Introduction
Monogenic diabetes accounts for 1–6% of pediatric
diabetes patients with the highest incidence among patients
manifesting non-type 1 diabetes mellitus in childhood or
adolescence (1).
A large, clinically heterogeneous group of
dominantly inherited disorders linked to primary β-cell dysfunction
is classified as maturity onset diabetes in the young (MODY). To
date, 13 genes causative of 13 types of MODY are known (2). MODY is typically diagnosed before 25
years of age; it is non-insulin dependent and its symptoms are
usually mild. However, due to the variety of clinical forms caused
by a wide spectrum of mutations in MODY-related genes, different
treatment strategies are used: From appropriate diet and physical
activity to oral and/or insulin therapy.
Monogenic diabetes also includes a number of
non-MODY transient or permanent neonatal forms occurring under 6
months of age. More than 20 genes are known to be related to
congenital neonatal diabetes (3).
Depending on the gene involved, neonatal diabetes may follow
patterns of dominant or recessive inheritance and may be isolated
or associated with a variety of syndromic features (4). However, due to a very early onset of
diabetes, hyperglycemia is often diagnosed prior to other syndromic
features. The treatment strategy for non-MODY neonatal diabetes
depends on the specific genetic defect causing the diabetic
phenotype.
Molecular genetic testing is highly recommended for
patients suspected of monogenic diabetes as it allows tailoring
treatments to specific etiological mechanisms. Up until recently,
search for diabetes-related mutations was usually performed by
Sanger sequencing and was therefore limited to only a few genes,
leaving a considerable proportion of cases without a known cause.
Moreover, a number of studies have demonstrated that frequencies of
certain monogenic diabetes subtypes vary strongly among different
populations (5), challenging the
development of unified recommendations for the target gene choice.
An efficient technology to detect previously known and to reveal
novel mutations related to monogenic diabetes is next-generation
sequencing. This technique allows for a rapid analysis of an
unlimited number of genes and may provide valuable knowledge on the
genetic variants causative of monogenic diabetes in different
populations.
Here, using targeted whole-exome sequencing (WES),
we studied the frequency and the spectrum of genetic variants
causative of monogenic diabetes in a cohort of Russian children
with non-type 1 diabetes mellitus.
Materials and methods
Study group
A total of 60 unrelated patients with diabetes and
impaired glucose tolerance (pre-diabetes) were prospectively
included in the study. All the patients were of Russian ethnicity
and resided in Northwest Russia. In accordance with the guidelines
of the American Diabetes Association (6), the diagnoses were based on plasma
glucose criteria, either the fasting plasma glucose (FPG) and/or
the 2-h plasma glucose (2-h PG) value after a 75-g oral glucose
tolerance test (OGTT) and/or the HbA1C criteria. All the patients
had an onset of diabetes before the age of 18 years and a
detectable C-peptide secretion (or a detectable insulin level in
the absence of insulin therapy) and were negative for insulin-,
islet-cell-, tyrosinphosphatase IA2-, and glutamate
decarboxylase-autoantibodies. The exclusion criterion was the
presence of the already confirmed syndrome associated with impaired
glucose metabolism (such as Prader-Willi syndrome). In 59 cases,
family history was available, and in 41 of them, it was positive
for diabetes. All the patients were referred to the study by their
medical supervisors.
Sample preparation and whole-exome
sequencing
Genomic DNA was extracted from whole blood by Magna
Pure System (Roche) using the standard protocol. Exome DNA
libraries were prepared from 100 ng DNA using TruSeq®
Exome Sample Preparation kit (Illumina, Inc.), following the
manufacturer's instructions. Libraries were sequenced on Illumina
HiSeq 2500 in 2×100 PE mode. An average of 63.6 million sequencing
reads were obtained for each sample, yielding ~50× mean coverage of
CDS regions and an average of 89% of CDS bases covered at least
10×.
Bioinformatic analysis
Bioinformatic analysis of the WES data was done
using a pipeline based on bwa v.0.7.12-r1044 aligner, Picard tools
v.2.0.1, and Genome Analysis Tool kit (GATK) v.3.5 software with
all the necessary preprocessing steps required by the GATK Best
Practices workflow (https://software.broadinstitute.org/gatk/best-practices/)
(7,8). Target enrichment metrics were
collected using the Picard CalculateHsMetrics tool. Variant calling
was done using GATK HaplotypeCaller in the cohort genotyping mode
with 250 samples included into the cohort (samples with a similar
ethnical background from St. Petersburg State University Biobank
were used for cohort padding). Variants were filtered using variant
quality score recalibration (VQSR) and annotated with SnpEff and
SnpSift tools (version 4.2). Additional annotations included the
following information: rsID of known variants from dbSNP (build
146), allele frequency (AF) from large sequencing consortia-1000
Genomes (9), Exome Aggregation
Consortium (ExAC) (10), and
ESP6500 (11); and pathogenicity
predictions by Polyphen-2 (12),
SIFT (13), PROVEAN (14) obtained from dbNSFP database
(15) and by Human Splicing Finder
(16) and DDIG (17). For additional prediction of protein
stability changes caused by missence mutations with uncertain
significance, I-Mutant 2.0 (18)
was used. Variant ranking was done using a custom scoring metric.
Reference minor allele presence in target genes was analyzed using
RMA Hunter (19).
To check the possible presence of copy-number
variants (CNVs), we analyzed the sequencing coverage across all
targeted exons of interest. To this end, we calculated coverage for
each interval using GATK, and then normalized the coverage matrix
across samples and intervals. We then used z-score value of the
normalized coverage to assess the statistical significance of the
results.
Verification of the WES results and
family analysis
Verification of the WES results in probands and
subsequent family analyses were performed by PCR-direct sequencing.
Specific primers were designed for verification of each case. The
PCR products were purified with 5M NH4Ac and 96% ethanol and then
with 70% ethanol, dried at 60°C, and dissolved in 10 µl of
deionized water. After purification, the PCR products were
sequenced using an ABI PRISM BigDyeTerminator 3.1 kit reagent
(Applied Biosystems). Then, a capillary electrophoresis was
performed in a GA3130×l Genetic Analyzer (Applied Biosystems).
Sequences were analyzed using the Sequence Scanner software
(Applied Biosystems).
Analysis of the GCK promoter for
c.-71G>C genetic variant
A single-base substitution c.-71G>C in the
GCK promoter is known to be linked to MODY2 phenotype
(20). However, WES did not allow
for analysis of the GCK promoter for c.-71G>C. For this
reason, the GCK promoter was analyzed for c.-71G>C
genetic variant by PCR-direct sequencing as described above with
the use of Hae III endonuclease and the following primers:
F-5′-GCATGGCAGCTCTAATGACAGG-3′ and
R-5′-CATCCTAGCCTGCTTCCCTGG-3′.
