Introduction
Globally, prostate cancer (PCa) is the second most
frequent cancer. It is the fifth leading cause of cancer-related
deaths in males, and is the most frequently diagnosed cancer among
men in more than half of all countries (1). It has been established that age,
race, and family history are risk factors associated with PCa
(2). Previous studies have
implicated the initiation and development of PCa as a multistep
complex process, driven by changes in the expression of a large
number of genes with epigenetic alterations influencing the
expression of crucial genes (3,4).
Among the epigenetic modifications, methylation,
phosphorylation, acetylation, and ubiquitination have been
identified (5,6). A study by De Carvalho et al
(7) revealed that DNA methylation
can genetically alter gene expression without a change in the DNA
sequence. Hypermethylation of a promoter may downregulate gene
expression and influence the progression of human cancer (8). Recently, studies have revealed that
DNA methylation can identify invasive lesions and silence tumor
suppressor genes in PCa, providing a new direction for the
treatment of PCa (9,10).
Bioinformatics analysis based on high-throughput
platform microarray technology has been extensively used to predict
biomarkers of cancers over the last few decades (11–13).
Numerous gene expression microarrays have been used to identify
potential target genes and their functions in PCa (14–16).
However, the aforementioned studies focused on gene expression
microarrays, the number of which is limited, preventing the
accurate identification of target genes and their functions in PCa.
Therefore, an approved approach includes the combination of gene
expression and gene methylation microarray data.
The purpose of this study was to identify aberrantly
methylated-differentially expressed genes based on gene expression
and gene methylation microarray datasets. The important node genes
were screened by integrated analysis with the goal of identifying a
novel therapeutic target for the treatment of PCa. The screening
steps for determining the aberrantly methylated-differentially
expressed genes in PCa are summarized in Fig. 1.
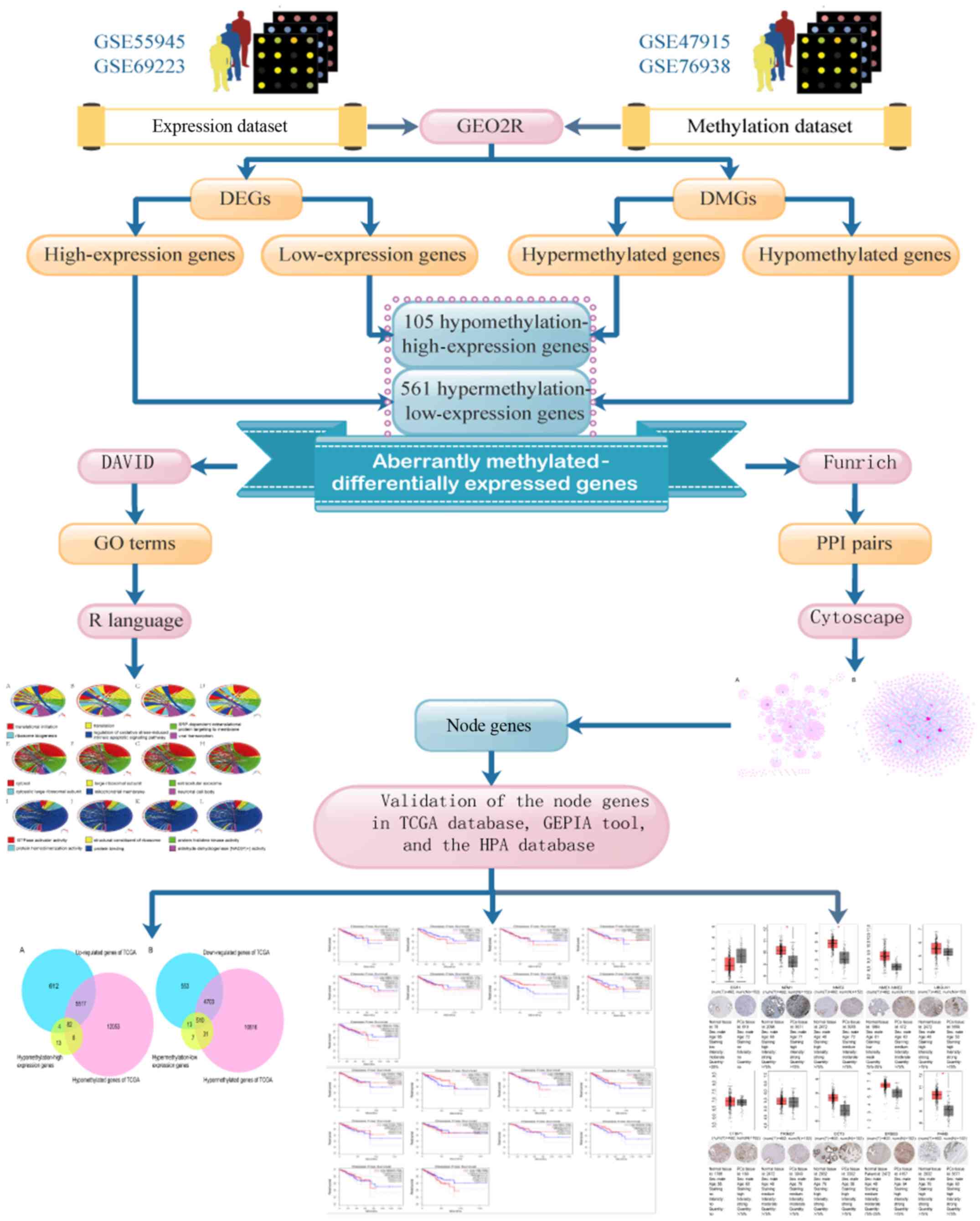 | Figure 1.Flow chart of aberrantly
methylated-differentially expressed genes in prostate cancer. DEGs,
differentially expressed genes; DMGs, differentially methylated
genes; GO, Gene Ontology; PPI, protein-protein interactions; DAVID,
