Introduction
Acute kidney injury (AKI) is a clinical syndrome
characterized by the rapid loss of kidney function and abrupt
kidney damage. Septic AKI is the most common complication of
sepsis, which substantially increases the risk of mortality
(1–3). The incidence of septic AKI is
increasing, and mortality rates remain high. Septic AKI occurs in
16–25% of patients with sepsis, with a mortality rate of 50–60% in
the USA and in Europe (3).
Although the clinical management of septic AKI has improved, no
specific or effective treatments are available to prevent or
improve this disease, as its exact mechanism remains unclear
(4,5). An understanding of the underlying
molecular mechanisms is therefore important for the development of
novel therapies to treat septic AKI.
Lipopolysaccharide (LPS), the endotoxin of
gram-negative bacteria, is one of the major triggers of
inflammatory responses in sepsis (4,6,7). LPS
not only triggers a large number of inflammatory cytokines, but can
also directly induce renal tubular epithelial cell injury through
the activation of Toll-like receptor 4 (TLR4). Tubular epithelial
cell apoptosis is one of the characteristic changes that occurs in
LPS-induced AKI (1,4,7,8), and
accumulating evidence has demonstrated that tubular epithelial cell
apoptosis has an important role in LPS-induced AKI (6,9–11).
Inhibiting tubular epithelial cell apoptosis could be an effective
strategy to prevent or treat LPS-induced AKI. However, the
mechanisms of LPS-induced tubular epithelial cell apoptosis are not
fully understood.
Receptor-interacting protein kinases (RIPKs) are
serine/threonine protein kinases that act as sensors of
intracellular and extracellular stress. In total, seven isoforms
that share a homologous kinase domain with different functional
domains have been identified (12). These isoforms have been shown to be
involved in inflammation and the cell death signaling pathway
(12–14). RIPK3 was originally cloned from
human fetal brain and the aortic endothelium, and subsequently
characterized as an N-terminal serine/threonine kinase. RIPK3 is
composed of an N-terminal kinase domain similar to other RIPKs, a
receptor-interacting protein homotypic interaction motif that
mediates protein-protein interactions and a unique C-terminal
domain (12,15,16).
Previous studies have shown that the overexpression of RIPK3
induces apoptosis in certain cell lines (15,17–20).
RIPK3 expression in renal tubular epithelial cells has been shown
to be higher in murine models of septic and non-septic AKI
(21,22). In addition, plasma RIPK3 is
elevated in critically ill trauma patients with AKI (23). These previous studies indicated
that RIPK3 may be involved in LPS-induced tubular epithelial cell
apoptosis. However, the role of RIPK3 in LPS-induced AKI is not
well defined. The aim of the present study was to investigate the
potential involvement of RIPK3 in LPS-induced tubular epithelial
cell apoptosis.
Materials and methods
Animals
All animal procedures conformed to the protocols of
the Guangdong Academy of Medical Sciences and were approved by the
ethics committee for the experimental use of animals at Guangdong
Provincial People's Hospital. C57BL/6 mice were purchased from the
Nanjing Biomedical Research Institute of Nanjing University and
housed at the animal center of Guangzhou Forevergen Biosciences
Co., Ltd. All mice were administered a standard diet and water
ad libitum, and were maintained at a temperature of 23±2°C
and an humidity of 55±5% in a controlled room with a 12 h
light/dark cycle. When the mice reached an age of 8–12 weeks and a
weight of 18–22 g, the 18 male mice were randomly divided into
three groups (n=6 per group): i) Control group, mice were
intraperitoneally injected with vehicle (saline); ii) LPS group,
mice were intraperitoneally injected with 10 mg/kg LPS; iii) LPS +
GSK′872 group, mice were intraperitoneally injected with 5 µM/kg
GSK′872 for 30 min prior to LPS treatment. All mice were observed
for 24 h after LPS/saline treatment and were then euthanized. Blood
was collected through a puncture in the inferior vena cava and the
kidneys were harvested.
Renal function test
Renal function was assessed by detecting serum
creatinine (SCr) levels using a QuantiChrom™ Creatinine Assay kit
(BioAssay Systems) and blood urea nitrogen (BUN) using a
QuantiChrom™ Urea Assay kit (BioAssay Systems), according to the
manufacturer's protocols.
Periodic acid-Schiff (PAS) staining of
kidney tissues
Kidney tissues were fixed with 4% paraformaldehyde
at 4°C for 24 h and embedded in paraffin. Paraffin blocks were cut
into 4 µm thick sections. Paraffin tissue sections were
deparaffinized using xylene and descending ethanol series (100, 95,
85 and 70%). Each incubation was performed for 10 min at room
temperature. The sections were washed for 5 min in distilled water
at room temperature, and the PAS Stain Kit (cat. no. ab150680;
Abcam) was used. Sections were immersed in the periodic acid
solution (included in the PAS kit), for 10 min at 20°C. Tissue
sections were then washed four times with distilled water and
immersed in Schiff's solution (included in the PAS kit) for 30 min
at room temperature and then washed using tap water for 15 min at
room temperature. Slides were stained with hematoxylin for 2–3 min
at room temperature. Slide were washed with distilled water,
dehydrated through ascending ethanol series (80, 95 and 100%) for 5
min at room temperature, cleared by xylene for 5 min at room
temperature and sealed with neutral balsam (cat. no. G8590; Beijing
Solarbio Science & Technology Co., Ltd.). Tubular epithelial
cell vacuolar deformation/hypertrophy and lumen occlusion occurred
during renal tubular injury. The areas of the tubular epithelial
cell vacuolar deformation, loss of brush border, tubular dilation,
cast formation and cell lysis were calculated. A score of 0
indicated no change; scores of 1, 2, 3 or 4 indicated an injury
section involving <25, 25–50, 50–75 or >75% of the area,
respectively. A minimum of six different fields of vision were
randomly selected using a light microscope (magnification ×400) and
an average score was calculated (24).