Results
Genetic variants causative of
monogenic diabetes in Russian children with non-type 1 diabetes
mellitus
Using whole-exome sequencing followed by PCR-direct
sequencing, we identified the frequency and the spectrum of genetic
variants causative of monogenic diabetes in 60 Russian children
with non-type 1 diabetes mellitus. Genetic variants were screened
for a total of 35 genes: 13 genes causative of MODY [HNF4A
(MODY1), GCK (MODY2), HNF1A (MODY3), PDX1
(MODY4), HNF1B (MODY5), NEUROD1 (MODY6), KLF11
(MODY7), CEL (MODY8), PAX4 (MODY9), INS
(MODY10), BLK (MODY11), ABCC8 (MODY12), and
KCNJ11 (MODY13)] and 22 genes causative of transient or
permanent neonatal diabetes, including the ones related to specific
syndromes (EIF2AK3, RFX6, WFS1, ZFP57, FOXP3, AKT2, PPARG,
APPL1, PTF1A, GATA4, GATA6, GLIS3, IER3IP1, LMNA, NEUROG3, PAX6,
PLAGL1, SLC19A2, SLC2A2, SH2B1, SERPINB4, and MADD).
Overall, 33 out of 60 patients (55%) had genetic
variants in the target genes (Table
I; 21–40). For 12 patients, parents were available for genetic
testing and origins of genetic variants were determined. In 11
cases, genetic variants had been inherited from the parents, and in
one case, a de novo genetic variant was confirmed. Of 33
patients, 27 (81.8%) had genetic variants in MODY-related genes.
The majority of these patients (19 out of 27) had genetic variants
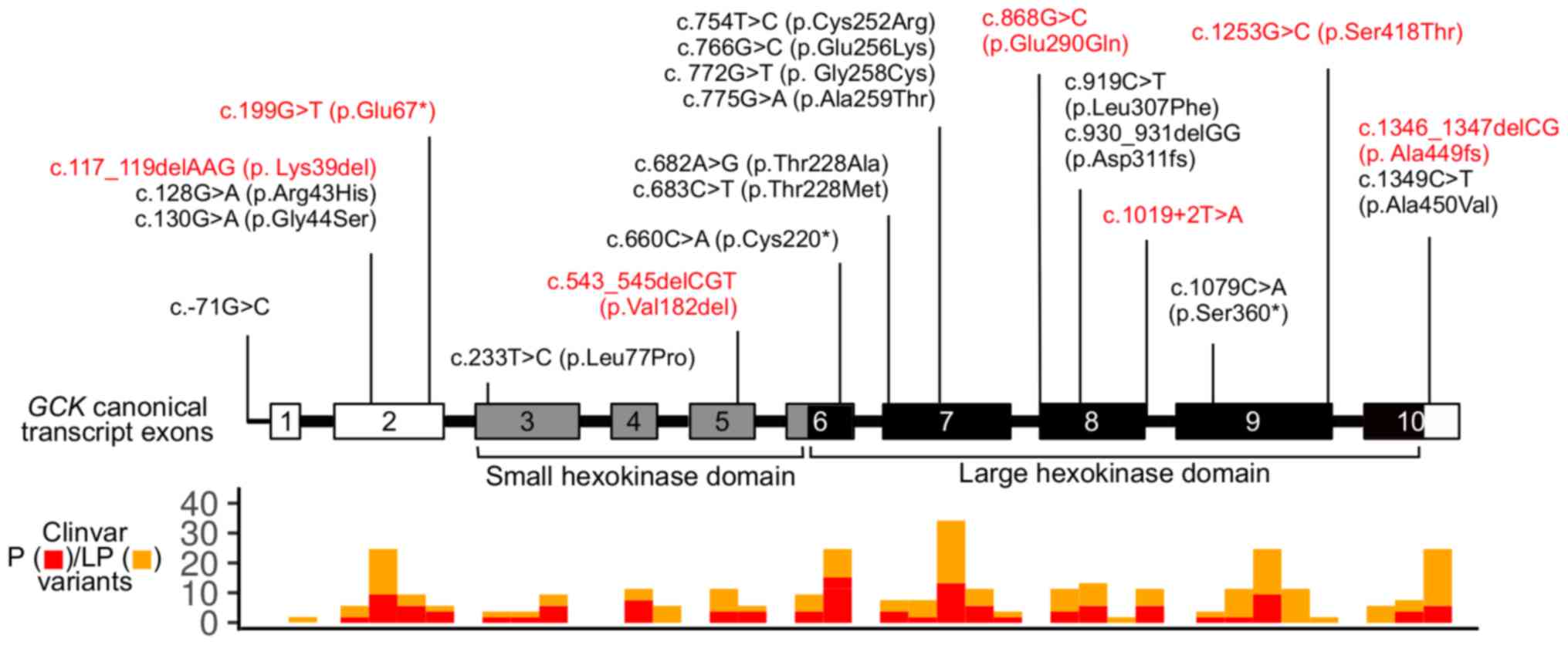
in GCK (MODY2). The spectrum of GCK genetic variants
included 13 missense mutations, 3 nonsense mutations, 1 in-frame
and 3 frameshift deletions, and 1 single-base substitution in the
promoter. In two GCK mutation-positive patients, two genetic
variants were present: Missense mutation along with a single-base
substitution in the promoter (patient #27) and missense mutation
along with a nonsense mutation (patient #78). The spectrum of the
identified GCK genetic variants is shown in Fig. 1. Missense mutations in HNF1A
(MODY3) were registered in two patients. The other MODY-related
genetic variants included three cases of missense mutations: In
PAX4 (MODY 9), in ABCC8 (MODY12), and in
KCNJ11 (MODY 13).
 | Table I.Genetic variants identified in
Russian children with non-type 1 diabetes mellitus. |
Table I.
Genetic variants identified in
Russian children with non-type 1 diabetes mellitus.