Database for Annotation, Visualization, and Integrated Discovery;
TCGA, The Cancer Genome Atlas; GEPIA, Gene Expression Profiling
Interactive Analysis; HPA, Human Protein Atlas. |
Materials and methods
Data sources
In the present study, the raw data were selected
from the Gene Expression Omnibus (GEO), which is an international
public repository that can be found on the National Center for
Biotechnology Information (NCBI) home page (https://www.ncbi.nlm.nih.gov/geo/). Microarray gene
expression data found at accession GSE55945 involved data from 13
PCa samples and eight normal samples, and accession GSE69223
encompassed 15 PCa samples and 15 normal samples, with the platform
GPL570 of the two datasets ([HG-U133_Plus_2] Affymetrix Human
Genome U133 Plus 2.0 Array). Methylation profile data in GSE47915
comprised four PCa samples and four normal samples, while GSE76938
contained 73 PCa samples and 63 normal samples. The platform of
both datasets (GSE47915 and GSE76938) was based on GPL13534
(Illumina HumanMethylation450 BeadChip).
Data processing
The raw data analysis was carried out using GEO2R,
which can separately screen differentially methylated genes (DMGs)
and differentially expressed genes (DEGs) between normal and cancer
prostate sample datasets (17).
DMGs and DEGs were obtained using the criteria|t|>2 and
P<0.05. The intersection of DMGs and DEGs was derived using the
FunRich Venn function (http://www.funrich.org) (18), followed by obtaining the
hypomethylation-high expression genes and hypermethylation-low
expression genes.
Gene ontology (GO) term enrichment
analysis
The GO terms, including the hypomethylation-high
expression genes and hypermethylation-low expression genes, were
enriched using the Database for Annotation, Visualization, and
Integrated Discovery (DAVID, http://david.niaid.nih.gov), and P-values <0.05
were considered statistically significant. The chord plots from the
GO results were created using R language with ggplot2 and GOplot
packages (19).
Construction of PPI networks
Protein-protein interactions (PPI) are critical
events in signaling pathways, especially when interpreting the
molecular mechanisms of cellular activities during carcinogenesis.
The PPI relationships of the hypomethylation-high expression genes
and hypermethylation-low expression genes were obtained by FunRich,
and their interactive and visual networks were created using
Cytoscape v3.5.0 software (https://cytoscape.org/) (20).