Electron microscopy
Renal tissue specimens were fixed with 2.5%
glutaraldehyde, 4% paraformaldehyde and 0.02% picric acid in 0.1 M
cacodylate buffer (pH 7.2) overnight at 4°C. Following washing
three times in cacodylate buffer, specimens were post-fixed with 1%
osmium tetroxide and 1.5% potassium ferrocyanide for 2 h at 4°C and
then washed three times in cacodylate buffer. The specimens were
dehydrated using a graded series of ethanol (50, 70, 80, 90 and
100%) and 100% propylene oxide for 15–20 min at each step, and then
embedded in SPI-PON 812 (Structure Probe, Inc.) for 12 h at 37°C,
45°C for 12 h and 60°C for 48 h. Ultrathin sections were cut into
50–60 nm using Accu-Edge high-profile blades (cat. no. 4685; Sakura
Finetek Japan Co., Ltd.) on a Microm HM 355s (Thermo Fisher
Scientific, Inc.). Sections were stained with 3% uranium acetate
and lead citrate at 4°C overnight. Renal tissue ultrastructure was
observed using a transmission electron microscope (JEM-100CX; JEOL
Ltd.). Images were recorded using a Veleta 2K digital camera
(Olympus-SIS; Olympus Corporation).
TUNEL staining
TUNEL staining was performed using the in
situ Cell Death Detection kit (Roche Diagnostics GmbH). Frozen
tissue sections were placed on coverslips and fixed in 4%
paraformaldehyde for 20 min at room temperature. Following washing
in PBS, frozen tissue sections were blocked with 3%
H2O2 in methanol for 10 min at room
temperature and then permeabilized with 0.1% Triton X-100 in 0.1%
sodium citrate for 10 min at room temperature. The tissue sections
were incubated with TUNEL reaction mixture for 60 min at 37°C in a
humidified atmosphere in the dark. Following washing three times
with PBS, tissue sections were stained with 1 µg/ml DAPI for 5 min
at room temperature. Using a confocal fluorescent microscope (Nikon
Eclipse Ni-E; Nikon Corporation), 10–20 fields were captured for
each kidney section (n=6 in each group) and positively stained
cells were quantified. Images were analyzed using ImageJ software
(version 1.49; National Institutes of Health).
Reverse transcription-quantitative PCR
(RT-qPCR)
Total RNA was extracted from cultured cells using
TRIzol® reagent (Invitrogen; Thermo Fisher Scientific,
Inc.), according to the manufacturer's instructions. RT was carried
out using a PrimeScript RT Reagent kit (Takara Bio, Inc.) according
to the manufacturer's protocol. The cDNAs were subjected to qPCR
using a Power SYBR Green PCR Master Mix (Takara Bio, Inc.). Data
were analyzed using the 2−ΔΔCq method (25) and GAPDH was used as the internal
control. Primer sequences are listed in Table I.
 | Table I.Primers for reverse transcription
quantitative PCR. |
Table I.
Primers for reverse transcription
quantitative PCR.
| Gene | Sequence
(5′-3′) |
|---|
| GAPDH | Forward
AGGTCGGTGTGAACGGATTTG |
|
| Reverse
TGTAGACCATGTAGTTGAGGTCA |
| RIPK3 | Forward
TGGGCCTGCTAAGATGGCT |
|
| Reverse
CTGCCAGAGTGTGGATTTGGT |
Cell culture and reagents
Mouse proximal renal tubular epithelial primary
cells were purchased from Cell Biologics, Inc. The cells were
cultured in DMEM/F12 medium (Gibco; Thermo Fisher Scientific, Inc.)
supplemented with 10% FBS (Gibco; Thermo Fisher Scientific, Inc.)
in a humidified atmosphere of 5% CO2 at 37°C and
passaged every 3–4 days. The cells were treated with 40 µg/ml LPS
at different times (6, 12, 24 and 48 h). Cells were treated with
0.5 µM GSK′872 (RIPK3 inhibitor; Selleck Chemicals) 1 h prior to
the addition of 40 µg/ml LPS for 24 h. Control cells were treated
with an equal volume of DMSO. Cells were transfected with 50 nM
negative control small interfering RNA (siRNA) or 50 nM RIPK3 siRNA
(Guangzhou RiboBio Co., Ltd.) by Lipofectamine 2000 (Thermo Fisher
Scientific, Inc.) for 48 h before being exposed to LPS. The
sequences of the siRNAs used in the present study are listed in
Table II.
| Table II.siRNA sequences. |
Table II.
siRNA sequences.
| Name | Sequence
(5′-3′) |
|---|
| RIPK3 siRNA-1 |
GCAGGAAATTTCAGGCCAA |
| RIPK3 siRNA-2 |
CCTGCTGAATTTCAAGAAA |
Western blot analysis
Total protein was extracted from kidney tissue using
RIPA lysate buffer (Beyotime Institute of Biotechnology)
supplemented with protease inhibitor cocktail tablets (Roche
Diagnostics GmbH). The mouse proximal renal tubular epithelial
cells were lysed using RIPA lysate buffer supplemented with
protease inhibitor mixture (Beyotime Institute of Biotechnology).
Protein concentrations were determined using a bicinchoninic acid
protein assay kit (Thermo Fisher Scientific, Inc.). Proteins were
separated by SDS-PAGE on 10–15% gels, with loaded with 30 µg of
total protein. Gels were transferred onto PVDF membranes. The
membranes were blocked with 5% non-fat dry milk in TBS-Tween 20
(TBST) for 1 h at room temperature. PVDF membranes were incubated
overnight at 4°C with antibodies against β-actin (1:1,000; cat. no.
8457; Cell Signaling Technology, Inc.), RIPK3 (1:1,000; cat. no.
ab56164; Abcam), cleaved caspase-3 (C-caspase-3; 1:1,000; cat. no.
9664; Cell Signaling Technology, Inc.) and Bax (1:1,000; cat. no.
