Introduction
Disabling hearing loss affects more than 5% of the
world's population (466 million individuals). It is estimated that
by 2050, more than 900 million people will be affected by disabling
hearing loss (http://www.who.int/en/news-room/fact-sheets/detail/deafness-and-hearing-loss).
Sensorineural hearing loss (SNHL) is a common disease worldwide
that accounts for most hearing loss. Currently, there are no
effective treatment options available. Factors that may lead to
SNHL include aging, exposure to loud noise, heredity, and some
medications (1). All of these
factors may result in the apoptosis or loss of cochlear hair cells.
Because hair cells cannot regenerate once they are lost (2,3), how
to protect them against apoptosis, either by inhibiting processes
that lead to damage or by enhancing their survival, is obviously an
important research goal.
The mitochondrion plays an important role in cell
apoptosis; it is also the main site of intracellular oxidative
phosphorylation and adenosine triphosphate (ATP) synthesis
(4). Mitochondria have their own
nucleic acid, mitochondrial deoxyribonucleic acid (mtDNA). As
reported (5,6) a variation in the mtDNA copy number
can affect cell apoptosis. Transgenic mice with mtDNA deletions
cannot survive the embryonic period, and a large number of
apoptotic cells have been found in such embryos (5). Our previous research also found that
decreased mtDNA copy number sensitizes cancer cells to
chemotherapeutic drugs (6).
However, its effect on hair cells has not been elucidated.
Increased mtDNA copy number may enhance the survival of hair cells
and protect them against apoptosis. In the present study, the mtDNA
copy number of mouse cochlear hair cells was assessed and it was
found that normal cochlear hair cells were relatively lacking in
mtDNA. Then the mtDNA copy number of hair cells and House Ear
Institute-Organ of Corti 1 (HEI-OC1) cells was increased by
mitochondrial transcription factor A (TFAM) transfection to
ascertain whether it had a protective effect. Finally, the
mechanisms by which increased mtDNA copy number protects against
apoptosis were explored.
Materials and methods
Cochlear basal membrane and HEI-OC1
cell culture
A total of 50 newborn C57BL/6J mice (P1-P3), each
weighing ~1.5–2.5 grams, were used in this experiment, and their
gender selection was random. International Council for Laboratory
Animal Science (ICLAS) Ethical Guideline (https://iclas.org/guidelines-for-researchers) and
the ethical requirements of experimental animals in Fudan
University (http://labanimal.fudan.edu.cn/95/6f/c5213a38255/page.htm)
were followed. Our laboratory animals were cared for by a full-time
breeder; our experiment was approved by the Animal Care and Use
Committee of the Affiliated Eye and ENT Hospital, Fudan University
(no. 201526). Mouse pups were decapitated and dissections were
performed under a light microscope in a Petri dish containing
ice-cold phosphate-buffered saline (PBS). The cochleae were removed
from the skulls and opened with forceps. The basal membranes were
detached and transferred into pre-prepared culture dishes
containing Dulbecco's modified Eagle's medium-Ham's F-12 (DMEM/F12;
Thermo Fisher Scientific, Inc.) supplemented with N2 and B27
(Thermo Fisher Scientific, Inc.), and cultured at 37°C with 5%
CO2. To induce apoptosis, neomycin (Sigma-Aldrich; Merck
KGaA) was added at a final concentration of 1 mM, and then a
terminal deoxynucleotidyl transferase (TdT)-mediated dUTP nick end
labeling (TUNEL) kit (Thermo Fisher Scientific, Inc.) was utilized
to mark the apoptotic cells.
HEI-OC1 is a widely used progenitor hair cell line
derived from mouse auditory organs (7). The HEI-OC1 cells were grown in DMEM
(Gibco Laboratories; Thermo Fisher Scientific, Inc.) containing 10%
fetal bovine serum (Gibco Laboratories; Thermo Fisher Scientific,
Inc.) at 33°C with 10% CO2 and subcultured at 80%
confluence using 0.25% trypsin/ethylenediaminetetraacetic acid
(EDTA; Thermo Fisher Scientific, Inc.). To induce apoptosis,
cisplatin (DDP; Sigma-Aldrich; Merck KGaA) was added at a final
concentration of 0.07 mM.
Measurement of mtDNA copy number and
transcription of mtDNA genes using real-time quantitative
polymerase chain reaction (qPCR)
Total RNA and DNA were extracted with a DNA/RNA
Isolation Kit (Qiagen). Reverse transcription was performed using a
PrimeScript RT Reagent Kit with gDNA Eraser (Takara) per the
manufacturer's protocols. The mitochondrial gene ND1 was
used to represent mtDNA copy number. The relative mtDNA copy number
was defined as the total amount of mtDNA divided by the total
amount of nuclear DNA.
qPCR was performed using the Applied Biosystems 7500
Real-Time PCR System (Thermo Fisher Scientific, Inc.) using GoTaq
qPCR Master Mix (Promega). We designed validated primers for each
target messenger RNA (mRNA) or DNA: TFAM forward,
ATGGCGTTTCTCCGAAGCAT and reverse, CAGATGAAAACCACCTCGGTAA;
COI forward, ACTCCTACCACCATCATTTCTCC and reverse,
GGCTAGATTTCCGGCTAGAGG; COII forward, CTTGGTCTACAAGACGCCAC
and reverse, CTATTGGCAGAACGACTCGG; 12SrRNA forward,
AATGAAGTACGCACACACCG and reverse, GGGTGTAGGCCAGATGCTTT; ATP6
forward, AAGCTCACTTGCCCACTTCC and reverse. GTAAGCCGGACTGCTAATGC;
GAPDH forward, TGCGACTTCAACAGCAACTC and reverse,
CTTGCTCAGTGTCCTTGCTG; ND1 forward, ACCATTTGCAGACGCCATAA and
reverse, TGAAATTGTTTGGGCTACGG; ACTB forward,
TCCCAGTTGGTAACAATGCCA and reverse, TGTTCCCTTCCACAGGGTGT. qPCR
conditions consisted of an initial denaturing step of 30 sec at
95°C followed by 35–40 cycles of 5 sec of denaturation at 95°C, 20
sec of annealing at 60°C and 20 sec of extension at 72°C. The
results were calculated using the comparative-cycle threshold
(ΔΔCq) method (8).
Adeno-associated virus (AAV)
transfection
An AAV vector containing human TFAM (Hanbio)
was used in this experiment. The control encoded a scrambled 150-bp
nucleotide sequence and expressed green fluorescent protein (GFP)
(Hanbio). When cochlear membranes had been cultured for 24 h or
HEI-OC1 cells cultivated to 80% confluence, 4 µl AAV-TFAM or
the control [2×1012 viral genomes per ml (vg/ml)] was
added into 1 ml culture medium for 24 h, and tissues or cells were
collected at the appropriate time points and then fixed in 4%
paraformaldehyde.
Fluorescence in situ hybridization
(FISH)
The probe was prepared using the FISH Tag DNA Kit
(Thermo Fisher Scientific, Inc.), and the spectrum of the intended
probe was from 278 to 1435 bases (NCBI Reference Sequence:
NC_005089.1) in mouse mtDNA.
The cochlear basal membranes or HEI-OC1 cells were
cultured, collected at proper time points on slides, and then fixed
with 4% polyoxymethylene and permeabilized with 0.5% Triton X-100.
Then, the FISH procedure was conducted as previously described
(6).