Node gene validation
Gene expression profiling data (HTSeq-FPKM) and
methylation sequencing data (Illumina Human Methylation 450) were
downloaded from the Cancer Genome Atlas (TCGA) database (https://cancergenome.nih.gov/). DMGs and DEGs were
obtained using the criteria |t|>2 and P<0.05. The
hypomethylated-high expression genes and hypermethylated-low
expression genes obtained from the GEO database were respectively
validated in TCGA database, and the methylation and gene expression
status of the top ten node genes were also validated. Subsequently,
the expression status, translational levels and the mRNA levels of
the top ten node genes were validated using the Gene Expression
Profiling Interactive Analysis (GEPIA; http://gepia.cancer-pku.cn) and The Human Protein
Atlas database (HPA; http://www.proteinatlas.org/), respectively. In
addition, the Disease-Free Survival (RFS) method of the GEPIA
online tool was used to analyze 20 aberrantly-methylated genes
including hypomethylated-high expression genes and
hypermethylated-low expression genes. Group cutoff was set as
Quartile (Cutoff-High (%) was 75% and Cutoff-Low (%) was 25%). The
confidence interval (CU) was 95%. High and low expression genes
were respectively represented in red and blue colour (21,22).
Results
Methylated DEGs in PCa screening
results
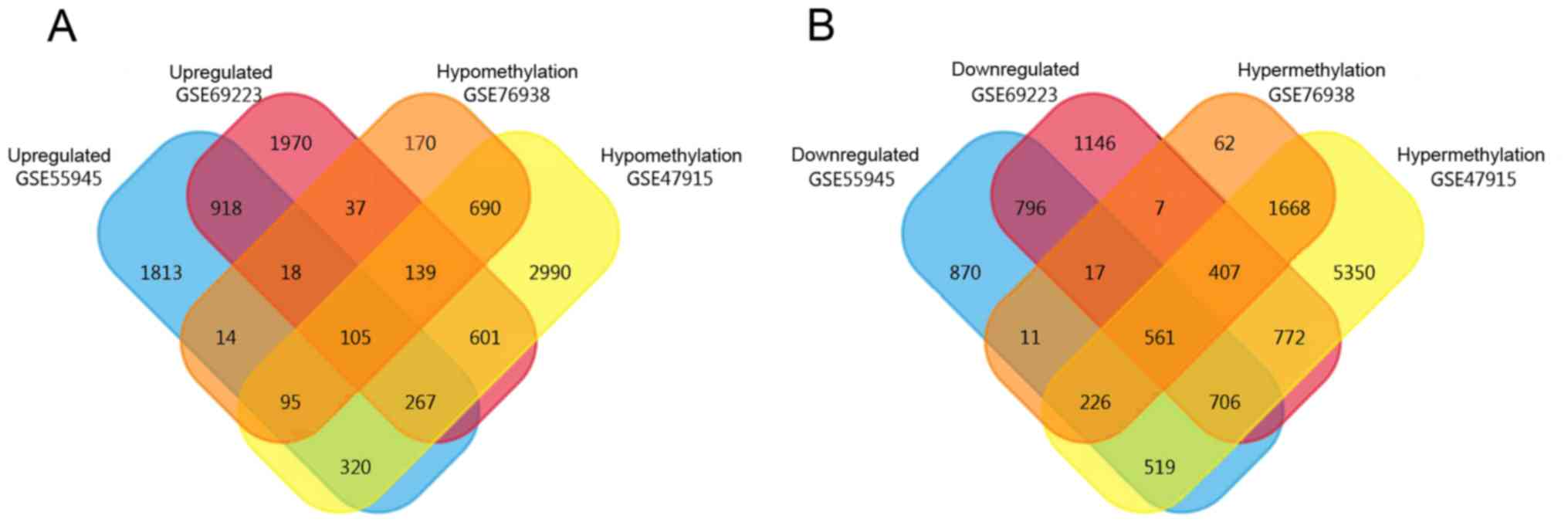
A total of 3,550 high-expression and 3,706
low-expression genes were obtained from GSE55945, while 4,055
high-expression and 4,412 low-expression genes were obtained from
GSE69223. In addition, 10,209 hypermethylated and 5,207
hypomethylated genes were obtained from GSE47915 and 2,959
hypermethylated and 1,268 hypomethylated genes were obtained from
GSE76938 by GEO2R. Using the FunRich Venn function, 105
hypomethylation-high expression genes and 561 hypermethylation-low
expression genes were identified, as revealed in Fig. 2.
