Introduction
Fanconi anemia (FA) is a genetic disorder that is
caused by an abnormality in genes encoding Fanconi anemia proteins
(FA proteins), which comprise a multi-protein nuclear complex that
has an essential role in DNA inter-strand crosslink repair
(1,2). Dysfunction of FA proteins leads to
cytogenetic instability, chromosomal breakage, defective DNA repair
and cellular hypersensitivity to DNA crosslinking agents (3), and these effects are known to cause
developmental defects, bone marrow failure, aplastic anemia and
increased cancer risk in individuals with FA (4–6). To
a lesser extent, metabolic disorders associated with FA, including
diabetes mellitus (DM), hyperglycemia and insulin disorders, have
been reported in ~40% of patients with FA (7–9).
FA complementation group C (FANCC) is a component of
the FA multiprotein nuclear complex (2). However, it has been demonstrated that
instead of FANCC being an isoform targeted to the nucleus, FANCC
localization in cytoplasm is essential for the correction of
enhanced cytotoxicity of crosslinks in cells with predominant
defects in the FANCC gene (10). Additionally, cytoplasmic FANCC has
a role in prevention of oxidative DNA damage (11), and protection against oxidative
stress-induced apoptosis (12).
Reactive oxygen species (ROS) accumulation and cell apoptosis have
been identified in many cell types with FANCC depletion. These cell
types include hematopoietic progenitor cells, hepatocytes, and
murine embryonic fibroblasts isolated from FA model mice (13–16).
It was suggested that FANCC counteraction of oxidative stress is
mediated through glutathione S-transferase P1 (GSTP1) and
nicotinamide adenine dinucleotide phosphate-cytochrome P450
reductase (17,18). FANCC significantly increased the
catalytic activity of GSTP1, which is an inducible antioxidant
enzyme that has a major role in the intracellular defense of toxin
and ROS (19). Fancc-/- cells were
hypersensitive to oxidant stimuli and underwent enhanced
oxidant-mediated apoptosis compared with the wild type controls as
a result of oxidative stress activating the redox-dependent
apoptosis signal-regulating kinase 1 pathway (15). Wang et al (20) reported that overexpression of
FANCC protected hematopoietic progenitors from death induced
by Fas-mediated apoptosis. A previous study demonstrated that FANCC
associated with uncoordinated-5A protein, a pro-apoptotic dependent
receptor, and delayed apoptosis in neuroblastoma SH-SY5Y cell line
(21). These findings indicate
that oxidative stress is a contributor to FA pathogenesis, and that
FANCC is a key contributor to oxidative stress response mechanisms
in various cell types.
Oxidative stress has also been implicated as an
important factor in the progression of DM via interference with
insulin signal transduction, insulin production and induction of
β-cell apoptosis (22,23). Oxidative stress and DM are commonly
observed in patients with FA, and high levels of ROS were detected
in insulin-sensitive tissues of Fancc-/- mice (24). Furthermore, insulin receptor and
insulin receptor substrate 1 tyrosine phosphorylation were impaired
in Fancc knockout mice treated with tumor necrosis factor
(TNF)-α compared with their wild-type littermates (24). It was concluded that defects in
Fancc led to ROS accumulation in insulin target cells, thus
interfering with the insulin signaling pathway, leading to insulin
resistance (24).
While most studies in the literature focused on the
actions of FANCC on signal transduction in insulin-responsive
cells, none has explored the role of FANCC in β-cells with limited
expression of antioxidant enzymes [reviewed by Robertson and Harmon
(25)]. In the present study, it
was hypothesized that depletion of FANCC causes pancreatic β-cell
hypersensitivity to oxidative stress-induced apoptosis.
Accordingly, the aim of the present study was to investigate the
role of FANCC in pancreatic β-cell response to oxidative
stress.
Materials and methods
Study approval
The present study was conducted at the Faculty of
Medicine Siriraj Hospital, Mahidol University (Bangkok, Thailand).
Siriraj Hospital is Thailand's largest university-based national
tertiary referral center. The protocol for the present study was
approved by the Siriraj Institutional Review Board (COA no.
Si491/2014).
Cell culture
Human 1.1B4 pancreatic β-cell line (American Type
Culture Collection, Manassas, VA, USA) was maintained in RPMI-1640
medium (Invitrogen; Thermo Fisher Scientific, Inc., Waltham, MA,
USA) containing 10% fetal bovine serum (Gibco; Thermo Fisher
Scientific, Inc.), 100 IU/ml penicillin, and 100 mg/ml streptomycin
at 37°C in a 95% humidified atmosphere containing 5% CO2.
Small interfering RNA (siRNA)
transfection
Human FANCC SMARTpool ON-TARGETplus siRNA
(siRNA-FANCC; cat. no. L-011033-00-0005) and siRNA ON-TARGETplus
Non-targeting Pool (siRNA-control; cat. no. D-001810-10-20) were
purchased from Dharmacon (GE Healthcare; Dharmacon, Inc.,
Lafayette, CO, USA). The day prior to transfection, 1.6×105
cells/well were seeded into 6-well plates. Knockdown was performed
at 24 and 48 h after seeding. siRNA-FANCC and siRNA-control were
transfected at a concentration of 100 pmol using
Lipofectamine® 2000 Transfection Reagent (Invitrogen;
Thermo Fisher Scientific, Inc.) according to the manufacturer's
protocol. At 48 h after transfection, the cells were harvested and
FANCC expression levels were determined by western blot
analysis.
Plasmid construct and
transfection
To generate FANCC recombinant plasmids, FANCC coding
sequence NM_000136.2 and FLAG-tag sequence DYKDDDDK were amplified
with the following primer sequences: Forward,
5′-CGGGATCCATGGCTCAAGATTCAGTAG-3′ and reverse,
5′-GCTCTAGACTACTTATCGTCGTCATCCTTGTAATCGACTTGAGTTCGCAGCTCTTTAAGG-3′.
The product was cloned into pcDNA3.1 plasmids (Invitrogen; Thermo
Fisher Scientific, Inc.). All plasmids were amplified in
Escherichia coli (DH5α) and purified using QIAamp DNA Mini
Kit (Qiagen GmbH, Hilden, Germany). The day prior to transfection,
2×105 cells/well were seeded into 6-well plates. After 24 h, the
cells were transfected with 500 ng FANCC-constructed plasmid or
pcDNA3.1 empty plasmid using the same conditions as the ones
described for siRNA transfection. At 48 h after transfection, the
cells were harvested and FANCC protein expression levels were
determined by western blot analysis.
