Introduction
Lung cancer is a common type of cancer and the main
cause of cancer-associated mortality worldwide (1,2). The
classification of lung cancer is divided into two major categories,
namely small cell lung cancer and non-small cell lung cancer
(NSCLC) (3). The two main
histological types of NSCLC are lung adenocarcinoma (LUAD) and lung
squamous carcinoma (LUSC). It has been shown that the prognosis of
LUAD patients is worse compared with that of LUSC (4). In the majority of cases, the type of
cancer (LUAD or LUSC) is distinguished based on standard
morphological criteria; however, the definition of glandular and/or
squamous carcinoma features is subtle and/or localized to the
tissue area. Thus, it is difficult to distinguish between these two
types of cancer in several poorly differentiated tumors.
Previous studies have focused on the genome,
epigenome, transcriptome and proteome of lung cancer in order to
identify new cancer-driven factors that may be targeted clinically
(5–7). In the last decade, researchers have
found that the changes noted in certain transcriptional regulators
can significantly improve the overall survival rate of a small
subset of patients with lung cancer (8,9).
Transcriptional and post-transcriptional levels are included in
transcriptional regulators. Since transcription factors (TFs) are
essential for the regulation of gene expression, they are present
in all living organisms. Small non-coding RNA molecules containing
approximately 22 nucleotides, termed microRNAs (miRNAs), have been
identified in plants, animals and certain viruses, and are involved
in RNA silencing and post-transcriptional regulation of gene
expression (10). In addition,
long-non-coding RNAs (lncRNAs) with a length of >200 nucleotides
are distributed throughout the genome, and the majority of human
diseases, including cancer, are associated with lncRNA expression
disorders (11,12). A number of studies have also
reported that the associations between TFs, lncRNAs and miRNAs may
serve a role in lung cancer development. For instance, a recent
study constructed a TF-lncRNA-gene network to illustrate how TFs
regulate lncRNA and gene expression in LUAD (13). Another study systematically
analyzed the interaction of co-expressed mRNAs, miRNAs and lncRNAs
activated by transforming growth factor-β1, and detected a total of
24 mRNAs, 11 miRNAs and 33 lncRNAs that interacted with one another
(14). Therefore, the regulatory
network information of TFs, miRNAs and lncRNAs can be utilized in
order to reveal disease-modulating genes and pathways, and to
identify new therapeutic targets. In particular, TFs and miRNAs can
coordinate to regulate common target genes (15), while transcription of miRNAs is
regulated by TFs, and a common set of target lncRNAs are regulated
by both TFs and miRNAs. This type of motif is defined as a
TF-miRNA-lncRNA (TML) network motif. However, it is insufficient to
analyze the difference between LUAD and LUSC at the molecular level
based on the application of regulatory network motifs.
In the present study, the TML network motif was
defined as a motif that includes a TF, miRNA and lncRNA, in which
the TF regulates the expression of the miRNA, and both the TF and
miRNA regulate a common set of target lncRNAs. A global TML network
was constructed, and then its features were characterized, and a
computational approach was designed by integrating the interaction
and expression data of miRNAs, TFs and lncRNAs. Dysregulated TML
network motifs that were common and specific with regard to LUAD
and LUSC were identified. The dysregulated TML network motifs were
associated with cancer-related functions and pathways. It is
important to note that several miRNAs in these TML network motifs
may be potential drug targets. The results of the present study
elucidated the roles of TML network motifs in LUAD and LUSC, which
may be beneficial for understanding lung cancer pathogenesis and
treatment. The applications of these findings require further
investigation in future studies.
Materials and methods
Collecting high-throughput expression
data for miRNAs, TFs and lncRNAs
The expression data of lncRNAs, TFs and miRNAs were
obtained from LUSC and LUAD tissues and their corresponding
adjacent normal tissues as previously described (16). The raw read counts for each exon
were downloaded from The Cancer Genome Atlas (TCGA) (https://portal.gdc.cancer.gov/) level 3 dataset,
and the human TF and lncRNA annotations from GENCODE were used to
map these exons (17). In order to
obtain expression data of human TFs and lncRNAs, the reads per
kilobase of transcript per million mapped reads were recalculated
for the TFs and lncRNAs. miRNA sequencing data (Illumina HiSeq
miRNA Seq; Illumina, Inc., San Diego, CA, USA) of LUAD and LUSC
were also downloaded from TCGA (level 3) (7). Finally, a total of 166 LUAD samples
and 9 matched normal samples were selected, whereas 120 LUSC
samples and 9 matched normal samples were selected.
Establishing a genome-wide TML
network
TML network motifs comprise a TF, an miRNA and their
common target lncRNAs. In these motifs, the TF strictly regulates
the expression of the miRNA, and these two cooperatively regulate a
common set of target lncRNAs. Three types of regulatory
interactions are required in order to construct a global TML
network, including miRNA-lncRNA, TF-miRNA and TF-lncRNA
interactions. In the present study, the CLIP-Seq experimentally
supported miRNA-lncRNA interaction data were downloaded from
starBase version 2.0 (18). In
addition, the SNP@lincTFBS
database provided global TF binding sites for lncRNAs, and these
data were used for the TF-lncRNA regulatory interactions (19). Finally, a large number of
experimental data on TF-miRNA interactions were extracted from the
TransmiR database by literature and publication searches (20).