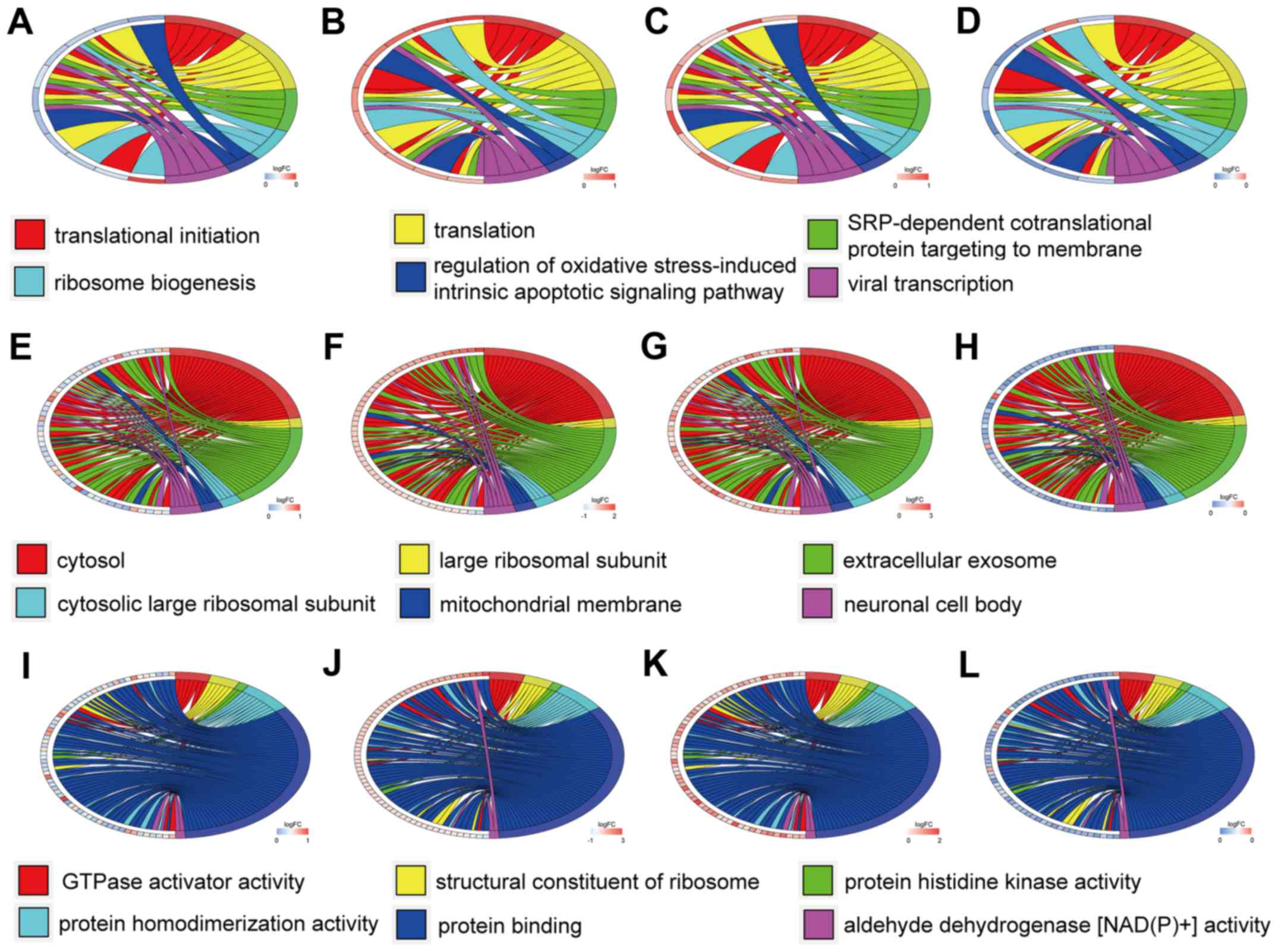
GO term enrichment analysis
The top six GO annotation results of aberrantly
methylated-differentially expressed genes are summarized in
Table I. The ontology enrichment
analysis of hypomethylation-high expression genes is presented in
Fig. 3 in the form of cluster
chord diagrams. The diagrams of cluster chord A-D, E-F and I-L
respectively show the biological processes, cellular components and
molecular functions of GO analysis. The four columns of chord
diagrams include the first column including A, E and I, the second
column including B, F and J, the third column including C, G and K,
and the fourth column including D, H and L respectively enriched
the GO of GSE47915, GSE55945, GSE69223 and GSE769938 datasets.