RNA isolation and RT-qPCR
Total RNA from 1.1B4 cells (5×105) was isolated
using TRIzol Reagent (Invitrogen; Thermo Fisher Scientific, Inc.).
Subsequently, RNA was reverse transcribed into cDNA using a
RevertAid First Strand cDNA Synthesis Kit (Thermo Fisher
Scientific, Inc.) Gene-specific primer pairs were designed using
Primer3Plus program (www.bioinformatics.nl/cgi-bin/primer3plus/primer3plus.cgi)
or derived from previous studies (26–28).
All primers (Table I) were checked
against National Center for Biotechnology Information Primer-BLAST.
RT-qPCR was performed in a LightCycler 480 Instrument (Roche
Diagnostics GmbH, Mannheim, Germany) using LightCycler®
480 SYBR Green I Master (Roche Diagnostics GmbH). Initial enzyme
activation proceeded at 95°C for 10 min, followed by 45 cycles at
95°C for 30 sec, 60–62°C for 20 sec, and 72°C for 20 sec. β-actin
was used as an internal control to normalize input cDNA. Relative
mRNA expression levels were calculated using the 2-ΔΔCq method
(29). All assays were performed
in triplicate.
 | Table I.Primers and conditions for reverse
transcription-quantitative polymerase chain reaction. |
Table I.
Primers and conditions for reverse
transcription-quantitative polymerase chain reaction.
| First author,
year | Gene | Primer (5′-3′) | Annealing
temperature (°C) | Product size
(bp) | (Refs.) |
|---|
| Present study | FANCC
(NM_000136) | F:
TCATCGCTGCCTCAAGC | 62 | 352 | – |
|
|
| R:
GGAACCAGCTCTAAAGGG |
|
|
|
| Robertson,
2007 | GCK
(NM_033508) | F:
TGGACCAAGGGCTTCAAGGCC | 60 | 207 | (25) |
|
|
| R:
CATGTAGCAGGCATTGCAGCC |
|
|
|
| Robertson,
2007 | INS
(NM_000207) | F:
TACCAGCATCTGCTCCCTCT | 60 | 120 | (25) |
|
|
| R:
TGCTGGTTCAAGGGCTTTAT |
|
|
|
| Vasu, 2013 | BCL-2
(NM_000657) | F:
TTTGAGTTCGGTGGGGTCAT | 62 | 275 | (26) |
|
|
| R:
TGACTTCACTTGTGGCCCAG |
|
|
|
| Vasu, 2013 | BAX
(NM_138763) | F:
TGGCAGCTGACATGTTTTCTGAC | 62 | 195 | (26) |
|
|
| R:
TCACCCAACCACCCTGGTCTT |
|
|
|
| Floros, 2006 | DKK1
(NM_012242) | F:
TCACGCTATGTGCTGCCCCG | 62 | 223 | (27) |
|
|
| R:
TGAGGCACAGTCTGATGACCGGA |
|
|
|
| Present study | β-actin
(NM_001101) | F:
AGAAAATCTGGCACCACACC | 62 | 395 | – |
|
|
| R:
CTCCTTAATGTCACGCACGA |
|
|
|
Western blot analysis
Total protein was extracted by lysing cells in
radioimmunoprecipitation assay lysis buffer (Thermo Fisher
Scientific Inc.). Protein concentrations were quantified by
Bradford protein assay (Bio-Rad Laboratories, Inc., Hercules, CA,
USA) using a NanoDrop 2000 spectrophotometer (Thermo Fisher
Scientific, Inc.). Proteins (50–100 µg) were separated onto 10% SDS
polyacrylamide gel, and then electroblotted onto nitrocellulose
membrane (Bio-Rad Laboratories, Inc.) using a semi-dry
electrophoretic transfer cell (Bio-Rad Laboratories, Inc.). Primary
goat polyclonal antibody against human FANCC (C-14; cat. no.
sc-18110; 1:500; Santa Cruz Biotechnology Inc., Dallas, TX, USA),
mouse monoclonal antibody against FLAG-tag (1:2,000; cat. no.
F3165; Sigma-Aldrich; Merck KGaA, Darmstadt, Germany), and mouse
monoclonal antibody against β-actin (1:1,000; cat. no. s-47778;
Santa Cruz Biotechnology, Inc.) were incubated with the membranes
for 2 h at room temperature. Following this, membranes were
incubated with horseradish peroxidase-linked rabbit anti-goat or
goat anti-mouse secondary antibody (1:1,000; cat. no. P044701-2;
Dako; Agilent Technologies, Inc., Santa Clara, CA, USA) for 1 h at
room temperature. Binding antibodies were visualized using an
enhanced chemiluminescence detection kit (SuperSignal West Pico
PLUS Chemiluminescent Substrate; Roche Diagnostics GmbH), and bands
were detected by biomolecular imager (ImageQuant LAS 4010; GE
Healthcare Life Sciences, Little Chalfont, UK). β-actin staining
was used as an internal control in all experiments. Expressed bands
were quantified by computer-assisted scanning densitometry using
ImageJ version 1.4.3.x (National Institutes of Health, Bethesda,
MD, USA).
Flow cytometry
Following knockdown or overexpression of
FANCC in 1.1B4 β-cells for 48 h, 1.4×106 cells were treated
with 0.5 mM H2O2 for 4 h. Subsequently, Annexin V-fluorescein
isothiocyanate (FITC) and propidium iodide (PI) staining was
performed using Annexin V-FITC/PI Apoptosis Detection Kit
(ImmunoTools GmbH, Friesoythe, Germany) according to the
manufacturer's protocol. Apoptosis was analyzed using a BD
FACSVerse flow cytometer (BD Biosciences, San Jose, CA, USA) and
FlowJo software version 10.4 (FlowJo LLC, Ashland, OR, USA). Each
experiment was independently performed three times.