Dissecting topological features for
the TML network
The total TML network features were obtained by four
measurements, as follows: Degrees, topological coefficient,
connectivity and clustering coefficient of nodes.
Identifying dysregulated TML motifs in LUAD and
LUSC. A comprehensive pipeline was subsequently established in
order to identify dysregulated TML network motifs in LUAD and LUSC,
based on the integration of the TML network and the expression
profiling of the data. Initially, a Student's t-test was performed,
and the derived P-values were used to evaluate the differences in
TF, lncRNA and miRNA expression levels between the lung cancer
samples and the corresponding normal samples in each single TML
network motif. Each interaction pair (TF-lncRNA, TF-miRNA and
miRNA-lncRNA regulatory interactions) in the TML network motif, the
study calculated the Pearson correlation coefficients (PCCs) and
the levels of difference between these coefficients with regard to
the lung cancer and normal samples. The associations between
regulatory interactions were represented by the absolute
differences of PCCs between lung cancer and normal samples.
Subsequently, the differential expression P-values and PCCs were
merged in order to calculate two comprehensive scores (namely
Scoredif and Scorepcc) for TML as
follows:
Scoredif=PTFPmiRNAPlncRNA
Scorepcc=|(LTm-NTm)(LTl-NTl)(Lml-Nml)|
where PTF, PmiRNA and
PlncRNA represent the differential expression P-values
of TF, miRNA and lncRNA, respectively, in each TML network motif.
Scoredif corresponds to the difference in expression of
a TML network motif between the lung cancer and the normal samples.
LTm, LTl and Lml correspond to the
PCCs of the three regulatory regulations, namely the TF-miRNA,
TF-lncRNA and miRNA-lncRNA interaction pairs, respectively, for the
lung cancer samples. NTm, NTl, and
Nml represent the PCCs for the TF and miRNA, TF and
lncRNA, and miRNA and lncRNA pairs, respectively, for the control
samples. Scorepcc is the absolute distinction of the PCC
score between the lung cancer and control samples in the total TML
motif.
An equally-weighted multidimensional approach was
then used to rank all the TML network motifs for lung cancer based
on Scoredif and Scorepcc (21). Subsequent to obtaining two ranked
lists based on the two aforementioned scores respectively, the
ranking position of the two lists was integrated to calculate the
final ranking score for each TML network motif. The higher ranking
score represented higher dysregulated motif levels in lung cancer.
Furthermore, each final motif ranking score was compared with the
permutation-based score list, which was generated by randomly
disturbing all sample labels in the expression profile for 1,000
times. This was used to produce a significant P-value for each TML
motif. Finally, the significant dysregulated TML motifs were
obtained for LUAD and LUSC (P<0.05).
Gene set enrichment analysis
lncRNAs from dysregulated TML network motifs were
used to perform functional enrichment using default parameters on
the Enrichr tool online web server (22). Enriched Gene Ontology (GO) terms
were obtained that were selected at a P-value of <0.05, which
indicated a statistically significant difference.
The drug targets analyses for
dysregulated TML network motifs
We determined the associations between drugs and
miRNAs using SM2miR (23), a
database of the experimentally validated small molecules that
affect the expression of miRNAs. Then, a drug-miRNA network for
miRNAs in dysregulated TML network motifs was generated based on
data from SM2miR.
Results
Topological characteristics of the TML
network
Multiple data sources were integrated in order to
identify >600 TML network motifs. Each motif consisted of a TF,
an miRNA and their common target lncRNAs. A global TML network
containing 240 nodes (96 lncRNAs, 17 TFs and 127 miRNAs) and 878
edges was constructed (Fig. 1A).
As expected, this transcriptional regulatory network was similar to
the scale-free network topology (Fig.
1B), which has a degree distribution that follows a power law,
at least asymptotically. In addition, the connectivity, topological
coefficient and clustering coefficient of nodes were computed and
analyzed. All the aforementioned analyzed characteristics indicated
scale-free distributions (Fig.
1C-E), suggesting that the TML network was a small-world
network (24). The mean level of
the neighborhood connectivity with k neighbors was defined as the
neighborhood connectivity distribution (k=0, 1…n). The network
highlighted a hierarchical modularity phenomenon due to the
stepwise increase of the degrees of connectivity following the
topological coefficient decrease.