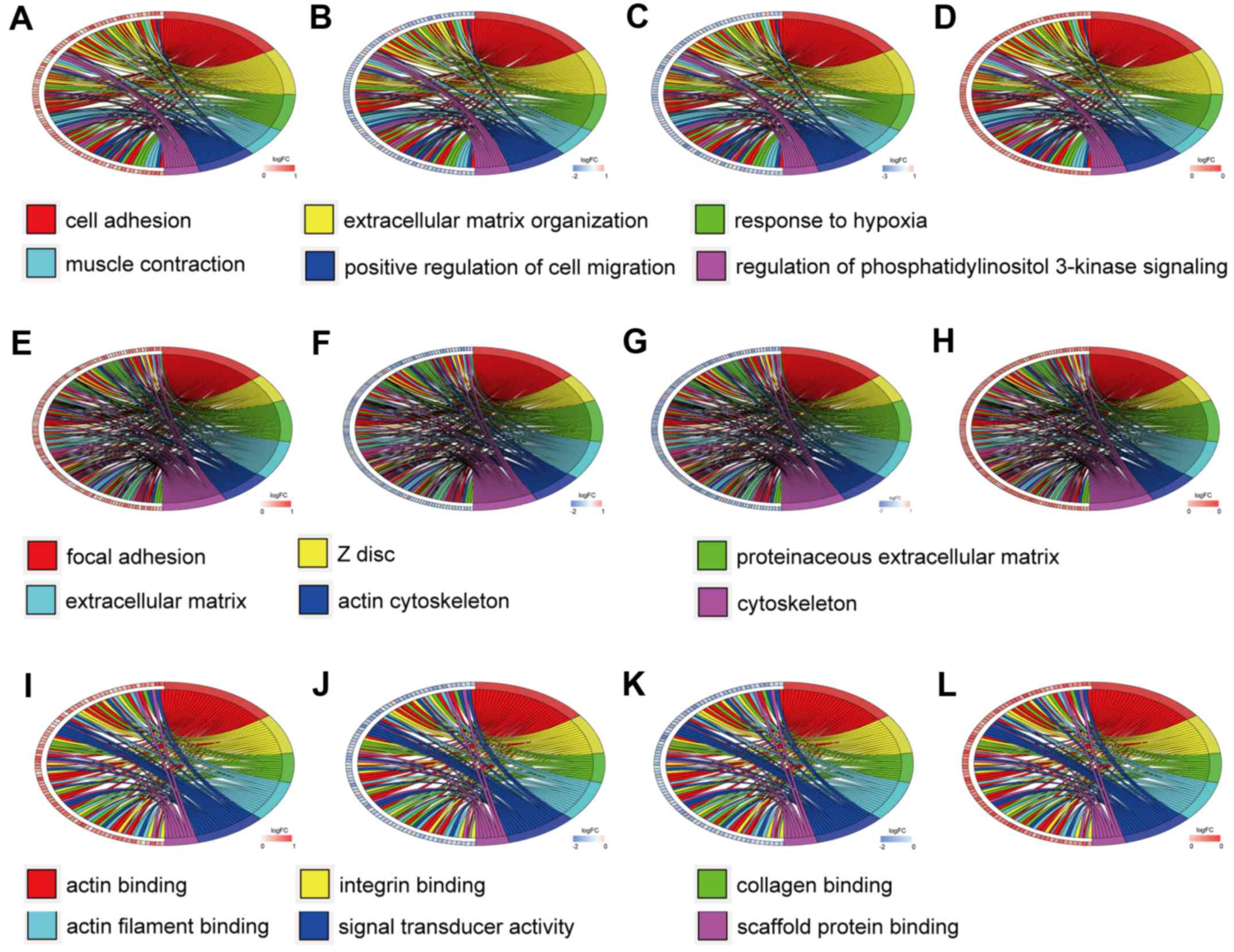
Similarly, Fig. 4 reveals the
enrichment of hypermethylation-low expression genes. Among the
hypomethylation-high expression genes, the biological processes
(BP) were mainly associated with translational initiation,
translation, SRP-dependent co-translational protein targeting to
membrane, ribosome biogenesis, regulation of oxidative
stress-induced intrinsic apoptotic signaling pathways, and viral
transcription. The major cell components (CC) included: Cytosol,
large ribosomal subunit, extracellular exosome, cytosolic large
ribosomal subunit, mitochondrial membrane, and neuronal cell body,
with the most important being the cytosol. The molecular functions
(MF) were primarily focused on GTPase activator activity, ribosome
structure, protein histidine kinase activity, protein
homodimerization activity, protein binding, and aldehyde
dehydrogenase [NAD(P)+] activity.
 | Table I.GO analysis of aberrantly
methylated-differentially expressed genes in prostate cancer. |
Table I.
