Introduction
Disorders of sex development (DSDs) are congenital
medical conditions in which there is no correlation between the
chromosomal, gonadal, and phenotypic characteristics (1). The term DSDs encompasses a broad
clinical spectrum that can be diagnosed at a range of ages, from
the neonatal period to late adulthood, the latter of which is often
the case with infertility. This condition varies clinically from
genital ambiguity to attenuated forms, such as mild hypospadias or
unilateral cryptorchidism; this makes it difficult to classify
patients with similar or almost identical phenotypes based on
different etiologies and molecular processes (2,3). The
Lawson Wilkins Pediatric Endocrine Society (LWPES) and the Chicago
Consensus in 2006 classified DSDs into three distinct groups: i)
DSD 46,XY; ii) DSD 46, and iii) XX DSD, with a certain degree of
overlap between the groups (4,5).
Although truly ambiguous genitalia are relatively
rare, their prevalence is estimated to be approximately 1 in
4,500-5,500 worldwide (6).
However, if we take into account all congenital genital anomalies,
including cryptorchidism and hypospadias, then the prevalence
ranges from 1:200 to 1:300 (7). In
Colombia, the prevalence is 1.7 per 10,000 births (8).
Sex-specific gonadal development starts with
formation of the bipotential gonad, which then differentiates into
either testicular or ovarian tissue. This process is dependent on
activation of either the testis- or ovary-specific pathway, with
parallel repression of the opposite pathway.
For the activation of these sexual differentiation
pathways to occur, the transcription factors that regulate the
expression of tissue-specific genes and signaling molecules must be
expressed (4). Alterations in the
molecular processes of these signaling pathways lead to the
development of DSDs.
The activation of the male sexual differentiation
cascade in mammals depends on the expression of certain genes, such
as SRY (Y sex-determining region), NR5A1, SOX9, and
DAX1 (in hemicigosis), among others (9); whereas in the activation of the
female differentiation cascade, genes including WNT4, DAX1
(in homozygosis), and RSPO1 can intervene (10). Given the importance of the correct
regulation of these genes, mutations in unidentified enhancer
sequences and in the coding regions of these genes, as well as
other anomalies, such as total loss of the gene, are important
factors that can contribute to the onset of DSDs in humans
(11). However, despite advances
in their molecular diagnosis, the etiology of DSDs has been
established in less than 20% of cases (12–14),
due to both the complexity of the signaling cascades that determine
sexual differentiation and the technical limitations of etiological
studies.
The etiological diagnosis of DSDs generally requires
a broad spectrum of endocrinological tests, radiological images,
and genetic tests. Karyotyping is an initial test that allows for
the classification of the disorder, according to Lawson Wilkins,
2006 (3,15). Providing patients with DSDs with a
molecular diagnosis allows for the establishment of clinical
management techniques, including providing patients with
information regarding the risks of neoplasia associated with some
types of DSDs (16), as well as
offering advice to the patients and their family members regarding
recurrence risks.
In Colombia, as well as on a global scale, there are
few cytogenetic and molecular studies on patients with DSDs that
include stepwise analyses using different genetic techniques.
Characterizing the chromosomal and/or molecular alterations and
analyzing their contribution to the phenotypes of patients with
DSDs could improve our understanding of the causes of these
clinical conditions, thus helping in improving diagnosis techniques
to allow for specific treatments and medical advice to be given to
patients and their families. In this study, we present the
cytogenetic and molecular results for 43 Colombian patients with
non-syndromic DSDs. The genetic analyses included karyotyping, FISH
for SRY, and evaluation of copy number variation in SF1,
DAX1, SOX9, SRY, and WNT4 genes in the blood and gonadal
tissue. Using this multistep experimental approach, a diagnostic
percentage of 25.58% was obtained, which highlights the importance
of the histological and cytogenetic study of gonadal biopsies. Our
protocol contributed to the diagnosis of 11 out of the 43 patients,
for whom the etiology or histological findings allowed for the
definition of medical management techniques.
Materials and methods
Patients
A cohort of 43 individuals with non-syndromic DSDs,
aged between 2 days and 49 years, were evaluated by the
transdisciplinary board of the Hospital San Ignacio, Bogotá
(Colombia). The team consisted of pediatric urologists, pediatric
endocrinologists, psychiatrists, clinical geneticists, and
cytogeneticists. The cohort consisted of patients with genital
ambiguity or discordance between chromosomal, gonadal, and/or
phenotypic sex, or genital abnormalities, such as cryptorchidism
and hypospadias. Each patient underwent classical cytogenetic
analysis, FISH (fluorescent in situ hybridization), and the
molecular tests described below.
All evaluations were performed using a step-by-step
clinical diagnostic approach. In addition, we provided
psychological counseling and advice on the risk of recurrence to
both the patients and their families. The genetic analyses included
karyotyping, FISH for SRY, and evaluation of the copy number
variation in the SF1, DAX1, SOX9, SRY, and WNT4 genes
by MLPA in the blood and gonadal tissue. According to the suspected
diagnosis, direct sequencing of specific genes was also performed
in some individuals.
Declaration of ethics
This research protocol was approved by the Ethics
Committee of the Hospital Universitario San Ignacio (FM-CIE-8540)
and the School of Medicine of the Pontificia Universidad Javeriana.
Patients and their guardians or family members were only included
in the study after they had received information regarding the
study and signed an informed consent form.
Cytogenetic analysis
Cytogenetic analysis was carried out on all
individuals using phytohemagglutinin-stimulated peripheral blood
lymphocyte cultures of patients and their parents, according to
standard laboratory protocols (17). Gonadal karyotyping was only
performed under the treating physician's order. For this, a
fragment of the biopsy was processed in culture with RPMI and 20%
SFB for cell growth. Chromosome preparations were treated with HCl
and stained with Wright for G-banding. A total of 50 metaphase
cells were analyzed at the 550-band resolution level. Molecular
cytogenetics using fluorescence in situ hybridization (FISH)
was performed with a probe specific for the SRY gene (SRY
Probe, Cytocell Aquarius). The probe mix was obtained according the
manufacturer's standard protocol and contained the following: A SRY
probe, labelled in red; a control probe for the X centromere
(DXZ1), labelled in blue; a control probe for chromosome Y (DYZ1,
the heterochromatic block at Yq12) labelled in green; and a
centromeric probe for the Y-chromosome (DYZ3 α-satellite; Cytocell
Aquarius).
