Introduction
Chronic obstructive pulmonary disease (COPD) is
mainly a consequence of persistent airway inflammation caused by
cigarette smoke (CS) and genetic predisposition (1). The exposure to cigarette smoke (CS)
activates the hedgehog (Hh) signaling pathway in bronchial
epithelial cells. Hh activation occurs in BEAS2B cells when
repeatedly exposed to smoke (2).
Hh signaling is one of the most conserved pathways,
that regulates morphogenesis of multiple organs, including lung,
intestines, and nervous system during embryogenesis. In the adult,
Hh signaling also maintains organ homeostasis and modulates tissue
repair (3). The Hh pathway is
comprised of a complex network of molecules. Three Hh ligands have
been identified: sonic hedgehog (Shh) for human, Indian hedgehog
(Ihh) and desert hedgehog (Dhh). Hh ligands bind to their
repressive receptor, Patched (Ptc), a twelve-transmembrane protein;
it relieves the suppression of the signaling transducer, Smoothened
(Smo), a seven-transmembrane protein, thereby activating the Hh
pathway. The unleashed Smo passages from the vesicles to the
primary cilium on the cell membrane, leading to translocation of
the transcription factors and glioma-associated oncoproteins (Gli1,
Gli2, Gli3), especially Gli1, and subsequently promotes the
transcription of the target genes.
The first identified Hh blocker is cyclopamine. It
is a steroid alkaloid and inhibits the Hh pathway by directly
binding Smo, which causes developmental abnormalities in animals
(4).
Previous data implicated a role of Hh in postnatal
lung development; the interruption of Hh signaling at earlier time
points leads to enlarged alveolar airspaces. In the adult lung, the
Hh pathway maintains mesenchymal quiescence and is dysregulated in
disease such as COPD (5). These
results suggest that the Hh pathways underlie the initiation,
maintenance, proliferation, and survival of CSE-transformed
epithelial cells. However, the role of Hh signaling pathway in
cigarette-induced airway inflammation remains unclear.
Materials and methods
Cell culture and reagents
Human alveolar epithelial cells (A549) were
purchased from the Shanghai Institutes for Biological Sciences and
cultured in a humidified incubator with 5% CO2 at 37°C
in RPMI 1640 medium (Gibco Life Technologies, Gaithersburg, MD,
USA) supplemented with 10% fetal bovine serum (FBS), penicillin,
and streptomycin. After reaching early confluency, the cells were
trypsinized and plated for experiments.
We generated three A549 cell populations. We defined
the primary cultures as untreated control cells (A0 group), A1
group represented A549 cells stimulated with nicotine
(Sigma-Aldrich St. Louis, MO, USA) at appropriate concentrations
evaluated by Cell Counting Kit-8 (CCK-8; Dojindo Laboratories,
Kumamoto, Japan) assay for 12, 24 and 48 h, and A2 group
represented A549 cells treated simultaneously with nicotine and
cyclopamine (Sigma-Aldrich) at a concentration of 10 µM according
to published studies (6–8).
Cell viability assay
The proliferation A549 cells was measured using the
CCK-8 according to the manufacturer's instructions in order to
determine the appropriate concentrations of nicotine treatment in
the experiment. Cells (2,000/100 µl/well) were plated in triplicate
into 96-well plates and cultured before treatment with nicotine at
0.1, 1, 10, and 100 µM for 12, 24 and 48 h, respectively.
Subsequently, the cells were incubated with CCK-8 reagent (10 µl)
for an additional 2 h. The absorbance was determined at 450 nm, and
the cell viability was calculated as a percentage of the OD value
in the control cells. The most appropriate concentration of
nicotine was determined and used in downstream experiments.
ELISA
The total concentrations of specific inflammatory
mediators (IL-6, IL-8, TNF-α) in A0, A1 and A2 cells stimulated by
nicotine with and without cyclopamine (at a concentration of 10 µM)
were assessed by commercial ELISA kits (Anogen, Canada), according
to the manufacturer's instructions. The total concentration of
IL-10, known as the cytokine synthesis inhibitory factor (CSIF),
which inhibits the production of IL-6, IL-8, TNF-α and
downregulates inflammation was also measured by ELISA immunoassay.
The results were expressed as pictogram of cytokine per milliliter
plasma (pg/ml).
Western blot analysis
The expression levels of Shh, Smo, and Gli1 proteins
in control A0, in A1 cells treated with 10 µM nicotine, and in A2
cells cultured simultaneously with nicotine (10 µM) and cyclopamine
(10 µM) for 24 and 48 h were assessed respectively using Western
blot. We also measured the expressions of IL-6, IL-8, and TNF-α at
protein levels in A0, A1, and A2 groups at 12, 24 and 48 h using
the same methods. The cells were lysed in RIPA lysis buffer, and
the lysates incubated on ice and centrifuged at 14,000 rpm, 4°C for
15 min. Total protein concentrations were determined by Bradford
assay. The protein samples were subjected to SDS-PAGE and
transferred to polyvinyliene difluoride membranes. The non-specific
binding sites on the membranes were blocked with skimmed milk for 1
h, and the blots were incubated with primary antibody overnight at
4°C (1:1,000, Proteinch, USA). Subsequently, the membranes were
probed with secondary antibodies (1:2,000, Santa Cruz, USA) for 1 h
at room temperature. Finally, the relative protein levels were
determined with Image J software.
Statistical analysis
All statistical analyses were performed using SPSS
(version 20.0; IBM Corp., Armonk, NY, USA). The values are
expressed as the mean ± standard deviation. Analysis of variance
was performed to evaluate the differences between the groups,
followed by Scheffe post hoc test for multiple comparisons. A
P-value <0.05 was considered as statistically significant.
