR-33
(Fig. 6A), NLRP3 (Fig. 6B) and IL-1β (Fig. 6C) levels when compared with
monocytes from healthy donors. In addition, the activity of
caspase-1 and the levels of plasma IL-1β were significantly
increased in the RA-associated monocytes compared with control
monocytes (Fig. 6D and E). The
results of the present study indicated a potential role for the
miR-33/NLRP3 inflammasome/IL-1β axis in the development of RA.
Discussion
To the best of the author's knowledge, the present
study is the first to suggest that miR-33 may function as a
positive regulator of the NLRP3 inflammasome by repressing
mitochondrial OCRs and inducing ROS accumulation in macrophages. In
addition, the results demonstrated that the expression of miR-33
and the activity of the NLRP3 inflammasome were significantly
increased in the blood monocytes of patients with RA when compared
with healthy donors. The results of the present study revealed a
potential role for miR-33 in chronic inflammatory diseases, such as
RA.
In the present study, miR-33 was observed to repress
the mitochondrial OCR and increase ROS production. A previous study
demonstrated that anti-miR-33 de-repressed peroxisome
proliferator-activated receptor γ coactivator 1-α (PGC1-α),
pyruvate dehydrogenase kinase 4 and solute carrier family 25 member
25 target genes, and enhanced mitochondrial respiration (21). It was hypothesized that miR-33
inhibited the OCR rate by repressing PGC1-α expression, as PGC1-α
is a well-documented stimulator of mitochondrial biogenesis
(26). Therefore, mitochondrial
biogenesis-associated genes require further analysis in future
studies to verify this hypothesis. The results of the present study
demonstrated that ROS accumulation was due to decreased oxygen
consumption in the mitochondria, which was consistent with results
from previous studies (5,27).
miR-33 may regulate proinflammatory cytokine
production via a number of different signaling pathways. For
instance, cholesterol is an important inducer of inflammatory
responses (28), and miR-33
regulates cholesterol efflux in macrophages (19). In addition, the 5′-adenosine
monophosphate-activated protein kinase signaling pathway, which is
regulated by miR-33 (22),
regulates macrophage polarization (29). Ho et al (30) reported that miR-33 repressed
receptor-interacting protein 140-dependent proinflammatory cytokine
production, which contradicts the results of the present study.
There are a number of differences in the treatment conditions
between this previous study and the present study, including the
macrophage cell type (Raw264.7 cells vs. primary macrophages) and
LPS treatment conditions (4 vs. 24 h; 20 ng/ml vs. 50 ng/ml). It is
possible that miR-33 may regulate toll-like receptor 4 (TLR-4)
signaling, thereby further potentiating NLRP3 expression and IL-1β
production. However, in the present study it was observed that
miR-33 induced IL-1β protein levels independently of TLR-4
expression (data not shown). The results presented indicated that
miR-33 may regulate a complex signaling network. However, the
respective contribution of each pathway downstream of miR-33 in
proinflammatory cytokine production remains to be elucidated. The
results of the present study reflected a long-term effect of
miR-33, based on its roles in regulating mitochondrial function and
ROS production. The miR-33-regulated cholesterol signaling pathway
may be important for the development of atherosclerosis, whereas
the NLRP3 inflammasome pathway may be associated with the
pathogenesis of RA.
In the present study, it was demonstrated that the
NLRP3 inflammasome was activated in RA-associated monocytes in
human subjects, which is consistent with the results from a recent
clinical study (31). In addition,
the present study demonstrated that miR-33 levels were
significantly increased in the blood monocytes of patients with RA
when compared with healthy control subjects, suggesting that the
miR-33/NLRP3 inflammasome axis may regulate RA progression and that
miR-33 may be a novel marker for the disease. In order to further
verify the results of the present study, future animal model and
clinical studies are required.
In conclusion, miR-33 was identified to be a
positive regulator of the NLRP3 inflammasome in macrophages, and
the miR-33/NLRP3 inflammasome signaling pathway may be associated
with the development of RA.
Acknowledgements
The present study was supported in part by the
National Nature Science Foundation of China (grant no. 81001336),
the Sichuan Provincial Health Department Foundation (grant nos.
130320 and 130322) and the Chengdu Military General Hospital
Foundation (grant no. 2013YG-B037/B096). NPG Language Editing
provided an English language service for the present study.