Multiplex ligation-dependent probe
amplification (MLPA)
The deletions and duplications of the SOX9,
DAX-1, SF-1, SRY, and WNT4 genes were studied using MLPA
SALSA P185-B2 Intersex (version 08; May 07, 2015) (MRC Holland),
following the manufacturer's instructions. Information on probe
sequences can be freely accessed on the MRC Holland website
(www.mlpa.com).
After 35 cycles of PCR amplification, the PCR
products were separated using an ABI 3100 genetic analyzer. Row
data was analyzed using Coffalyser software (MRC
Holland®). For each sample, the peak areas corresponding
to each probe were normalized to the average of the peak areas in
the three controls. DNA samples showing a reduction or increase in
the MLPA peak area values were reanalyzed using the same MLPA
procedure, and only the samples showing consistent results between
the two experiment replicates were considered positive for copy
number alteration.
Sequencing
The identification of point mutations in the DNA of
the AR and AMH genes was performed using PCR followed
by direct sequencing. Sanger sequencing was performed using the
BigDye Terminator v1.1 cycle sequencing kit (Thermo Fisher
Scientific, Inc.) in a 3500×l genetic analyzer (Thermo Fisher
Scientific, Inc.). The direct and inverse sequences were analyzed
and compared with each gene's mRNA reference sequence: AR
(NM_000044.2) and AMH (NM_000479).
Histological study in gonadal
tissue
Based on the recommendations made by the
multidisciplinary board, a gonadal biopsy (or gonadectomy) was
performed on patients with non-concordant cytogenetic findings, an
altered gonadal hormonal study, or whose gonads could not be
visualized in diagnostic images. In total, gonadal biopsy samples
were obtained from 12 patients.
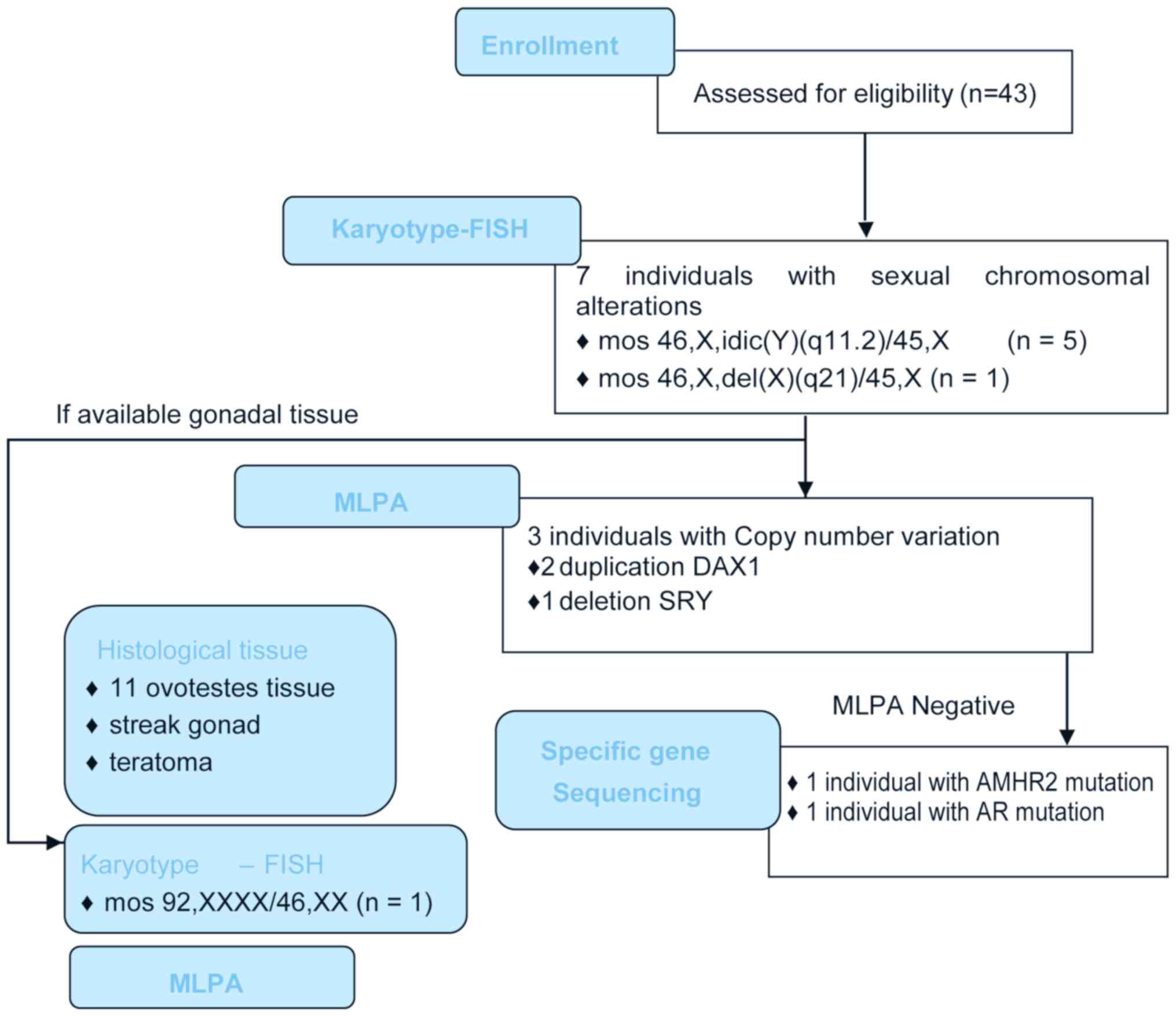
Results
Classification of patients with
DSD
Cytogenetic, clinical, and molecular techniques were
used to classify each patient by syndromic, etiological, and
cytogenetic factors (Table I;
Fig. 1). Seven patients had a
46,XX karyotype, 30 patients had a 46,XY karyotype, and 6 had a
karyotype with sex chromosome aneuploidies that included numerical
and/or structural anomalies. The detection of the absence or
presence of the SRY gene in the XY and XX individuals,
respectively, did not identify any alterations in relation to the
chromosomal complement. Based on additional clinical investigations
of the 43 patients, 17 patients (39.53%) were classified as having
gonadal development disorders, most of which were ovotesticular
disorders with numerical and/or structural alterations of the sex
chromosomes; 9 patients (20.93%) were classified as having
testicular DSDs with a 46,XY karyotype; 3 patients (6.97%) were
classified as having ovarian DSDs with a 46,XX karyotype, and 14
patients (32.55%) individuals were classified as ‘others’ since
they could not grouped into a specific class of gonadal
development, corresponding to hypospadias and multiple congenital
anomalies.