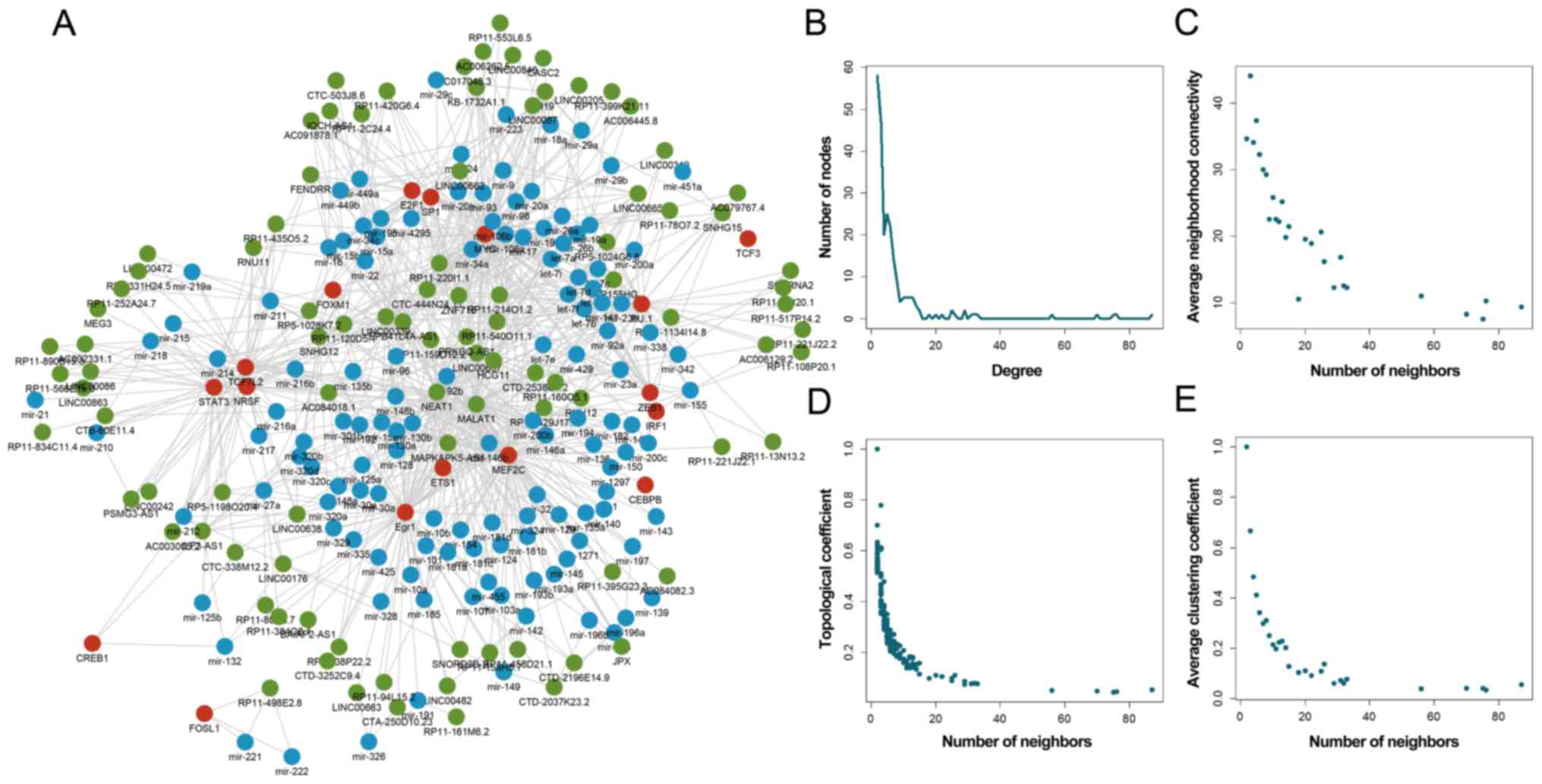 | Figure 1.Global topological characteristics of
the TML network. (A) Global TML network, including TFs (red),
miRNAs (blue) and lncRNAs (green). Basic characteristics of the
network are displayed, including (B) degree, (C) connectivity, (D)
topological coefficient and (E) clustering coefficient of TFs,
miRNAs and lncRNAs. TML, TF-miRNA-lncRNA; miRNA, microRNA; TF,
transcription factor; lncRNA, long non-coding RNA. |
Specific TML motifs are significantly
dysregulated in LUAD and LUSC
Dysregulated TML network motifs that were
significantly different with regard to the LUAD and LUSC in the TML
network were identified, and the functional significance of the TML
network motifs was further analyzed. These dysregulated motifs were
used to construct significantly dysregulated TML sub-networks for
LUAD and LUSC (Fig. 2). A total of
11 TML network motifs, including 7 lncRNAs, 5 TFs and 8 miRNAs,
were obtained in LUAD (Fig. 2A),
while a total of 15 TML network motifs with 8 lncRNAs, 5 TFs and 13
miRNAs were obtained in LUSC (Fig.
2B). In these dysregulated TML network motifs, a number of
specific TFs, miRNAs and lncRNAs have been reported to serve
essential roles in lung cancer. For instance, a previous study
reported that mutation of the zinc finger protein family member 718
may be a potential germline mutation of lung cancer (25). In addition, let-7c, let-7d and
let-7f were all present in dysregulated TML motifs in the present
analysis, whereas it has been reported that the let-7 miRNA family
suppressed NSCLC development (26). Notably, a study demonstrated that
the MYC-regulated long non-coding RNA H19 was associated with poor
prognosis and affected cell proliferation in NSCLC (27). Similarly, this association was also
identified in the present analysis.
Common TML motifs between LUAD and
LUSC reveal a lung cancer subtype-specific mechanism
The performance of the TML network motif in
distinguishing lung cancer subtypes was investigated in the present
study. LUAD and LUSC are two major histological subtypes of NSCLC.