| Patient number | Gene | Nucleotide change
(protein change) | Mutation type | Mutation
origin | Pathogenicity
according to ACMG | (Refs.) |
|---|
| 59 | GCK | c.772G>T
(p.Gly258Cys) | Missense | Unknown | Likely
pathogenic | (21) |
| 62 | GCK | c.930_931delGG
(p.Asp311fs) | Frameshift | Unknown | Pathogenic | (22) |
| 83 | GСK | c.930_931delGG
(p.Asp311fs) | Frameshift | Unknown | Pathogenic | (22) |
| 95 | GCK | c.130G>A
(p.Gly44Ser) | Missense | Father | Likely
pathogenic | (23) |
| 167 | GCK | c.128G>A
(p.Arg43His) | Missense | Mother | Likely
pathogenic | (24) |
| 197 | GCK | c.233T>C
(p.Leu77Pro) | Missense | Father | Likely
pathogenic | (25) |
| 426 | GCK | c.683C>T
(p.Thr228Met) | Missense | Unknown | Likely
pathogenic | (26) |
| 460 | GCK | c.682A>G
(p.Thr228Ala) | Missense | Mother | Likely
pathogenic | (21) |
| 580 | GCK | c.775G>A
(p.Ala259Thr) | Missense | Unknown | Likely
pathogenic | (27) |
| 663 | GCK | c.1079C>A
(p.Ser360*) | Nonsense | Unknown | Pathogenic | (28) |
| 665 | GCK | c.660C>A
(p.Cys220*) | Nonsense | Unknown | Pathogenic | (24) |
| 176 | GCK | c.1349C>T
(p.Ala450Val) | Missense | Unknown | Likely
pathogenic | (29) |
| 661 | GCK | c.1349C>T
(p.Ala450Val) | Missense | Unknown | Likely
pathogenic | (29) |
| 118 | GCK | c.117_119delAAG
(p.Lys39del) | In-frame
deletion | Unknown | Uncertain
significance | Novel |
| 119 | GCK | c.1346_1347delCG
(p.Ala449fs) | Frameshift | Unknown | Pathogenic | Novel |
| 434 | GCK | c.868G>C
(p.Glu290Gln) | Missense | Mother | Uncertain
significance | Novel |
| 578 | GCK | c.1253G>C
(p.Ser418Thr) | Missense | Unknown | Pathogenic | Novel |
| 27 | GCK | c.754T>C
(p.Cys252Arg) | Missense | Unknown | Likely
pathogenic | (30) |
|
|
| c.-71G>C | Promoter | Unknown | Likely
pathogenic | (20) |
| 78 | GCK | c.199G>T
(p.Glu67*) | Nonsense | Mother | Pathogenic | Novel |
|
|
| c.766G>C
(p.Glu256Lys) | Missense | Mother | Likely
pathogenic | (31) |
| 153 | HNF1A | c.709A>G
(p.Asn237Asp) | Missense | Unknown | Uncertain
significance | (32) |
| 422 | HNF1A | c.485T>G
(p.Leu162Arg) | Missense | Unknown | Uncertain
significance | Novel |
| 215 | PAX4 | c.574C>A
(p.Arg192Ser) | Missense | Unknown | Uncertain
significance | (33) |
| 114 | ABCC8 | c.4139G>A
(p.Arg1380His) | Missense | Unknown | Likely
pathogenic | (34) |
| 134 | KCNJ11 | c.406C>A
(p.Arg136Ser) | Missense | Unknown | Uncertain
significance | (35) |
| 68 | GATA6 | c.1477C>T
(p.Arg493*) | Nonsense | De novo | Pathogenic | (36) |
| 266 | WFS1 | c.2452C>T
(p.Arg818Cys) | Missense | Mother | Likely benign | (37) |
| 408 | WFS1 | c.2327A>T
(p.Glu776Val) | Missense | Mother | Likely benign | (38) |
| 133 | WFS1 | c.1124G>A
(p.Arg375His) | Missense | Unknown | Uncertain
significance | Novel |
| 411 | EIF2AK3 | c.1912C>T
(p.Arg638*) | Nonsense | From | Pathogenic | Novel |
|
| EIF2AK3 | c.1912C>T
(p.Arg638*) | Nonsense | parents |
|
|
| 432 | SLC19A2 | c.164delC
(p.Pro55fs) | Frameshift | Mother | Pathogenic | Novel |
|
| SLC19A2 | c.161C>A
(p.Thr54Asn) | Missense | Father | Uncertain
significance | Novel |
| 226 | GCK | c.543_545delCGT
(p.Val182del) | In-frame
deletion | Unknown | Uncertain
significance | Novel |
|
| HNF1A | c.92G>A
(p.Gly31Asp) | Missense | Unknown | Likely
pathogenic | (39) |
| 529 | BLK | c.939G>C
(p.Glu313Asp) | Missense | Unknown | Uncertain
significance | Novel |
|
| GCK | c.919C>T
(p.Leu307Phe) | Missense | Unknown | Uncertain
significance | Novel |
| 662 | GCK | c.1019+2T>A | Splicing
defect | Unknown | Pathogenic | Novel |
|
| BLK | c.1148G>A
(p.Arg383Gln) | Missense | Unknown | Uncertain
significance | Novel |
|
| WFS1 | c.1957C>T
(p.Arg653Cys) | Missense | Unknown | Likely
pathogenic | (40) |
The presence of genetic variants in different target
genes was detected in three patients. In one of them, a GCK
in-frame deletion was accompanied by an HNF1A missense
mutation (patient #226). In another one, two missense mutations
were present: In GCK and in BLK (patient #529). In
the third patient (#662), a splicing defect in GCK and
missense mutations in BLK and WFS1 were present.
Genetic variants causative of non-MODY monogenic
diabetes were found in 6 out of 33 mutation-positive patients
(18.2%). These included a nonsense mutation in GATA6, three
cases of missense mutations in WFS1, one case of a
homozygous EIF2AK3 nonsense mutation (patient #411), and one
case of missense mutation and a frameshift deletion present in
SLC19A2 (c.164delC and c.161C>A) (patient #432). The
EIF2AK3 nonsense mutations had been inherited from
consanguineous parents who were heterozygous carriers of the same
mutation. The SLC19A2 mutations also appeared to have been
inherited from the parents: C.164delC from the mother and
c.161C>A from the father, indicating that both SLC19A2
alleles in patient #432 were affected.
Considering that monogenic diabetes may be
associated with deletions and duplications, we analyzed the
possible presence of CNVs in the target genes. We found no evidence
for CNVs in the target genes in either sample. However, it should
be noted that the limitations of WES technology do not allow for
confident detection of small-scale CNVs.
Relationship between genetic variants
and diabetic phenotypes
We analyzed the relationship of the detected genetic
variants to the patients' diabetic phenotypes. Among the 38
detected genetic variants, 23 had been previously reported as
linked to monogenic diabetes and 15 were novel ones (Table I). According to the American
College of Medical Genetics and Genomics (ACMG) guidelines
(41), most of the detected
genetic variants (18 previously reported and 6 novel ones) were
classified as pathogenic or likely pathogenic and thus were
considered as causative of the diabetic phenotypes in the studied
patients. However, the relationship of the detected KCNJ11
missense mutation to the diabetic phenotype was not apparent,
because earlier it had been shown to be associated with
hyperinsulinism (35), which was
not present in patient #134.
Three previously reported and 9 novel genetic
variants were classified as those of uncertain significance, and
two genetic variants were likely benign (Table I). These variants included 12
missense mutations; for them, we performed an additional in
silico analysis using I-Mutant 2.0 (18) (Table
II). In all but one case, the in silico modeling
attested to a decrease of protein stability, thus suggesting the
pathogenic effect of the checked genetic variants. Of special
interest were two novel WFS1 genetic variants, initially
classified as likely benign. Patient #266 inherited the genetic
variant from a non-diabetic mother, while patient #408 inherited
the genetic variant from a mother with diabetes. Homozygous
mutations in WFS1 lead to the development of Wolfram
syndrome, an autosomal recessive disorder characterized by a list
of clinical signs including a bilateral progressive optic atrophy,
deafness, and diabetes mellitus (42). Heterozygous carriers of WFS1
mutations have been reported to have risk of early-onset diabetes
mellitus (43). The latter cannot
be excluded in our patients. However, an intriguing point is that
the WFS1 genetic variant in patient #408, who inherited it
from a diabetic mother, appeared to not decrease the protein
stability according to I-Mutant, which makes its pathogenicity
questionable.
| Table II.In silico prediction of
increase/decrease in the protein stability caused by missense
mutations with uncertain significance and by benign missense
mutations. |
Table II.