2772; Cell Signaling Technology, Inc.). Membranes were then
incubated with a horseradish peroxidase-conjugated goat anti-rabbit
secondary antibody (1:2,000; cat. no. 7074; Cell Signaling
Technology, Inc.) for 1 h at room temperature. Following washing in
TBST, protein bands were visualized using Pierce™ ECL Western
Blotting Substrate (Thermo Fisher Scientific, Inc.) and band
intensity was quantified using Tanon software (version 5200; Tanon
Science and Technology Co., Ltd.).
Cell apoptosis detection
An Annexin V/propidium iodide (PI) double staining
assay (Nanjing KeyGen Biotech Co., Ltd.) was used to determine the
level of apoptosis, according to the manufacturer's instructions.
Medium was removed from the cells following the different
treatments for 24 h. Cells were washed three times with cold PBS
and digested with 0.25% EDTA-free trypsin. Cells were centrifuged
for 5 min at 805 × g at 4°C and the supernatant was discarded.
Cells were resuspended in 500 µl 1X binding buffer and 5 µl Annexin
V-FITC was added. The mixture was incubated for 15 min in the dark
and 5 µl PI was subsequently added at 4°C. The solution was
incubated for a further 5 min in the dark at 4°C. Cells were
analyzed using a flow cytometer (BD Biosciences).
Statistical analysis
Results are presented as the mean ± SEM. All
experiments were performed at least in triplicate. Statistical
analysis of the data was performed using SPSS 20.0 (IBM Corp.).
Comparisons among multiple groups were performed using one-way
ANOVA and the least square difference was used as a post hoc test.
P<0.05 was considered to indicate a statistically significant
difference.
Results
Effects of LPS on renal function and
pathology
After 6 h of LPS intervention, mice exhibited
symptoms of decreased food intake and increased salivation or tear
secretion, consistent with sepsis. These symptoms markedly
increased over time (data not shown). Renal function and pathology
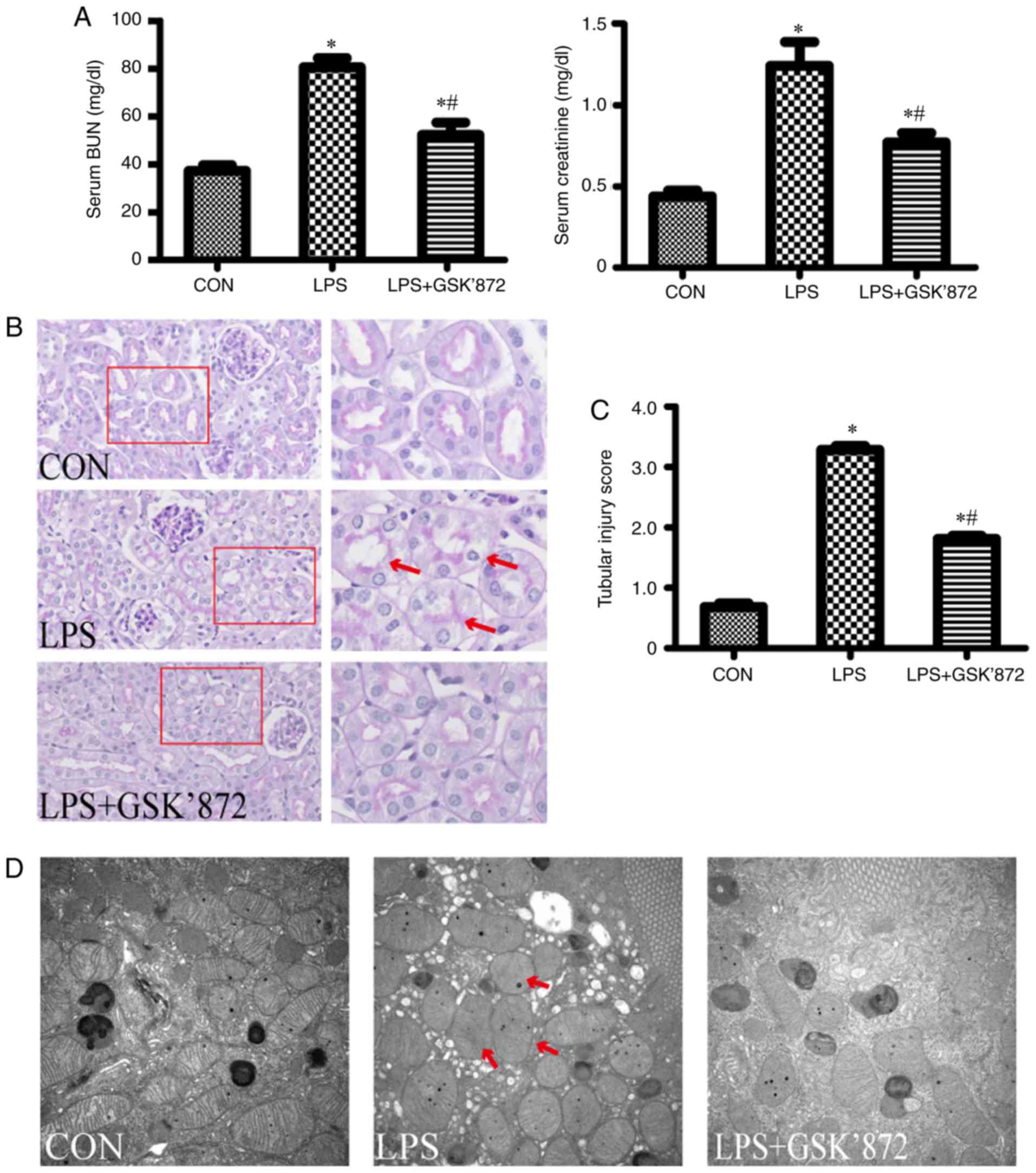
were detected 24 h after LPS intervention. As shown in Fig. 1A, compared with the control group,
LPS-treated mice showed significantly higher levels of SCr
(1.26±0.13 mg/dl in the LPS group vs. 0.44±0.03 mg/dl in the
control group; P<0.001) and BUN (80.59±3.74 mg/dl in the LPS
group vs. 37.22±2.50 mg/dl in the control group; P<0.001). As
shown in Fig. 1B, the pathological
injury of the kidney, including vacuolar deformation, swelling of
tubular epithelial cells and tubular lumen stenosis, mainly
occurred in the cortex and outer stripe of the outer medulla in the
LPS group. The tubular injury score of the LPS group was
significantly higher than that in the control group (3.29±0.06 in
the LPS group vs. 0.68 ±0.06 in the control group; P<0.001;
Fig. 1C). In addition,
mitochondrial injury in tubular epithelial cells, demonstrated by
the disorder or disappearance of crista, and mitochondrial swelling
and deformation, was aggravated in the LPS group compared with the
control group (Fig. 1D).