Measurement of mitochondrial mass and
immunofluorescence
MitoTracker Green FM (Thermo Fisher Scientific,
Inc.) was used to determine mitochondrial mass, and MitoSOX Red
(Thermo Fisher Scientific, Inc.) was used to determine reactive
oxygen species (ROS) levels. Cochlear basal membranes were grown in
culture medium for a certain period of time, the culture medium was
removed and the samples were washed with PBS. We then added a
pre-warmed (37°C) solution containing MitoTracker Green FM or
MitoSOX Red and the cells were incubated at 37°C for 20 min. After
staining, the samples were washed in PBS and imaged (living-cell
imaging) under a Leica SP8 confocal microscope (magnification, ×40;
Leica Biosystems). To label hair cells, anti-myosin VIIA rabbit
polyclonal antibody (ab3481, Abcam) was added and the cells were
incubated for 8 h (4°C) at a dilution of 1:200 after cochlear basal
membranes had been fixed with 4% polyoxymethylene and permeabilized
with 0.5% Triton X-100. We washed the samples 3 times with PBS and
incubated them for 2 h with fluorescence-conjugated goat
anti-rabbit immunoglobulin G (IgG) secondary antibody (ab150077,
Abcam) at room temperature (RT).
Since GFP expression was extremely low on the second
day after TFAM transfection and signals could not be
detected after FISH treatment, we used anti-GFP (ab290, Abcam) to
enhance the signals. The immunofluorescence procedure was the same
as mentioned above.
Flow cytometry (FCM)
To measure mitochondrial permeability, we conducted
FCM using a MitoProbe Transition Pore Assay kit (M34153, Thermo
Fisher Scientific, Inc.) according to the manufacturer's protocols.
For each sample, we prepared 3 aliquots: 1 contained
calcein-acetoxymethyl (calcein AM) only; 1 contained calcein AM and
CoCl2; and 1 contained calcein AM, CoCl2 and
ionomycin. The samples were incubated at 37°C in the dark for 15
min. Detailed information can be found in the manufacturer's
protocols.
To determine mitochondrial membrane potential (MMP)
and analyze reactive oxygen species (ROS), the HEI-OC1 cells were
cultured, trypsinized and collected; and then resuspended in a
pre-warmed (37°C) solution containing MitoTracker Red CMXRos or
MitoSOX Red (Thermo Fisher Scientific, Inc.) for 10 min. The
HEI-OC1 cells were then washed with PBS; and they were analyzed by
FCM. For apoptosis analysis, a TUNEL assay kit was utilized. First,
the cells were collected and fixed with paraformaldehyde, and then
centrifuged for 5 min at 300 × g and then the supernatant was
discarded. Next, the cells were washed in PBS, pelleted by
centrifugation and then added to 5 ml ice-cold 70% (v/v) ethanol.
Finally, the cells were analyzed by FCM. A Dead Cell Apoptosis Kit
with Annexin V-FITC and PI (Thermo Fisher Scientific, Inc.) was
used. The cells were washed twice with cold PBS and then
resuspended at a concentration of 1×106 cells/ml. We
added Annexin V/PI, gently mixed it with the cells, incubated the
mixture at RT in the dark and then analyzed the cells by FCM as
soon as possible. All tests were repeated at least 3 times.
Statistical analysis
For all experiments, values for the normal controls
were set to 1, except for the experiments of flow cytometry.
Therefore, most of our histograms show relatively quantified (RQ)
values. We made cross-sectional reconstruction after confocal
microscope scanning in all our hair cell imaging except for the
apoptosis experiments. Al fluorescence quantification was performed
with Adobe Photoshop CC (Adobe Systems Inc.). The targeted area
(hair cell) was selected and the average brightness of each cell in
gray mode was calculated. At least 50 cells in each group were
selected. We repeated this procedure at least 3 times for all our
experiments. Statistical analyses were conducted with SigmaPlot
software version 12.3 (Systat Software, Inc.). A 2-tailed t test
was used to compare values between groups. Data are presented as
mean ± standard deviation (SD). The difference was considered
significant when P<0.05.
Results
Relative deficiency of mtDNA copy
number and high levels of ROS in hair cells
Mitochondria are the powerhouses of cells. As
auditory receptors and mechano-electrical transducers, hair cells
need a large number of mitochondria, which we confirmed in this
experiment. After culturing mouse cochlear basal membranes in
vitro for 24 h, we measured their mitochondrial mass using
MitoTracker Green, for which the location in mitochondria is not
affected by MMP. We observed that the fluorescence of hair cells
was extremely intense compared with that of supporting cells,
indicating that the mitochondrial mass of hair cells was
significantly higher (Fig. 1A and
D; t=14.024; n=5; P=0.000000649). We also performed FISH
on mtDNA in cochlear basal membranes after culturing them for 24 h
in vitro. In this experiment, we found that the mtDNA copy
number of hair cells was also higher than that of supporting cells
(Fig. 1B and E; t=4.083; n=6;
P=0.00274), but the difference in fluorescence intensity between
hair cells and supporting cells was smaller than the difference in
mitochondrial mass between both types of cells. This demonstrated
that the copy number per mitochondrion was relatively deficient in
hair cells compared with that of supporting cells (Fig. 1G; t=9.366; n=5; P=0.0000138), which
may have influenced normal mitochondrial function. Finally, we
measured the ROS level using MitoSOX Red and observed that it was
higher in hair cells than that in supporting cells (Fig. 1C and F; t=4.6422; n=5; P=0.00166),
which may have been caused in part by relative mtDNA copy number
deficiency.