In silico prediction of
increase/decrease in the protein stability caused by missense
mutations with uncertain significance and by benign missense
mutations.
| Patient number | Gene | Genetic variant
(amino acid change) | Pathogenicity
according to ACMG | Protein stability
predicted by I-Mutant |
|---|
| 434 | GCK | c.868G>C
(p.Glu290Gln) | Uncertain
significance | Decrease |
| 153 | HNF1A | c.709A>G
(p.Asn237Asp) | Uncertain
significance | Decrease |
| 422 | HNF1A | c.485T>G
(p.Leu162Arg) | Uncertain
significance | Decrease |
| 215 | PAX4 | c.574C>A
(p.Arg192Ser) | Uncertain
significance | Decrease |
| 134 | KCNJ11 | c.406C>A
(p.Arg136Ser) | Uncertain
significance | Decrease |
| 266 | WFS1 | c.2452C>T
(p.Arg818Cys) | Likely benign | Decrease |
| 408 | WFS1 | c.2327A>T
(p.Glu776Val) | Likely benign | Increase |
| 133 | WFS1 | c.1124G>A
(p.Arg375His) | Uncertain
significance | Decrease |
| 432 | SLC19A2 | c.161C>A
(p.Thr54Asn) | Uncertain
significance | Decrease |
| 529 | BLK | c.939G>C
(p.Glu313Asp) | Uncertain
significance | Decrease |
|
| GCK | c.919C>T
(p.Leu307Phe) | Uncertain
significance | Decrease |
| 662 | BLK | c.1148G>A
(p.Arg383Gln) | Uncertain
significance | Decrease |
Clinical picture in patients with
multiple genetic variants
Finally, we analyzed the clinical picture in
patients with more than one genetic variant in one or different
target genes (Table III). A
simultaneous presence of two GCK genetic variants in patient
#27 raised the question of their location in one or both alleles.
The parents were not available for analysis. The clinical picture
was mild and typical for MODY2. It contrasted with the severe one
usually reported in patients with both GCK alleles affected
(44,45), suggesting that, in patient #27,
both genetic variants were present in the same allele and thus had
no accumulative effect. In patient #78, who was also a carrier of
two GCK genetic variants, the clinical picture was typical
for MODY2. As both genetic variants were inherited from the mother,
we concluded that only one allele was affected. Moreover, only
nonsense mutation c.199G>T seemed to be clinically significant,
because the resulting stop-codon terminates translation before the
c.766G>C site. The clinical picture in patient #226, who had
genetic variants in GCK and HNF1A, was more typical
for MODY2 than for MODY3: He had mild fasting and postprandial
hyperglycemia, had no glucosuria, and was successfully being
treated by a diet. Patient #411 had a homozygous EIF2AK3
nonsense mutation, inherited from consanguineous parents and
associated with Wolcott-Rallison syndrome, which, in turn, has been
reported to be the most common genetic cause of permanent neonatal
diabetes in consanguineous families (46). Patient #432 had two novel genetic
variants affecting both SLC19A2 alleles. Homozygous
mutations in SLC19A2 cause Rogers syndrome:
Thiamine-responsive megaloblastic anaemia associated with diabetes
mellitus and deafness (47). Among
other clinical signs are congenital heart defects, retinal
degeneration, ketonuria, dwarfism, and neurological symptomatology
(42). Of note, patient #432 had
only diabetes mellitus, retinal degeneration, ketonuria, and
neurological symptomatology and thus did not manifest a typical
clinical picture. Both patients #529 and #662 had typical clinical
signs of GCK-MODY rather than BLK-MODY, suggesting an
absence of strong accumulation of the pathogenic effect of the
detected genetic variants.
| Table III.Clinical characteristics of the
patients with multiple genetic variants in monogenic
diabetes-related genes. |
Table III.
Clinical characteristics of the
patients with multiple genetic variants in monogenic
diabetes-related genes.
| Patient number | Gene Nucleotide
change Amino acid change | Age at diagnosis
months | Diabetic
ketoacidosis | C-peptide
ng/ml | HbA1C % | SDS BMI | Treatment |
|---|
| 27 | GCK | 3 | No | 0.7 | 6 | −0.63 | Diet |
|
|
c.754T>C (p.Cys252Arg) |
|
|
|
|
|
|
|
| GCK |
|
|
|
|
|
|
|
| c.-71G>C |
|
|
|
|
|
|
| 78 | GCK | 39 | No | 0.63 | 6.4 | +0.83 | Diet |
|
|
c.199G>T (p.Glu67*) |
|
|
|
|
|
|
|
| GCK |
|
|
|
|
|
|
|
| c.766G>C
(p.Glu256Lys) |
|
|
|
|
|
|
| 226 | GCK | 36 | No | 1.1 | 6 | −1.69 | Diet |
|
| c.543_545delCGT
(p.Val182del) |
|
|
|
|
|
|
|
| HNF1A |
|
|
|
|
|
|
|
|
c.92G>A (p.Gly31Asp) |
|
|
|
|
|
|
| 411 | EIF2AK3 | 3 | Ketonuria | 0.2 | 9.2 | −0.72 | Insulin |
|
| c.1912C>T
(p.Arg638*) |
|
|
|
|
|
|
|
| EIF2AK3 |
|
|
|
|
|
|
|
| c.1912C>T
(p.Arg638*) |
|
|
|
|
|
|
| 432 | SLC19A2 | 48 | Ketonuria | 1.1 | 5.3 | −1.0 | Insulin for |
|
| c.164delC
(p.Pro55fs) |
|
|
|
|
| a few days/ |
|
| SLC19A2 |
|
|
|
|
| diet |
|
| c.161C>A
(p.Thr54Asn) |
|
|
|
|
|
|
| 529 | BLK | 10 | No | 0.43 | 6.7 | −0.46 | Diet |
|
| c.939G>C
(p.Glu313Asp) |
|
|
|
|
|
|
|
| GCK |
|
|
|
|
|
|
|
| c.919C>T
(p.Leu307Phe) |
|
|
|
|
|
|
| 662 | GCK | 22 | No | 1.1 | 6.82 | −1.32 | Diet |
|
| c.1019+2T>A |
|
|
|
|
|
|
|
| BLK |
|
|
|
|
|
|
|
| c.1148G>A
(p.Arg383Gln) |
|
|
|
|
|
|
|
| WFS1 |
|
|
|
|
|
|
|
| c.1957C>T
(p.Arg653Cys) |
|
|
|
|
|
|
Discussion
In 1974, Tattersall reported on three families
suffering from mild non-insulin dependent diabetes with Mendelian
dominant inheritance (48). The
disease was diagnosed in children and young adults and was later
defined as maturity-onset type diabetes of young people (MODY)
(49). The discovery of mutations
in the genes encoding HNF4A (50),
HNF1A (51), HNF1B (52), IPF (PDX1) (53), and GCK (54,55)
as the causes of MODY provided evidence for genetic heterogeneity
of familial diabetes. To date, MODY-causing mutations are
identified in a total of 13 genes, and mutations in more than 20
genes are known to be associated with neonatal hyperglycemia
(56). Because of such a variety
of genetic causes, many cases of monogenic diabetes remain without
a genetic diagnosis, and its frequency remains underestimated.