 | Table I.Syndromic classification according to
cytogenetic and molecular studies of patients with DSD. |
Table I.
Syndromic classification according to
cytogenetic and molecular studies of patients with DSD.
|
|
|
|
|
|
| Molecular
finding |
|
|---|
|
|
|
|
|
|
|
|
|
|---|
| Syndromic
diagnosis | Etiologic
diagnosis | Karytoype | Case | Assigned sex | MLPA | Sequence | Cases, n |
|---|
| Disorders of
gonadal development | Ovotesticular
DSD | Numerical
aneuploidy | 46,XX.ish
Yp11.31(SRY-) | 1a | M | Negative | Normal SRY
Negative for pathogenic variants | 1 |
|
|
| Y chromosome
anomalies | mos
45,X[4]/46,XY[96]. ish idic(Y)(SRY++,DYZ1-) | 2a | M | Negative | It was not
performed | 4 |
|
|
|
| mos
46,X,idic(Y)(q11.2)[42]/45, X[8].ish idic(Y)(q11.2)(SRY++,
DYZ3++,DYZ1-) | 3a | M | It was not
performed | It was not
performed |
|
|
|
|
| mos
46,X,idic(Y)(q11.2)[23]/47,X,idic(Y) (q11.2),+idic(Y) (q11.2)[8]/
45,X[19] idic(Y) (q11.2)(SRY++,DYZ3++,DYZ1-) | 4 | M | It was not
performed | It was not
performed |
|
|
|
|
| mos
45,X[15]/46,XY[35].ish Yp11.31 (SRY-) mos 46,X,idic(Y)(q11.2)
[241]/45,X[8].ish idic(Y)(q11.2) (SRY++,DYZ3++,DYZ1-) | 5a | F | It was not
performed | Normal SRY
Negative for pathogenic variants |
|
|
|
| X chromosome
anomalies | mos
46,X,del(X)(q21)[71]/45,X[29]. ish Yp11.31(SRY-) | 6 | F | It was not
performed | It was not
performed | 1 |
|
|
|
| 46,XX.ish
Yp11.31(SRY-) | 7 | F | Negative | It was not
performed | 1 |
|
|
|
| 46,XX.ish
Yp11.31(SRY-) | 8 | F | Negative | It was not
performed | 1 |
|
|
|
| 46,XY.ish
Yp11.31(SRY+) | 9 | M | Negative | It was not
performed | 1 |
|
|
|
| 46,XY.ish
Yp11.31(SRY+) | 10a | F | Negative | It was not
performed | 1 |
|
|
|
| 46,XY.ish
Yp11.31(SRY+) | 11 | M | Negative | It was not
performed | 1 |
|
|
| Total |
|
|
|
|
| 11 |
|
| Testicular
regression syndrome |
| 46,XY | 12 | F | Negative | Normal AR
Negative for pathogenic variants | 1 |
|
| Streak gonad |
| 46,XY.ish
Yp11.31(SRY+) | 13a | F | Negative | It was not
performed | 1 |
|
| Complete gonadal
dysgenesis | Duplication
DAX1 | 46,XY.ish
Yp11.31(SRY+) | 14a | F | rsa DAX1 (SALSA
P185-B2 Intersex)×3 | Normal SRY
Negative for pathogenic variants | 1 |
|
|
|
| 46,XY.ish
Yp11.31(SRY+) | 15a | F | rsa DAX1 (SALSA
P185-B2 Intersex)×3 | It was not
performed | 1 |
|
| SRY | Deletion?SRY (SALSA
P185-B2 Intersex)×1? | 46,XY.ish
Yp11.31(SRY+) | 16 | F | rsa SRY | It was not
performed | 1 |
|
| Mixed gonadal
dysgenesis |
|
46,X,idic(Y)(q11.2)[38]/45,X[12]. ish
idic(Y) (SRYx2,DYZ1×0) | 17a | M | It was not
performed | It was not
performed | 1 |
|
| Total |
|
|
|
|
|
| 17 |
|
| Androgen |
| 46,XY.ish
Yp11.31(SRY+) | 18 | F | Negative | It was not
performed | 1 |
|
| Insensitivity
partial |
| 46,XY.ish
Yp11.31(SRY+) | 19a | M | Negative | It was not
performed | 1 |
| 46,XY testicular
DSD | Androgen
insensitivity total |
| 46,XY.ish
Yp11.31(SRY+) | 20 | F | Negative | AR c.231-239
delgcafcagca (p.gln78_gln80del) homozygous | 1 |
|
| Muller duct |
| 46,XY.ish
Yp11.31(SRY+) | 21 | M | It was not
performed | AMHR2
c.916delC (p.Leu306Cysfs*29) | 1 |
|
| idiopathic |
| 46,XY.ish
Yp11.31(SRY+) | 22a | F | Negative | It was not
performed | 1 |
|
|
|
| 46,XY.ish
Yp11.31(SRY+) | 23a | F | Negative | It was not
performed | 1 |
|
|
|
| 46,XY.ish