Results
Cell viability assay
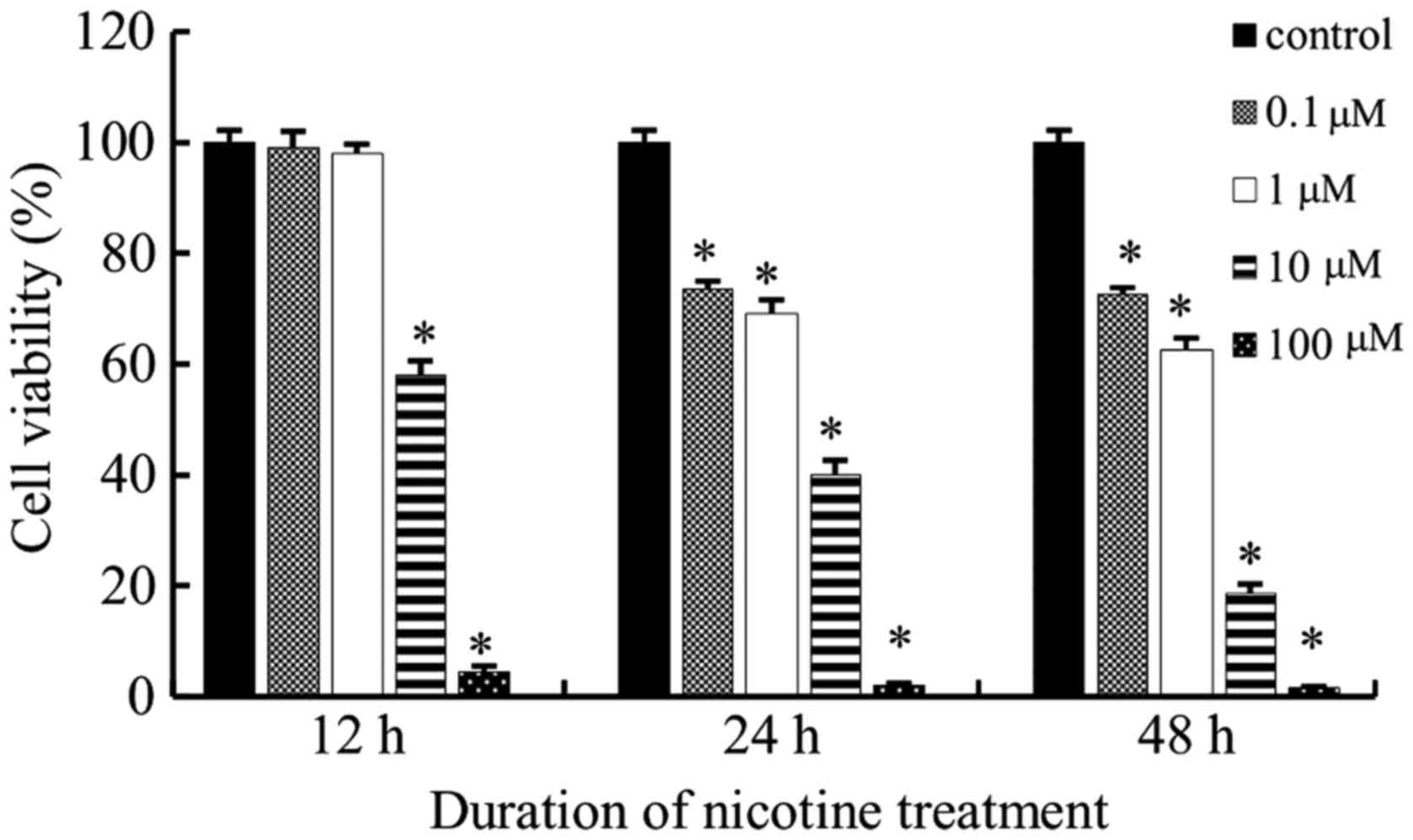
Cell viability curves were plotted as viable cell
percentage based on the CCK-8 assay. As shown in Fig. 1, nicotine inhibited the
proliferation of the tested cell lines in a time and dose dependent
manner. According to the results of the CCK-8 assay, with nicotine
treatment, a concentration of 0.1 and 1 µM were not sufficient to
stimulate cells at all the three time points; however, cells failed
to survive at 100 µM. Therefore, 10 µM was the most appropriate
nicotine concentration used in the subsequent study.
Expression levels of IL-6, IL-8,
TNF-α, and IL-10 in cells by ELISA
A0 were cultured as control cells, A1 cells were
stimulated with nicotine at a concentration of 10 µM for 12, 24 and
48 h, respectively, and A2 cells were treated simultaneously with
nicotine (10 µM) and cyclopamine (10 µM) for 12, 24 and 48 h,
respectively. Nicotine induced the production of the inflammatory
mediators in A549 cells.