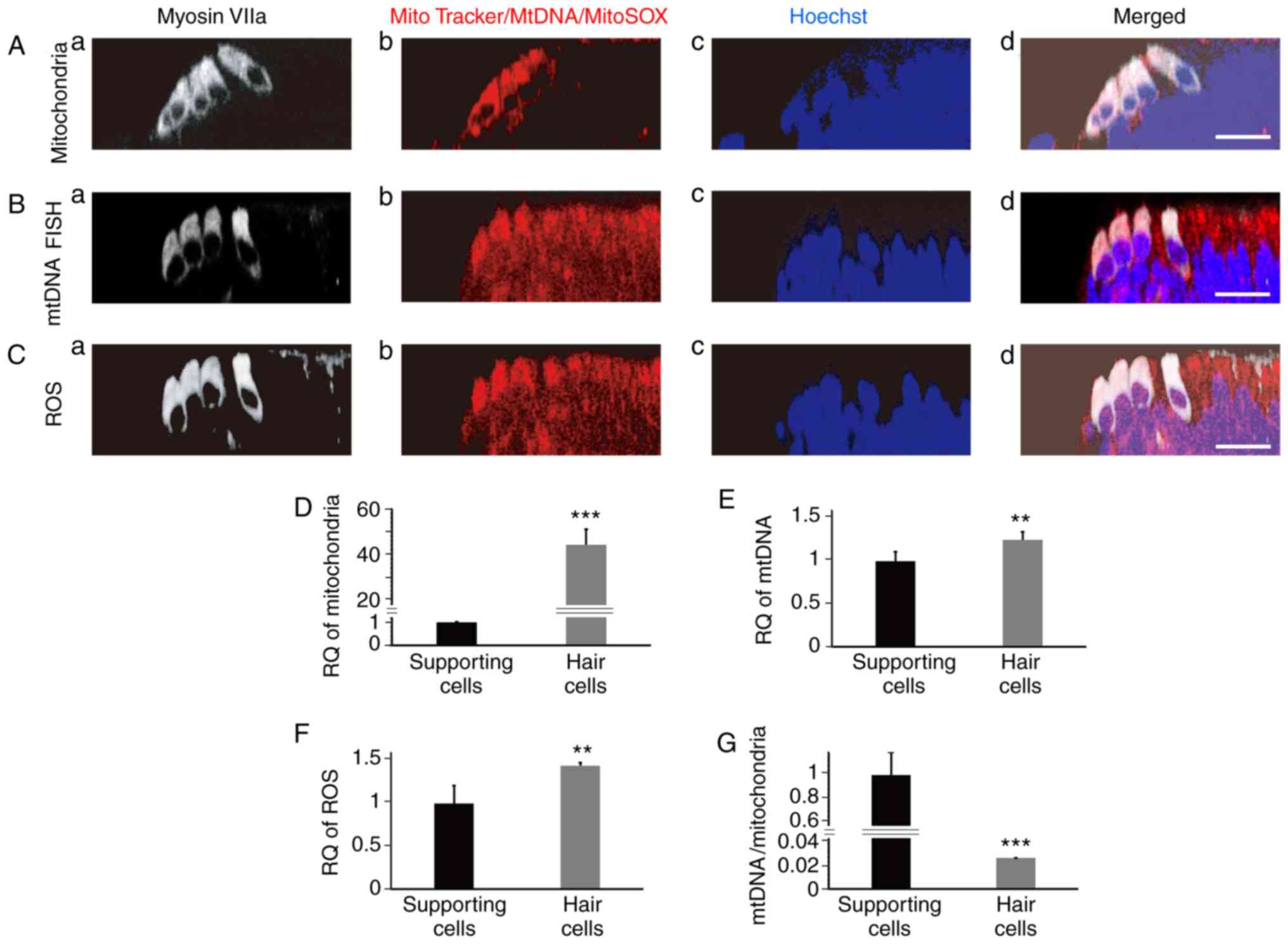 | Figure 1.Measurement of mitochondrial mass,
mtDNA and ROS in newborn mouse cochlear hair cells. (A)
Mitochondria were marked in cochlear basal membranes using
MitoTracker Green (cross-sectional reconstruction). The
mitochondrial mass of hair cells was significantly higher than that
of the supporting cells (a, marked hair cells; b, mitochondrial
staining; c, nucleus staining; d, the merged image). (B) FISH of
mtDNA in cochlear basal membrane, in which the intensity of red
fluorescence is indicative of mtDNA copy number (cross-sectional
reconstruction). The mtDNA copy number of hair cells was higher
than that of the supporting cells, but the difference in mtDNA copy
number between hair cells and supporting cells was obviously
smaller than the difference in mitochondrial mass (a, marked hair
cells; b, mtDNA staining; c, nucleus staining; d, the merged
image). (C) The ROS level was measured using MitoSOX Red
(cross-sectional reconstruction). The ROS level in hair cells was
higher than that in the supporting cells (a, marked hair cells; b,
ROS staining; c, nucleus staining; d, the merged image). (D)
Quantitative analysis of fluorescence intensity in supporting cells
and hair cells in A (1±0.198; 44.177±6.881; t=14.024; n=5;
P=0.000000649). (E) Quantitative analysis of fluorescence intensity
in supporting cells and hair cells in B (1±0.110; 1.252±0.091;
t=4.083; n=6; P=0.00274). (F) Quantitative analysis of fluorescence
intensity in supporting cells and hair cells in C (1±0.210;
1.443±0.040; t=4.6422; n=5; P=0.00166). (G) Quantitative analysis
of fluorescence intensity in supporting cells and hair cells in A
and B (1±0.232; 0.027±0.004; t=9.366; n=5; P=0.0000138).
Fluorescence intensity of mtDNA was divided by that of
mitochondrial mass. Scale bars, 20 µm. **P<0.01, ***P<0.001.
ROS, reactive oxygen species; RQ, relatively quantified values. |
Protective effect of increased
mitochondrial DNA copy number on hair and HEI-OC1 cells
TFAM is an important gene that regulates the
copy number of mitochondrial DNA, and its overexpression has no
effect on mitochondrial mass (9).
We used AAV-TFAM GFP to increase hair cell mtDNA copy number
in this experiment (see our ‘Materials and methods’ section for
detailed information). After culturing mouse cochlear basal
membranes in vitro for 24 h, AAV-TFAM was added into
the medium, and FISH was performed on mtDNA and qPCR on TFAM
RNA. It was found that the mtDNA copy number was significantly
increased in some hair cells at the second day after transfection
(Fig. 2A, B and E; t=4.44; n=9;
P=0.000412). The result was confirmed in our qPCR experiment
(Fig. 2F; t=5.052; n=3;
P=0.00722). We also found that mRNA levels of TFAM were
significantly increased (Fig. 3A;
t=13.698, 19.14, 17.638; n=3, 3, 3; P=0.000165, 0.0000439,
0.0000607). At the fifth day after transfection, it was found that
the GFP signal remained in some hair cells, but the mtDNA copy
number of transfected hair cells was no longer higher than the
non-transfected cells (Fig. 3B-D).
The results indicated that the increase in mtDNA copy number
occurred only early on after TFAM transfection. As time
progressed, TFAM overexpression may have blocked mtDNA
replication and offset its effect of increasing mtDNA copy
number.
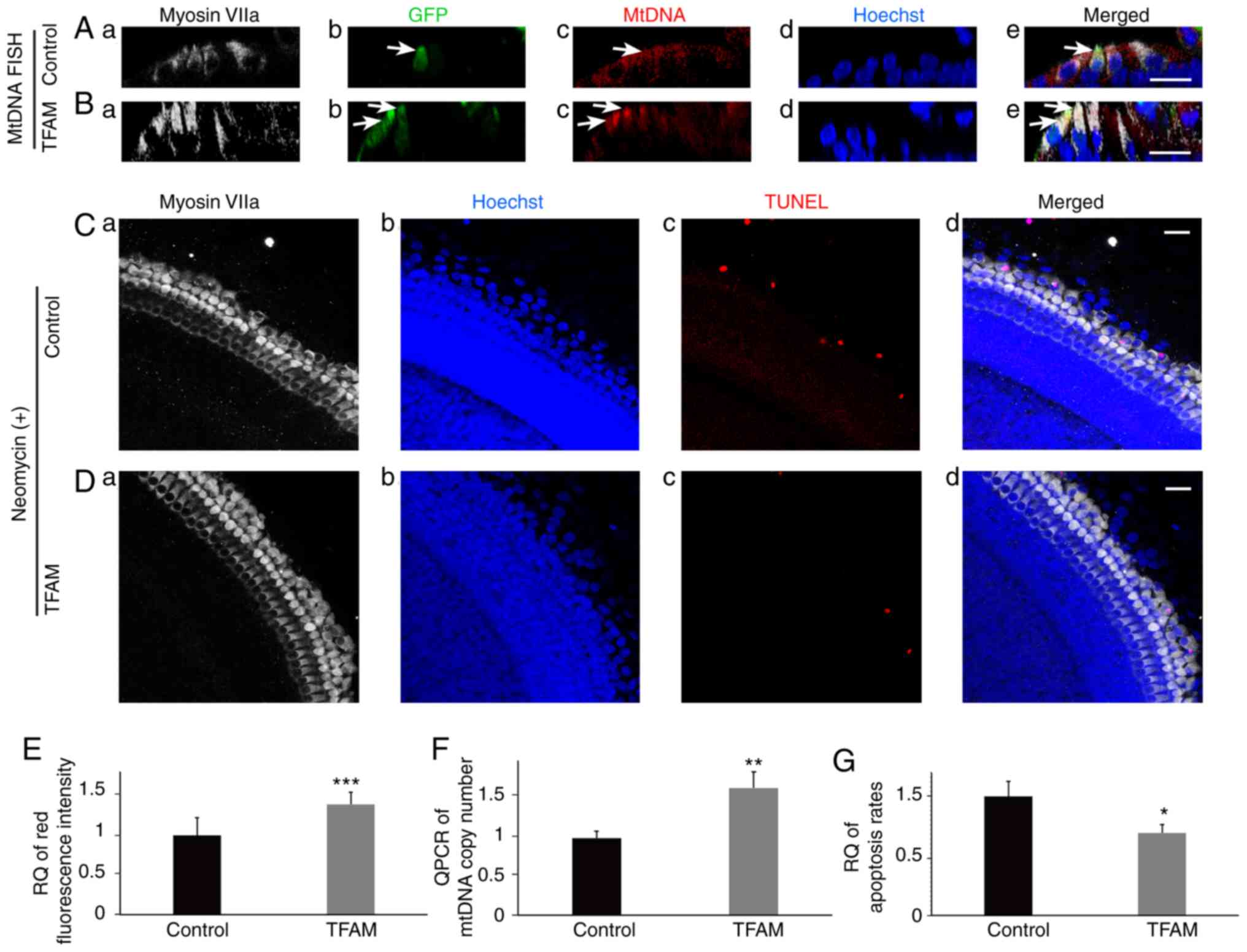 | Figure 2.Increased mtDNA copy number has a
protective effect on hair cells. (A) FISH of mtDNA in control
cochlear basal membrane (a, marked hair cells; b, transfected
cells; c, mtDNA staining; d, nucleus staining; e, the merged
image), in which the intensity of red fluorescence is indicative of
the mtDNA copy number, and the green fluorescence is indicative of
the transfected cell (white arrow; cross-sectional reconstruction).