The development of high throughput sequencing became
a milestone in the search for diabetes-related mutations. Allowing
for simultaneous testing of an unlimited number of genes (i.e. of
all known genetic etiology in monogenic diabetes), the method
increased the mutation detection rate significantly (57). In our study, we detected genetic
variants causative of monogenic diabetes and hyperglycemia-related
syndromes in 33 out of 60 children (55%) with non-type 1 diabetes
mellitus. This frequency is considerably higher than that detected
by Sanger sequencing, which is usually restricted to the analysis
of several MODY-related genes and confirms approximately 15% of the
cases tested for MODY (58). The
higher mutation detection rate in our study is achieved by
increasing the number of genes tested and a thorough clinical
selection of patients with possible monogenic diabetes. In this
regard, one more advantage of WES should be mentioned: DNA
sequencing data may be easily stored for further analysis of newly
discovered candidate genes.
Ethnic differences play an important role in
determining the epidemiology of monogenic diabetes, especially of
MODY. Large population studies in European Caucasians showed a
general trend of increased HNF1A-MODY frequency in Northern
Europe, while GCK-MODY is prevalent in Southern European
populations (5). Here, we report
GCK-MODY in 19 and HNF1A-MODY in only 2 out of 27
MODY-positive Russian patients. These mutation rates appeared to be
closer to those in Southern European populations than to those in
Northern Europe residents. Our finding may indicate the
population-specific frequency MODY types in Russian patients. The
recently shown high prevalence of GCK-MODY cases among
Russian patients with diabetes in pregnancy supports this
suggestion (59). However, it
should be also considered that our study was performed on children
who developed diabetes before the age of 18 years. In the previous
observations, it was noticed that the relative proportion of
GCK-MODY is higher in cases ascertained through pediatric
clinics, in contrast to HNF1A-MODY, which predominates in
cases from adult clinics (58,60).
Thus, considering this information, our results are in good
accordance with those reported in Spain, Italy, France, Germany,
and the Czech Republic, where mostly pediatric cases were tested
(25,61). The prevalence of GCK
variants (57.6%) in our study suggests that genetic analysis in
Russian children with suspected monogenic diabetes may start with
testing for MODY2, which may not necessarily be performed by WES.
However, other cases amount to 42.4% and are linked to 9 different
genes, which attests to the efficiency of using WES for the search
of genetic causes of diabetes in non-GCK-MODY cases.
Our results show that the spectrum of monogenic
diabetes-related genetic variants in Russian children includes
missense and nonsense mutations, in-frame and frameshift deletions,
and a promoter mutation. Generally, these data do not contrast with
results obtained in other populations, which also demonstrated a
wide spectrum of mutations (62–64).
Among genetic variants detected in our study, 60.5% had already
been reported in diabetic patients and 39.5% were novel ones. On
the one hand, these results point towards a significant recurrent
variation within monogenic-diabetes-related genes. On the other
hand, they suggest that, in spite of the multitude of monogenic
diabetes studies, many variants still remain unidentified.
Identification of novel genetic variants as well as accumulating
data on previously known causes of monogenic diabetes is of high
importance, both for fundamental understanding of the disease
pathogenesis and for clinical practice.
Interpretation of genetic variants, especially novel
ones, may be challenging. In this study, only 63.2% of the detected
genetic variants (18 previously reported and 6 novel ones) were
unambiguously considered as causative of the diabetes in the
studied patients. The remaining 36.8% variants, including 9 novel
ones, were initially classified as those of uncertain significance
(n=12) or likely benign (n=2). Additional in silico
predictions performed for missense mutations among these variants
indicate that, with the exception of one variant, they all likely
have an adverse effect on protein stability. Considering these
results and the patients' phenotypes, the assumption that the
abovementioned variants may be causative of monogenic diabetes can
be made. Importantly, the detected genetic variants are absent in
non-diabetic Russian population resided in Northwest Russia
(65). However, to make a strong
conclusion on the pathogenic effect of each novel variant, more
data are required, including functional characterization and
reports of a specific genetic variant in multiple patients with
similar phenotypes. The latter highlights the importance of our
results for future studies of monogenic diabetes-related genetic
variants.
Noteworthy, our analysis of the clinical picture in
the patients simultaneously having BLK+GCK (patient
#529 and #662) and GCK+HNF1A (patient #226) genetic
variants suggests no accumulation of adverse effect: All these
patients had a typical MODY2 phenotype. The most plausible
explanation for this is the specific age of development of
different MODY types. Patients suffering from GCK-MODY have
an impaired glucose metabolism since birth (66). In contrast, carriers of
HNF1A genetic variants may develop diabetes by the age of 35
years or even by the age of 55 years, although most of them have
diabetes before 25 years of age (67). In the study by López-Garrido et
al (68), the co-inheritance
of GCK and HNF1A genetic variants was reported in two
patients and was associated with a typical MODY3 phenotype in an
adult patient and only impaired fasting glucose in a younger
patient with the same genotype. In addition, HNF1A genetic
variant detected in patient #226 in this study (c.92G>A) was
previously reported in a diabetic proband and his non-diabetic
sister of 43 years of age (69).
Similarly, affected carriers of BLK genetic variants usually
develop diabetes at the middle age (70). Thus, it is likely that patients
#529, #662, and #226, who were all involved in our study before the
age of 4 years, have not developed the clinical picture of
HNF1A-MODY and BLK-MODY yet. The possibility of a
late manifestation of HNF1A-MODY and BLK-MODY in the
children who already have GCK-MODY strongly suggests the
necessity of their strict medical supervision in order to timely
modify their therapy. Additional studies, including functional
ones, on the pathogenicity of the novel BLK genetic variants
detected in patients #529 and #662 will also facilitate the
development of the most effective treatment strategies for
them.
To summarize, our data show a high rate of genetic
variants causative of monogenic diabetes in Russian children with
non-type 1 diabetes mellitus. The use of a WES-based panel allowed
us to identify a variety of previously known and novel genetic
variants in MODY-related and unrelated genes, including multiple
variants in a number of patients. The revealed variety is
characterized by a prevalence of GCK genetic variants
(MODY2) and also includes variants in HNF1A, PAX4, KCNJ11, BLK,
ABCC8, GATA6, WFS1, EIF2AK3, and SLC19A2. These results,
on the one hand, suggest that genetic analysis for monogenic
diabetes in Russian children may start with testing for GCK
variants, which may not necessarily be performed by WES. On the
other hand, non-GCK variants are linked to 9 different
genes, which attests to the efficiency of using WES while searching
for genetic causes of diabetes in non-GCK-MODY cases.
Notably, the detection of genetic variants in the genes linked to
specific syndromes with recessive inheritance-WFS1, EIF2AK3,
and SLC19A2-is essential for appropriate genetic counseling
and family planning. Our study highlights the importance of using
WES for monogenic diabetes testing and provides new information on
the diabetes-related genetic variants in the Russian
population.
Acknowledgements
The authors would like to thank Mrs Ksenia
Khudadyan (Logrus LLC) for helpful advice during the preparation of
the manuscript.
Funding
The current study was supported by the Ministry of
Science and Higher Education of the Russian Federation within the
framework of the Basic Research Program in 2019–2021 (grant no.
AAAA-A19-119021290033-1), the alpha-Endo program, the Charities Aid
Foundation Foundation (grant nos. 65/315 and 133/315), and the
Russian Science Foundation (grant no. 14-50-00069).
Availability of data and materials
The datasets used and/or analyzed during the
current study are available from the corresponding author on
reasonable request.