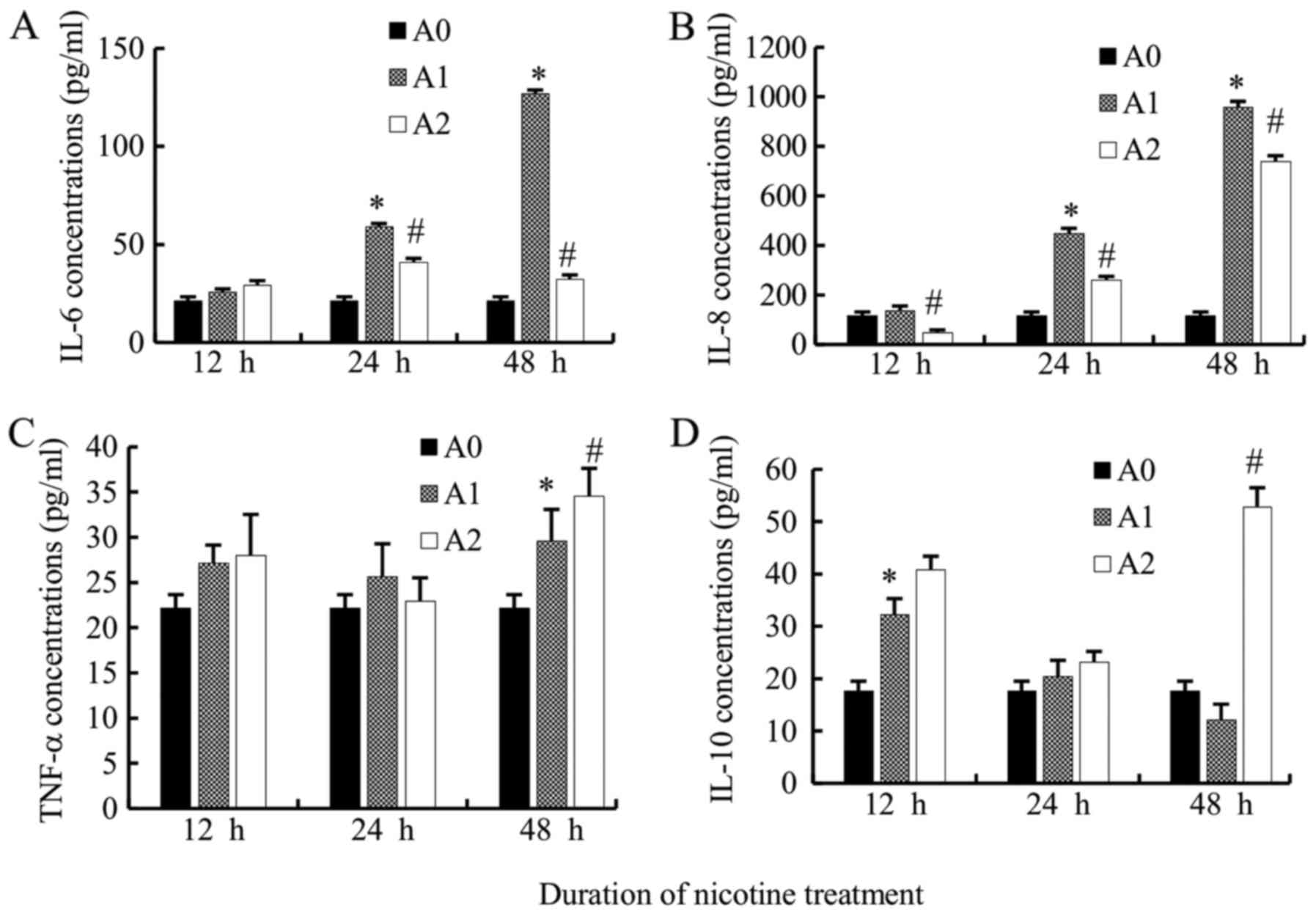
We compared the IL-6 levels produced by A549 cells
among the three groups. IL-6 levels in A1 cells were increased
significantly as compared to A0 cells at 24 and 48 h (all
P<0.05) but not at 12 h. Supposedly, the 12 h time point was
extremely short to induce the secretion of IL-6. Additionally, the
levels of IL-6 in A2 group were significantly lower than A1 group
at 24 and 48 h (all P<0.05) (Fig.
2A).
The levels of IL-8 also increased significantly in
A1 cells at 24 and 48 h as compared to the A0 group (all
P<0.05). The levels of IL-8 in the A2 group were significantly
lower than that in the A1 group (12, 24, 48 h) (all P<0.05)
(Fig. 2B).
The TNF-α levels in A1 group were significantly
higher than that in A0 at 48 h (P<0.05); however, that in A2
decreased non-significantly as compared to A1 when treated
simultaneously with cyclopamine only at 24 h, while increased at 12
and 48 h (P<0.05 at 48 h) (Fig.
2C). Therefore, the variation trend of TNF-α levels appears to
be different from that of IL-6 and IL-8. The underlying mechanism
necessitates further investigation.
The level of the anti-inflammatory mediator, IL-10
in A1 group, increased at 12 and 24 h as compared to the control
cells (P<0.05 at 12 h), while a decreased level was observed at
48 h. When treated with cyclopamine, in contrary to IL-6 and IL-8,
the IL-10 levels increased in A2 cells as compared to the A1 cells
at 12, 24, 48 h, but significantly at 48 h (P<0.05) (Fig. 2D).