(B) FISH of mtDNA in cochlear basal membrane on the second day
after TFAM transfection (cross-sectional reconstruction) (a, marked
hair cells; b, transfected cells; c, mtDNA staining; d, nucleus
staining; e, the merged image). The outer two hair cells were
transfected, and the mtDNA copy number was higher (white arrows).
(C) We treated the control hair cells with neomycin (a, marked hair
cells; b, nucleus staining; c, apoptotic cells; d, the merged
image). Many outer hair cells were lost or apoptotic (apoptotic
cells are labeled red). (D) We treated hair cells in the
TFAM-transfected group with neomycin for the same length of time
(a, marked hair cells; b, nucleus staining; c, apoptotic cells; d,
the merged image). Few hair cells were lost. (E) Quantitative
analysis of red fluorescence intensity in hair cells in A and B
(1±0.220; 1.392±0.147; t=4.44; n=9; P=0.000412). (F) qPCR of mtDNA
copy number. Cochlear basal membrane mtDNA copy number was
increased after TFAM transfection (1±0.084; 1.649±0.206; t=5.052;
n=3; P=0.00722). (G) Quantitation of C and D (1±0.127; 0.694±0.071;
t=3.638; n=3; P=0.022). Apoptosis rate of TFAM-transfected hair
cells was lower than that of the control. Scale bars, 20 µm.
*P<0.05, **P<0.01, ***P<0.01. TFAM, mitochondrial
transcription factor A; RQ, relatively quantified values. |
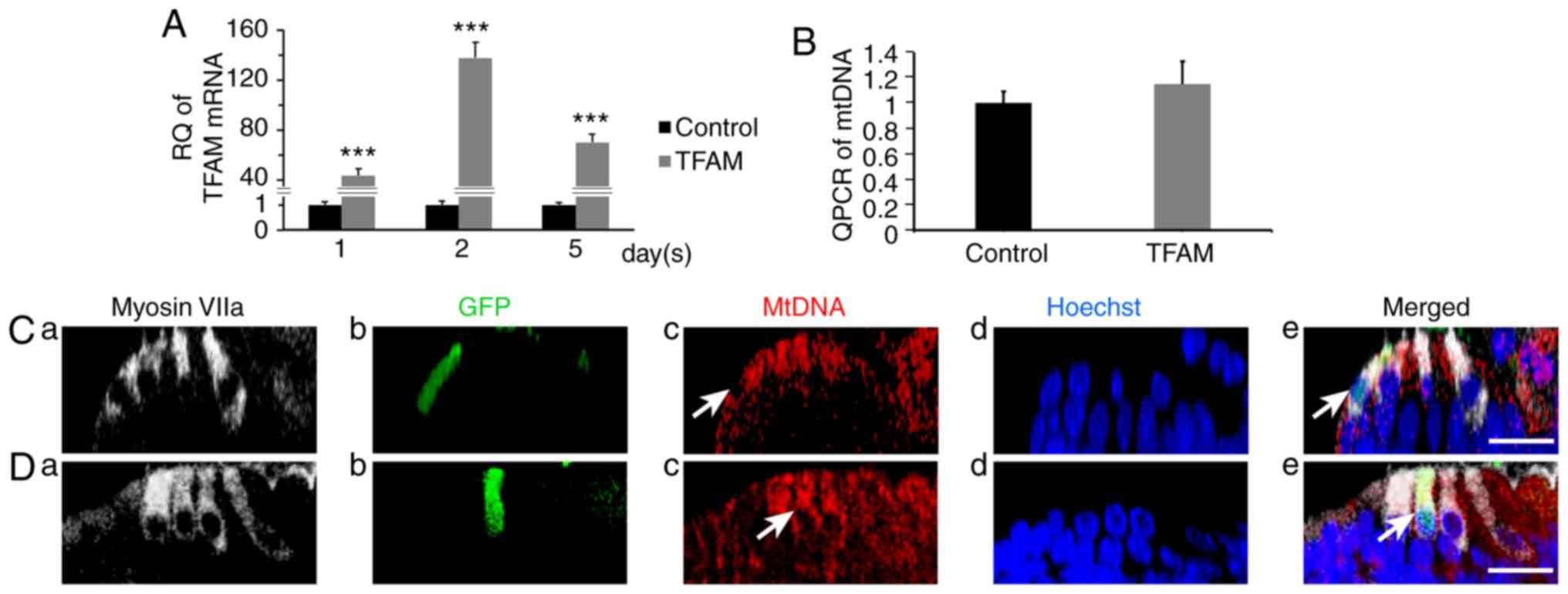 | Figure 3.Measurement of the transcription of
TFAM and the mtDNA copy number of cochlear hair cells at the fifth
day after AAV-TFAM-GFP transfection. (A) qPCR of the mRNA levels of
TFAM. At the first, second and fifth day, we extracted the RNA of
both control and TFAM-transfected cochlear basal membranes and
performed PCR analysis. We found the mRNA levels were statistically
higher in the TFAM-transfected group (1±0.159/43.615±5.386,
1±0.259/137.804±12.377, 1±0.131/69.955±6.770; t=13.698, 19.14,
17.638; n=3, 3, 3; P=0.000165, 0.0000439, 0.0000607). (B) qPCR of
mtDNA copy number. At the fifth day, we extracted the DNA of both
control and TFAM-transfected cochlear basal membranes and performed
PCR analysis. No statistically significant differences were found
(1±0.091; 1.149±0.180; t=1.282; n=3; P=0.269). At the fifth day,
the transfected hair cells had bright-green fluorescence, but its
mtDNA copy number (red fluorescence) was not higher than those of
the other 2 outer hair cells: (C) control group and (D) TFAM group
(cross-sectional reconstruction; a, marked hair cells; b,
transfected cells; c, mtDNA staining; d, nucleus staining; e, the
merged image). Scale bar, 20 µm. ***P<0.01. TFAM, mitochondrial
transcription factor A; RQ, relatively quantified values. |
In the apoptosis induction experiment, we added
neomycin into the medium at a final concentration of 1 mM on the
second day after TFAM transfection. Twelve hours later, we
collected the basal membranes and compared hair cell loss or
apoptosis rates between the TFAM transfection and control
groups using a TUNEL kit. It was demonstrated that the basal
membranes of the TFAM transfection group had an obviously
lower apoptosis rate than did those of the control group (Fig. 2C, D and G; t=3.638; n=3,
P=0.022).