LPS-induced AKI increases tubular
epithelial cell apoptosis and expression of RIPK3
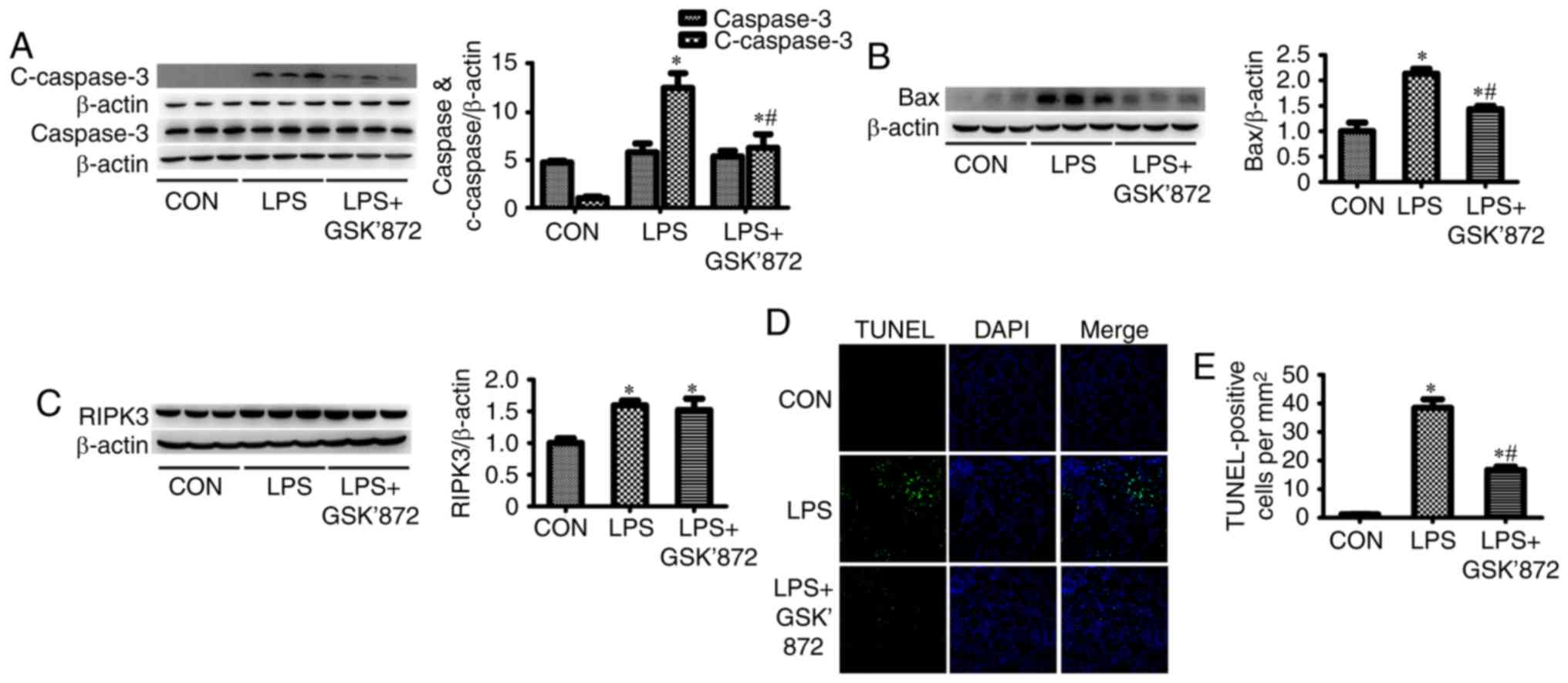
C-caspase-3, a marker of apoptosis, was
significantly increased in the kidney tissues of the LPS group
(Fig. 2A). The expression of Bax,
a proapoptotic member of the Bcl-2 family, was also significantly
increased in the LPS group (Fig.
2B). RIPK3 expression was significantly increased in the kidney
tissues of LPS-treated mice (Fig.
2C). In addition, the percentage of TUNEL-positive tubular
epithelial cells in the LPS group was significantly higher than in
the control group (7.28±0.39% in the LPS group vs. 1.60±0.12% in
the control group; P<0.001; Fig. 2D
and E). These results indicated that LPS induced tubular
epithelial cell apoptosis.
Inhibition of RIPK3 reduces the level
of apoptosis in tubular epithelial cells and improves renal
function in mice with LPS-induced AKI
To establish the potential effect of RIPK3 on
LPS-induced tubular epithelial cell apoptosis and AKI, GSK′872, a
selective RIPK3 inhibitor, was administered to mice following LPS
intervention. As shown in Fig.
2A-C, although LPS + GSK′872-treated mice did not exhibit
reduced RIPK3 expression compared with the LPS group, GSK′872
significantly inhibited the expression of C-caspase-3 and Bax.
Immunofluorescent staining showed that the number of TUNEL-positive
cells were signficantly decreased in the proximal renal tubular
epithelial cells of mice in the LPS + GSK′872 group compared with
those in the LPS group (4.97±0.22% in the LPS + GSK′872 group vs.
7.28±0.39% in the LPS group; P<0.001; Fig. 2D and E). Mice treated with
LPS+GSK′872 showed a visible improvement in histology (Fig. 1B). A semiquantitative evaluation of
kidney sections showed that the tubular injury score decreased in
the LPS + GSK′872 group (1.81±0.05 in the LPS + GSK′872 group vs.