Authors' contributions
OSG, EAS, MET, OAE, EBB and VSB designed the study.
OSG, EAS, MET, ASG, YAN, DEP, MAF, IVP, TEI, NYS, ESS, AVT, OVR,
AMS, AAP, SGS, EVM, AVPK, LRL, LVD, LAZ, LVT, OSB, ENS and EBB
recruited the patients and performed experimental procedures. YAB,
AVP and RKS performed bioinformatic analysis. OSG, EAS, MET, OAE,
AAP, TEI, LRL, EBB and VSB analyzed result and performed literature
search. OAE wrote the manuscript. All authors read and approved the
final manuscript.
Ethics approval and consent to
participate
The study was approved by the Ethics Committee of
D.O. Ott Research Institute of Obstetrics, Gynecology and
Reproductology. All the patients/patients' representatives gave
written informed consent to participate in the study. The study was
performed in accordance with the Declaration of Helsinki.
Patient consent for publication
All the patients/patients' representatives gave
written informed consent for publication of the study results.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Hattersley AT, Greeley SAW, Polak M,
Rubio-Cabezas O, Njølstad PR, Mlynarski W, Castano L, Carlsson A,
Raile K, Chi DV, et al: ISPAD clinical practice consensus
guidelines 2018: The diagnosis and management of monogenic diabetes
in children and adolescents. Pediatr Diabetes. 19 (Suppl
27):S47–S63. 2018. View Article : Google Scholar
|
|
2
|
Barbetti F and D'Annunzio G: Genetic
causes and treatment of neonatal diabetes and early childhood
diabetes. Best Pract Res Clin Endocrinol Metab. 32:575–591. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Lemelman MB, Letourneau L and Greeley SAW:
Neonatal diabetes mellitus: An update on diagnosis and management.
Clin Perinatol. 45:41–59. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Greeley SA, Naylor RN, Philipson LH and
Bell GI: Neonatal diabetes: An expanding list of genes allows for
improved diagnosis and treatment. Curr Diab Rep. 11:519–532. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kleinberger JW and Pollin TI: Undiagnosed
MODY: Time for Action. Curr Diab Rep. 15:1102015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
American Diabetes Association: 2.
Classification and diagnosis of diabetes: Standards of medical care
in diabetes-2018. Diabetes Care. 41 (Suppl 1):S13–S27. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
DePristo MA, Banks E, Poplin R, Garimella
KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA,
Hanna M, et al: A framework for variation discovery and genotyping
using next-generation DNA sequencing data. Nat Genet. 43:491–498.
2011. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Van der Auwera GA, Carneiro MO, Hartl C,
Poplin R, Del Angel G, Levy-Moonshine A, Jordan T, Shakir K, Roazen
D, Thibault J, et al: From FastQ data to high-confidence variant
calls: The genome analysis toolkit best practices pipeline. Curr
Protoc Bioinform. 43:11.10.1–33. 2013.
|
|
9
|
1000 Genomes Project Consortium, ; Auton
A, Brooks LD, Durbin RM, Garrison EP, Kang HM, Korbel JO, Marchini
JL, McCarthy S, McVean GA and Abecasis GR: A global reference for
human genetic variation. Nature. 526:68–74. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lek M, Karczewski KJ, Minikel EV, Samocha
KE, Banks E, Fennell T, O'Donnell-Luria AH, Ware JS, Hill AJ,
Cummings BB, et al: Analysis of protein-coding genetic variation in
60,706 humans. Nature. 536:285–291. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Fu W, O'Connor TD, Jun G, Kang HM,
Abecasis G, Leal SM, Gabriel S, Rieder MJ, Altshuler D, Shendure J,
et al: Analysis of 6,515 exomes reveals the recent origin of most
human protein-coding variants. Nature. 493:216–220. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Adzhubei IA, Schmidt S, Peshkin L,
Ramensky VE, Gerasimova A, Bork P, Kondrashov AS and Sunyaev SR: A
method and server for predicting damaging missense mutations. Nat
Methods. 7:248–249. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ng PC and Henikoff S: Predicting
deleterious amino acid substitutions. Genome Res. 11:863–874. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Choi Y and Chan AP: PROVEAN web server: A
tool to predict the functional effect of amino acid substitutions
and indels. Bioinformatics. 31:2745–2747. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Liu X, Jian X and Boerwinkle E: dbNSFP
v2.0: A database of human non-synonymous SNVs and their functional
predictions and annotations. Hum Mutat. 34:E2393–E2402. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Desmet FO, Hamroun D, Lalande M,
Collod-Béroud G, Claustres M and Béroud C: Human splicing finder:
An online bioinformatics tool to predict splicing signals. Nucleic
Acids Res. 37:e672009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhao H, Yang Y, Lin H, Zhang X, Mort M,
Cooper DN, Liu Y and Zhou Y: DDIG-in: Discriminating between
disease-associated and neutral non-frameshifting micro-indels.
Genome Biol. 14:R232013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Capriotti E, Fariselli P and Casadio R:
I-Mutant2.0: Predicting stability changes upon mutation from the
protein sequence or structure. Nucleic Acids Res. 33:W306–W310.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Barbitoff YA, Bezdvornykh IV, Polev DE,
Serebryakova EA, Glotov AS, Glotov OS and Predeus AV: Catching
hidden variation: Systematic correction of reference minor allele
annotation in clinical variant calling. Genet Med. 20:360–364.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Gasperíková D, Tribble ND, Staník J,
Hucková M, Misovicová N, van de Bunt M, Valentínová L, Barrow BA,
Barák L, Dobránsky R, et al: Identification of a novel beta-cell
glucokinase (GCK) promoter mutation (−71G>C) that modulates GCK
gene expression through loss of allele-specific Sp1 binding causing
mild fasting hyperglycemia in humans. Diabetes. 58:1929–1935. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Mantovani V, Salardi S, Cerreta V, Bastia
D, Cenci M, Ragni L, Zucchini S, Parente R and Cicognani A:
Identification of eight novel glucokinase mutations in Italian
children with maturity-onset diabetes of the young. Hum Mutat.
22:3382003. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Bacon S, Kyithar MP, Schmid J, Rizvi SR,
Bonner C, Graf R, Prehn JH and Byrne MM: Serum levels of pancreatic
stone protein (PSP)/reg1A as an indicator of beta-cell apoptosis
suggest an increased apoptosis rate in hepatocyte nuclear factor 1
alpha (HNF1A-MODY) carriers from the third decade of life onward.
BMC Endocr Disord. 12:132012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Gragnoli C, Cockburn BN, Chiaramonte F,
Gorini A, Marietti G, Marozzi G and Signorini AM: Early-onset Type
II diabetes mellitus in Italian families due to mutations in the
genes encoding hepatic nuclear factor 1 alpha and glucokinase.
Diabetologia. 44:1326–1329. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ziemssen F, Bellanné-Chantelot C,
Osterhoff M, Schatz H and Pfeiffer AF: To: Lindner T, cockburn BN,
Bell GI (1999) Molecular genetics of MODY in Germany. Diabetologia.