Glossary
Abbreviations
Abbreviations:
|
miR
|
microRNA
|
|
RA
|
rheumatoid arthritis
|
|
NLRP3
|
nucleotide binding domain and
leucine-rich repeat pyrin 3 domain
|
|
IL-1β
|
interleukin-1β
|
|
OCR
|
oxygen consumption rate
|
|
ROS
|
reactive oxygen species
|
References
|
1
|
Strowig T, Henao-Mejia J, Elinav E and
Flavell R: Inflammasomes in health and disease. Nature.
481:278–286. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Eisenbarth SC, Colegio OR, O'Connor W,
Sutterwala FS and Flavell RA: Crucial role for the Nalp3
inflammasome in the immunostimulatory properties of aluminium
adjuvants. Nature. 453:1122–1126. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Schroder K, Zhou R and Tschopp J: The
NLRP3 inflammasome: A sensor for metabolic danger? Science.
327:296–300. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Zhou R, Yazdi AS, Menu P and Tschopp J: A
role for mitochondria in NLRP3 inflammasome activation. Nature.
469:221–225. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Miao H, Ou J, Ma Y, Guo F, Yang Z, Wiggins
M, Liu C, Song W, Han X, Wang M, et al: Macrophage CGI-58
deficiency activates ROS-inflammasome pathway to promote insulin
resistance in mice. Cell Rep. 7:223–235. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Siebert S, Tsoukas A, Robertson J and
McInnes I: Cytokines as therapeutic targets in rheumatoid arthritis
and other inflammatory diseases. Pharmacol Rev. 67:280–309. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Cunnane G, Madigan A, Murphy E, FitzGerald
O and Bresnihan B: The effects of treatment with interleukin-1
receptor antagonist on the inflamed synovial membrane in rheumatoid
arthritis. Rheumatology (Oxford). 40:62–69. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ma Z, Wang B, Wang M, Sun X, Tang Y, Li M,
Li F and Li X: TL1A increased IL-6 production on fibroblast-like
synoviocytes by preferentially activating TNF receptor 2 in
rheumatoid arthritis. Cytokine. 83:92–98. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Fontalba A, Martinez-Taboada V, Gutierrez
O, Pipaon C, Benito N, Balsa A, Blanco R and Fernandez-Luna JL:
Deficiency of the NF-kappaB inhibitor caspase activating and
recruitment domain 8 in patients with rheumatoid arthritis is
associated with disease severity. J Immunol. 179:4867–4873. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kastbom A, Verma D, Eriksson P, Skogh T,
Wingren G and Söderkvist P: Genetic variation in proteins of the
cryopyrin inflammasome influences susceptibility and severity of
rheumatoid arthritis (the Swedish TIRA project). Rheumatology
(Oxford). 47:415–417. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ben Hamad M, Cornelis F, Marzouk S,
Chabchoub G, Bahloul Z, Rebai A, Fakhfakh F, Ayadi H,
Petit-Teixeira E and Maalej A: Association study of CARD8 (p.C10X)
and NLRP3 (p.Q705K) variants with rheumatoid arthritis in French
and Tunisian populations. Int J Immunogenet. 39:131–136. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Walle LV, Van Opdenbosch N, Jacques P,
Fossoul A, Verheugen E, Vogel P, Beyaert R, Elewaut D, Kanneganti
TD, van Loo G and Lamkanfi M: Negative regulation of the NLRP3
inflammasome by A20 protects against arthritis. Nature. 512:69–73.
2014.PubMed/NCBI
|
|
13
|
Wen H, Gris D, Lei Y, Jha S, Zhang L,
Huang MT, Brickey WJ and Ting JP: Fatty acid-induced NLRP3-ASC
inflammasome activation interferes with insulin signaling. Nat
Immunol. 12:408–415. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
14
|
Heneka MT, Kummer MP, Stutz A, Delekate A,
Schwartz S, Vieira-Saecker A, Griep A, Axt D, Remus A, Tzeng TC, et
al: NLRP3 is activated in Alzheimer's disease and contributes to
pathology in APP/PS1 mice. Nature. 493:674–678. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wree A, Eguchi A, McGeough MD, Pena CA,
Johnson CD, Canbay A, Hoffman HM and Feldstein AE: NLRP3
inflammasome activation results in hepatocyte pyroptosis, liver
inflammation, and fibrosis in mice. Hepatology. 59:898–910. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Shalgi R, Lieber D, Oren M and Pilpel Y:
Global and local architecture of the mammalian
microRNA-transcription factor regulatory network. PLoS Comput Biol.