Specific differences were apparent with regard to the underlying
mechanisms, which suggested the association of specific TMLs with
LUAD and LUSC. Two subnetworks of significant TML network motifs in
LUAD and LUSC were integrated, and 10 common factors were detected
between the two lung cancer subtypes, including 4 TFs, 3 miRNAs and
3 lncRNAs (Fig. 3A and B). For
instance, the TF E2F1, lncRNA KB-1732A1.1 and miR-15b were
identified in LUAD and LUSC. Several of these common factors were
reported to be associated with lung cancer. Recently, it was
reported that mutations in the tyrosine kinase domain of the
epidermal growth factor receptor (EGFR) gene occurred in a subset
of patients with lung cancer, indicating a marked response to
treatment by EGFR tyrosine kinase inhibitors (28). A previous study revealed that
miR-195 was significantly downregulated in NSCLC samples and cell
lines compared with the corresponding normal counterparts, and that
it functioned as a tumor suppressor (29).
 | Figure 3.Common and specific TML network
motifs in LUAD and LUSC. (A) Sub-network of dysregulated TML
network motifs in LUSC, LUAD and the intersection of LUAD and LUSC.
(B) Venn diagram denoting the intersection of TFs, miRNAs and
lncRNAs between LUAD and LUSC. Six motifs are shown as examples.
(C) The cube represents three expression layers of lncRNAs, miRNAs
and TFs in LUAD and LUSC. Yellow nodes represent dysregulated TML
network motifs in LUAD samples and green nodes represent a
dysregulated TML network motif in LUSC samples. The expression
values of TFs, miRNAs and lncRNAs are shown along the x, y and z
axes, respectively. TML, TF-miRNA-lncRNA; miRNA, microRNA; TF,
transcription factor; lncRNA, long non-coding RNA; LUAD, lung
adenocarcinoma; LUSC, lung squamous carcinoma. |
In addition, each lung cancer subtype contained its
own specific factors. For instance, FOXM1 may serve an important
role in advancing LUAD progression. Aberrant FOXM1 expression
directly and constitutively activates SNAIL, thereby promoting LUAD
metastasis. Inhibition of FOXM1-SNAIL signaling may thus present an
ideal target for future cancer treatment (30).
The data also indicated that certain TML network
motifs may distinguish LUAD and LUSC samples by combining multiple
layers of TML network motif's expression (Fig. 3C). A TML network motif was
recognized as a node in three-dimensional space. The z-, y- and