To verify the previous results on hair cells, we
repeated the experiment on HEI-OC1 cells. After transfecting them
with TFAM, we collected the cells at different time points.
First, we analyzed TFAM RNA expression levels by qPCR and
found that the expression had significantly increased, peaking at
the second day (Fig. 4A;
t=21.516; n=3; P=0.0000276). Then, we analyzed the change in
mtDNA copy number in HEI-OC1 cells by qPCR. The mtDNA copy number
was also increased and peaked at the third day, lagging just behind
the change in RNA levels (Fig. 4B;
t=6.556; n=3; P=0.0028). However, the mtDNA copy number was
obviously decreased after the fifth day but had no statistically
significant difference from the control (Fig. 4B). The results were consistent with
those we found in hair cells. In the apoptosis induction
experiment, the HEI-OC1 cells were treated with 0.07 mM DDP for 24
h, and then the cells were collected and labeled with TUNEL or
Annexin V/PI. The apoptosis rates were analyzed by FCM and the
results revealed that the rates of the TFAM transfection
group were lower (Fig. 4C and D).
After quantification of the result, it was found that the
difference was statistically significant (Fig. 4E and F; t=4.797, 16.067;
n=4, 3; P=0.00301, 0.0000878).
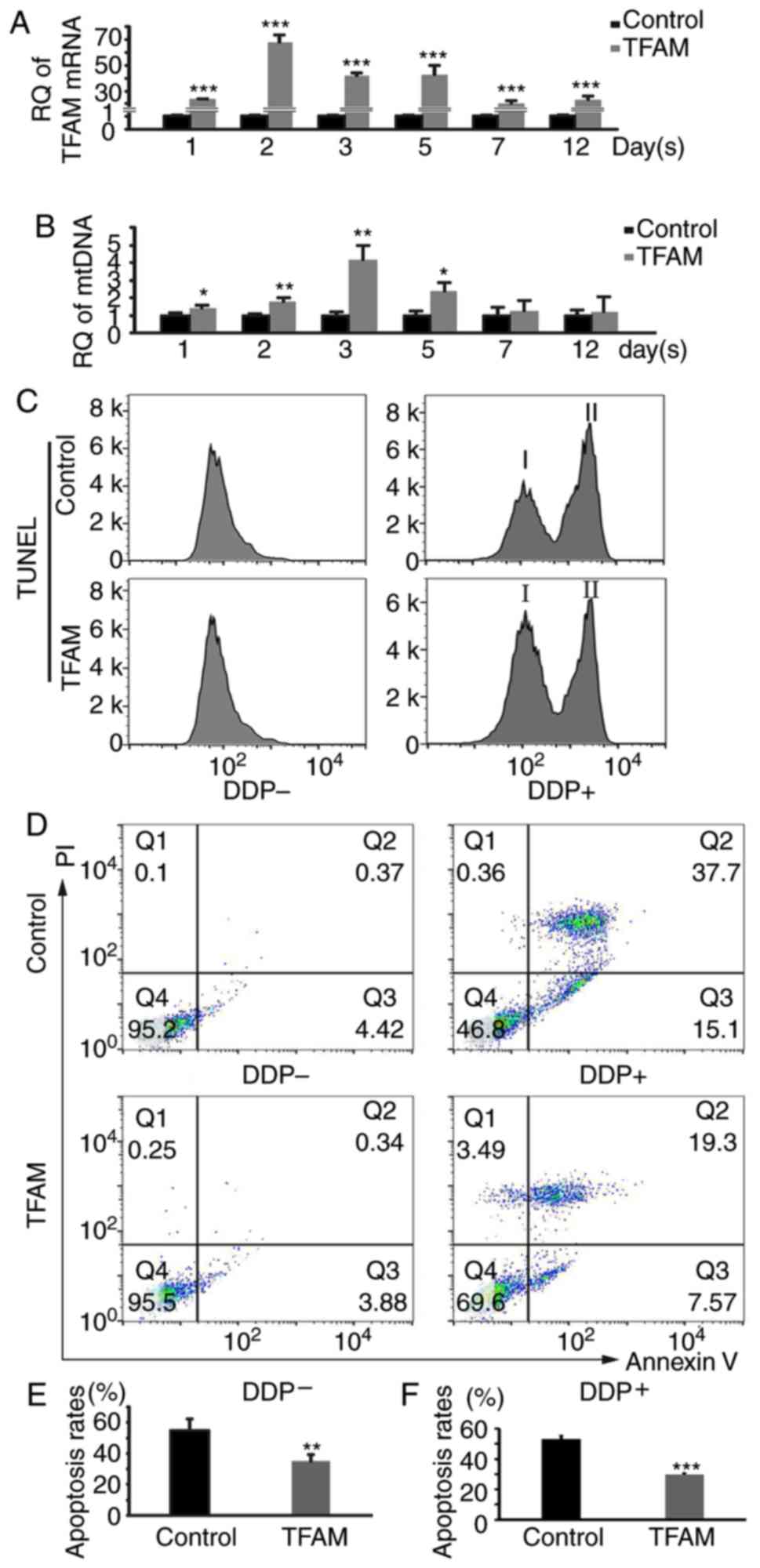 | Figure 4.Increased mtDNA copy number has a
protective effect on HEI-OC1 cells. (A) Expression levels of TFAM
RNA in HEI-OC1 cells. The expression was significantly increased,
peaked at the second day (1±0.059; 67.804±5.377; t=21.516; n=3;
P=0.0000276) and then decreased. (B) The mtDNA copy number of
HEI-OC1 cells as analyzed by qPCR. The number was increased
gradually after TFAM transfection, peaked at the third day
(1±0.199; 4.152±0.808; t=6.556; n=3; P=0.0028), and then fell back.
(C) Apoptosis analysis by FCM. We induced apoptosis in HEI-OC1
cells with DDP, and then analyzed the results using TUNEL. I,
normal cells; II, apoptotic cells. The apoptosis rate decreased
obviously after TFAM transfection. (D) Apoptosis analysis by FCM.
We induced apoptosis in HEI-OC1 cells with DDP, and then analyzed
the results using Annexin V/PI staining. The apoptosis rate
decreased obviously after TFAM transfection (the value of each
quadrant was a percentage). (E and F) Quantification of C and D.
The results were consistent with that for hair cells (0.587±0.035,
0.515±0.013; 0.46±0.034, 0.274±0.006; t=4.797, 16.067; n=4, 3;
P=0.00301, 0.0000878). *P<0.05, **P<0.01, ***P<0.001.