Expression levels of Shh, Gli1 and Smo
proteins
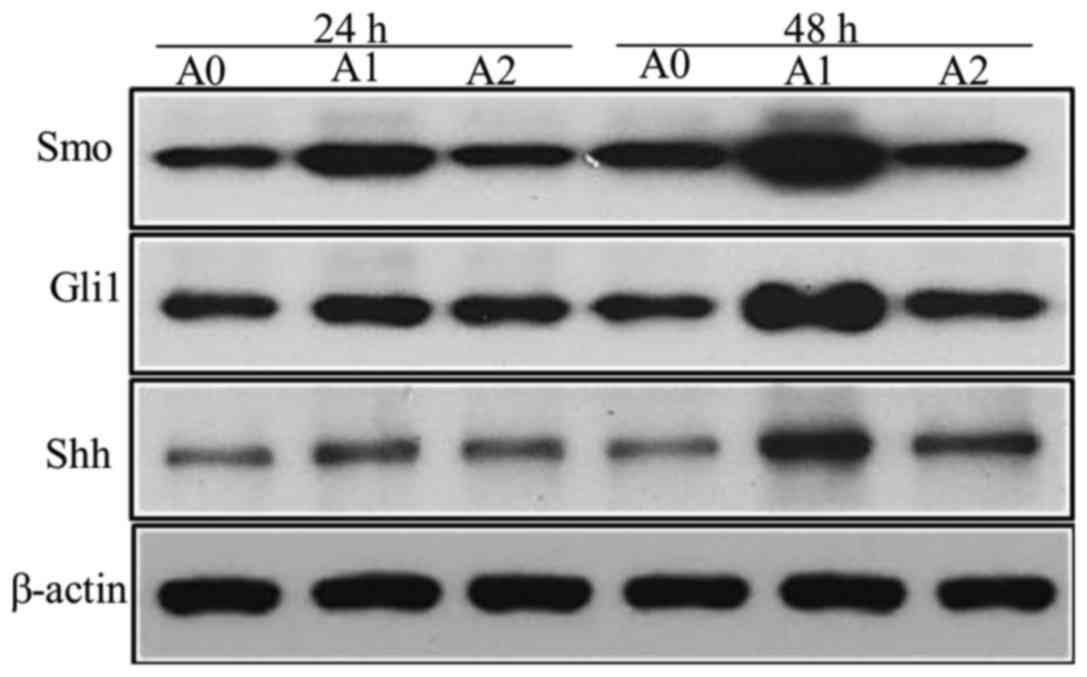
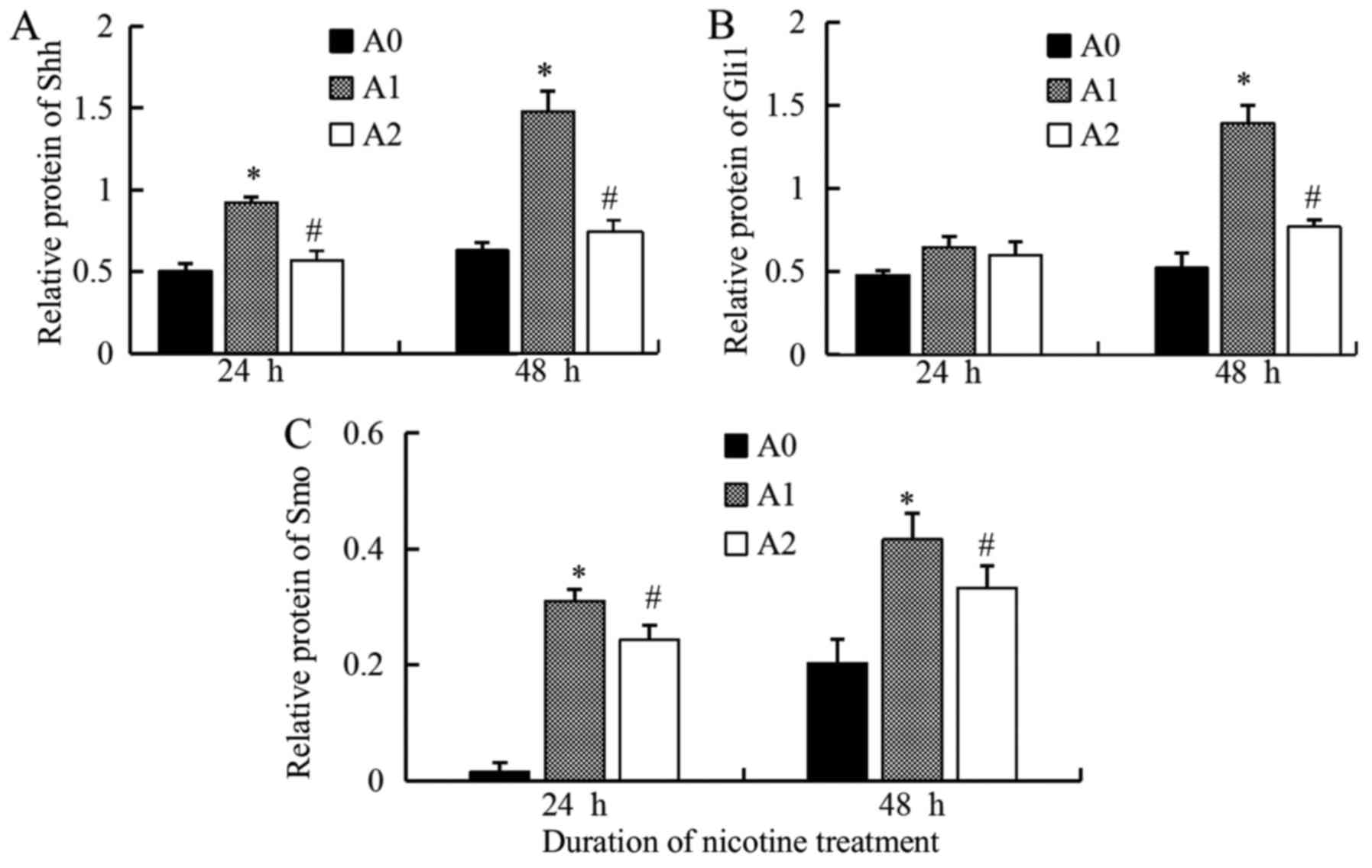
We detected the faint expression of Shh protein in
control cells (A0); however, when treated with nicotine (A1) at a
concentration of 10 µM for 24 and 48 h, the levels of Shh increased
significantly (P<0.05 at 24 and 48 h), while it decreased
significantly with cyclopamine (A2) as compared to the A1 cells
(Fig. 3). Since the expression of
Gli1 reflects the activation of Hh pathway, we also measured the
Gli1 levels. The results were similar to that of Shh. The
expression of the Gli1 protein, stimulated by nicotine (10 µM), was
significantly higher than the control but decreased significantly
with cyclopamine treatment for 48 h (P<0.05) (Fig. 3). The results of Smo protein were
also similar to that of Shh, the expressions were significantly
higher in A1 group than A0, but significantly lower in A2 compared
to A1 (P<0.05 at 24 and 48 h) (Fig.