Caspase activity
Luminescent type Caspase-Glo 3/7 Assay (Promega
Corporation, Madison, WI, USA) was used to measure caspase 3/7
activity. Cells were seeded in a Corning 96-well white flat bottom
plate (Corning Life Sciences, Tewksbury, MA, USA) at 8×103 and
1×104 cells per well for knockdown and overexpression,
respectively. Following 24 h, the cells were transfected with 500
ng plasmids (FANCC-pcDNA3.1 or empty vector) or 100 pmol siRNA
(siRNA-FANCC or siRNA-control). In order to increase the population
of cells successfully transfected with either FANCC-pcDNA3.1, empty
vector, siRNA-FANCC or siRNA-control, the transfected cells were
transfected twice with the same type and amount of plasmid 24 h
after initial transfection. At 24 h after the second transfection,
cells were treated with 0.5 mM H2O2 for 4 h. Following this, 100 µl
Caspase-Glo 3/7 Reagent (Promega Corporation) was added into each
well. The plate was then shaken at 300 to 500 rpm in a dark room
for 30 min at room temperature (~25°C). Luminescence was measured
using a multi-detection microplate reader (Synergy H1; BioTek
Instruments, Inc., Winooski, VT, USA).
Statistical analysis
Data analysis was performed using SPSS Statistics
version 18.0 (SPSS, Inc., Chicago, IL, USA). Student's t-test was
used to evaluate differences of the mean ± standard error of the
mean. P<0.05 was considered to indicate a statistically
significant difference.
Results
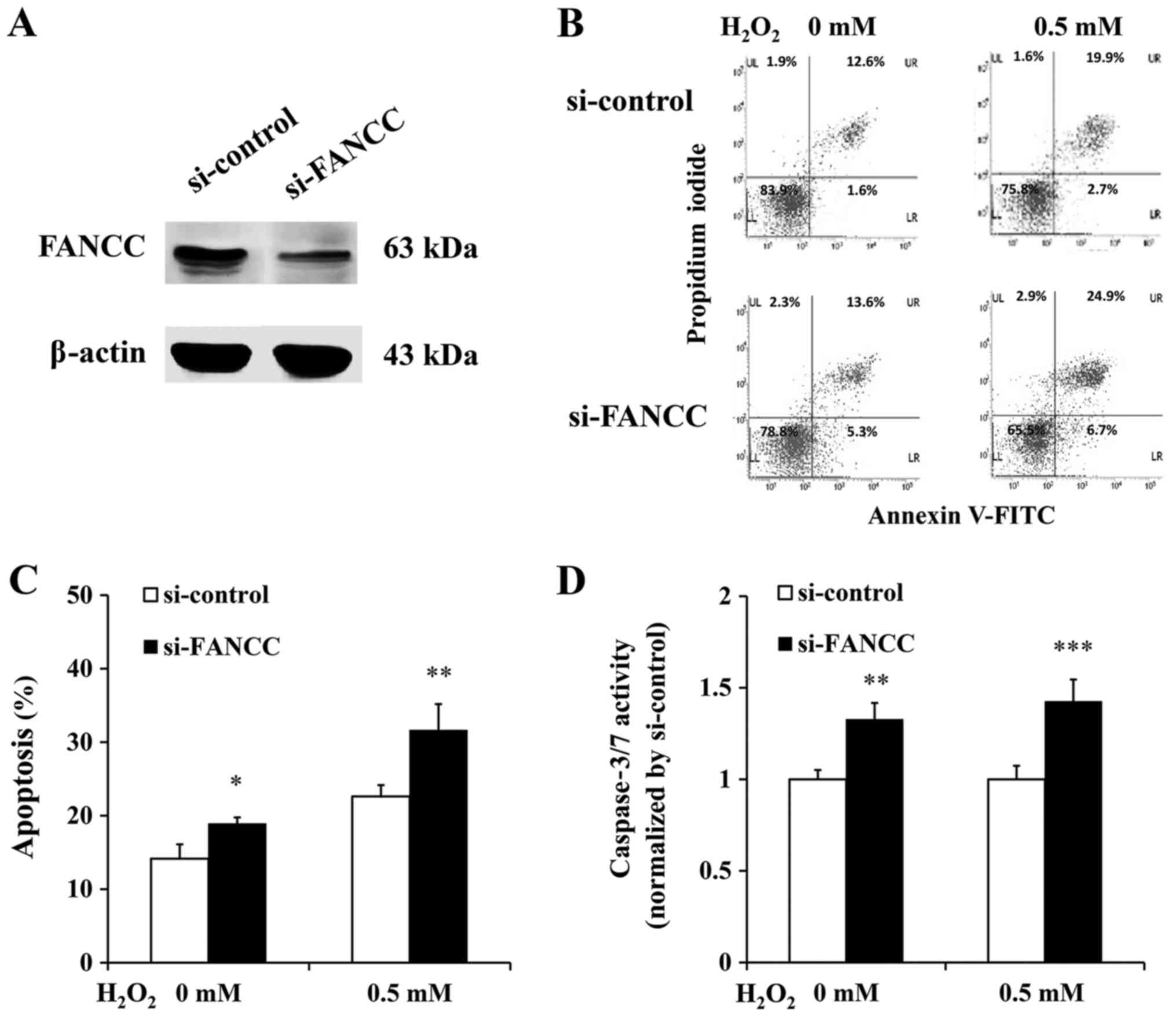
Increased apoptosis of FANCC-deficient
β-cells
In order to investigate the role of FANCC in
β-cell response to oxidative stress, FANCC-depleted 1.1b4 cells
were generated via siRNA-mediated transient silencing. Knockdown
efficiency is demonstrated in Fig.
1A. The results demonstrated that FANCC knockdown cells
exhibited increased apoptosis compared with si-control cells, in
non-induced and H2O2-induced oxidative stress conditions (Fig. 1B-D). In non-induced condition,
Annexin V-FITC/PI staining revealed 12.6 and 1.6% of control cells
to be in late and early apoptotic states, respectively; while, 13.6
and 5.3% of FANCC knockdown cells were in late and early apoptotic
states, respectively (Fig. 1B,
left). Under H2O2-induced oxidative stress induction, 19.9 and 2.7%
of control cells were in late and early apoptotic states,
respectively, while 24.9 and 6.7% of FANCC knockdown cells were in
late and early apoptotic states, respectively (Fig. 1B, right). Overall, in non-induced
condition, ~14.2 and ~18.9% of control cells and FANCC
knockdown cells, respectively, were apoptotic (P<0.05). Under
oxidative stress induction, the percentage of apoptotic cells in
control cells and FANCC knockdown cells increased to 22.6
and 31.3%, respectively (P<0.01; Fig. 1C). Caspase 3/7 activities of
FANCC knockdown cells were 1.33-fold and 1.43-fold higher
than control cells under non-induced and H2O2-induced oxidative
stress conditions, respectively (P<0.01 and P<0.001,
respectively; Fig. 1D).