x-axes dimensions respectively represented the expression of
lncRNA, miRNA and TF in a TML network motif. It was observed that a
single layer of expression, including miRNA, lncRNA or TF, was
unable to distinguish the LUAD and LUSC samples. However, the TML
network motifs were able to effectively distinguish between the two
different cancer subtypes by combining multiple layers of
expression. The results also indicated that the nodes of LUAD and
LUSC samples could clearly be separated in three-dimensional space.
Overall, the TML motif separated LUSC and LUAD samples more
efficiently as compared with the use of each single type of
molecules.
Functional analysis demonstrates the
roles of TML motifs in LUAD and LUSC
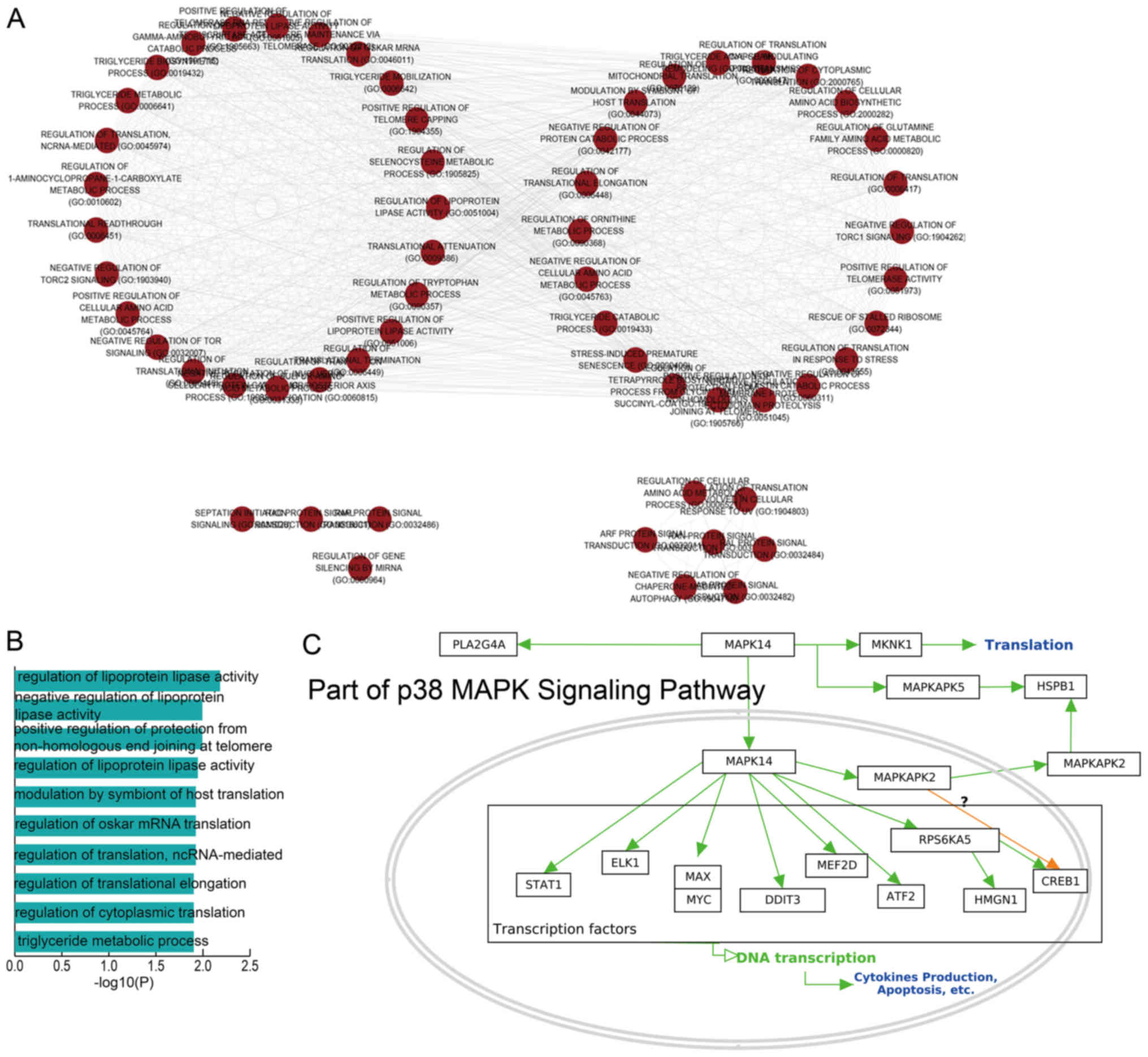
GO analysis based on the lncRNAs in the dysregulated
TML network motifs of LUAD and LUSC was also performed in the
present study. These dysregulated lncRNAs were enriched in various
GO terms, including negative regulation of lipoprotein lipase
activity, regulation of translation and lncRNA-mediated regulation
of translational elongation (Fig. 4A
and B). The function termed positive regulation of protection
from non-homologous end joining at the telomere sites was also
identified as a significant pathway. Telomerase is an attractive
cancer target as it appears to be required in essentially all
tumors for immortalization of a subset of cells, such as cancer
stem cells (31).
In addition, two dysregulated factors, namely
MAPKAPK5 and MYC, were identified as key genes in the p38
mitogen-activated protein kinase (MAPK) signaling pathway in the
current study (Fig. 4C).
Uncontrolled growth is a necessary step for the development of
cancer. In various types of cancer, a defect in the MAPK signaling
pathway leads to this uncontrolled growth. Several compounds can
inhibit key proteins in the MAPK pathway, and these may serve as
potential drugs for the treatment of cancer (32,33).
Taken together, functional analysis indicated that the dysregulated
TML network motifs identified by our method exhibited a strong
association with cancer.
TML network motifs contain potential
drug targets
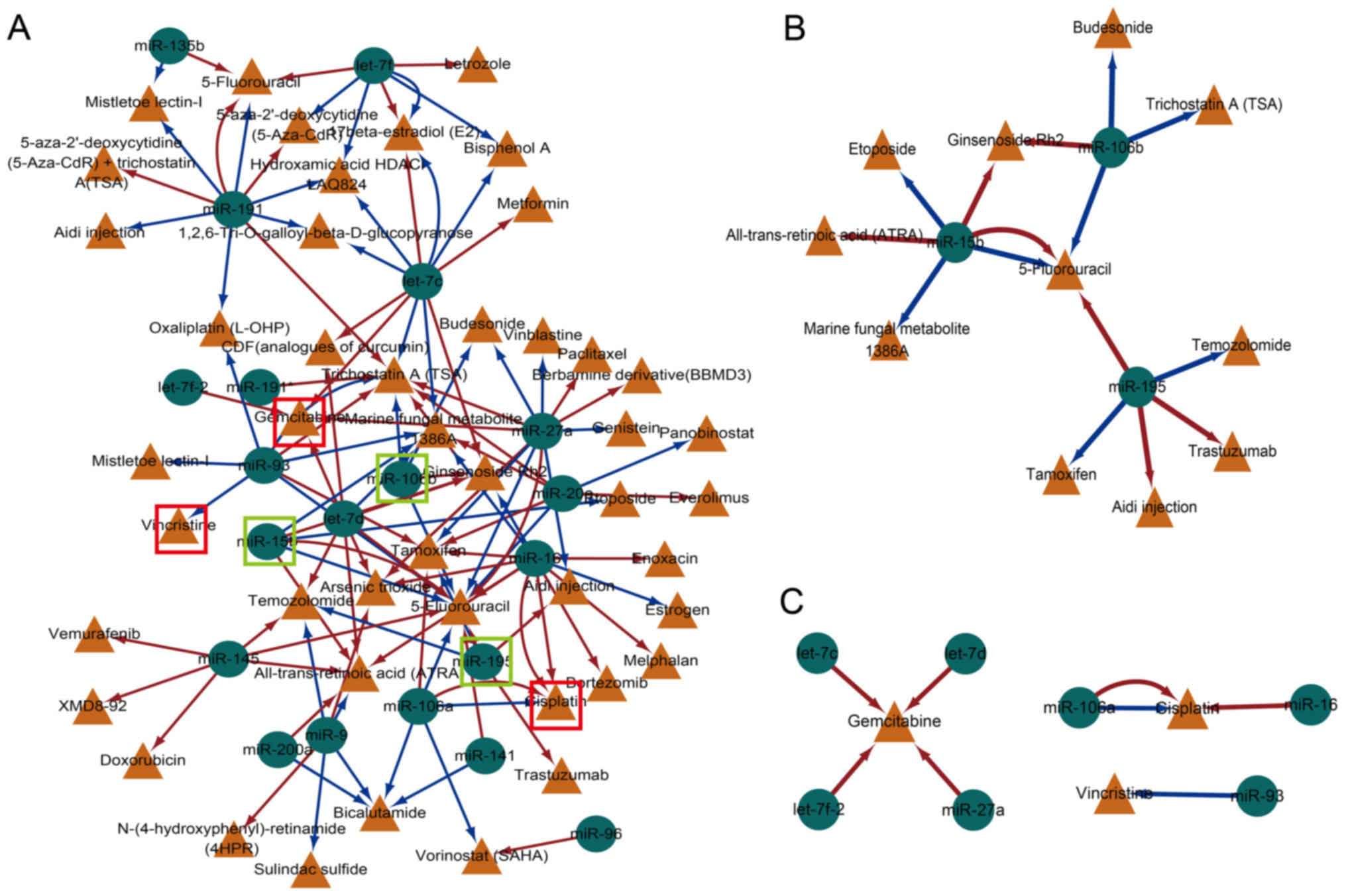
A cancer-associated drug-miRNA network based on
miRNAs in dysregulated TML network motifs was constructed, and the
data from SM2miR were used (33).