TFAM, mitochondrial transcription factor A; RQ, relatively
quantified values; DDP, cisplatin. |
Increased mtDNA copy number exerts a
protective effect by decreasing mitochondrial membrane
permeability
In this experiment, the protection mechanisms of
mtDNA copy number in HEI-OC1 cells were explored. As previously
mentioned, mtDNA copy number peaked at the third day after
TFAM transfection, whereupon we induced apoptosis in the
HEI-OC1 cells by DDP for 12 h. We then analyzed the mitochondrial
permeability of HEI-OC1 cells by FCM using a Mitochondrial
Permeability Transition Pore Assay Kit (Fig. 5A) and found that the fluorescence
signal was obviously stronger in the TFAM transfection group
(Fig. 5B-a). The result were
quantified and the difference was found to be statistically
significant, indicating that the mitochondrial permeability had
decreased (Fig. 5B-b; t=7.772;
n=3; P=0.00148). The increased mtDNA copy number may have blocked
the mitochondrial-permeability transition pore (mPTP) when
apoptosis was induced in the HEI-OC1 cells. The decrease in
mitochondrial permeability may have stabilized mitochondrial
function.
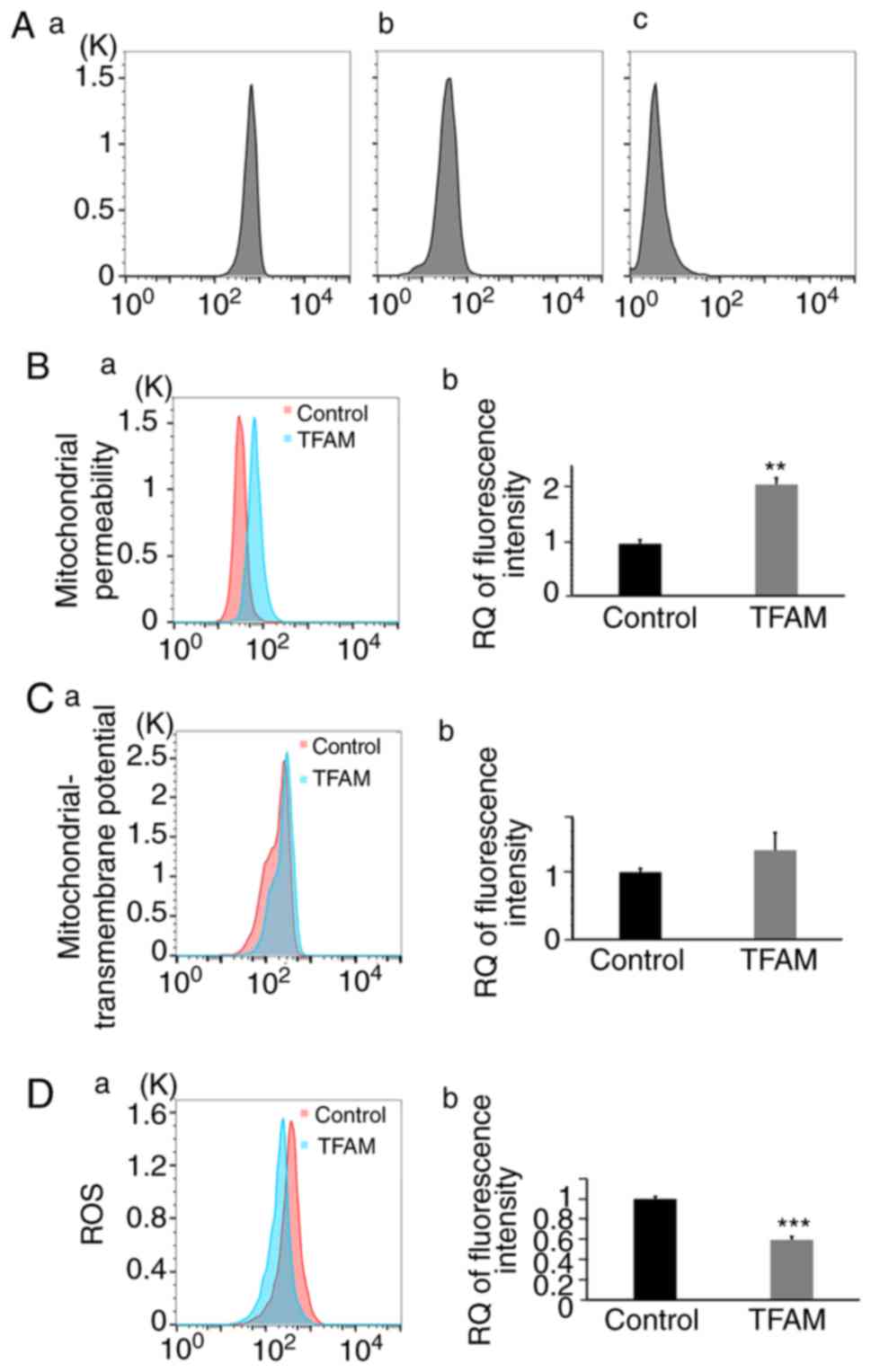 | Figure 5.mtDNA copy number affects
mitochondrial permeability, MMP, and ROS in HEI-OC1 cells. (A) mPTP
assay by FCM; (a) Fluorescent calcein was present in the cytosol as
well as the mitochondria, resulting in a bright signal. (b) In the
presence of CoCl2, only calcein in the mitochondria emitted a
signal and reflected cell mitochondrial permeability; (c) When
ionomycin, a calcium ionophore, and CoCl2 were added to the cells
at the same time, fluorescence signals were largely abolished. (B)
Mitochondrial permeability of HEI-OC1 cells, analyzed by FCM.
Fluorescence was obviously stronger in the TFAM group (1±0.078;
2.144±0.125; t=7.772; n=3; P=0.00148). (C) Mitochondrial
transmembrane potential analyzed by FCM. This was higher in the
TFAM group, but not to a statistically significant degree (1±0.053;
1.323±0.264; t=2.082, n=3, P=0.106). (D) ROS level analyzed by FCM.
The level was lower in the TFAM group (1±0.024; 0.594±0.034;
t=9.766; n=3; P=0.000616). **P<0.01, ***P<0.001. MMP,
mitochondrial membrane potential; ROS, reactive oxygen species;
TFAM, mitochondrial transcription factor A; mPTP, mitochondrial
permeability transition pore; RQ, relatively quantified values. |
Then, we analyzed the mitochondrial transmembrane
potential of HEI-OC1 cells by FCM using MitoTracker Red CMXRos at
the same time point after DDP treatment. Mitochondrial
transmembrane potential was higher in the TFAM transfection
group (Fig. 5C-a), but the
difference was not statistically significant (Fig. 5C-b). Meanwhile, we analyzed the ROS
level of HEI-OC1 cells by FCM using MitoSOX Red. The ROS level was
lower in the TFAM transfection group, and the difference was
statistically significant (Fig.
5D; t=9.766; n=3; P=0.000616). After TFAM
transfection, increased mtDNA copy number may have blocked the
mPTP, further inhibiting the decline of mitochondrial transmembrane
potential and suppressing the production of ROS. Ultimately, its
protective effect was exerted when apoptosis was induced in the
cells.