3). The densitometric analyses of the three proteins are shown
in Fig. 4.
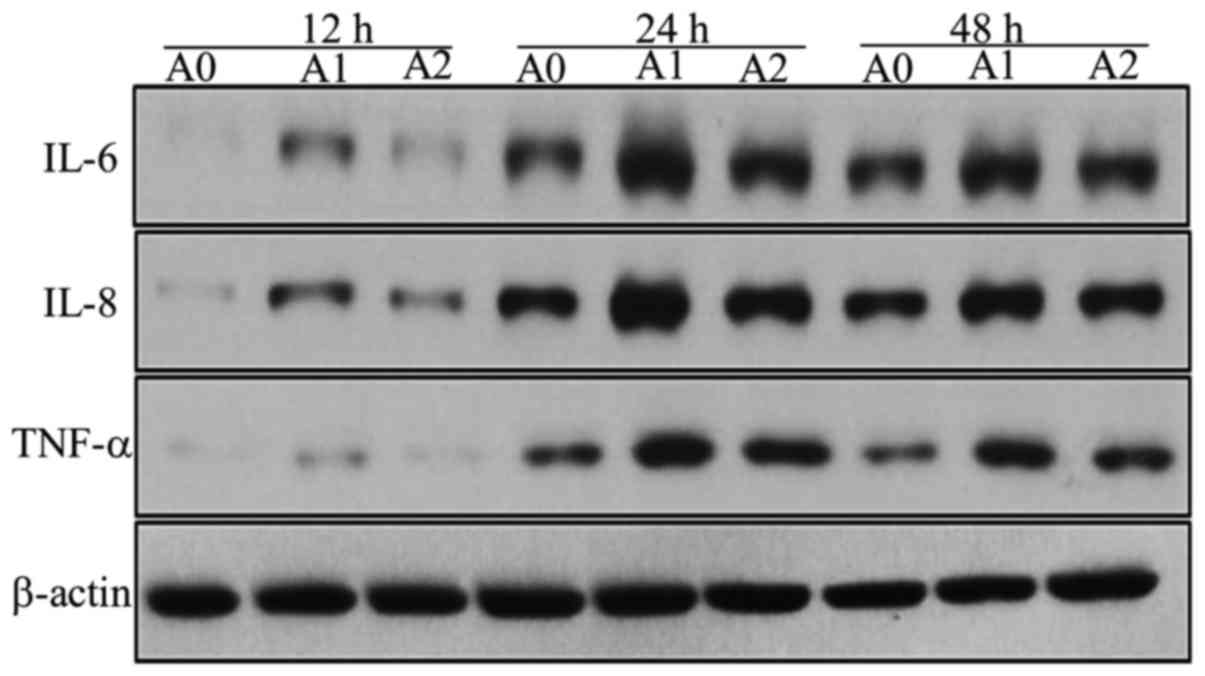
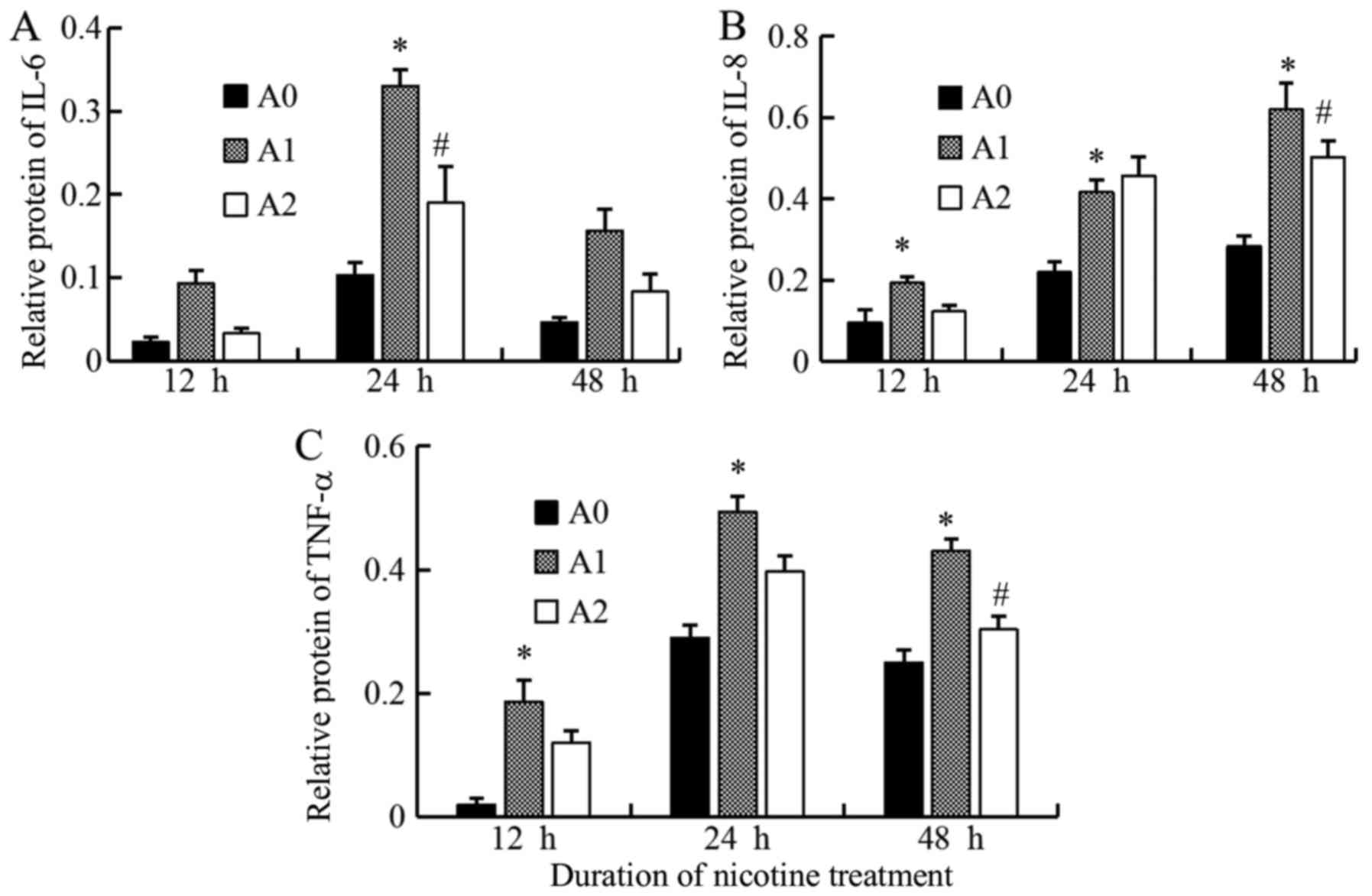
Expressions of IL-6, IL-8, TNF-α at
protein levels
We assessed the protein expressions of IL-6, IL-8,
and TNF-α in A0, A1 and A2 cells using Western blot. We compared
the expression levels of these inflammatory mediators produced by
A549 cells between the three groups and found that the expression
of IL-6 protein level in A1 cells increased compared to A0 at all
the times, but significantly at 24 h (P<0.05). On the other
hand, the treatment with cyclopamine decreased the IL-6 level in
the A2 group as compared to A1, the significant decrease was
observed only at 24 h (P<0.05) (Fig. 5). We suggest that 24 h is the most
appropriate times for change of IL-6 in inflammatory stimuli in the
current study.
The IL-8 protein level in the A1 group was
significantly higher than that for A0 group at 12, 24, and 48 h
(all P<0.05). The level was decreased with cyclopamine treatment
in the A2 group, significantly at 48 h (P<0.05) (Fig. 5). We suggest that 48h is the most
appropriate times for change of IL-8 in inflammatory stimuli in the
current study on Hh signaling.
The results of TNF-α protein were similar to that of
IL-8; the level was maximal in A1 cells (all P<0.05) and
decreased significantly in A2 cells as compared to A1 at 48 h
(P<0.05). Although these Western blot results were not
completely identical as those of ELISA owing to the differences in
the techniques, the results showed that the expression levels of
the inflammatory mediators tended to decrease with cyclopamine
treatment, when the Hh pathway was blocked (Fig. 5). The results of the densitometric
analyses are shown in Fig. 6.
Discussion
In the current study, we measured the expression
levels of some airway inflammatory mediators, such as IL-6, IL-8,
TNF-α and also an anti-inflammatory mediator, IL-10, produced by
A549 cells treated by nicotine with and without cyclopamine, the
inhibitor of Hh pathway. Next, we attempted to explore the effect
of Hh signaling on cigarette-induced airway inflammation, since few
studies focusing on the role of such chronic airway disease were
carried out.
The Hh pathway regulates organ homeostasis and stem
cell renewal in the adult (9).
This signaling is involved in early embryogenesis, where it