Yp11.31(SRY+) | 24 | M | Negative | It was not
performed | 1 |
|
|
|
| 46,XY.ish
Yp11.31(SRY+) | 25 | M | Negative | It was not
performed | 1 |
|
|
|
| 46,XY.ish
Yp11.31(SRY+) | 26 | F | Negative | It was not
performed | 1 |
|
| Total |
|
|
|
|
|
| 9 |
|
| Congenital adrenal
hyperplasia |
| 46,XX.ish
Yp11.31(SRY-) | 27 | F | Negative | It was not
performed | 1 |
| 46,XX | Idiopathic |
| 46,XY.ish
Yp11.31(SRY+) | 28 | M | Negative | It was not
performed | 1 |
| ovarian DSD |
|
| 46,XX.ish
Yp11.31(SRY-) | 29 | M | Negative | It was not
performed | 1 |
|
| Total |
|
|
|
|
|
| 3 |
| Others | Hipospadias |
| 46,XY.ish
Yp11.31(SRY+) | 30 | M | It was not
performed | It was not
performed | 1 |
|
|
|
| 46,XY.ish
Yp11.31(SRY+) | 31 | M | Negative | It was not
performed | 1 |
|
|
|
| 46,XY.ish
Yp11.31(SRY+) | 32 | M | Negative | It was not
performed | 1 |
|
|
|
| 46,XY.ish
Yp11.31(SRY+) | 33 | M | Negative | It was not
performed | 1 |
|
|
|
| 46,XY.ish
Yp11.31(SRY+) | 34 | M | Negative | It was not
performed | 1 |
|
|
|
| 46,XY | 35 | M | Negative | It was not
performed | 1 |
|
|
|
| 46,XY.ish
Yp11.31(SRY+) | 36 | M | It was not
performed | It was not
performed | 1 |
|
|
|
| 46,XY | 37 | M | Negative | It was not
performed | 1 |
|
|
|
| 46,XY.ish
Yp11.31(SRY+) | 38 | M | Negative | It was not
performed | 1 |
|
|
|
| 46,XY.ish
Yp11.31(SRY+) | 39 | M | Negative | It was not
performed | 1 |
|
|
|
| 46,XX.ish
Yp11.31(SRY-) | 40 | M | Negative | It was not
performed | 1 |
|
|
|
| 46,XX.ish
Yp11.31(SRY-) | 41 | M | Negative | It was not
performed | 1 |
|
| Total |
|
|
|
|
|
| 12 |
|
| Multiple |
| 46,XY.ish
Yp11.31(SRY+) | 42 | M | Negative | It was not
performed | 1 |
|
| malformations |
| 46,XY.ish
Yp11.31(SRY+) | 43 | M | Negative | It was not
performed | 1 |
|
| Total |
|
|
|
|
|
| 2 |
| Total |
|
|
|
|
|
|
| 43 |
Disorders of gonadal development
The histological findings indicated that 11
individuals with gonadal development alterations had ovotesticular
gonads (Fig. 1). Mosaicism was
identified in one individual with a male phenotype (case 1) by the
presence of 92,XXXX/46,XX tetraploidy in the gonadal tissue
(Fig. 2A). The presence of an
isodicentric Y chromosome was detected in 4 individuals (3 males
and 1 female: cases 2, 3, 4, and 5). Mosaicism with X chromosome
structural and numerical anomalies One was found in an individual
with the female phenotype (46,X,del(X) (q21) [71]/45,X[29].ish
Yp11.31 (SRY-) (case 6). Five individuals with ovotestis had XY or
XX chromosomal complement without alterations (cases 7, 8, 9, 10,
and 11). Two individuals had gonadal strip and testicular
regression syndrome with a 46,XY karyotype (cases 12 and 13). Two
sister patients with a female phenotype had a 46,XY karyotype with
duplication of DAX1 (Fig.
2B), and who had been diagnosed with teratoma at an early age
(cases 14 and 15). An adult patient with primary amenorrhea with a
46,XY karyotype had an alteration of SRY, as evidenced by
MLPA (case 16). Finally, one individual with a male sex phenotype
and a mosaic karyotype 46,X,idic(Y)(q11.2)[38]/45,X[12].ish idic(Y)
(SRYx2,DYZ1×0) (case 17) (Fig.
2C).
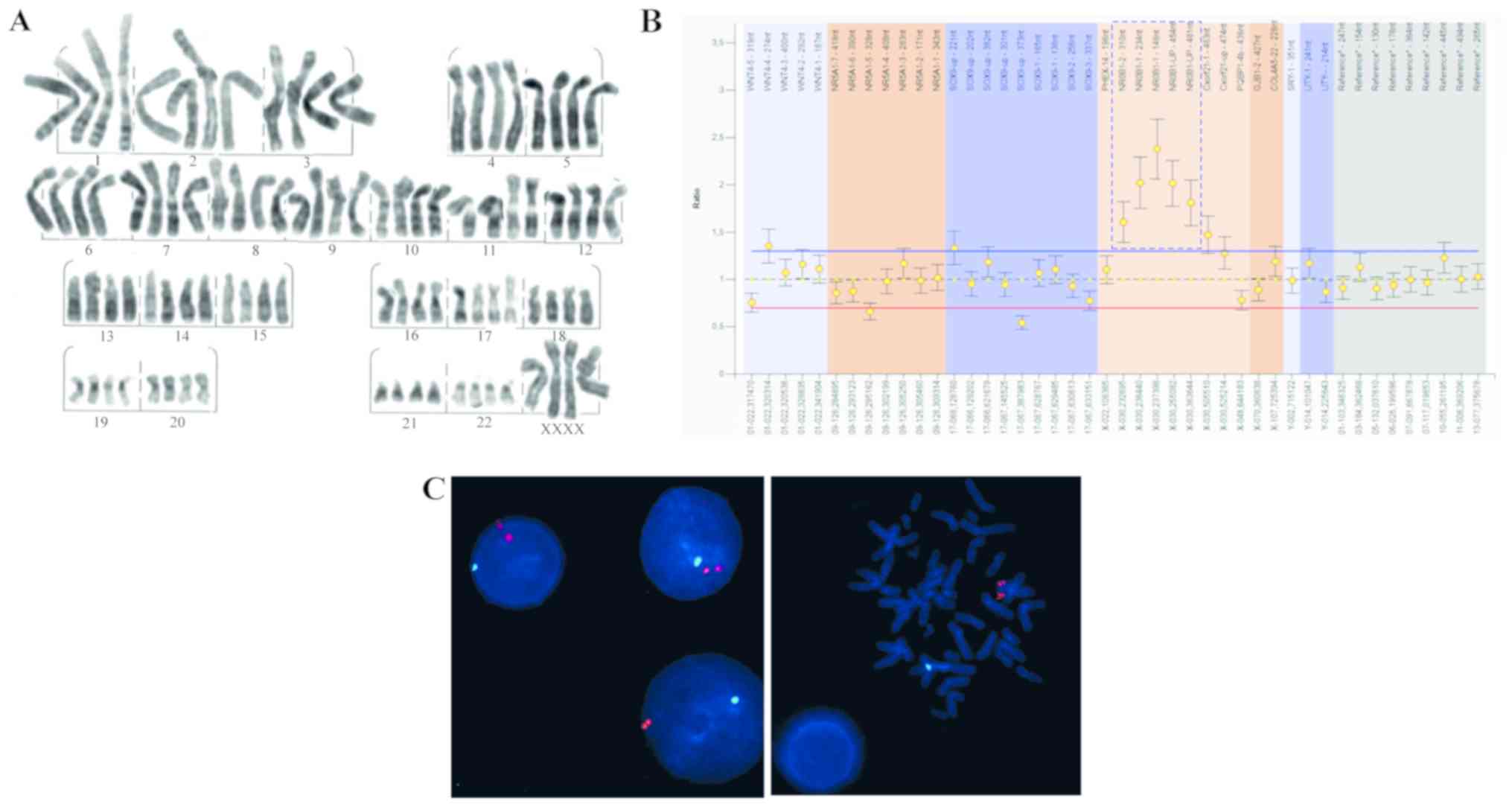 | Figure 2.Graphical representation of results
obtained through the proposed approach. (A) High resolution