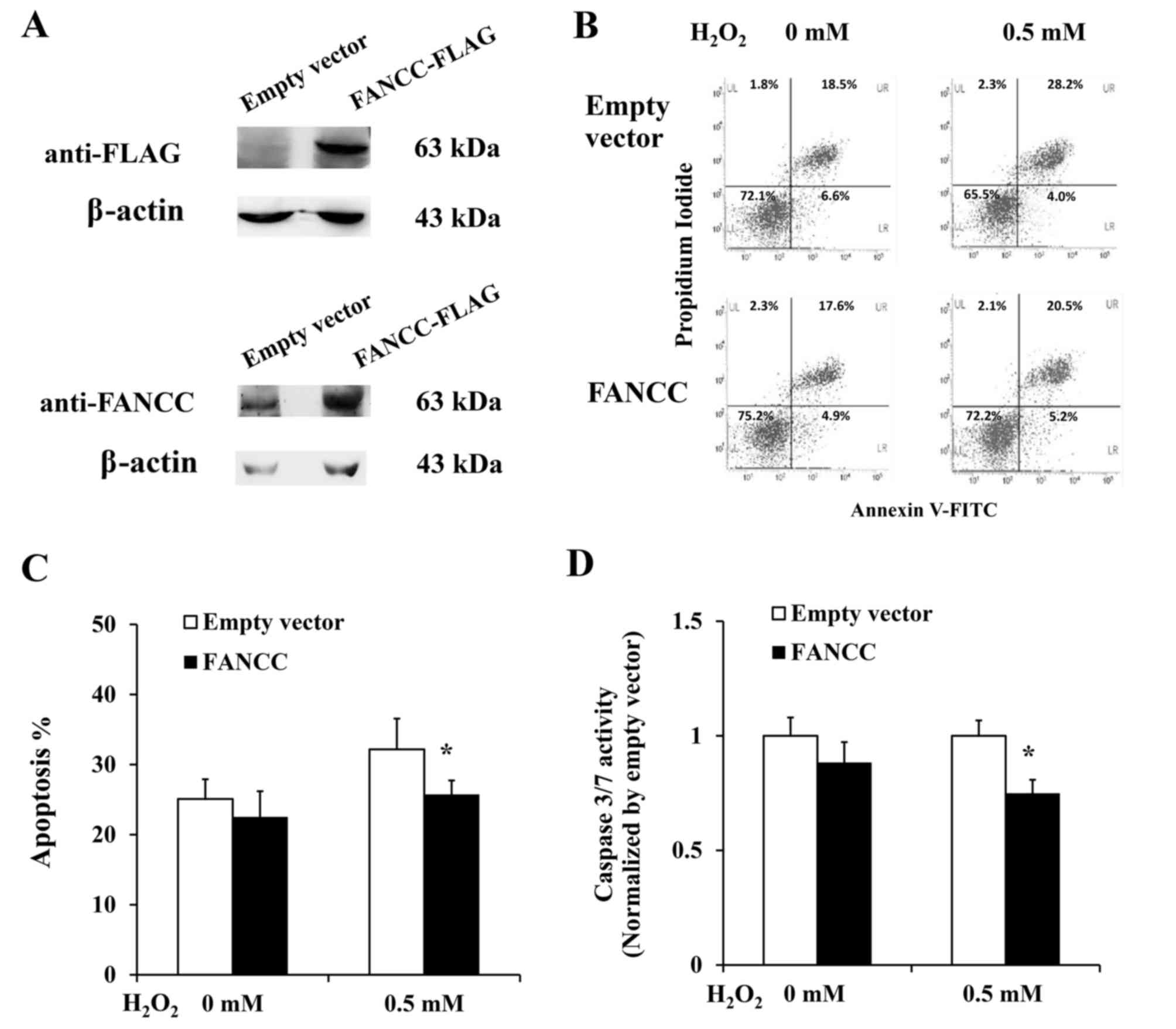
Overexpression of FANCC protects
β-cells from oxidative stress-induced apoptosis
To further elucidate the effect of FANCC in
attenuating β-cell apoptosis, plasmid construct for FANCC
overexpression was transfected into 1.1B4 β-cells, and the
percentage of apoptosis in cells overexpressing FANCC was
investigated. Annexin V-FITC/PI staining (Fig. 2A and B) demonstrated that 1.1B4
β-cells overexpressing FANCC had lower total percentage of
apoptotic cells than control cells transfected with empty vector
(22.5 vs. 25.1%, respectively; P=0.067). Additionally, under
oxidative stress induction, the percentage of apoptotic cells in
1.1B4 β-cells overexpressing FANCC was significantly lower
than that of control cells transfected with empty vector (25.7 vs.
32.2%, respectively; P<0.05; Fig.
2C). In addition, 1.1B4 β-cells overexpressing FANCC had
lower caspase 3/7 activity compared to control cells (0.75-fold;
P<0.05) under H2O2-induced oxidative stress conditions (Fig. 2D). However, the significant
protective effect of FANCC against apoptosis was not observed in
non-induced condition.
Depletion of FANCC alters expression
of genes involved in insulin expression, secretion and apoptosis
pathways
To confirm the anti-apoptotic effect of FANCC in the
human β-cell line, the expression of anti-apoptotic [BCL2
apoptosis regulator (BCL-2)] and pro-apoptotic genes
[BCL2 associated X, apoptosis regulator (BAX)] were
investigated. The results demonstrated that in FANCC-deficient
1.1b4 β-cells, BCL-2 mRNA expression decreased by 2.59 fold
(P<0.001), while BAX mRNA level increased by 1.93 fold
(P<0.01), compared to the expressions of control cells (Fig. 3). Depletion of FANCC also led to a
3.15-fold increase in dickkopf WNT signaling pathway inhibitor
1 (DKK1) expression (P<0.01). As DKK1 encodes
a negative regulator of Wnt signaling, a pathway implicated in
β-cell apoptotic defenders (30),
increased DKK1 expression level in FANCC-deficient cells
thereby reaffirms the crucial role of FANCC in protection against
β-cell apoptosis. Notably, the expression levels of insulin
(INS) and glucokinase (GCK) genes, which are
critical for insulin synthesis and secretion, were decreased
[0.77-fold (P<0.01) and 0.82-fold (P<0.05), respectively]
compared to cells transfected with siRNA-control.