promotes cell growth, differentiation, tissue patterning, and
vascularization. The canonical process of this pathway is that when
Shh is secreted and reaches its target cells, Smo is suppressed,
which results in the transcription of Gli1, Gli2, and Gli3.
Nonetheless, Gli1 exclusively acts as a transcription activator,
while Gli2 and Gli3 act either as repressors or activators
(10,11). In the nucleus, Gli1 modulates the
transcription of a multitude of genes including Fox, Myc and
cyclin D, involved in tissue development, differentiation,
epithelial-mesenchymal transition (EMT), and stem cell
maintenance.
Although the expression level of Shh is decreased at
birth, it is continually observed in epithelial cells. The Hh
pathway is also demonstrated to be involved in postnatal lung
maturation (12). In the healthy
adult lung, Hh signaling maintains adult lung quiescence and
regulates repair (13). The Shh
expression from the embryonic day is critical for branching and
growing bronchi. The interruption of Hh signaling at earlier time
points leads to defects in the wall structure of enlarged alveolar
airspaces and fewer secondary septa (5), as well as, in the regeneration and
repair after injury to adult lungs (10).
Some previous studies revealed that Hh signaling
pathway was dysregulated in diseases such as COPD and idiopathic
pulmonary fibrosis. The results of the study by Lemjabbar-Alaoui
et al (2) demonstrated that
CSE exposure activated Hh signaling in bronchial epithelial cells,
rendering that Hh plays a causal role in the initiation,
maintenance, proliferation, and survival of CSE-transformed
epithelial cells; however, its role in airway inflammation is yet
unclear.
The present study detected low Shh protein
expression in A0 control cells; however, the levels increased
significantly when treated with nicotine, and inhibited by
cyclopamine. This phenomenon demonstrated that Hh signaling is
lowly expressed in normal alveolar epithelial cells. We have also
shown that exposure to nicotine results in aberrant reactivation of
Hh pathway, as indicated by the marked expression of Shh. In
addition, the expression level of Gli1 reflects the activation of
Hh pathway significantly higher than control when stimulated by
nicotine; however, it was decreased significantly with cyclopamine
treatment. The results of Smo protein were similar to that of Gli1.