G-banding karyotype, 96,XXXX. Magnification, ×100. (B) Result of
the multiplex ligation-dependent probe amplification test showing
duplication of the DAX1, rsa DAX1(SALSA P185-B2 Intersex) ×3 gene.
(C) Fluorescence in situ hybridization for SRY in
interphase nuclei (left) and metaphase chromosomes (right) of a
patient exhibiting double signal for SRY (red). This probe
contained a blue probe that recognizes the DXZ1 region of the
centromere of the X, mos 45,X[4]/46,XY[96]
idic?(Y)(q11.2)(SRY++,DYZ1-). Magnification, ×100. DAX1,
dosage-sensitive sex reversal; SRY, sex determining region Y. |
46,XY testicular DSD
46,XY testicular DSD was diagnosed in two
individuals with partial androgen insensitivity by biochemistry,
without molecular confirmation (cases 18 and 19), as well as in a
male subject with total androgen insensitivity, with molecular
confirmation of a homozygous pathogenic variant not previously
reported c.231-239delgcafcagca (p.gln78_gln80del) in the AR
gene (case 20). An infertile male phenotype individual was also
diagnosed, in whom the persistence of Mullerian derivatives was
documented with molecular confirmation of a homozygous pathogenic
variant not previously reported c.916delC (p.Leu306Cysfs*29) in the
AMHR2 gene (case 21). On the other hand, for 5 of the 9
patients diagnosed with 46,XY testicular DSD, no genetic
alterations were found using the proposed approach (cases 22, 23,
24, 25, and 26).
46,XX ovarian DSD
46,XX ovarian DSD was diagnosed in a male patient
with atypical congenital adrenal hyperplasia using their
biochemical profile, without molecular confirmation (case 28). Two
twin brothers with assigned male sex were also diagnosed with 46,XX
ovarian DSD, with 46,XX and 46,XY karyotypes, but did not show any
molecular alterations using the proposed approach.
Other
We did not find any genetic alterations in 14
patients with hypospadias and multiple congenital anomalies (cases
27–43).
The step-by-step approach with which genetic
alterations were found in 11 of the 43 patients (25.58%) is shown
in Fig. 1.
Discussion
DSDs affect 1:150 individuals between the neonatal
period and adulthood, with various phenotypes and degrees of
severity, however all have a significant psychological and clinical
impact. It is often difficult to provide patients with DSDs with a
molecular diagnosis due to the great clinical heterogeneity of
these disorders, as well as our poor understanding of the genetic
mechanisms involved in sex development (18). The performances of previously
reported different diagnostic approaches range from 13 to 64%
(18–20). In this study, we presented a
multistep approach for the diagnosis of DSDs based on blood and
gonadal tissue analyses by means of basic and molecular
cytogenetics, as well as the detection of copy number variation,
which yielded a diagnostic rate of 25.58%. These results highlight
the importance of histological and cytogenetic studies in gonadal
biopsy, which helped to define the medical management of 11/43
patients based on the resulting etiological or histological
findings (Table II).
| Table II.Results of cytogenetic and molecular
studies in gonads of patients with developmental sex disorder. |
Table II.
Results of cytogenetic and molecular
studies in gonads of patients with developmental sex disorder.