Discussion
Mitochondria have their own genetic material, mtDNA,
which encodes 13 proteins, 22 transfer RNAs (tRNAs), and 2
ribosomal RNAs (rRNAs), which are involved in maintaining the
mitochondrial function. mtDNA copy number varies by cell (10) and by developmental stage (11,12),
and depends on the dynamic equilibrium between mtDNA synthesis and
degradation. As reported, while a decreased mtDNA copy number or
mtDNA depletion may break cellular mitochondrial function and
result in many diseases, an increased mtDNA copy number has a
protective effect (6,13–15).
Given the highly active and vulnerable nature of hair cells,
increasing their mtDNA copy number is a potential way to enhance
their survival.
In the present study, we measured the mitochondrial
mass, ROS level, and mtDNA copy number in newborn mouse cochlear
hair cells. It was found that the mitochondrial mass of hair cells
was significantly higher than that of the supporting cells, and the
same was true for their ROS level. These results were anticipated
as hair cells require a great deal of energy from mitochondria to
sustain their normal function, and the levels of by-product ROS
increase during the ATP production process. Then, the mtDNA copy
number in hair cells was then assessed and it was higher than that
of the supporting cells, but to a smaller degree than was true for
mitochondrial mass, meaning that mtDNA copy number was reduced
relative to mitochondrial mass in hair cells. This result was not
what we had expected. We think the reduced mtDNA copy may have
influenced the normal mitochondrial function to a certain extent,
giving rise to a high level of ROS and leading to the fragility of
hair cells. Therefore, increased mtDNA copy number may have a
protective effect on hair cells.
The regulation of intracellular mtDNA copy number is
complicated. The catalytic subunit of DNA polymerase γ and its
processivity factor (both encoded by the POLG2 gene),
together with the Twinkle helicase (encoded by the TWNK
gene), DNA replication helicase/nuclease 2 (encoded by the
DNA2 gene), single-stranded DNA binding protein 1 (encoded
by the SSBP1 gene), primase and polymerase (DNA-directed;
encoded by the PRIMPOL gene), mitochondrial genome
maintenance exonuclease 1 (encoded by the MGME1 gene), and
the mitochondrial transcription factor A (encoded by the
TFAM gene), play key roles in mitochondrial DNA maintenance
(13,16,17).
Of all the genes, TFAM has been the most researched and may
be the best choice for increasing the mtDNA copy number. In
addition, human TFAM overexpression can increase a mouse
cell mtDNA copy number (9,18). Therefore, we used an AAV vector
containing the human TFAM gene to increase murine cell mtDNA
copy number in our research. After TFAM transfection, we
collected the cochlear basal membranes and analyzed the mtDNA by
FISH. We found that the mtDNA copy number was increased obviously
in transfected hair cells, although the increase lasted just a few
days.
In the apoptosis induction experiment, we used
neomycin, which is an aminoglycoside antibiotic and can
specifically damage hair cells in the cochlear basal membrane but
does not damage the surrounding supporting cells (19–21).
We added neomycin and found that the hair cells had an obviously
lower apoptosis rate after TFAM transfection. The results
implied the protective effect of increased mtDNA copy number
against apoptosis.
To further confirm the protective effect of mtDNA
copy number, we repeated these experiments using HEI-OC1 cells. The
results were the same as for hair cells. The mtDNA copy number was
increased and remained higher for approximately 1 week after
TFAM transfection. During induction of apoptosis, we tried
many times but found that the cell line was not sensitive to
neomycin. Even at a high concentration of the drug, we found no
difference in apoptosis rates between the neomycin-induced and
control groups. HEI-OC1 cells originate from mouse cochleas, but
they are not real hair cells. Therefore, we finally chose
cisplatin. We also found that the TFAM-transfected HEI-OC1
cells had a lower apoptosis rate, consistent with that in hair
cells.
Increased mtDNA copy number may exert its protective
effect by enhancing the mitochondrial function of the cell.
However, the detailed mechanisms are not clear. When the mtDNA copy
number was increased in HEI-OC1 cells, we analyzed the
transcription of mtDNA genes (ND1, COI, COII, 12SrRNA, ATP6)
through PCR, but we found no significant increase (data not shown).
Therefore, mitochondrial genes are not activated and involved in
the protection against apoptosis.
Mitochondrial DNA is compacted into nucleoids with
numerous nucleoid-associated proteins, each nucleoid containing
just 1 copy of mtDNA (22–24). As previously reported, the
nucleoids attach to the inner membranes of mitochondria and
interact with many mitochondrial membrane-bound proteins, such as
ATPase family AAA domain-containing protein 3 (ATAD3) (22,25,26).
We therefore assume that increased mtDNA copy number may result in
a number of mitochondrial nucleoids attaching to the inner
membrane, which, together with their interaction with membrane
proteins, may further affect mitochondrial-membrane
permeability.
The mitochondrial permeability transition pore
(mPTP) is formed in the inner membrane of the mitochondrion, and it
plays an important role in the physiological regulation of
Ca2+ and in ROS homeostasis. The opening of the mPTP
initiates the production and release of ROS, which damages
mitochondrial and nuclear DNA, proteins, and phospholipids
(27–29). In addition, mPTP has been found to
be involved in cell death induced by hypoglycemia,
ischemia/reperfusion damage, and neurodegenerative and
neuromuscular disorders (30,31).
We analyzed the mitochondrial permeability of the
HEI-OC1 cells by FCM using a mitochondrial-permeability transition
pore assay kit and found that permeability was decreased in the
TFAM group, which had a higher mtDNA copy number. The
results confirmed our hypothesis that the change in the
mitochondrial permeability may have influenced the mitochondrial
function. We then analyzed the mitochondrial transmembrane
potential and the ROS level of the HEI-OC1 cells after TFAM
transfection, and found that the potential was higher in the
TFAM group (though not to a statistically significant
degree), whereas the ROS level was lower. Therefore, decreased
mitochondrial permeability enhances the mitochondrial function of
cells by stabilizing MMP and reducing ROS, exerting a protective
effect on the cells.
In summary, we found that the mtDNA copy number of
cochlear hair cells was reduced relative to mitochondrial mass,
which may contribute to the fragility of these cells. The mtDNA
copy number can be increased in vitro by TFAM
overexpression and may help protect the cells against apoptosis.
Although the detailed mechanisms, which we intend to study in the
future, remain unclear, it is apparent that increased mtDNA copy
number may interfere with the opening of the mPTP, reduce the
production of ROS and ultimately result in lower rates of
apoptosis. These findings provide a potential novel strategy for
research into the protection of cochlear hair cells.
Acknowledgements
Not applicable.
Funding
The present study was supported by grants from the
National Science Foundation for Young Scientists of China (no.
81500788), the National Key R&D Program of China (nos.
2017YFA0103900, 2016YFC0905200) and the National Natural Science
Foundation of China (nos. 81620108005, 81470687, 81830029,
81570913).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
HM, DM and HY performed the experiments, collected
the data and wrote the manuscript; SS performed the PCR analysis
and collected the data. YC and YZ performed the transfection
experiments and collected the data. RC performed the flow cytometry
analysis and collected the data. HL designed the research and gave
final approval of the version to be published. HL and SS directed
the research and provided supporting materials.