42:121–123, Diabetologia 45: 286–288. 2002.
|
|
25
|
Estalella I, Rica I, Perez de Nanclares G,
Bilbao JR, Vazquez JA, San Pedro JI, Busturia MA and Castaño L;
Spanish MODY Group, : Mutations in GCK and HNF-1alpha explain the
majority of cases with clinical diagnosis of MODY in Spain. Clin
Endocrinol (Oxf). 67:538–546. 2007.PubMed/NCBI
|
|
26
|
Stoffel M, Froguel P, Takeda J, Zouali H,
Vionnet N, Nishi S, Weber IT, Harrison RW, Pilkis SJ, Lesage S, et
al: Human glucokinase gene: Isolation, characterization, and
identification of two missense mutations linked to early-onset
non-insulin-dependent (type 2) diabetes mellitus. Proc Natl Acad
Sci USA. 89:7698–7702. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Hattersley AT, Beards F, Ballantyne E,
Appleton M, Harvey R and Ellard S: Mutations in the glucokinase
gene of the fetus result in reduced birth weight. Nat Genet.
19:268–270. 1998. View
Article : Google Scholar : PubMed/NCBI
|
|
28
|
Froguel P, Zouali H, Vionnet N, Velho G,
Vaxillaire M, Sun F, Lesage S, Stoffel M, Takeda J, Passa P, et al:
Familial hyperglycemia due to mutations in glucokinase. Definition
of a subtype of diabetes mellitus. N Engl J Med. 328:697–702. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Borowiec M, Fendler W, Antosik K,
Baranowska A, Gnys P, Zmyslowska A, Malecki M and Mlynarski W:
Doubling the referral rate of monogenic diabetes through a
nationwide information campaign-update on glucokinase gene
mutations in a Polish cohort. Clin Genet. 82:587–590. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Pruhova S, Ek J, Lebl J, Sumnik Z, Saudek
F, Andel M, Pedersen O and Hansen T: Genetic epidemiology of MODY
in the Czech republic: New mutations in the MODY genes HNF-4alpha,
GCK and HNF-1alpha. Diabetologia. 46:291–295. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Gidh-Jain M, Takeda J, Xu LZ, Lange AJ,
Vionnet N, Stoffel M, Froguel P, Velho G, Sun F, Cohen D, et al:
Glucokinase mutations associated with non-insulin-dependent (type
2) diabetes mellitus have decreased enzymatic activity:
Implications for structure/function relationships. Proc Natl Acad
Sci USA. 90:1932–1936. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Colclough K, Bellanne-Chantelot C,
Saint-Martin C, Flanagan SE and Ellard S: Mutations in the genes
encoding the transcription factors hepatocyte nuclear factor 1
alpha and 4 alpha in maturity-onset diabetes of the young and
hyperinsulinemic hypoglycemia. Hum Mutat. 34:669–685. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Plengvidhya N, Kooptiwut S, Songtawee N,
Doi A, Furuta H, Nishi M, Nanjo K, Tantibhedhyangkul W,
Boonyasrisawat W, Yenchitsomanus PT, et al: PAX4 mutations in Thais
with maturity onset diabetes of the young. J Clin Endocrinol Metab.
92:2821–2826. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Flanagan SE, Patch AM, Mackay DJ, Edghill
EL, Gloyn AL, Robinson D, Shield JP, Temple K, Ellard S and
Hattersley AT: Mutations in ATP-sensitive K+ channel
genes cause transient neonatal diabetes and permanent diabetes in
childhood or adulthood. Diabetes. 56:1930–1937. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Mohnike K, Wieland I, Barthlen W,
Vogelgesang S, Empting S, Mohnike W, Meissner T and Zenker M:
Clinical and genetic evaluation of patients with KATP channel
mutations from the German registry for congenital hyperinsulinism.
Horm Res Paediatr. 81:156–168. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Suzuki S, Nakao A, Sarhat AR, Furuya A,
Matsuo K, Tanahashi Y, Kajino H and Azuma H: A case of pancreatic
agenesis and congenital heart defects with a novel GATA6 nonsense
mutation: Evidence of haploinsufficiency due to nonsense-mediated
mRNA decay. Am J Med Genet A. 164A:476–479. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Gomez-Zaera M, Strom T, Meitinger T and
Nunes V: Wolframin mutations in Spanish families with Wolfram
syndrome. Meeting abstract. Am J Hum Genet. 65:16731999.
|
|
38
|
Smith CJ, Crock PA, King BR, Meldrum CJ
and Scott RJ: Phenotype-genotype correlations in a series of
wolfram syndrome families. Diabetes Care. 27:2003–2009. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Chèvre JC, Hani EH, Boutin P, Vaxillaire
M, Blanché H, Vionnet N, Pardini VC, Timsit J, Larger E,
Charpentier G, et al: Mutation screening in 18 Caucasian families
suggest the existence of other MODY genes. Diabetologia.
41:1017–1023. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Awata T, Inoue K, Kurihara S, Ohkubo T,
Inoue I, Abe T, Takino H, Kanazawa Y and Katayama S: Missense
variations of the gene responsible for Wolfram syndrome
(WFS1/wolframin) in Japanese: Possible contribution of the
Arg456His mutation to type 1 diabetes as a nonautoimmune genetic
basis. Biochem Biophys Res Commun. 268:612–616. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Richards S, Aziz N, Bale S, Bick D, Das S,
Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al:
Standards and guidelines for the interpretation of sequence
variants: A joint consensus recommendation of the American College
of Medical Genetics and Genomics and the Association for Molecular
Pathology. Genet Med. 17:405–424. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Shaw-Smith C, Flanagan SE, Patch AM,
Grulich-Henn J, Habeb AM, Hussain K, Pomahacova R, Matyka K,
Abdullah M, Hattersley AT and Ellard S: Recessive SLC19A2 mutations
are a cause of neonatal diabetes mellitus in thiamine-responsive
megaloblastic anaemia. Pediatr Diabetes. 13:314–321. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Fraser FC and Gunn T: Diabetes mellitus,
diabetes insipidus, and optic atrophy. An autosomal recessive
syndrome? J Med Genet. 14:190–193. 1977. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Bennett K, James C, Mutair A, Al-Shaikh H,
Sinani A and Hussain K: Four novel cases of permanent neonatal
diabetes mellitus caused by homozygous mutations in the glucokinase
gene. Pediatr Diabetes. 12:P192–P196. 2011. View Article : Google Scholar
|
|
45
|
Raimondo A, Chakera AJ, Thomsen SK,
Colclough K, Barrett A, De Franco E, Chatelas A, Demirbilek H,
Akcay T, Alawneh H, et al: Phenotypic severity of homozygous GCK
mutations causing neonatal or childhood-onset diabetes is primarily
mediated through effects on protein stability. Hum Mol Genet.
23:6432–6440. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Rubio-Cabezas O, Patch AM, Minton JA,
Flanagan SE, Edghill EL, Hussain K, Balafrej A, Deeb A, Buchanan
CR, Jefferson IG, et al: Wolcott-Rallison syndrome is the most
common genetic cause of permanent neonatal diabetes in
consanguineous families. J Clin Endocrinol Metab. 94:4162–4170.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Labay V, Raz T, Baron D, Mandel H,
Williams H, Barrett T, Szargel R, McDonald L, Shalata A, Nosaka K,
et al: Mutations in SLC19A2 cause thiamine-responsive megaloblastic
anaemia associated with diabetes mellitus and deafness. Nat Genet.