3.29±0.06 in the LPS group; P<0.001; Fig. 1C). Mitochondrial injury was also
alleviated by GSK′872 (Fig. 1D).
GSK′872 significantly attenuated the increase in BUN (52.32±5.15
mg/dl in the LPS + GSK′872 group vs. 80.59±3.74 mg/dl in the LPS
group; P=0.001) and SCr (0.78±0.05 mg/dl in the LPS + GSK′872 group
vs. 1.26±0.13 mg/dl in the LPS group; P=0.003) in mice that had
undergone LPS intervention (Fig.
1A).
RIPK3-mediates LPS-induced apoptosis
in cultured renal tubular epithelial cells
The effect of RIPK3 on LPS-induced tubular
epithelial cell apoptosis was assessed in vitro. It was
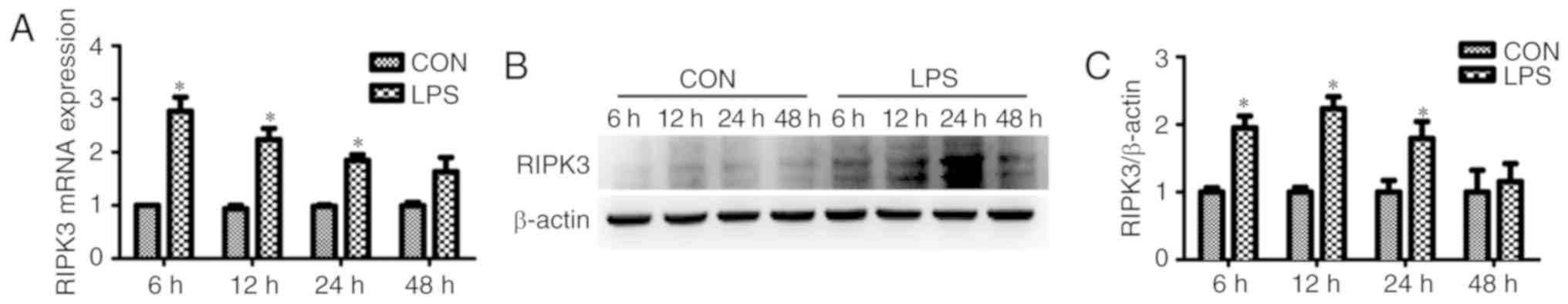
found that the mRNA expression of RIPK3 was significantly increased
in renal tubular epithelial cells as early as 6 h after LPS
intervention (Fig. 3A). The
protein expression of RIPK3 was induced 12 h after treatment with
LPS (Fig. 3B and C). Renal tubular
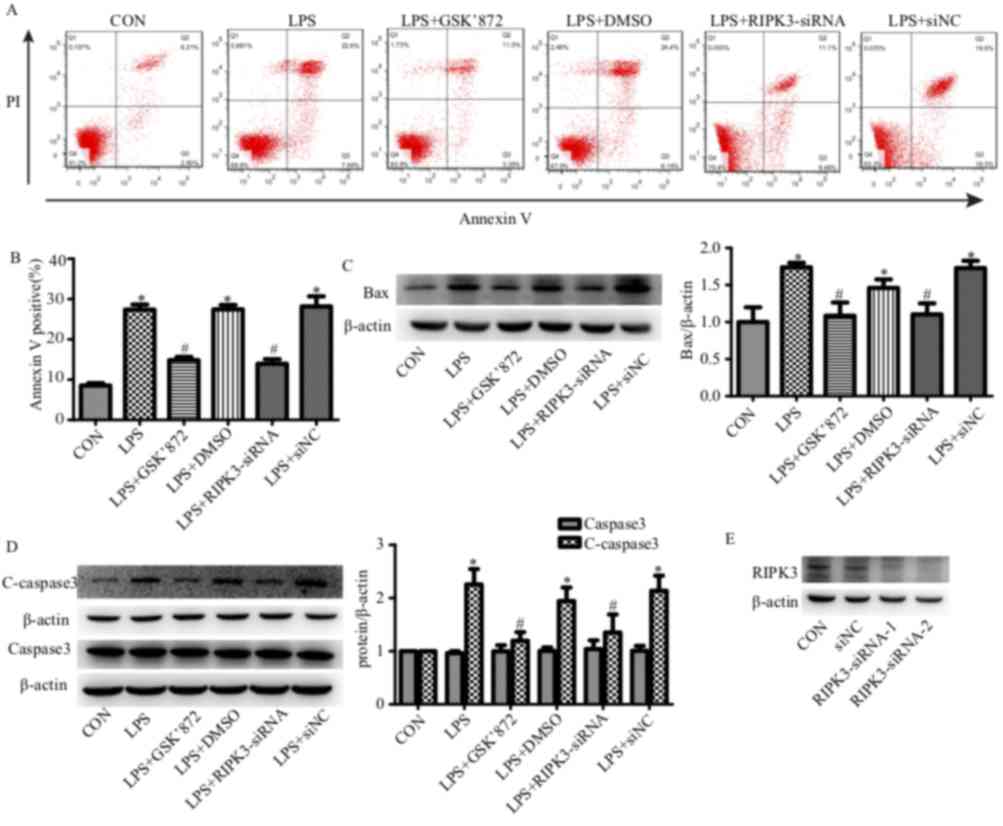
epithelial cell apoptosis was determined using flow cytometry. The
number of Annexin V-positive cells (apoptotic cells) was
significantly increased in the renal tubular epithelial cells that
had undergone LPS treatment for 24 h (27.38±1.12% in the LPS group
vs. 8.55±0.61% in the control group; P<0.001; Fig. 4A and B). siRNA reduced the
expression level of RIPK3 (Fig.
4E) and reduced tubular epithelial cell apoptosis (27.38±1.12%
in the LPS group vs. 13.95±1.12 in the LPS + RIPK3-siRNA group;
P<0.001; Fig. 4A and B).
LPS-induced apoptosis could also be inhibited by GSK′872
(27.38±1.12% in the LPS group vs. 14.87±0.77% in the LPS + GSK′872
group; P<0.001; Fig. 4A and B).