SM2miR is a database of the experimentally validated small
molecules that affect the expression of miRNAs (23). The cancer-associated drug-miRNA
network included 68 nodes (24 miRNAs and 44 drugs) and 118 edges
(Fig. 5A). Initially, the common
miRNAs in LUAD and LUSC, including miR-15b, miR-106b and miR-195,
were analyzed (Fig. 5B). These
three miRNAs were all associated with the drug 5-fluorouracil,
which is widely used in the treatment of cancer, including
colorectal and breast cancer (34). Although the miRNAs identified in
the dysregulated TML network motifs were influenced by cancer drug
treatment, the regulated direction (upregulated or downregulated)
was distinct and indicated that the drug effect to the miRNAs was
complex. In addition, certain drugs were associated with miR-15b,
such as etoposide, which is a chemotherapy medication used for the
treatment of a number of cancer types, including lung cancer
(35). Another such compound is
ginsenoside, a traditional Chinese medicine that exhibited an
inhibitory effect on the cell growth of various cancer cells and
animal models, which was observed to be associated with miR-15b and
miR-106b in the current study (Fig.
5C) (36). Furthermore, the
current results identified a number of lung cancer treatment drugs,
including gemcitabine, cisplatin and vincristine. Gemcitabine,
which is used in the treatment of NSCLC, was associated with the
let-7 miRNA family, including let-7c, let-7d and let-7f. Taken
together, the data of the current study indicated that the
dysregulated TML network motifs may be used to identify drug
targets for novel treatment strategies.
Discussion
Diverse types of regulatory transcripts (such as
lncRNAs, TFs and miRNAs) exhibit different types of interactions.
Recently, the application of computational modeling has been
employed for the prediction of lung cancer incidence. Certain
miRNAs and lncRNAs have been identified as potential biomarkers,
and interact with each other in lung cancer (37,38).
These different types of molecules form complex network motifs in
order to play specific biological roles and accordingly affect the
pathogenic mechanisms in several cancer types. Other types of TML
motifs have also been identified, such as motifs that describe the
regulatory action of miRNAs and lncRNAs on TFs or the regulatory
action of lncRNAs on miRNAs (39).
The present study concentrated on an important network motif,
namely the TML network motif, which included a TF, miRNA and
lncRNA. A computational pipeline was presented in order to study
the TML network motifs by integrating the interaction and
expression data of TFs, miRNAs and lncRNAs. The study identified
dysregulated TML network motifs in the two lung cancer types, LUAD
and LUSC. Furthermore, the results as to whether a TML motif i
specific in one type of cancer or common in both, were consistent
with previous studies on the tissue specificity of lncRNAs and
miRNAs. Cancer subtype-specific TML network motifs may aid the
development of drugs that minimize the side effects of patient
treatment. Functional and drug effect analyses conducted in the
present study suggested that certain TML network motifs may serve
as putative biomarkers for LUAD and LUSC. The data presented in the
current study are in agreement with previous studies that
highlighted the use of single miRNAs and lncRNAs as disease
biomarkers, since the TML network motif identified may be
considered as a significant pattern to study lung cancer.
In the present study, the clinical applications of
dysregulated TML network motifs were further explored for drug
development. The data revealed that the miRNAs in LUAD and
LUSC-associated TMLs were influenced by anticancer drug treatment.
The TML motifs may act as functional modules, providing the
potential mechanism of action of the corresponding drugs.
Furthermore, the results indicated that the same miRNAs had
distinct regulatory mechanisms with regard to the different drugs.
For example, Let-7c was up-regulated by Metformin and downregulated
by Trichostatin A. Similarly, the same drugs may also influence
different miRNA expression levels by distinct directions. Further
studies should explore the multiple changes in the transcriptome
that occur as a result of the treatment of the patients with
anti-cancer drugs.