Taken together, these results suggested that Hh signaling might be
implicated in cigarette-induced airway diseases; nonetheless, the
mechanism should be investigated further.
Systemic oxidative stress and inflammation have been
implicated in COPD. This process involves the pro-inflammatory
cytokines such as IL-6, IL-8, and TNF-α, as the main promoters of
the inflammation, which serve as markers of COPD progression.
Therefore, we measured the levels of IL-6, IL-8, and TNF-α in A549
cells treated with nicotine and with and without cyclopamine using
ELISA and Western blot. The alveolar epithelial cells distinctly
produced higher levels of IL-6, IL-8, and TNF-α when cultured with
nicotine as compared to control cells for 24 and 48 h. Although
Western blot results were not completely identical to that of
ELISA; the results showed that the expression levels of these
inflammatory mediators in A2 group tended to decrease with
cyclopamine treatment at different time points as compared to the
A1 group, IL-6 at 24 h, IL-8 at 48 h, and TNF-α at 48 h,
significantly respectively). These results showed a potential role
of Hh probably in cigarette-induced airway inflammation via
regulation of specific inflammatory mediators' expressions. Hence,
we suggested that smoking aberrantly reactivated the Hh pathway,
while IL-6, IL-8, and TNF-α levels increase progressively, which
partly enhances the airway inflammation. If the Hh signaling can be
blocked, the expressions of these mediators may decrease, thereby
reducing the inflammatory reaction.
Moreover, IL-10 was also tested in our study, along
with the effect of Hh on an anti-inflammatory mediator. Its
expression in A1 group was significantly higher than that in A0 at
the 12 h time point, while a decrease was noted at 48 h. Moreover,
the IL-10 levels increased significantly in cells as a result of
cyclopamine treatment at 48 h, which was contrary to that of IL-6,
IL-8, and TNF-α. This phenomenon could be explained by its role of
anti-inflammation.
Several studies revealed the role of Hh pathway in
tumors; the constitutive activation of Hh has been observed in
cancers, such as lung, stomach, and colon (14). Hitherto, few studies focused on the
effect of Hh on cigarette-induced airway inflammation. The
additional evidence further showed that the Hh pathway could
promote the inflammatory process not only in lung diseases but also
in other systems. The study by Li et al (15) showed that the inhibition of Hh
signaling in rats with arthritis using cyclopamine reduced the
expressions of TNF-α, IL-1β and IL-6.
Interestingly, the role of Hh in airway inflammation
effectuated via hedgehog interacting protein (HIP), a negative
regulator of the Hh pathway, exerts a protective role in COPD
pathogenesis (16). HIP
competitively binds to all three ligands: Shh, Ihh, and Dhh. Lao
et al (17) detected
approximately 32% reduced expression of the HIP gene in
human COPD lung tissues as compared to ex-smoker controls with
normal lung function. Mice with HIP haploinsufficiency
(HIP+/-) are susceptible to present more severe airspace
enlargement and emphysema induced by cigarette smoke than the
wild-type. Moreover, the study also demonstrated that numbers of
lymphoid aggregates, MCP1 (monocyte chemotactic protein 1), MMP-9
and MMP-12 levels increased in HIP+/- mice lung after
cigarette smoke exposure. In addition, a significantly increased
cell death was detected in lungs from HIP+/- mice. Our
previous study also identified some single nucleotide polymorphisms
on a HIP gene associated with the susceptibility of COPD in
the Chinese Han population (18).
Consequently, HIP plays a critical role in COPD
pathogenesis; reduction in HIP expression probably increases
susceptibility to develop emphysema. HIP represses the Hh signaling
by competitive binding with three Hh ligands. However, whether the
mechanism is linked to the Hh signaling pathway is yet to be
elucidated, which has been explored in this study.
In addition, other systems showed that Hh signaling
is critical in T-cell development (19), which plays a significant role in
gastric development, homeostasis, and neoplastic transformation
(20). Reportedly, Hh signaling
components are expressed in acute myeloid leukemia cells (21).
Nevertheless, the current study had some
limitations. First, additional experiments are essential to
accumulate evidence in order to understand the mechanisme
underlying the impact of Hh pathway on inflammatory mediators. Are
these inhibitions are recovered by Hh signaling activator? Second,
these inflammatory cytokines are also controlled by another
transcriptional factor such as NF-κB. Thus, the relationship of
NF-κB with the Hh pathway necessitates further studies. Third, the
current study focused on somking-related airway inflammation;
however, the impact of Hh signaling on non-smoking-related cases of
COPD remains unclear, further studies should be performed.
The current study showed that Hh signaling pathway
expressed at a low level in normal alveolar epithelial cells. It
can be aberrantly activated by nicotine, resulting in upregulation
of certain inflammatory mediators; however, the expression levels
of mediators decreased after blockage of the Hh pathway. In
conclusion, the Hh pathway might play a major role in
cigarette-induced airway inflammation via regulation of
inflammatory mediators. Therefore, blocking the Hh signal and
diminishing the airway inflammation reaction might be a new therapy
of somking-related COPD.
Acknowledgements
The abstract was presented at the 2017 ERS
International Congress on 9th to 13th September 2017 in Milan and
published as abstract no. PA4454 in European Respiratory Journal 50
(Suppl 61): 2017 (DOI: 10.1183/1393003.congress-2017).