The expression levels of the proapoptotic factor Bax and the
apoptotic marker C-caspase-3 were also determined. Bax and
C-caspase-3 were induced by LPS and reversed by GSK′872 or
RIPK3-siRNA (Fig. 4C and D).
Discussion
In the present study it was found that renal tubular
epithelial cell apoptosis, mitochondrial injury and RIPK3
upregulation were induced in LPS-induced AKI. Inhibiting RIPK3
using GSK′872 or reducing RIPK3 expression using siRNA reduced
LPS-induced renal tubular epithelial cell apoptosis. GSK′872
functions by inhibiting the kinase activity of RIPK3 and does not
reduce the expression of RIPK3. GSK′872 was found to alleviate
LPS-induced renal tubular epithelial cell injury in vivo and
in vitro by inhibiting the RIPK3 kinase activity. These
results suggested that RIPK3 may mediate renal tubular epithelial
cell apoptosis in LPS-induced AKI.
AKI is a well-documented complication of sepsis
(1–3). LPS, also known as endotoxin, is an
outer membrane component of gram-negative bacteria and has been
identified as a major factor associated with the development of
AKI. Renal tubular epithelial cell apoptosis is increasingly
recognized as having an important role in the pathogenesis of
septic or endotoxic AKI (1,4,8).
A previous study reported renal tubular epithelial cell apoptosis
in septic AKI patients (26). In
sepsis, LPS promotes the secretion of proinflammatory cytokines,
nitric oxide and eicosanoids by binding to the monocyte
differentiation antigen CD14/TLR4/lymphocyte antigen 96 receptor
complex in various cell types, especially immunocytes, resulting in
multiple organ dysfunction and AKI (1). Previous studies have shown that LPS
could directly lead to renal tubular cell injury and AKI by binding
to TLR4 (6,27–29).
Consistent with previous studies (6,9–11),
the present study found that LPS induced renal tubular epithelial
cell apoptosis in vitro and in vivo. However, the
mechanisms of LPS-induced tubular epithelial cell apoptosis are not
completely understood. In the present study, a novel mechanism of
LPS-induced tubular epithelial cell apoptosis was investigated.
RIPKs are a group of threonine/serine protein
kinases with a conserved kinase domain and distinct non-kinase
regions (12). RIPKs are known to
be important sensors of intracellular and extracellular stresses
and to participate in different biological processes, including
cell death signaling and inflammation pathways (12–14).
RIPK3 is known to mediate necroptosis, a type of regulated
necrosis. Activated RIPK3 could phosphorylate the mixed-lineage
kinase domain-like protein, leading to plasma membrane rupture
(30,31). RIPK3-mediated necroptosis has been
reported in ischemia/reperfusion and cisplatin-induced AKI
(21,32–34).
LPS activates RIPK3 by binding to TLR4 in fibroblasts (35). Consistent with a previous study on
septic AKI (22), RIPK3 was
upregulated in tubular epithelial cells in the present study.
However, it was found that RIPK3 mediated renal tubular epithelial
cell apoptosis. Several previous studies have also demonstrated the
proapoptotic effect of RIPK3 (36–41).
The effect of the RIPK3 inhibitor GSK′872 on apoptosis is
concentration-dependent. Similar to RIPK3 deficiency,
low-concentrations of GSK′872 (0.04–1 µM), which binds to the RIPK3
kinase domain and inhibits kinase activity with a high specificity,
prevents apoptosis in mouse bone-marrow-derived macrophages,
peritoneal macrophages and fibroblasts (35,42).
However, previous studies reported that high-concentrations of
GSK′872 (3–10 µM), resembling a RIPK3-kinase-inactive mutant
(D161N), could induce apoptosis in mouse cells by altering the
conformation of RIPK3 (15,18,19,36,42).
In the present study, a low concentration of GSK′872 (0.5 µM)
significantly attenuated renal tubular cell apoptosis in
vitro, consistent with the protective effect of RIPK3
knockdown. Therefore, the present study indicated that RIPK3 may be
a new target for reducing renal tubular epithelial cell apoptosis
and improving AKI.
To further explore the mechanisms of RIPK3-mediated
renal tubular cell apoptosis induced by LPS, the expression of Bax
was also investigated. Bax is a proapoptotic member of the Bcl-2
family. Previous studies have demonstrated that Bax translocates to
the mitochondria and forms hetero- and homo-oligomers within the
mitochondrial outer membrane, leading to the release of
apoptosis-inducing proteins (43–45).
In addition, Bax-deficient mice were found to be resistant to
apoptosis (46). RIPK3-induced Bax
upregulation has been reported recently (47–49).
In the present study, Bax protein expression was significantly
increased in renal tubular epithelial cells following LPS
stimulation and this effect was abrogated by the downregulation of
RIPK3 expression or the inhibition of RIPK3 kinase activity. It was
therefore speculated that RIPK3 may mediate renal tubular
epithelial cell apoptosis by upregulating the expression of
Bax.
In conclusion, the present study demonstrated that
the expression of RIPK3 was increased in LPS-induced AKI and that
this mediated LPS-induced renal epithelial cell apoptosis. The
proapoptotic effect of RIPK3 may function by upregulating the
expression of Bax. The present study also revealed that low
concentration GSK′872 inhibited LPS-induced renal epithelial cell
apoptosis and attenuated AKI. Therefore, inhibiting RIPK3 may be a
potential therapeutic strategy to treat septic AKI. However,
further studies are required to determine the molecular mechanism
underpinning the RIPK3/Bax pathway.
Acknowledgements
Not applicable.
Funding
The present study was supported by the National
Natural Science Foundation of China (grant nos. 81570609 and
81770667) and Natural Science Foundation of Guangdong Province
(grant nos. 2016A030313767 and 2018A0303130284).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
SZ, RL and XL designed the study and wrote the
manuscript. WD and YW performed the pathological evaluations. HY,
WW, CL and ZY performed the animal experiments. LZ, YC, XZ, ZL, MZ
and SL performed the in vitro experiments. All authors read
and approved the final manuscript.