In conclusion, the results of the present study
provided novel insights into the potential function of miRNAs,
lncRNAs and TFs in LUAD and LUSC. Dysregulated TML network motifs
and the common motifs in LUAD and LUSC were identified. The common
features between LUAD and LUSC indicated that diverse mechanisms
were involved and different clinical treatments may be suitable for
the two cancer subtypes. Functional and drug analyses further
indicated the fundamental action of the TML network motifs in lung
cancer. Taken together, the present study identified and
investigated TML network motifs, and the obtained data provide
further insight into the multi-level crosstalk regulation found in
LUAD and LUSC.
Acknowledgements
Not applicable.
Funding
This study was supported by the Research Fund of the
First Affiliated Hospital of Harbin Medical University (grant no.
2017Y013).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
SZ, HC and WZ conceived and designed the present
study. SZ, BD, JL, FL and KH performed the experiment and analyzed
the data. DZ, BY and YY verified and improved the computational
approaches conducted in the present study. HC and SZ were involved
in drafting the manuscript. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Saracci R and Wild CP: Fifty years of the
international agency for research on cancer (1965 to 2015). Int J
Cancer. 138:1309–1311. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Inamura K, Fujiwara T, Hoshida Y, Isagawa
T, Jones MH, Virtanen C, Shimane M, Satoh Y, Okumura S, Nakagawa K,
et al: Two subclasses of lung squamous cell carcinoma with
different gene expression profiles and prognosis identified by
hierarchical clustering and non-negative matrix factorization.
Oncogene. 24:7105–7113. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Li S, Huang S and Peng SB: Overexpression
of G protein-coupled receptors in cancer cells: Involvement in
tumor progression. Int J Oncol. 27:1329–1339. 2005.PubMed/NCBI
|
|
4
|
Suzuki K, Nagai K, Yoshida J, Nishimura M,
Takahashi K, Yokose T and Nishiwaki Y: Conventional
clinicopathologic prognostic factors in surgically resected
nonsmall cell lung carcinoma. A comparison of prognostic factors
for each pathologic TNM stage based on multivariate analyses.
Cancer. 86:1976–1984. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Cancer Genome Atlas Research Network:
Comprehensive molecular profiling of lung adenocarcinoma. Nature.
511:543–550. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Imielinski M, Berger AH, Hammerman PS,
Hernandez B, Pugh TJ, Hodis E, Cho J, Suh J, Capelletti M,
Sivachenko A, et al: Mapping the hallmarks of lung adenocarcinoma
with massively parallel sequencing. Cell. 150:1107–1120. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Cancer Genome Atlas Research Network:
Comprehensive genomic characterization of squamous cell lung
cancers. Nature. 489:519–525. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Shaw AT, Kim DW, Nakagawa K, Seto T, Crinó
L, Ahn MJ, De Pas T, Besse B, Solomon BJ, Blackhall F, et al:
Crizotinib versus chemotherapy in advanced ALK-positive lung
cancer. N Engl J Med. 368:2385–2394. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Mok TS, Wu YL, Thongprasert S, Yang CH,
Chu DT, Saijo N, Sunpaweravong P, Han B, Margono B, Ichinose Y, et
al: Gefitinib or carboplatin-paclitaxel in pulmonary
adenocarcinoma. N Engl J Med. 361:947–957. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ambros V: The functions of animal
microRNAs. Nature. 431:350–355. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
St Laurent G, Wahlestedt C and Kapranov P:
The landscape of long noncoding RNA classification. Trends Genet.
31:239–251. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Bassett AR, Akhtar A, Barlow DP, Bird AP,
Brockdorff N, Duboule D, Ephrussi A, Ferguson-Smith AC, Gingeras
TR, Haerty W, et al: Considerations when investigating lncRNA
function in vivo. Elife. 3:e030582014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Qiu M, Xu Y, Wang J, Zhang E, Sun M, Zheng
Y, Li M, Xia W, Feng D, Yin R and Xu L: A novel lncRNA, LUADT1,
promotes lung adenocarcinoma proliferation via the epigenetic
suppression of p27. Cell Death Dis. 6:e18582015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Liu H, Zhao X, Xiang J, Zhang J, Meng C,
Zhang J, Li M, Song X and Lv C: Interaction network of coexpressed
mRNA, miRNA, and lncRNA activated by TGF-β1 regulates EMT in human
pulmonary epithelial cell. Mol Med Rep. 16:8045–8054. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Shalgi R, Lieber D, Oren M and Pilpel Y:
Global and local architecture of the mammalian
microRNA-transcription factor regulatory network. PLoS Comput Biol.