Funding
The present study was supported by The Natural
Science Foundation of China (grant no. 81500027).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
YG performed ELISA and western blot analysis, and
drafted the manuscript. GS, HW and MZ participated in the design of
the study and revised the manuscript. All authors read and approved
the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
COPD
|
chronic obstructive pulmonary
disease
|
|
GWAS
|
genome-wide association study
|
|
Hh
|
Hedgehog
|
|
HIP
|
edgehog interacting protein
|
|
SNP
|
single-nucleotide polymorphisms
|
References
|
1
|
McCloskey SC, Patel BD, Hinchliffe SJ,
Reid ED, Wareham NJ and Lomas DA: Siblings of patients with severe
chronic obstructive pulmonary disease have significant risk of
airflow obstruction. Am J Respir Crit Care Med. 164:1419–1424.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Lemjabbar-Alaoui H, Dasari V, Sidhu SS,
Mengistab A, Finkbeiner W, Gallup M and Basbaum C: Wnt and hedgehog
are critical mediators of cigarette smoke-induced lung cancer. PLoS
One. 1:e932006. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Hanna A and Shevde LA: Hedgehog signaling:
Modulation of cancer properies and tumor mircroenvironment. Mol
Cancer. 15:242016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Cooper MK, Porter JA, Young KE and Beachy
PA: Teratogen-mediated inhibition of target tissue response to Shh
signaling. Science. 280:1603–1607. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kugler MC, Loomis CA, Zhao Z, Cushman JC,
Liu L and Munger JS: Sonic hedgehog signaling regulates
myofibroblast function during alveolar septum formation in murine
postnatal lung. Am J Respir Cell Mol Biol. 57:280–293. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Xu JH, Yang HP, Zhou XD, Wang HJ, Gong L
and Tang CL: Autophagy accompanied with
bisdemethoxycurcumin-induced apoptosis in non-small cell lung
cancer cells. Biomed Environ Sci. 28:105–115. 2015.PubMed/NCBI
|
|
7
|
Bermudez O, Hennen E, Koch I, Lindner M
and Eickelberg O: Gli1 mediates lung cancer cell proliferation and
sonic hedgehog-dependent mesenchymal cell activation. PLoS One.
8:e632262013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Bao J, Ma C, Ran J, Xiong Y, Yan S and Wu
L: Wnt/β-catenin and hedgehog pathways are involved in the
inflammatory effect of interleukin 18 on rat chondrocytes.
Oncotarget. 8:109962–109972. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Beachy PA, Karhadkar SS and Berman DM:
Tissue repair and stem cell renewal in carcinogenesis. Nature.
432:324–331. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Deng M, Li J, Gan Y, Chen Y and Chen P:
Changes in the number of CD31-CD45-Sca-1+ cells and Shh signaling
pathway involvement in the lungs of mice with emphysema and
relevant effects of acute adenovirus infection. Int J Chron
Obstruct Pulmon Dis. 12:861–872. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Fu L, Wu H, Cheng SY, Gao D, Zhang L and
Zhao Y: Set7 mediated Gli3 methylation plays a positive role in the
activation of sonic hedgehog pathway in mammals. ELife.
5:e156902016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Liu L, Kugler MC, Loomis CA, Samdani R,
Zhao Z, Chen GJ, Brandt JP, Brownell I, Joyner AL, Rom WN and
Munger JS: Hedgehog signaling in neonatal and adult lung. Am J
Respir Cell Mol Biol. 48:703–710. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Peng T, Frank DB, Kadzik RS, Morley MP,
Rathi KS, Wang T, Zhou S, Cheng L, Lu MM and Morrisey EE: Hedgehog
actively maintains adult lung quiescence and regulates repair and
regeneration. Nature. 526:578–582. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Abe Y and Tanaka N: The hedgehog signaling
networks in lung cancer: The mechanisms and roles in tumor
progression and implications for cancer therapy. Biomed Res Int.
2016:79692862016. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Li R, Cai L, Ding J, Hu CM, Wu TN and Hu
XY: Inhibition of hedgehog signal pathway by cyclopamine attenuates
inflammation and articular cartilage damage in rats with
adjuvant-induced arthritis. J Pharm Pharmacol. 67:963–971. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lao T, Jiang Z, Yun J, Qiu W, Guo F, Huang
C, Mancini JD, Gupta K, Laucho-Contreras ME, Naing ZZ, et al: Hhip
haploinsufficiency sensitizes mice to age-related emphysema. Proc
Natl Acad Sci USA. 113:E4681–E4687. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Lao T, Glass K, Qiu W, Polverino F, Gupta
K, Morrow J, Mancini JD, Vuong L, Perrella MA, Hersh CP, et al:
Haploinsufficiency of hedgehog interacting protein causes increased
emphysema induced by cigarette smoke through network rewiring.
Genome Med. 7:122015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Guo Y, Gong Y, Pan C, Qian Y, Shi G, Cheng
Q, Li Q, Ren L, Weng Q, Chen Y, et al: Association of genetic
polymorphisms with chronic obstructive pulmonary disease in the
chinese han population: A case-control study. BMC Med Genomics.
5:64–83. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Crompton T, Outram SV and
Hager-Theodorides AL: Sonic hedgehog signalling in T-cell
development and activation. Nat Rev Immunol. 7:726–735. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Merchant JL and Ding L: Hedgehog signaling
links chronic inflammation to gastric cancer precursor lesions.
Cell Mol Gastroenterol Hepatol. 3:201–210. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kakiuchi S, Minami Y, Miyata Y, Mizutani
Y, Goto H, Kawamoto S, Yakushijin K, Kurata K, Matsuoka H and
Minami H: NANOG Expression as a responsive biomarker during
treatment with hedgehog signal inhibitor in acute myeloid leukemia.
Int J Mol Sci. 18:4862017. View Article : Google Scholar :
|