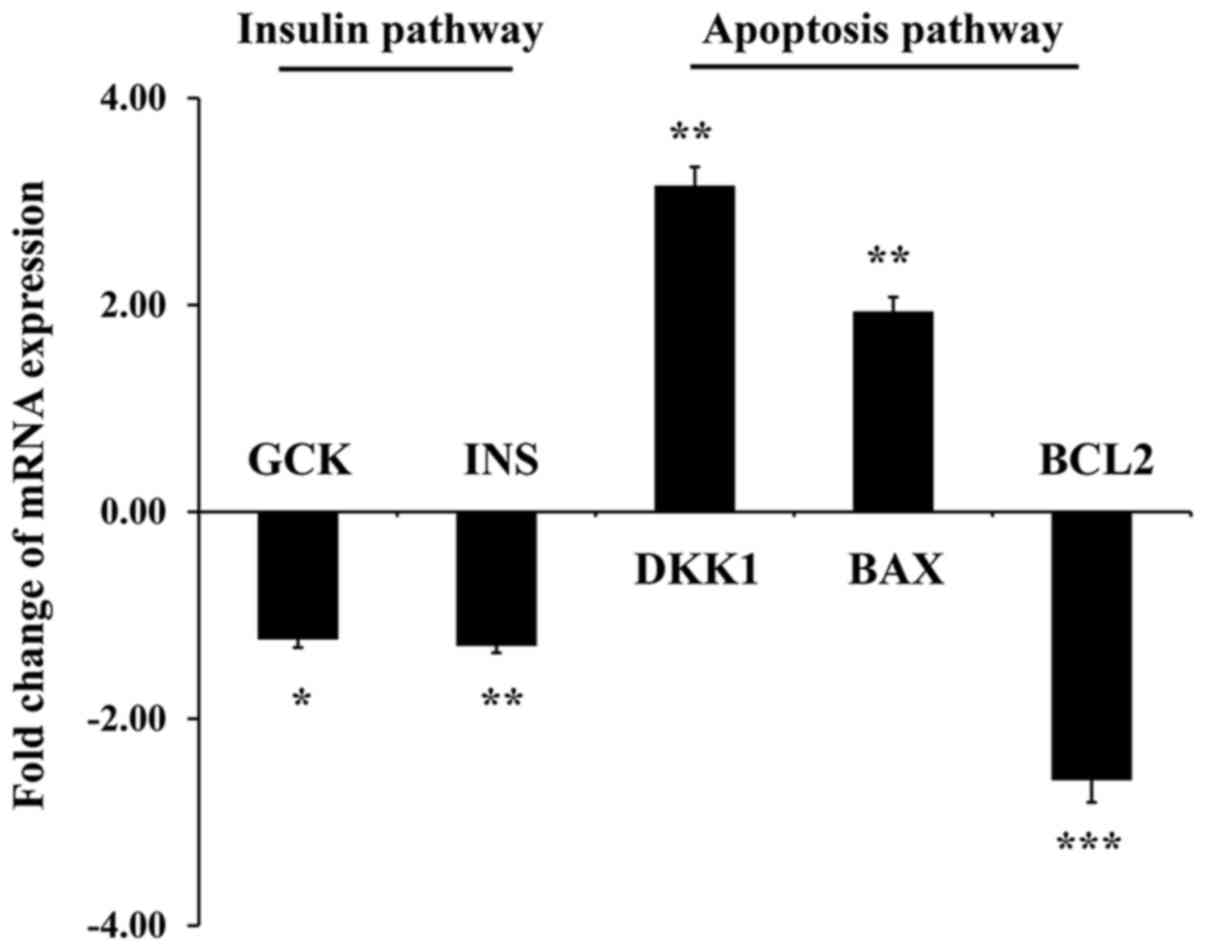 | Figure 3.Up and downregulation of genes
involved in insulin (GCK, INS) and apoptosis pathways (DKK1, BAX,
BCL2) investigated in FANCC-depleted 1.1B4 β-cells, as measured by
reverse transcription-quantitative polymerase chain reaction
experiments. Data are presented as mRNA expression fold change
(mean ± standard error) in FANCC-depleted cells relative to cells
expressing siRNA-control. *P<0.05, **P<0.01, ***P<0.001
vs. si-RNA control. FANCC, Fanconi anemia complementation group C;
siRNA, small interfering RNA; GCK, glucokinase; INS, insulin; DKK1,
dickkopf WNT signaling pathway inhibitor 1; BAX, BCL2 associated X,
apoptosis regulator; BCL2, BCL2 apoptosis regulator. |
Discussion
DM and abnormalities of glucose and insulin
metabolism are common among patients with FA (7–9).
However, little is known regarding the role of FA proteins in
maintenance of glucose homeostasis. While previous studies focused
on the link between ROS accumulation and insulin resistance in
FANCC-deficient state (24), the
results of the current study provide evidence linking FANCC
insufficiency to DM via deterioration of β-cell ability to defend
against oxidative stress-induced apoptosis.
Pancreatic β-cells have inherently low levels of
antioxidants (31), which may be
due to the need to maintain a low level of ROS to facilitate
insulin production. Accordingly, this specialized cell type is
extremely susceptible to oxidative stress. Previous studies
addressed the deleterious effects of oxidative stress on β-cell
function and survival (24,32),
and a link between inability of β-cells to adequately secrete
insulin and the development of type 2 DM was demonstrated (33,34).