|
|
|
| Blood |
|---|
|
|
|
|
|
|---|
| Case | Karyotype and
FISH | MLPA | Normal | Abnormal |
|---|
| 1 | GD: mos 92,
XXXX[8]/46, XX[42] GI: | It was not
performed | Table I |
|
| 2 | mos
45,X[97]/46,XY[3].ish idic(Y)(SRY++,DYZ1-) | It was not
performed |
| Table I |
| 10 | 46,XY.ish
Yp11.31(SRY+) | Negative | Table I |
|
| 22 | 46,XY.ish
Yp11.31(SRY+) | It was not
performed | Table I |
|
| 14 | Not performed | Dup Dax1 rsa DAX1
(SALSA P185-B2 Intersex)×3 | Table I |
|
| 15 | Not performed | Dup Dax1 rsa DAX1
(SALSA P185-B2 Intersex)×3 | Table I |
|
| 13 | 46,XY.ish
Yp11.31(SRY+) | Negative | Table I |
|
| 23 | GI:
46,XY[50]SRY(+) | It was not
performed | Table I |
|
| 19 | GD:
46,XY[36]/92,XXYY[14] |
|
|
|
|
| GI:
46,XY[40]92,XXYY[10] | It was not
performed | Table I |
|
| 3 | mos
46,X,idic(Y)(q11.2)[42]/45,X[8].ish idic | It was not
performed |
| Table I |
|
|
(Y)(q11.2)(SRY++,DYZ3++,DYZ1-) |
|
|
|
| 5 | mos
45,X[15]/46,XY[35].ish Yp11.31(SRY-) | It was not
performed |
| Table I |
|
| mos
46,X,idic(Y)(q11.2)[241]/45,X[8].ish idic |
|
|
|
|
|
(Y)(q11.2)(SRY++,DYZ3++,DYZ1-) |
|
|
|
| 17 | mos
45,X[15]/46,XY[35].ish Yp11.31(SRY-) | It was not
performed |
| Table I |
|
| mos
46,X,idic(Y)(q11.2)[241]/45,X[8].ish idic |
|
|
|
|
|
(Y)(q11.2)(SRY++,DYZ3++,DYZ1-) |
|
|
|
Eleven patients (11/43, 25.58%) were clinically
diagnosed with hypospadias. In most cases, the genetic etiology of
this pathology is not established (21). Hypospadias are genital
malformations that occur predominantly in individuals with a 46,XY
karyotype. The urinary meatus and urethra in individuals with
hypospadias is anomalously located in relation to the normal male
genital phenotype (22). In
patients with this condition, it is necessary to combine all the
diagnostic strategies in order to establish their etiology.
However, previous reports have shown that in over 50% of cases it
was not possible to prove the existence of any genetic or
chromosomal cause, such that these patients were classified as
having an unknown, or idiopathic, etiology (22,23).
Our findings correlate with these results, such that genetic
analysis of another series of genes was carried out for this group
of patients, in order to determine each patient's phenotype. We
chose to investigate genes that are known to play an important role
in the development of the urogenital system, such as WT1 and
SF1 (22). In 2001, Köhler
et al (24) reported the
presence of mutations in SF1 to be the cause of severe
penoscrotal hypospadias and cryptorchidism. Then, in 2004, Wang
et al (25) demonstrated
that mutations in WT1 were responsible for the development
of certain sex development disorders, including penoscrotal
hypospadias and micropenis.
The karyotyping performed in this study demonstrated
that 7 out of 43 patients (16.27%) had an abnormality in their
mosaic sex chromosomes. When confirming the FISH results using
commercial probes for the SRY (Yp11.31), DYZ1 (Yq12), DYZ3
(Xp11.1-q11.1), and DXZ1 (Xp11.1-q11.1) regions, we found that the
mosaicisms included 45,X, 46,XY, and 47,XYY cell lines and the
presence of isodicentric Y chromosomes. It is worth noting that the
phenotypic effect of these mosaics is highly variable, which can be
explained by: (1) Difference in
the Y chromosome breakpoints, which creates variability in the
genetic information that is lost; (2) idic(Y), an unstable chromosome, which
is prone to loss during the division of mitotic cells; and
(3) different percentages of
tissue distribution of mosaicism (26). In addition, a 10–24% risk of
gonadoblastomas has been previously demonstrated in idic(Y)
patients with dysgenetic gonads, which, together with the results
described here, justifies the implementation of cytogenetic
analyses (both conventional and molecular) as first-line tests,
since they are able to detect the frequent genetic causes of DSDs
(18).
MLPA analysis detected copy number variations in 3
patients: 2 patients, who were sisters, showed duplication of
DAX1 and a 46,XY karyotype, suggesting that the causal
alteration for DSDs in these patients was inherited from a parent
with gonadal mosaicism; although this is a very rare
characteristic, it has been previously described in a patient with
DSDs, with copy number variation in another chromosomal region
(27,28). In the third patient, the deletion
of the SRY gene region with a 46,XY karyotype was
demonstrated; in this case, the presence of SRY, as detected
by FISH, suggests the probable alteration of the gene by mutation
in the annealing region, which has an effect on the function of the
gene, and is likely to be the cause of the DSDs phenotype (29).
Sequencing analysis of specific genes resulted in
the identification of a pathogenic variant in the AR gene,
which caused total androgen insensitivity, and a homozygous
pathogenic variant in the AMHR2 gene in an infertile
male-phenotype individual with persistent Mullerian derivatives. In
both cases, the clinical, endocrine, histopathological, and
cytogenetic data were consistent with the results of the molecular
study. This suggests that the de novo mutations detected in
the AR and AMHR2 genes are etiopathogenic factors in
several DSDs, including partial androgen insensitivity syndrome
(MIM 312300) and persistent Müllerian duct syndrome (MIM
261550).
In 6 of the 9 patients with 46,XY testicular DSD and
2 of the 3 patients with 46,XX ovarian DSD, no genetic alteration
was found using the proposed diagnostic approach. This suggests
that other molecular mechanisms were responsible for the DSDs in
these patients. Unexplained cases of SRY-negative XX male
reversal in humans may arise from loss-of-function mutations in the
pro-ovarian pathway (such as in canonical WNT signaling components)
or gain-of-function mutations in genes whose products mimic SRY
activity, of which members of the SOX gene family are good
candidates (30,31).
Additionally, a histological study of the gonadal
tissues from 13 patients was performed. Eleven of the 13 patients
were diagnosed with ovotesticular DSD, which is associated with
different anomalies in the mosaic sex chromosomes, including
mosaicism in gonadal tissue (Table
II). One out of the 13 patients was diagnosed with streak
gonad. In general, this type of gonadal abnormality is observed in
patients with pure and syndromic gonadal dysgenesis, which is
associated with the development of gonadoblastoma (32–34).