Ethics approval and consent to
participate
All animal procedures conformed to the protocols of
the Guangdong Academy of Medical Sciences and were approved by the
ethics committee for the experimental use of animals at Guangdong
Provincial People's Hospital.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Zarjou A and Agarwal A: Sepsis and acute
kidney injury. J Am Soc Nephrol. 22:999–1006. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Emlet AD, Shaw AD and Kellum JA:
Sepsis-associated AKI: Epithelial cell dysfunction. Semin Nephrol.
35:85–95. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Mårtensson RB and Bellomo R:
Sepsis-induced acute kidney injury. Crit Care Clin. 31:649–660.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Zarbock A, Gomez H and Kellum JA:
Sepsis-induced acute kidney injury revisited: Pathophysiology,
prevention and future therapies. Curr Opin Crit Care. 20:588–595.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Gotts JE and Matthay MA: Sepsis:
Pathophysiology and clinical management. BMJ. 353:i15852016.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Stasi A, Intini A, Divella C, Franzin R,
Montemurno E, Grandaliano G, Ronco C, Fiaccadori E, Pertosa GB,
Gesualdo and LCastellano G: Emerging role of lipopolysaccharide
binding protein in sepsis-induced acute kidney injury. Nephrol Dial
Transplant. 32:24–31. 2017.PubMed/NCBI
|
|
7
|
Plotnikov EY, Brezgunova AA, Pevzner IB,
Zorova LD, Manskikh VN, Popkov VA, Silachev DN and Zorov DB:
Mechanisms of LPS-induced acute kidney injury in neonatal and adult
rats. Antioxidants (Basel). 7(pii): E1052018. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Havasi A and Borkan SC: Apoptosis and
acute kidney injury. Kidney Int. 80:29–40. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zhao H, Liu Z, Shen H, Jin S and Zhang S:
Glycyrrhizic acid pretreatment prevents sepsis-induced acute kidney
injury via suppressing inflammation, apoptosis and oxidative
stress. Eur J Pharmacol. 781:92–99. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Rousta AM, Mirahmadi SM, Shahmohammadi A,
Nourabadi D, Khajevand-Khazaei MR, Baluchnejadmojarad T and Roghani
M: Protective effect of sesamin in lipopolysaccharide-induced mouse
model of acute kidney injury via attenuation of oxidative stress,
inflammation, and apoptosis. Immunopharmacol Immunotoxicol.
40:423–429. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Su Z, Yu P, Sheng L, Ye J and Qin Z:
Fangjifuling ameliorates lipopolysaccharide-induced renal injury
via inhibition of inflammatory and apoptotic response in mice. Cell
Physiol Biochem. 49:2124–2137. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zhang D, Lin J and Han J:
Receptor-interacting protein (RIP) kinase family. Cell Mol Immunol.
7:243–249. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Declercq W, Vanden Berghe T and
Vandenabeele P: RIP kinases at the crossroads of cell death and
survival. Cell. 138:229–232. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Witt A and Vucic D: Diverse ubiquitin
linkages regulate RIP kinases-mediated inflammatory and cell death
signaling. Cell Death Differ. 24:1160–1171. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Meylan ET and Tschopp J: The RIP kinases:
Crucial integrators of cellular stress. Trends Biochem Sci.
30:151–159. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Sun X, Lee J, Navas T, Baldwin DT, Stewart
TA and Dixit VM: RIP3 a novel apoptosis inducing kinase. J Biol
Chem. 274:16871–16875. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Pazdernik NJ, Donner DB, Goebl MG and
Harrington MA: Mouse receptor interacting protein 3 does not
contain a caspase recruiting or a death domain but induces
apoptosis and activates NF-kappaB. Mol Cell Biol. 19:6500–6508.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kasof GM, Prosser JC, Liu D, Lorenzi MV
and Gomes BC: The RIP-like kinase, RIP3, induces apoptosis and
NF-kappaB nuclear translocation and localizes to mitochondria. FEBS
Lett. 473:285–291. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Feng S, Ma L, Yang Y and Wu M: Truncated
RIP3 (tRIP3) acts upstream of FADD to induce apoptosis in the human
hepatocellular carcinoma cell line QGY-7703. Biochem Biophys Res
Commun. 347:558–565. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Xu Y, Ma H, Shao J, Wu J, Zhou L, Zhang Z,
Wang Y, Huang Z, Ren J, Liu S, et al: A role for tubular
necroptosis in cisplatin-induced AKI. J Am Soc Nephrol.
26:2647–2658. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Sureshbabu A, Patino E, Ma KC, Laursen K,
Finkelsztein EJ, Akchurin O, Muthukumar T, Ryter SW, Gudas L, Choi
AMK and Choi ME: RIPK3 promotes sepsis-induced acute kidney injury
via mitochondrial dysfunction. JCI Insight. 3(pii): 984112018.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Shashaty MG, Reilly JP, Sims CA, Holena
DN, Qing D, Forker CM, Hotz MJ, Meyer NJ, Lanken PN, Feldman HI, et
al: Plasma levels of receptor interacting protein kinase-3 (RIP3),
an essential mediator of necroptosis, are associated with acute
kidney injury in critically Ill trauma patients. Shock. 46:139–143.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Brooks C, Wei Q, Cho SG and Dong Z:
Regulation of mitochondrial dynamics in acute kidney injury in cell
culture and rodent models. J Clin Invest. 119:1275–1285. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Schmittgen TD and Livak KJ: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Lerolle N, Nochy D, Guérot E, Bruneval P,
Fagon JY, Diehl JL and Hill G: Histopathology of septic shock
induced acute kidney injury: Apoptosis and leukocytic infiltration.