Furthermore, hypersensitivity to oxidative agents and enhancement
of oxidant-mediated apoptosis were evident in FANCC-deficient cells
isolated from mice (35). In
addition, Zhang et al (36)
demonstrated that TNF-α-induced senescence was associated with the
accumulation of ROS and oxidative DNA damage in Fancc−/−
mice compared with wild-type littermates. In the present study, the
role of FANCC in antioxidant defense mechanisms of the human 1.1b4
β-cell line was investigated. It was also demonstrated that without
H2O2-induced oxidative stress, siRNA-mediated FANCC
suppression led to a significant increase in β-cell apoptosis,
while transient overexpression of FANCC decreased β-cell
apoptosis. However, the significant protective effect of FANCC
overexpression and empty vector against apoptosis was not observed
in non-induced conditions. This may be due to the presence of
endogenous FANCC expression in 1.1b4 cells, which may have been
sufficient enough to prevent apoptosis in physiological conditions.
These results suggest the necessity of a physiological level of
FANCC as a requirement for β-cell survival. Under oxidative
stress-induced conditions, apoptosis was more pronounced in
FANCC-depleted cells, while FANCC-overexpressed cells were
resistant to H2O2-induced oxidative stress. These data reaffirm the
critical role of FANCC in protection against oxidative
stress-induced β-cell apoptosis. However, the data indicated a
minor increase of apoptosis level in FANCC knockdown cells as
compared with control cells. Potentially, other pathways besides
FANCC are involved in cell apoptosis or there may be compensatory
mechanisms that counteract the apoptosis. Additionally, the
increased or decrease in caspase 3/7 activities were marginal,
although statistically significant. BCL2 expression was decreased
and BAX expression was increased >2-fold in FANCC knockdown
cells compared with controls. The difference in apoptotic indicator
expression is not surprising, as BCL2 and BAX were detected at the
mRNA level while caspase 3/7 activity was determined at the protein
level; furthermore, numerous molecules and signaling pathways are
involved. However, it can be concluded that lack of FANCC increases
caspase 3/7 activity, increases BAX and decreases BCL-2 RNA
level.
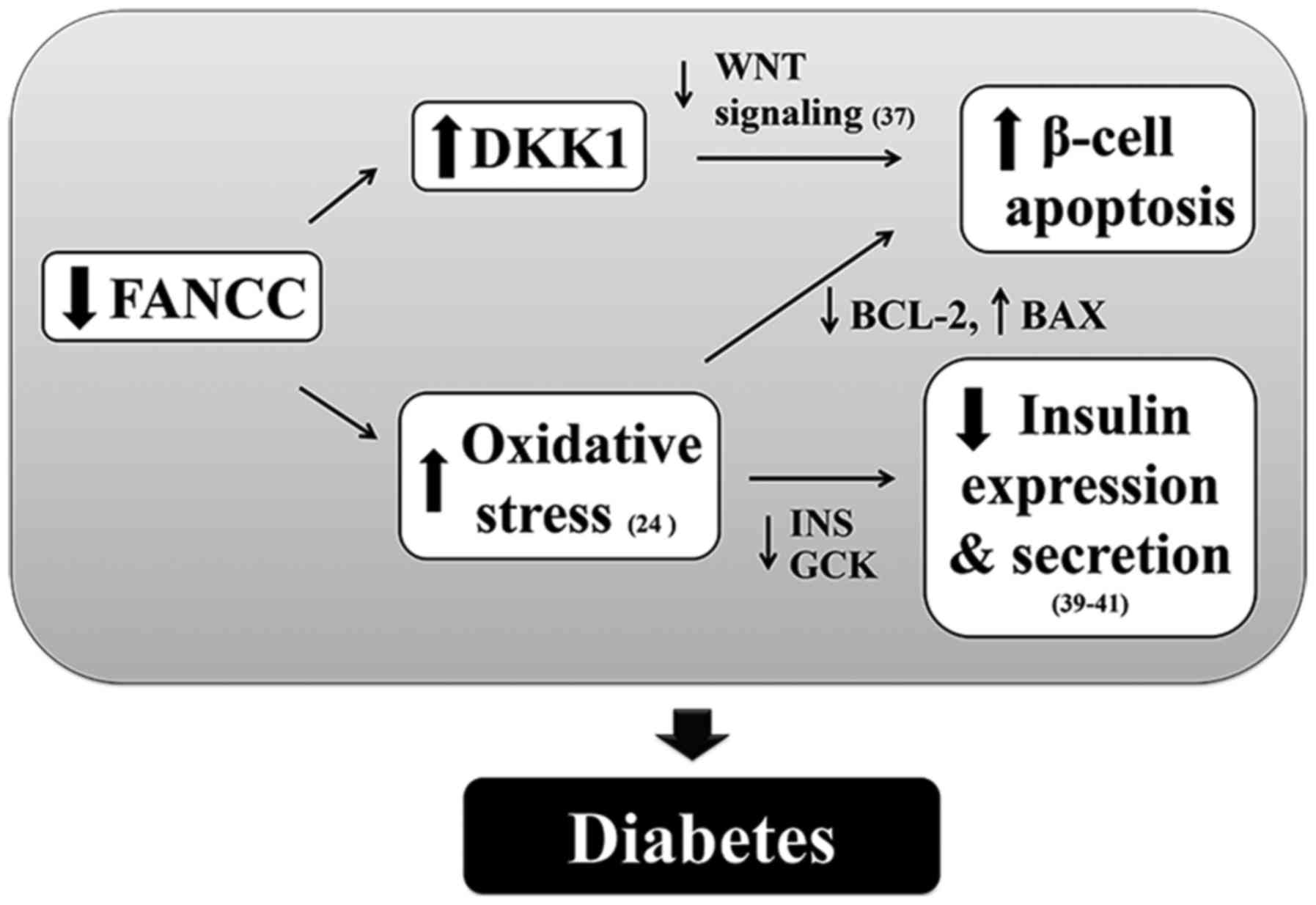
Although a comprehensive and conclusive mechanism
that explains how FANCC facilitates β-cell survival was not
elucidated in the present study, the finding that DKK1
expression level was increased in FANCC-depleted β-cells suggests
the possibility of Wnt signal transduction involvement, which
required further investigation. DKK1 is known to be a Wnt
signaling antagonist that sequesters Wnt receptors (30,37).
Upregulation of DKK1 inhibits Wnt signal transduction, thus
promoting apoptosis (38), and
DKK1 upregulation was observed in FANCC-depleted cells
(39). FANCC forms a complex with
C-terminal-binding protein-1 and β-catenin, and acts as a
transcriptional repressor that directly inhibits DKK1
expression (40). It is,
therefore, possible that FANCC modulates DKK1 expression via
nuclear activity. The exact mechanism by which FANCC regulates
DKK1 requires further investigation.