It is highlighted, despite the group of patients
that had undergone gonadectomy and subsequent histopathologic
examination the clinical decision was not based on the our propose
algorithm, in the near future this diagnostic algorithm will help
to focus genetics investigation and in some case could improve
clinical management. For example, in case of detection of an
abnormal Y chromosome with classic cytogenetic analysis
complemented with FISH techniques or detection of gene dosage
imbalances using MLPA in patients with 46,XY gonadal dysgenesis
with implications in sex determination and development of gonadal
tumors, the identification of the molecular cause can be very
helpful in decision-making such as prophylactic gonadectomy
(35).
Although an etiological diagnosis may not affect the
clinical management of many cases of DSDs, diagnosis allows
patients with rare chronic disorders to anticipate the
health-related and psychological effects of their condition to
optimize their quality of life (36). Additionally, the diagnosis of
genetic etiologies plays an important role in the genetic
counseling of parents, children, and other family members (37). The clinical management of all
patients with DSDs requires a multidisciplinary team, since the
issues associated with DSDs are multidimensional and thus require
the cooperation of a number of disciplines in order to provide an
effective diagnosis, treatment, and support. An ideal management
team includes pediatric subspecialists in endocrinology, surgery
and/or urology, psychology or psychiatry, gynecology, genetics,
neonatology, and, if available, social workers and nurses, among
others. This multidisciplinary team plays a critical role in the
provision of care and has as its aim the physical and psychological
well-being of individuals with DSDs and their families (37,38).
Karyotyping serves as an initial test, the results
of which lead to additional tests, as it allows for zeroing in on a
specific diagnosis from possible diagnoses. In this context, the
study of sequence variants and copy number variations is necessary,
wherein a sequencing approach with an algorithm for the detection
of copy number variants would be a good option. However, it is
worth bearing in mind that DNA modifications are not the only
possible explanation for DSDs, and thus epigenetic modifications
should be taken into account in the approach. Finally, to determine
tissue-specific expression, this type of study must be performed
using gonadal tissue.
Our step-by-step approach in individuals with DSDs
combines the cost-effectiveness of molecular cytogenetics with the
study of copy number variation to provide an effective algorithm
for the genetic diagnosis of patients with DSDs to improve our
understanding of the genetic etiology in a heterogeneous cohort of
patients. Our findings demonstrate the utility of a gonadal biopsy
in male- and female-phenotype individuals with DSDs.
Acknowledgements
Not applicable.
Funding
The present study was supported by COLCIENCIAS
(grant no. 711/2015; project no. 120371150037). MGA also received a
Young Researcher grant from COLCIENCIAS (grant no. 761/2016).
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
MGA, MM, OMN and AR designed and performed the
experiments and analyzed the data. MCM, FS, JCP, CF, CC, JP and NF
provided technical and conceptual advice and analyzed the results.
AR and OMN supervised the research and analyzed the data. MGA, MCM
and AR wrote the main parts of the manuscript.
Ethics approval and consent to
participate
This research protocol was approved by the Ethics
Committee of the Hospital Universitario San Ignacio (approval no.
FM-CIE-8540) and the School of Medicine of the Pontificia
Universidad Javeriana.
Patient consent for publication
The data and results presented in the present study
were anonymized and patients provided consent for publication.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
MLPA
|
multiplex ligation-dependent probe
amplification
|
|
FISH
|
fluorescence in situ
hybridization
|
References
|
1
|
Woodward MN and Patwardhan N: Disorders of
sex development. Surg. 28:396–401. 2010.
|
|
2
|
Dreger AD, Chase C, Sousa A, Gruppuso PA
and Frader J: Changing the nomenclature/taxonomy for intersex: A
scientific and clinical rationale. J Pediatr Endocrinol Metab.
18:729–33. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Hughes IA: Disorders of Sexual
Differentiation. Horm Res Paediatr. 67:91–95. 2007. View Article : Google Scholar
|
|
4
|
Lee PA, Nordenström A, Houk CP, Ahmed SF,
Auchus R, Baratz A, Baratz Dalke K, Liao LM, Lin-Su K, Looijenga LH
III, et al: Global disorders of sex development update since 2006:
Perceptions, approach and care. Horm Res Paediatr. 85:158–180.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Hughes IA, Houk CP, Ahmed SF and Lee PA;
LWPES Consensus Group; ESPE Consensus Group, : Consensus statement
on management of intersex disorders. Arch Dis Child. 91:554–563.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Sax L: How common is lntersex? A response
to Anne Fausto-Sterling. J Sex Res. 39:174–178. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Nordenvall AS, Frisén L, Nordenström A,
Lichtenstein P and Nordenskjöld A: Population based nationwide
study of hypospadias in sweden, 1973 to 2009: Incidence and risk
factors. J Urol. 191:783–789. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zarante I, Franco L, López C and Fernández
N: Frecuencia de malformaciones congénitas: Evaluación y pronóstico
de 52.744 nacimientos en tres ciudades colombianas. Biomédica.
30:65–71. 2010. View Article : Google Scholar
|
|
9
|
Biason-Lauber A: Control of sex
development. Best Pract Res Clin Endocrinol Metab. 24:163–186.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Houmard B, Small C, Yang L,
Naluai-Cecchini T, Cheng E, Hassold T and Griswold M: Global gene
expression in the human fetal testis and Ovary1. Biol Reprod.
81:438–443. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Eggers S, Sadedin S, van den Bergen JA,
Robevska G, Ohnesorg T, Hewitt J, Lambeth L, Bouty A, Knarston IM,
Tan TY, et al: Disorders of sex development: Insights from targeted
gene sequencing of a large international patient cohort. Genome
Biol. 17:2432016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
McClelland K, Bowles J and Koopman P: Male
sex determination: Insights into molecular mechanisms. Asian J
Androl. 14:164–171. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Arboleda VA, Sandberg DE and Vilain E:
DSDs: Genetics, underlying pathologies and psychosexual
differentiation. Nat Rev Endocrinol. 10:603–615. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Baxter RM and Vilain E: Translational
genetics for diagnosis of human disorders of sex development. Annu
Rev Genomics Hum Genet. 14:371–392. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Rajfer J and Walsh PC: The incidence of
intersexuality in patients with hypospadias and cryptorchidism. J
Urol. 116:769–770. 1976. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
van der Zwan YG, Biermann K, Wolffenbuttel
KP, Cools M and Looijenga LH: Gonadal maldevelopment as risk factor
for germ cell cancer: Towards a clinical decision model. Eur Urol.