3:e1312007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wang P, Ning S, Zhang Y, Li R, Ye J, Zhao
Z, Zhi H, Wang T, Guo Z and Li X: Identification of
lncRNA-associated competing triplets reveals global patterns and
prognostic markers for cancer. Nucleic Acids Res. 43:3478–3489.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kersey PJ, Lawson D, Birney E, Derwent PS,
Haimel M, Herrero J, Keenan S, Kerhornou A, Koscielny G, Kähäri A,
et al: Ensembl Genomes: extending Ensembl across the taxonomic
space. Nucleic Acids Res. 38:(Database Issue). D563–D569. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Li JH, Liu S, Zhou H, Qu LH and Yang JH:
starBase v2.0: Decoding miRNA-ceRNA, miRNA-ncRNA and protein-RNA
interaction networks from large-scale CLIP-Seq data. Nucleic Acids
Res. 42:(Database Issue). D92–D97. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ning S, Zhao Z, Ye J, Wang P, Zhi H, Li R,
Wang T, Wang J, Wang L and Li X: SNP@lincTFBS: An integrated
database of polymorphisms in human LincRNA transcription factor
binding sites. PLoS One. 9:e1038512014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wang J, Lu M, Qiu C and Cui Q: TransmiR: A
transcription factor-microRNA regulation database. Nucleic Acids
Res. 38:(Database Issue). D119–D122. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Aerts S, Lambrechts D, Maity S, Van Loo P,
Coessens B, De Smet F, Tranchevent LC, De Moor B, Marynen P, Hassan
B, et al: Gene prioritization through genomic data fusion. Nat
Biotechnol. 24:537–544. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kuleshov MV, Jones MR, Rouillard AD,
Fernandez NF, Duan Q, Wang Z, Koplev S, Jenkins SL, Jagodnik KM,
Lachmann A, et al: Enrichr: A comprehensive gene set enrichment
analysis web server 2016 update. Nucleic Acids Res. 44:W90–W97.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Liu X, Wang S, Meng F, Wang J, Zhang Y,
Dai E, Yu X, Li X and Jiang W: SM2miR: A database of the
experimentally validated small molecules' effects on microRNA
expression. Bioinformatics. 29:409–411. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Amaral LA, Scala A, Barthelemy M and
Stanley HE: Classes of small-world networks. Proc Natl Acad Sci
USA. 97:11149–11152. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kanwal M, Ding XJ, Ma ZH, Li LW, Wang P,
Chen Y, Huang YC and Cao Y: Characterization of germline mutations
in familial lung cancer from the Chinese population. Gene.
641:94–104. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kumar MS, Erkeland SJ, Pester RE, Chen CY,
Ebert MS, Sharp PA and Jacks T: Suppression of non-small cell lung
tumor development by the let-7 microRNA family. Proc Natl Acad Sci
USA. 105:3903–3908. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhang E, Li W, Yin D, De W, Zhu L, Sun S
and Han L: c-Myc-regulated long non-coding RNA H19 indicates a poor
prognosis and affects cell proliferation in non-small-cell lung
cancer. Tumour Biol. 37:4007–4015. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kosaka T, Yatabe Y, Endoh H, Kuwano H,
Takahashi T and Mitsudomi T: Mutations of the epidermal growth
factor receptor gene in lung cancer: Biological and clinical
implications. Cancer Res. 64:8919–8923. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Yongchun Z, Linwei T, Xicai W, Lianhua Y,
Guangqiang Z, Ming Y, Guanjian L, Yujie L and Yunchao H:
MicroRNA-195 inhibits non-small cell lung cancer cell
proliferation, migration and invasion by targeting MYB. Cancer
Lett. 347:65–74. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Wei P, Zhang N, Wang Y, Li D, Wang L, Sun
X, Shen C, Yang Y, Zhou X and Du X: FOXM1 promotes lung
adenocarcinoma invasion and metastasis by upregulating SNAIL. Int J
Biol Sci. 11:186–198. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Guittat L, Alberti P, Gomez D, De Cian A,
Pennarun G, Lemarteleur T, Belmokhtar C, Paterski R, Morjani H,
Trentesaux C, et al: Targeting human telomerase for cancer
therapeutics. Cytotechnology. 45:75–90. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Downward J: Targeting RAS signalling
pathways in cancer therapy. Nat Rev Cancer. 3:11–22. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Sebolt-Leopold JS: Advances in the
development of cancer therapeutics directed against the
RAS-mitogen-activated protein kinase pathway. Clin Cancer Res.
14:3651–3656. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Longley DB, Harkin DP and Johnston PG:
5-fluorouracil: Mechanisms of action and clinical strategies. Nat
Rev Cancer. 3:330–338. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Takada M, Fukuoka M, Kawahara M, Sugiura
T, Yokoyama A, Yokota S, Nishiwaki Y, Watanabe K, Noda K, Tamura T,
et al: Phase III study of concurrent versus sequential thoracic
radiotherapy in combination with cisplatin and etoposide for
limited-stage small-cell lung cancer: Results of the Japan clinical
oncology group study 9104. J Clin Oncol. 20:3054–3060. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Lü JM, Yao Q and Chen C: Ginseng
compounds: An update on their molecular mechanisms and medical
applications. Curr Vasc Pharmacol. 7:293–302. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Chen X, Xie D, Wang L, Zhao Q, You ZH and
Liu H: BNPMDA: BNPMDA: Bipartite network projection for
MiRNA-disease association prediction. Bioinformatics. 34:3178–3186.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Chen X, Yan CC, Zhang X and You ZH: Long
non-coding RNAs and complex diseases: From experimental results to
computational models. Brief Bioinform. 18:558–576. 2017.PubMed/NCBI
|
|
39
|
Jiang W, Mitra R, Lin CC, Wang Q, Cheng F
and Zhao Z: Systematic dissection of dysregulated transcription
factor-miRNA feed-forward loops across tumor types. Brief
Bioinform. 17:996–1008. 2016. View Article : Google Scholar : PubMed/NCBI
|