In addition to loss of protection against oxidative
stress-induced apoptosis in FANCC-depleted 1.1B4 β-cells, the
expression of genes involved in insulin synthesis (such as,
INS), a key hormone for regulating glucose uptake into
peripheral cells, and secretion (such as, GCK), a
rate-limiting enzyme in glycolysis that is a sensor of
glucose-stimulated insulin secretion (41), was decreased. Reduction in
INS and GCK expression is common in pancreatic
β-cells exposed to oxidative stress, and in islets isolated from DM
subjects (42–44). Taken together, the data from the
present study reveal an association between FANCC depletion and
loss of oxidative defense in pancreatic β-cells and reduction in
insulin production (Fig. 4).
Furthermore, the mechanism underpinning the association of
FANCC knockdown with upregulation of DKK1 in human β-cells
will be explored in future studies. These findings may explain the
development of DM and abnormal glucose metabolism in patients with
FA (7). Finally, the finding that
FANCC overexpression reduced β-cell apoptosis advances the
possibility of an alternative approach to the treatment of DM
caused by FANCC defects.
Acknowledgements
We thank the members of Siriraj Center of Research
Excellence for Diabetes and Obesity (SiCORE-DO) for their
assistance.
Funding
The present study was supported by Mahidol
University Grant, Siriraj Research Grant for Research and
Development, Faculty of Medicine, Siriraj Hospital R015810001 (to
NP), Thailand Research Fund IRG5980006 (to PY), Mahidol University
Postdoctoral Fellowship Grant (to SK) and Chalermprakiat Grants and
Research Fund (R016034011) from the Faculty of Medicine Siriraj
Hospital (to PJ and PY).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
SK, PTY and NP conceived and designed the study. SK
performed the cell experiments and molecular biology analysis. PJ
and WT analyzed and interpreted the data. SK was a major
contributor in writing the manuscript. All authors read and
approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Pagano G and Youssoufian H: Fanconi
anaemia proteins: Major roles in cell protection against oxidative
damage. Bioessays. 25:589–595. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Pace P, Johnson M, Tan WM, Mosedale G, Sng
C, Hoatlin M, de Winter J, Joenje H, Gergely F and Patel KJ: FANCE:
The link between Fanconi anaemia complex assembly and activity.
EMBO J. 21:3414–3423. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kalb R, Neveling K, Nanda I, Schindler D
and Hoehn H: Fanconi anemia: Causes and consequences of genetic
instability. Genome Dyn. 1:218–242. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Bagby GC Jr: Genetic basis of Fanconi
anemia. Curr Opin Hemato. 10:68–76. 2003. View Article : Google Scholar
|
|
5
|
de Winter JP and Joenje H: The genetic and
molecular basis of Fanconi anemia. Mutat Res. 668:11–19. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Mathew CG: Fanconi anaemia genes and
susceptibility to cancer. Oncogene. 25:5875–5884. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Elder DA, D'Alessio DA, Eyal O, Mueller R,
Smith FO, Kansra AR and Rose SR: Abnormalities in glucose tolerance
are common in children with fanconi anemia and associated with
impaired insulin secretion. Pediatr Blood Cancer. 51:256–260. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Swift M, Sholman L and Gilmour D: Diabetes
mellitus and the gene for Fanconi's anemia. Science. 178:308–310.
1972. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Giri N, Batista DL, Alter BP and Stratakis
CA: Endocrine abnormalities in patients with Fanconi anemia. J Clin
Endocrinol Metab. 92:2624–2631. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Youssoufian H: Localization of Fanconi
anemia C protein to the cytoplasm of mammalian cells. Proc Natl
Acad Sci USA. 91:7975–7979. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lackinger D, Ruppitsch W, Ramirez MH,
Hirsch-Kauffmann M and Schweiger M: Involvement of the Fanconi
anemia protein FA-C in repair processes of oxidative DNA damages.
FEBS Lett. 440:103–106. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Youssoufian H: Cytoplasmic localization of
FAC is essential for the correction of a prerepair defect in
Fanconi anemia group C cells. J Clin Inves. 97:2003–2010. 1996.
View Article : Google Scholar
|
|
13
|
Cumming RC, Liu JM, Youssoufian H and
Buchwald M: Suppression of apoptosis in hematopoietic
factor-dependent progenitor cell lines by expression of the FAC
gene. Blood. 88:4558–4567. 1996.PubMed/NCBI
|
|
14
|
Hadjur S, Ung K, Wadsworth L, Dimmick J,
Rajcan-Separovic E, Scott RW, Buchwald M and Jirik FR: Defective
hematopoiesis and hepatic steatosis in mice with combined
deficiencies of the genes encoding Fancc and Cu/Zn superoxide
dismutase. Blood. 98:1003–1011. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Saadatzadeh MR, Bijangi-Vishehsaraei K,
Hong P, Bergmann H and Haneline LS: Oxidant hypersensitivity of
Fanconi anemia type C-deficient cells is dependent on a
redox-regulated apoptotic pathway. J Biol Chem. 279:16805–16812.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Rathbun RK, Faulkner GR, Ostroski MH,
Christianson TA, Hughes G, Jones G, Cahn R, Maziarz R, Royle G,
Keeble W, et al: Inactivation of the Fanconi anemia group C gene
augments interferon-gamma-induced apoptotic responses in
hematopoietic cells. Blood. 90:974–985. 1997.PubMed/NCBI
|
|
17
|
Cumming RC, Lightfoot J, Beard K,
Youssoufian H, O'Brien PJ and Buchwald M: Fanconi anemia group C
protein prevents apoptosis in hematopoietic cells through redox
regulation of GSTP1. Nat Med. 7:814–820. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kruyt FA, Hoshino T, Liu JM, Joseph P,
Jaiswal AK and Youssoufian H: Abnormal microsomal detoxification
implicated in Fanconi anemia group C by interaction of the FAC
protein with NADPH cytochrome P450 reductase. Blood. 92:3050–3056.
1998.PubMed/NCBI
|
|
19
|
Pinkus R, Weiner LM and Daniel V: Role of
quinone-mediated generation of hydroxyl radicals in the induction
of glutathione S-transferase gene expression. Biochemistry.