Ethics approval and consent to
participate
Experimental protocols and procedures were reviewed
and approved by the Institutional Animal Care and Use Committee of
Affiliated Eye and ENT Hospital, Fudan University (Shanghai,
China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
DDP
|
cisplatin
|
|
HEI-OC1
|
House Ear Institute-Organ of Corti
1
|
|
mtDNA
|
mitochondrial deoxyribonucleic
acid
|
|
mPTP
|
mitochondrial permeability transition
pore
|
|
ROS
|
reactive oxygen species
|
|
TFAM
|
mitochondrial transcription factor
A
|
|
MMP
|
mitochondrial membrane potential
|
|
FCM
|
flow cytometry
|
|
TUNEL
|
TdT-mediated dUTP nick-end
labeling
|
|
FITC
|
fluorescein isothiocyanate
|
|
PI
|
propidium iodide
|
References
|
1
|
Zhang W, Kim SM, Wang W, Cai C, Feng Y,
Kong W and Lin X: Cochlear gene therapy for sensorineural hearing
loss: Current status and major remaining hurdles for translational
success. Front Mol Neurosci. 11:2212018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Monroe JD, Rajadinakaran G and Smith ME:
Sensory hair cell death and regeneration in fishes. Front Cell
Neurosci. 9:1312015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Atkinson PJ, Huarcaya Najarro E, Sayyid ZN
and Cheng AG: Sensory hair cell development and regeneration:
Similarities and differences. Development. 142:1561–1571. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hardie DG: Keeping the home fires burning:
AMP-activated protein kinase. J R Soc Interface. 15:201707742018.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Cerritelli SM, Frolova EG, Feng C,
Grinberg A, Love PE and Crouch RJ: Failure to produce mitochondrial
DNA results in embryonic lethality in Rnaseh1 null mice. Mol Cell.
11:807–815. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Mei H, Sun S, Bai Y, Chen Y, Chai R and Li
H: Reduced mtDNA copy number increases the sensitivity of tumor
cells to chemotherapeutic drugs. Cell Death Dis. 6:e17102015.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kalinec GM, Webster P, Lim DJ and Kalinec
F: A cochlear cell line as an in vitro system for drug ototoxicity
screening. Audiol Neurotol. 8:177–189. 2003. View Article : Google Scholar
|
|
8
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ekstrand MI, Falkenberg M, Rantanen A,
Park CB, Gaspari M, Hultenby K, Rustin P, Gustafsson CM and Larsson
NG: Mitochondrial transcription factor A regulates mtDNA copy
number in mammals. Hum Mol Genet. 13:935–944. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Goodall-Copestake WP: nrDNA:mtDNA copy
number ratios as a comparative metric for evolutionary and
conservation genetics. Heredity (Edinb). 121:105–111. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Hashimoto S, Morimoto N, Yamanaka M,
Matsumoto H, Yamochi T, Goto H, Inoue M, Nakaoka Y, Shibahara H and
Morimoto Y: Quantitative and qualitative changes of mitochondria in
human preimplantation embryos. J Assist Reprod Genet. 34:573–580.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Birket MJ, Orr AL, Gerencser AA, Madden
DT, Vitelli C, Swistowski A, Brand MD and Zeng X: A reduction in
ATP demand and mitochondrial activity with neural differentiation
of human embryonic stem cells. J Cell Sci. 124:348–358. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Rusecka J, Kaliszewska M, Bartnik E and
Tońska K: Nuclear genes involved in mitochondrial diseases caused
by instability of mitochondrial DNA. J Appl Genet. 59:43–57. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Foote K, Reinhold J, Yu EPK, Figg NL,
Finigan A, Murphy MP and Bennett MR: Restoring mitochondrial DNA
copy number preserves mitochondrial function and delays vascular
aging in mice. Aging Cell. 17:e127732018. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Warren EB, Aicher AE, Fessel JP and
Konradi C: Mitochondrial DNA depletion by ethidium bromide
decreases neuronal mitochondrial creatine kinase: Implications for
striatal energy metabolism. PLoS One. 12:e01904562017. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Gustafsson CM, Falkenberg M and Larsson
NG: Maintenance and expression of mammalian mitochondrial DNA. Annu
Rev Biochem. 85:133–160. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
King GA, Hashemi Shabestari M, Taris KH,
Pandey AK, Venkatesh S, Thilagavathi J, Singh K, Krishna Koppisetti
R, Temiakov D, Roos WH, et al: Acetylation and phosphorylation of
human TFAM regulate TFAM-DNA interactions via contrasting
mechanisms. Nucleic Acids Res. 46:3633–3642. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kukat C, Davies KM, Wurm CA, Spåhr H,
Bonekamp NA, Kühl I, Joos F, Polosa PL, Park CB, Posse V, et al:
Cross-strand binding of TFAM to a single mtDNA molecule forms the
mitochondrial nucleoid. Proc Natl Acad Sci USA. 112:11288–11293.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Lin SCY, Thorne PR, Housley GD and
Vlajkovic SM: Resistance to neomycin ototoxicity in the extreme
basal (hook) region of the mouse cochlea. Histochem Cell Biol.
150:281–289. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Jiang M, Karasawa T and Steyger PS:
Aminoglycoside-induced cochleotoxicity: A review. Front Cell
Neurosci. 11:3082017. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Chen Y, Yu H, Zhang Y, Li W, Lu N, Ni W,
He Y, Li J, Sun S, Wang Z and Li H: Cotransfection of Pax2 and
Math1 promote in situ cochlear hair cell regeneration after
neomycin insult. Sci Rep. 3:29962013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Lee SR and Han J: Mitochondrial nucleoid:
Shield and switch of the mitochondrial genome. Oxid Med Cell
Longev. 2017:80609492017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Gilkerson R, Bravo L, Garcia I, Gaytan N,
Herrera A, Maldonado A and Quintanilla B: The mitochondrial
nucleoid: Integrating mitochondrial DNA into cellular homeostasis.
Cold Spring Harb Perspect Biol. 5:a0110802013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kukat C, Wurm CA, Spåhr H, Falkenberg M,
Larsson NG and Jakobs S: Super-resolution microscopy reveals that
mammalian mitochondrial nucleoids have a uniform size and
frequently contain a single copy of mtDNA. Proc Natl Acad Sci USA.
108:13534–13539. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Gerhold JM, Cansiz-Arda Ş, Lõhmus M,
Engberg O, Reyes A, van Rennes H, Sanz A, Holt IJ, Cooper HM and
Spelbrink JN: Human mitochondrial DNA-protein complexes attach to a
cholesterol-rich membrane structure. Sci Rep. 5:152922015.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Desai R, Frazier AE, Durigon R, Patel H,
Jones AW, Dalla Rosa I, Lake NJ, Compton AG, Mountford HS, Tucker
EJ, et al: ATAD3 gene cluster deletions cause cerebellar
dysfunction associated with altered mitochondrial DNA and
cholesterol metabolism. Brain. 140:1595–1610. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Rottenberg H and Hoek JB: The path from
mitochondrial ROS to aging runs through the mitochondrial
permeability transition pore. Aging Cell. 16:943–955. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Treulen F, Uribe P, Boguen R and Villegas
JV: Mitochondrial permeability transition increases reactive oxygen
species production and induces DNA fragmentation in human
spermatozoa. Hum Reprod. 30:767–776. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zorov DB, Juhaszova M and Sollott SJ:
Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS
release. Physiol Rev. 94:909–950. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Baines CP and Gutiérrez-Aguilar M: The
still uncertain identity of the channel-forming unit(s) of the
mitochondrial permeability transition pore. Cell Calcium.
73:121–130. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Bernardi P and Di Lisa F: The
mitochondrial permeability transition pore: Molecular nature and
role as a target in cardioprotection. J Mol Cell Cardiol.
78:100–106. 2015. View Article : Google Scholar : PubMed/NCBI
|