67:692–701. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Moorhead PS, Nowell PC, Mellman WJ,
Battips DM and Hungerford DA: Chromosome preparations of leukocytes
cultured from human peripheral blood. Exp Cell Res. 20:613–616.
1960. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Laino L, Majore S, Preziosi N, Grammatico
B, De Bernardo C, Scommegna S, Rapone AM, Marrocco G, Bottillo I
and Grammatico P: Disorders of sex development: A genetic study of
patients in a multidisciplinary clinic. Endocr Connect. 3:180–192.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Arboleda VA, Lee H, Sánchez FJ, Délot EC,
Sandberg DE, Grody WW, Nelson SF and Vilain E: Targeted massively
parallel sequencing provides comprehensive genetic diagnosis for
patients with disorders of sex development. Clin Genet. 83:35–43.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Dong Y, Yi Y, Yao H, Hu H, Liu J, Gao C,
Zhang M, Zhou L, Asan, Yi X and Liang Z: Targeted next-generation
sequencing identification of mutations in patients with disorders
of sex development. BMC Med Genet. 17:232016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Marrocco G, Grammatico P, Vallasciani S,
Gulia C, Zangari A, Marrocco F, Bateni ZH, Porrello A and
Piergentili R: Environmental, parental and gestational factors that
influence the occurrence of hypospadias in male patients. J Pediatr
Urol. 11:12–19. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Audí L and Fernández-Cancio MF:
Etiopatogenia del hipospadias. Rev Esp Endocrinol Pediatr. 5
(Suppl):S72014.
|
|
23
|
Si YM, Dong Y, Wang W, Qi KY and Wang X:
Hypospadias in a male infant with an unusual mosaic 45,X/46,X,psu
idic(Y)(p11.32)/46,XY and haploinsufficiency of SHOX: A case
report. Mol Med Rep. 16:201–207. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Köhler B, Schumacher V, L'Allemand D,
Royer-Pokora B and Grüters A: Germline Wilms tumor suppressor gene
(WT1) mutation leading to isolated genital malformation without
Wilms tumor or nephropathy. J Pediatr. 138:421–424. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wang Y, Li Q, Xu J, Liu Q, Wang W, Lin Y,
Ma F, Chen T, Li S and Shen Y: Mutation analysis of five candidate
genes in Chinese patients with hypospadias. Eur J Hum Genet.
12:706–712. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Beaulieu Bergeron M, Brochu P, Lemyre E
and Lemieux N: Correlation of intercentromeric distance, mosaicism,
and sexual phenotype: Molecular localization of breakpoints in
isodicentric Y chromosomes. Am J Med Genet A 155A. 2705–2712. 2011.
View Article : Google Scholar
|
|
27
|
Haines B, Hughes J, Corbett M, Shaw M,
Innes J, Patel L, Gecz J, Clayton-Smith J and Thomas P:
Interchromosomal insertional translocation at Xq26.3 Alters SOX3
expression in an individual with XX male sex reversal. J Clin
Endocrinol Metab. 100:E815–E820. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
García-Acero M, Molina M, Moreno O,
Ramirez A, Forero C, Céspedes C, Prieto JC, Pérez J, Suárez-Obando
F and Rojas A: Gene dosage of DAX-1, determining in sexual
differentiation: Duplication of DAX-1 in two sisters with gonadal
dysgenesis. Mol Biol Rep. 46:2971–2978. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
McElreavy K, Vilain E, Abbas N, Costa JM,
Souleyreau N, Kucheria K, Boucekkine C, Thibaud E, Brauner R,
Flamant F, et al: XY sex reversal associated with a deletion 5′ to
the SRY ‘HMG box’ in the testis-determining region. Proc Natl Acad
Sci USA. 89:11016–11020. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Sarkar A and Hochedlinger K: The Sox
family of transcription factors: Versatile regulators of stem and
progenitor cell fate. Cell Stem Cell. 12:15–30. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
XIA XY, ZHANG C, LI TF, Wu QY, Li N, Li
WW, Cui YX, Li XJ and Shi YC: A duplication upstream of SOX9 was
not positively correlated with the SRY-negative 46,XX testicular
disorder of sex development: A case report and literature review.
Mol Med Rep. 12:5659–5664. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Hashimoto K, Horibe YU, Ezaki J, Kanno T,
Takahashi N, Akizawa Y, Matsui H, Yamamoto T and Shibata N:
Laparoscopically removed streak gonad revealed gonadoblastoma in
frasier syndrome. Anticancer Res. 37:3975–3979. 2017.PubMed/NCBI
|
|
33
|
Mandelberger A, Mathews S, Andikyan V and
Chuang L: Laparoscopic removal of streak gonads in turner syndrome.
J Minim Invasive Gynecol. 23:10252016. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Lepais L, Morel Y, Mouriquand P, Gorduza
D, Plotton I, Collardeau-Frachon S and Dijoud F: A novel
morphological approach to gonads in disorders of sex development.
Mod Pathol. 29:1399–1414. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Achermann JC, Domenice S, Bachega TA,
Nishi MY and Mendonca BB: Disorders of sex development: Effect of
molecular diagnostics. Nat Rev Endocrinol. 11:478–488. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Délot EC, Papp JC, Délot EC; DSD-TRN
Genetics Workgroup, ; Sandberg DE and Vilain E: Genetics of
Disorders of Sex Development: The DSD-TRN experience. Endocrinol
Metab Clin North Am. 46:519–537. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Anthony E, Baratz AB, Boney C, et al:
Clinical Guidelines for the Management of Disorders of Sex
Development in Childhood. 1st. Intersex Society of North America;
Rohnert Park, CA: 2006
|
|
38
|
Gomez-Lobo V: Multidisciplinary care for
individuals with disorders of sex development. Curr Opin Obstet
Gynecol. 26:366–371. 2014. View Article : Google Scholar : PubMed/NCBI
|