34:81–88. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wang J, Otsuki T, Youssoufian H, Foe JL,
Kim S, Devetten M, Yu J, Li Y, Dunn D and Liu JM: Overexpression of
the fanconi anemia group C gene (FAC) protects hematopoietic
progenitors from death induced by Fas-mediated apoptosis. Cancer
Res. 58:3538–3541. 1998.PubMed/NCBI
|
|
21
|
Huang F, Ben Aissa M, Magron A, Huard CC,
Godin C, Lévesque G and Carreau M: The Fanconi anemia group C
protein interacts with uncoordinated 5A and delays apoptosis. PLoS
One. 9:e928112014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Rains JL and Jain SK: Oxidative stress,
insulin signaling, and diabetes. Free Radic Biol Med. 50:567–575.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Bloch-Damti A and Bashan N: Proposed
mechanisms for the induction of insulin resistance by oxidative
stress. Antioxid Redox Signal. 7:1553–1567. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Li J, Sipple J, Maynard S, Mehta PA, Rose
SR, Davies SM and Pang Q: Fanconi anemia links reactive oxygen
species to insulin resistance and obesity. Antioxid Redox Signal.
17:1083–1098. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Robertson RP and Harmon JS: Pancreatic
islet beta-cell and oxidative stress: The importance of glutathione
peroxidase. FEBS Lett. 581:3743–3748. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Vasu S, McClenaghan NH, McCluskey JT and
Flatt PR: Cellular responses of novel human pancreatic β-cell line,
1.1B4 to hyperglycemia. Islets. 5:170–177. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Floros KV, Thomadaki H, Florou D, Talieri
M and Scorilas A: Alterations in mRNA expression of
apoptosis-related genes BCL2, BAX, FAS, caspase-3, and the novel
member BCL2L12 after treatment of human leukemic cell line HL60
with the antineoplastic agent etoposide. Ann N Y Acad Sci.
1090:89–97. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zhou Y, Li W, Xu Q and Huang Y: Elevated
expression of Dickkopf-1 increases the sensitivity of human glioma
cell line SHG44 to BCNU. J Exp Clin Cancer Res. 29:1312010.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Niida A, Hiroko T, Kasai M, Furukawa Y,
Nakamura Y, Suzuki Y, Sugano S and Akiyama T: DKK1, a negative
regulator of Wnt signaling, is a target of the beta-catenin/TCF
pathway. Oncogene. 23:8520–8526. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Tiedge M, Lortz S, Drinkgern J and Lenzen
S: Relation between antioxidant enzyme gene expression and
antioxidative defense status of insulin-producing cells. Diabetes.
46:1733–1742. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Keane KN, Cruzat VF, Carlessi R, de
Bittencourt PI Jr and Newsholme P: Molecular events linking
oxidative stress and inflammation to insulin resistance and β-cell
dysfunction. Oxid Med Cell Longev. 2015:1816432015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Maechler P, Jornot L and Wollheim CB:
Hydrogen peroxide alters mitochondrial activation and insulin
secretion in pancreatic beta cells. J Biol Chem. 274:27905–27913.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Zraika S, Aston-Mourney K, Laybutt DR,
Kebede M, Dunlop ME, Proietto J and Andrikopoulos S: The influence
of genetic background on the induction of oxidative stress and
impaired insulin secretion in mouse islets. Diabetologia.
49:1254–1263. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Bijangi-Vishehsaraei K, Saadatzadeh MR,
Werne A, McKenzie KA, Kapur R, Ichijo H and Haneline LS: Enhanced
TNF-alpha-induced apoptosis in Fanconi anemia type C-deficient
cells is dependent on apoptosis signal-regulating kinase 1. Blood.
106:4124–4130. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Zhang X, Sejas DP, Qiu Y, Williams DA and
Pang Q: Inflammatory ROS promote and cooperate with the Fanconi
anemia mutation for hematopoietic senescence. J Cell Sci.
120:1572–1583. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Zhang QG, Wang R, Khan M, Mahesh V and
Brann DW: Role of Dickkopf-1, an antagonist of the Wnt/beta-catenin
signaling pathway, in estrogen-induced neuroprotection and
attenuation of tau phosphorylation. J Neurosci. 28:8430–8441. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Gui S, Yuan G, Wang L, Zhou L, Xue Y, Yu
Y, Zhang J, Zhang M, Yang Y and Wang DW: Wnt3a regulates
proliferation, apoptosis and function of pancreatic NIT-1 beta
cells via activation of IRS2/PI3K signaling. J Cell Biochem.
114:1488–1497. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Huard CC, Tremblay CS, Helsper K, Delisle
MC, Schindler D, Lévesque G and Carreau M: Fanconi anemia proteins
interact with CtBP1 and modulate the expression of the Wnt
antagonist Dickkopf-1. Blood. 121:1729–1739. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Huard CC, Tremblay CS, Magron A, Lévesque
G and Carreau M: The Fanconi anemia pathway has a dual function in
Dickkopf-1 transcriptional repression. Proc Natl Acad Sci USA.
111:2152–2157. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Gloyn AL, Odili S, Buettger C, Njolstad
PR, Shiota C, Matschinsky FM and Magnuson MA: Glucokinase and
Glycemic Disease: From Basics to Novel Therapeutics. 16. Karger;
Basel: pp. 92–109. 2004
|
|
42
|
Vasu S, McClenaghan NH and Flatt PR:
Molecular mechanisms of toxicity and cell damage by chemicals in a
human pancreatic beta cell line, 1.1B4. Pancreas. 45:1320–1329.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Iype T, Francis J, Garmey JC, Schisler JC,
Nesher R, Weir GC, Becker TC, Newgard CB, Griffen SC and Mirmira
RG: Mechanism of insulin gene regulation by the pancreatic
transcription factor Pdx-1: Application of pre-mRNA analysis and
chromatin immunoprecipitation to assess formation of functional
transcriptional complexes. J Biol Chem. 280:16798–16807. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Zhang C, Moriguchi T, Kajihara M, Esaki R,
Harada A, Shimohata H, Oishi H, Hamada M, Morito N, Hasegawa K, et
al: MafA is a key regulator of glucose-stimulated insulin
secretion. Mol Cell Biol. 25:4969–4976. 2005. View Article : Google Scholar : PubMed/NCBI
|