22:300–304. 1999. View
Article : Google Scholar : PubMed/NCBI
|
|
48
|
Tattersall RB: Mild familial diabetes with
dominant inheritance. Q J Med. 43:339–357. 1974.PubMed/NCBI
|
|
49
|
Tattersall RB and Fajans SS: A difference
between the inheritance of classical juvenile-onset and
maturity-onset type diabetes of young people. Diabetes. 24:44–53.
1975. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Yamagata K, Furuta H, Oda N, Kaisaki PJ,
Menzel S, Cox NJ, Fajans SS, Signorini S, Stoffel M and Bell GI:
Mutations in the hepatocyte nuclear factor-4alpha gene in
maturity-onset diabetes of the young (MODY1). Nature. 384:458–460.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Yamagata K, Oda N, Kaisaki PJ, Menzel S,
Furuta H, Vaxillaire M, Southam L, Cox RD, Lathrop GM, Boriraj VV,
et al: Mutations in the hepatocyte nuclear factor-1alpha gene in
maturity-onset diabetes of the young (MODY3). Nature. 384:455–458.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Horikawa Y, Iwasaki N, Hara M, Furuta H,
Hinokio Y, Cockburn BN, Lindner T, Yamagata K, Ogata M, Tomonaga O,
et al: Mutation in hepatocyte nuclear factor-1 beta gene (TCF2)
associated with MODY. Nat Genet. 17:384–385. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Stoffers DA, Ferrer J, Clarke WL and
Habener JF: Early-onset type-II diabetes mellitus (MODY4) linked to
IPF1. Nat Genet. 17:138–139. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Froguel P, Vaxillaire M, Sun F, Velho G,
Zouali H, Butel MO, Lesage S, Vionnet N, Clément K, Fougerousse F,
et al: Close linkage of glucokinase locus on chromosome 7p to
early-onset non-insulin-dependent diabetes mellitus. Nature.
356:162–164. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Hattersley AT, Turner RC, Permutt MA,
Patel P, Tanizawa Y, Chiu KC, O'Rahilly S, Watkins PJ and Wainscoat
JS: Linkage of type 2 diabetes to the glucokinase gene. Lancet.
339:1307–1310. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Timsit J, Saint-Martin C, Dubois-Laforgue
D and Bellanné-Chantelot C: Searching for Maturity-Onset diabetes
of the Young (MODY): When and What for? Can J Diabetes. 40:455–461.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Ellard S, Lango Allen H, De Franco E,
Flanagan SE, Hysenaj G, Colclough K, Houghton JA, Shepherd M,
Hattersley AT, Weedon MN and Caswell R: Improved genetic testing
for monogenic diabetes using targeted next-generation sequencing.
Diabetologia. 56:1958–1963. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Shields BM, Hicks S, Shepherd MH,
Colclough K, Hattersley AT and Ellard S: Maturity-onset diabetes of
the young (MODY): How many cases are we missing? Diabetologia.
53:2504–2508. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Zubkova N, Burumkulova F, Plechanova M,
Petrukhin V, Petrov V, Vasilyev E, Panov A, Sorkina E, Ulyatovskaya
V, Makretskaya N and Tiulpakov A: High frequency of pathogenic and
rare sequence variants in diabetes-related genes among Russian
patients with diabetes in pregnancy. Acta Diabetol. 56:413–420.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Lorini R, Klersy C, d'Annunzio G, Massa O,
Minuto N, Iafusco D, Bellannè-Chantelot C, Frongia AP, Toni S,
Meschi F, et al: Maturity-onset diabetes of the young in children
with incidental hyperglycemia: A multicenter Italian study of 172
families. Diabetes Care. 32:1864–1866. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Schober E, Rami B, Grabert M, Thon A,
Kapellen T, Reinehr T and Holl RW: Phenotypical aspects of
maturity-onset diabetes of the young (MODY diabetes) in comparison
with Type 2 diabetes mellitus (T2DM) in children and adolescents:
Experience from a large multicentre database. Diabet Med.
26:466–473. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Ağladıoğlu SY, Aycan Z, Çetinkaya S, Baş
VN, Önder A, Peltek Kendirci HN, Doğan H and Ceylaner S: Maturity
onset diabetes of youth (MODY) in Turkish children: Sequence
analysis of 11 causative genes by next generation sequencing. J
Pediatr Endocrinol Metab. 29:487–496. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Globa E, Zelinska N, Elblova L, Dusatkova
P, Cinek O, Lebl J, Colclough K, Ellard S and Pruhova S: MODY in
Ukraine: Genes, clinical phenotypes and treatment. J Pediatr
Endocrinol Metab. 30:1095–1103. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Johansson BB, Irgens HU, Molnes J,
Sztromwasser P, Aukrust I, Juliusson PB, Søvik O, Levy S,
Skrivarhaug T, Joner G, et al: Targeted next-generation sequencing
reveals MODY in up to 6.5% of antibody-negative diabetes cases
listed in the Norwegian Childhood Diabetes Registry. Diabetologia.
60:625–635. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Barbitoff YA, Skitchenko RK, Poleshchuk
OI, Shikov AE, Serebryakova EA, Nasykhova YA, Polev DE, Shuvalova
AR, Shcherbakova IV, Fedyakov MA, et al: Whole-exome sequencing
provides insights into monogenic disease prevalence in Northwest
Russia. Mol Genet Genomic Med. Sep 3;e9642019.doi: 10.1002/mgg3.964
(Epub ahead of print). PubMed/NCBI
|
|
66
|
Chakera AJ, Steele AM, Gloyn AL, Shepherd
MH, Shields B, Ellard S and Hattersley AT: Recognition and
management of individuals with hyperglycemia because of a
heterozygous glucokinase mutation. Diabetes Care. 38:1383–1392.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Murphy R, Ellard S and Hattersley AT:
Clinical implications of a molecular genetic classification of
monogenic beta-cell diabetes. Nat Clin Pract Endocrinol Metab.
4:200–213. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
López-Garrido MP, Herranz-Antolín S,
Alija-Merillas MJ, Giralt P and Escribano J: Co-inheritance of
HNF1a and GCK mutations in a family with maturity-onset diabetes of
the young (MODY): Implications for genetic testing. Clin Endocrinol
(Oxf). 79:342–347. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Thanabalasingham G, Pal A, Selwood MP,
Dudley C, Fisher K, Bingley PJ, Ellard S, Farmer AJ, McCarthy MI
and Owen KR: Systematic assessment of etiology in adults with a
clinical diagnosis of young-onset type 2 diabetes is a successful
strategy for identifying maturity-onset diabetes of the young.
Diabetes Care. 35:1206–1212. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Borowiec M, Liew CW, Thompson R,
Boonyasrisawat W, Hu J, Mlynarski WM, El Khattabi I, Kim SH,
Marselli L, Rich SS, et al: Mutations at the BLK locus linked to
maturity onset diabetes of the young and beta-cell dysfunction.
Proc Natl Acad Sci USA. 106:14460–14465. 2009. View Article : Google Scholar : PubMed/NCBI
|