3:e1312007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ryazansky SS, Gvozdev VA and Berezikov E:
Evidence for post-transcriptional regulation of clustered microRNAs
in Drosophila. BMC Genomics. 12:3712011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Krützfeldt J and Stoffel M: MicroRNAs: A
new class of regulatory genes affecting metabolism. Cell Metab.
4:9–12. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Rayner KJ, Suárez Y, Dávalos A, Parathath
S, Fitzgerald ML, Tamehiro N, Fisher EA, Moore KJ and
Fernández-Hernando C: MiR-33 contributes to the regulation of
cholesterol homeostasis. Science. 328:1570–1573. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Esau CC, Hussain FN, McDaniel AL, Marshall
SM, van Gils JM, Ray TD, Sheedy FJ, Goedeke L, Liu X, Khatsenko OG,
et al: Inhibition of miR-33a/b in non-human primates raises plasma
HDL and lowers VLDL triglycerides. Nature. 478:404–407. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Karunakaran D, Thrush AB, Nguyen MA,
Richards L, Geoffrion M, Singaravelu R, Ramphos E, Shangari P,
Ouimet M, Pezacki JP, et al: Macrophage mitochondrial energy status
regulates cholesterol efflux and is enhanced by Anti-miR33 in
atherosclerosis. Circ Res. 117:266–278. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ouimet M, Ediriweera HN, Gundra UM, Sheedy
FJ, Ramkhelawon B, Hutchison SB, Rinehold K, van Solingen C,
Fullerton MD, Cecchini K, et al: MicroRNA-33-dependent regulation
of macrophage metabolism directs immune cell polarization in
atherosclerosis. J Clin Invest. 125:4334–4348. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Eklund KK, Leirisalo-Repo M, Ranta P, Mäki
T, Kautiainen H, Hannonen P, Korpela M, Hakala M, Järvinen P and
Möttönen T: Serum IL-1beta levels are associated with the presence
of erosions in recent onset rheumatoid arthritis. Clin Exp
Rheumatol. 25:684–689. 2007.PubMed/NCBI
|
|
25
|
Ruscitti P, Cipriani P, Carubbi F,
Liakouli V, Zazzeroni F, Di Benedetto P, Berardicurti O, Alesse E
and Giacomelli R: The role of IL-1β in the bone loss during
rheumatic diseases. Mediators Inflamm. 2015:7823822015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Spiegelman BM: Transcriptional control of
mitochondrial energy metabolism through the PGC1 coactivators.
Novartis Found Symp. 287:60–63. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Haemmerle G, Moustafa T, Woelkart G,
Büttner S, Schmidt A, van de Weijer T, Hesselink M, Jaeger D,
Kienesberger PC, Zierler K, et al: ATGL-mediated fat catabolism
regulates cardiac mitochondrial function via PPAR-α and PGC-1. Nat
Med. 17:1076–1085. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
28
|
Chen FY, Zhou J, Guo N, Ma WG, Huang X,
Wang H and Yuan ZY: Curcumin retunes cholesterol transport
homeostasis and inflammation response in M1 macrophage to prevent
atherosclerosis. Biochem Biophys Res Commun. 467:872–878. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Chan KL, Pillon NJ, Sivaloganathan DM,
Costford SR, Liu Z, Théret M, Chazaud B and Klip A: Palmitoleate
reverses high fat-induced proinflammatory macrophage polarization
via AMP-activated protein kinase (AMPK). J Biol Chem.
290:16979–16988. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Ho PC, Chang KC, Chuang YS and Wei LN:
Cholesterol regulation of receptor-interacting protein 140 via
microRNA-33 in inflammatory cytokine production. FASEB J.
25:1758–1766. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Choulaki C, Papadaki G, Repa A, Kampouraki
E, Kambas K, Ritis K, Bertsias G, Boumpas DT and Sidiropoulos P:
Enhanced activity of NLRP3 inflammasome in peripheral blood cells
of patients with active rheumatoid arthritis. Arthritis Res Ther.
17:2572015. View Article : Google Scholar : PubMed/NCBI
|