Intensive Care Med. 36:471–478. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Shi M, Zeng X, Guo F, Huang R, Feng Y, Ma
L, Zhou L and Fu P: Anti-inflammatory pyranochalcone derivative
attenuates LPS-induced acute kidney injury via inhibiting
TLR4/NF-kappaB pathway. Molecules. 22(pii): E16832017. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Nakano D, Doi K, Kitamura H, Kuwabara T,
Mori K, Mukoyama M and Nishiyama A: Reduction of tubular flow rate
as a mechanism of oliguria in the early phase of endotoxemia
revealed by intravital imaging. J Am Soc Nephrol. 26:3035–3044.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Watts BA III, George T, Sherwood ER and
Good DW: Monophosphoryl lipid a induces protection against LPS in
medullary thick ascending limb through a TLR4-TRIF-PI3K signaling
pathway. Am J Physiol Renal Physiol. 313:F103–F115. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Sun L, Wang H, Wang Z, He S, Chen S, Liao
D, Wang L, Yan J, Liu W, Lei X and Wang X: Mixed lineage kinase
domain-like protein mediates necrosis signaling downstream of RIP3
kinase. Cell. 148:213–227. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Cho YS, Challa S, Moquin D, Genga R, Ray
TD, Guildford M and Chan FK: Phosphorylation-driven assembly of the
RIP1-RIP3 complex regulates programmed necrosis and virus-induced
inflammation. Cell. 137:1112–1123. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Linkermanna A, Bräsen JH, Dardingd M, Jin
MK, Sanz AB, Heller JO, De Zen F, Weinlich R, Ortiz A, Walczak H,
et al: Two independent pathways of regulated necrosis mediate
ischemia-reperfusion injury. Proc Natl Acad Sci USA.
110:12024–12029. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Liu W, Chen B, Wang Y, Meng C, Huang H,
Huang XR, Qin J, Mulay SR, Anders HJ, Qiu A, et al: RGMb protects
against acute kidney injury by inhibiting tubular cell necroptosis
via an MLKL-dependent mechanism. Proc Natl Acad Sci USA.
115:E1475–E1484. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
von Mässenhausen A, Tonnus W, Himmerkus N,
Parmentier S, Saleh D, Rodriguez D, Ousingsawat J, Ang RL, Weinberg
JM, Sanz AB, et al: Phenytoin inhibits necroptosis. Cell Death Dis.
9:3592018. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Kaiser WJ, Sridharan H, Huang C, Mandal P,
Upton JW, Gough PJ, Sehon CA, Marquis RW, Bertin J and Mocarski ES:
Toll-like receptor 3-mediated necrosis via TRIF, RIP3, and MLKL. J
Biol Chem. 288:31268–31279. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Newton K, Dugger DL, Wickliffe KE, Kapoor
N, de Almagro MC, Vucic D, Komuves L, Ferrando RE, French DM,
Webster J, et al: Activity of protein kinase RIPK3 determines
whether cells die by necroptosis or apoptosis. Science.
343:1357–1360. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Yabal M, Muller N, Adler H, Knies N, Groß
CJ, Damgaard RB, Kanegane H, Ringelhan M, Kaufmann T, Heikenwälder
M, et al: XIAP restricts TNF- and RIP3-dependent cell death and
inflammasome activation. Cell Rep. 7:1796–1808. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Lawlor KE, Khan N, Mildenhall A, Gerlic M,
Croker BA, D'Cruz AA, Hall C, Kaur Spall S, Anderton H, Masters SL,
et al: RIPK3 promotes cell death and NLRP3 inflammasome activation
in the absence of MLKL. Nat Commun. 6:62822015. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Wicki S, Gurzeler U, Wei-Lynn Wong, Jost
PJ, Bachmann D and Kaufmann T: Loss of XIAP facilitates switch to
TNFα-induced necroptosis in mouse neutrophils. Cell Death Dis.
7:e24222016. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Upton JW, Kaiser WJ and Mocarski ES: Virus
inhibition of RIP3-dependent necrosis. Cell Host Microbe.
7:302–313. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Vandenabeele P, Declercq W, Van Herreweghe
F and Vanden Berghe T: The role of the kinases RIP1 and RIP3 in
TNF-induced necrosis. Sci Signal. 3:re42010. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Mandal P, Berger SB, Pillay S, Moriwaki K,
Huang C, Guo H, Lich JD, Finger J, Kasparcova V, Votta B, et al:
RIP3 induces apoptosis independent of pronecrotic kinase activity.
Mol Cell. 56:481–495. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Kroemer D: The pathophysiology of
mitochondrial cell death. Science. 305:626–629. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Bhatt K, Feng L, Pabla N, Liu K, Smith S
and Dong Z: Effects of targeted Bcl-2 expression in mitochondria or
endoplasmic reticulum on renal tubular cell apoptosis. Am J Physiol
Renal Physiol. 294:F499–F507. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Bleicken S, Classen M, Padmavathi PV,
Ishikawa T, Zeth K, Steinhoff HJ and Bordignon E: Molecular details
of Bax activation, oligomerization, and membrane insertion. J Biol
Chem. 285:6636–6647. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Wei Q, Dong G, Franklin J and Dong Z: The
pathological role of Bax in cisplatin nephrotoxicity. Kidney Int.
72:53–62. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Wei Q, Dong G, Chen JK, Ramesh G and Dong
Z: Bax and bak have critical roles in ischemic acute kidney injury
in global and proximal tubule-specific knockout mouse models.
Kidney Int. 84:138–148. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Zhou Y, Li Y, Zhou B, Chen K, Lyv Z, Huang
D, Liu B, Xu Z, Xiang B, Jin S, et al: Inflammation and apoptosis:
Dual mediator role for toll-like receptor 4 in the development of
necrotizing enterocolitis. Inflamm Bowel Dis. 23:44–56. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Liu PY, Chang DC, Lo YS, His YT, Lin CC,
Chuang YC, Lin SH, Hsieh MJ and Chen MK: Osthole induces human
nasopharyngeal cancer cells apoptosis through fas-fas ligand and
mitochondrial pathway. Environ Toxicol. 33:446–453. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Zielinska E, Zauszkiewicz-Pawlak A, Wojcik
M and Inkielewicz-Stepniak I: Silver nanoparticles of different
sizes induce a mixed type of programmed cell death in human
pancreatic ductal adenocarcinoma. Oncotarget. 9:4675–4697.
2017.PubMed/NCBI
|