GO analysis of aberrantly
methylated-differentially expressed genes in prostate cancer.
| A, Hypomethylation
and high expression |
|---|
|
|---|
| GO analysis | Term | Gene count | Percentage (%) | P-value |
|---|
| BP | Translational
initiation | 5 | 4.76 |
5.15×10−3 |
| BP | Translation | 6 | 5.71 |
9.21×10−3 |
| BP | SRP-dependent
co-translational protein targeting to membrane | 4 | 3.81 |
1.21×10−2 |
| BP | Ribosome
biogenesis | 3 | 2.86 |
1.43×10−2 |
| BP | Regulation of
oxidative stress-induced intrinsic apoptotic signaling pathway | 2 | 1.90 |
1.51×10−2 |
| BP | Viral
transcription | 4 | 3.81 |
1.92×10−2 |
| CC | Cytosol | 34 | 32.38 |
1.94×10−5 |
| CC | Large ribosomal
subunit | 3 | 2.86 |
2.07×10−3 |
| CC | Extracellular
exosome | 25 | 23.81 |
3.56×10−3 |
| CC | Cytosolic large
ribosomal subunit | 4 | 3.81 |
4.49×10−3 |
| CC | Mitochondrial
membrane | 4 | 3.81 |
1.10×10−2 |
| CC | Neuronal cell
body | 6 | 5.71 |
1.91×10−2 |
| MF | GTPase activator
activity | 7 | 6.67 |
2.67×10−3 |
| MF | Structural
constituent of ribosome | 6 | 5.71 |
5.00×10−3 |
| MF | Protein histidine
kinase activity | 2 | 1.90 |
9.93×10−3 |
| MF | Protein
homodimerization activity | 10 | 9.52 |
1.03×10−2 |
| MF | Protein
binding | 55 | 52.38 |
1.56×10−2 |
| MF | Aldehyde
dehydrogenase [NAD(P)+] activity | 2 | 1.90 |
2.95×10−2 |
|
| B,
Hypermethylation and low expression |
|
| BP | Cell adhesion | 39 | 6.96 |
2.49×10−8 |
| BP | Extracellular
matrix organization | 23 | 4.11 |
1.46×10−7 |
| BP | Response to
hypoxia | 18 | 3.21 |
2.09×10−5 |
| BP | Muscle
contraction | 14 | 2.50 |
2.24×10−5 |
| BP | Positive regulation
of cell migration | 18 | 3.21 |
4.97×10−5 |
| BP | Regulation of
phosphatidylinositol 3-kinase signaling | 11 | 1.96 |
1.25×10−4 |
| CC | Focal adhesion | 53 | 9.46 |
6.76×10−20 |
| CC | Z disc | 22 | 3.93 |
3.87×10−11 |
| CC | Proteinaceous
extracellular matrix | 32 | 5.71 |
1.08×10−10 |
| CC | Extracellular
matrix | 30 | 5.36 |
2.00×10−8 |
| CC | Actin
cytoskeleton | 25 | 4.46 |
3.70×10−8 |
| CC | Cytoskeleton | 32 | 5.71 |
2.49×10−7 |
| MF | Actin binding | 28 | 5.00 |
1.03×10−7 |
| MF | Integrin
binding | 15 | 2.68 |
3.34×10−6 |
| MF | Collagen
binding | 11 | 1.96 |
1.15×10−5 |
| MF | Actin filament
binding | 15 | 2.68 |
4.73×10−5 |
| MF | Signal transducer
activity | 17 | 3.04 |
4.92×10−4 |
| MF | Scaffold protein
binding | 8 | 1.43 |
5.61×10−4 |
For the hypermethylation-low expression genes, cell
adhesion, extracellular matrix organization, response to hypoxia,
muscle contraction, positive regulation of cell migration, and
regulation of phosphatidylinositol 3-kinase signaling were the
predominant BP. The CC of these genes were primarily distributed in
focal adhesions, Z discs, proteinaceous extracellular matrices,
other extracellular matrices, and the cytoskeleton. The MF mainly
included actin binding, integrin binding, collagen binding, actin
filament binding, signal transducer activity, and scaffold protein
binding.
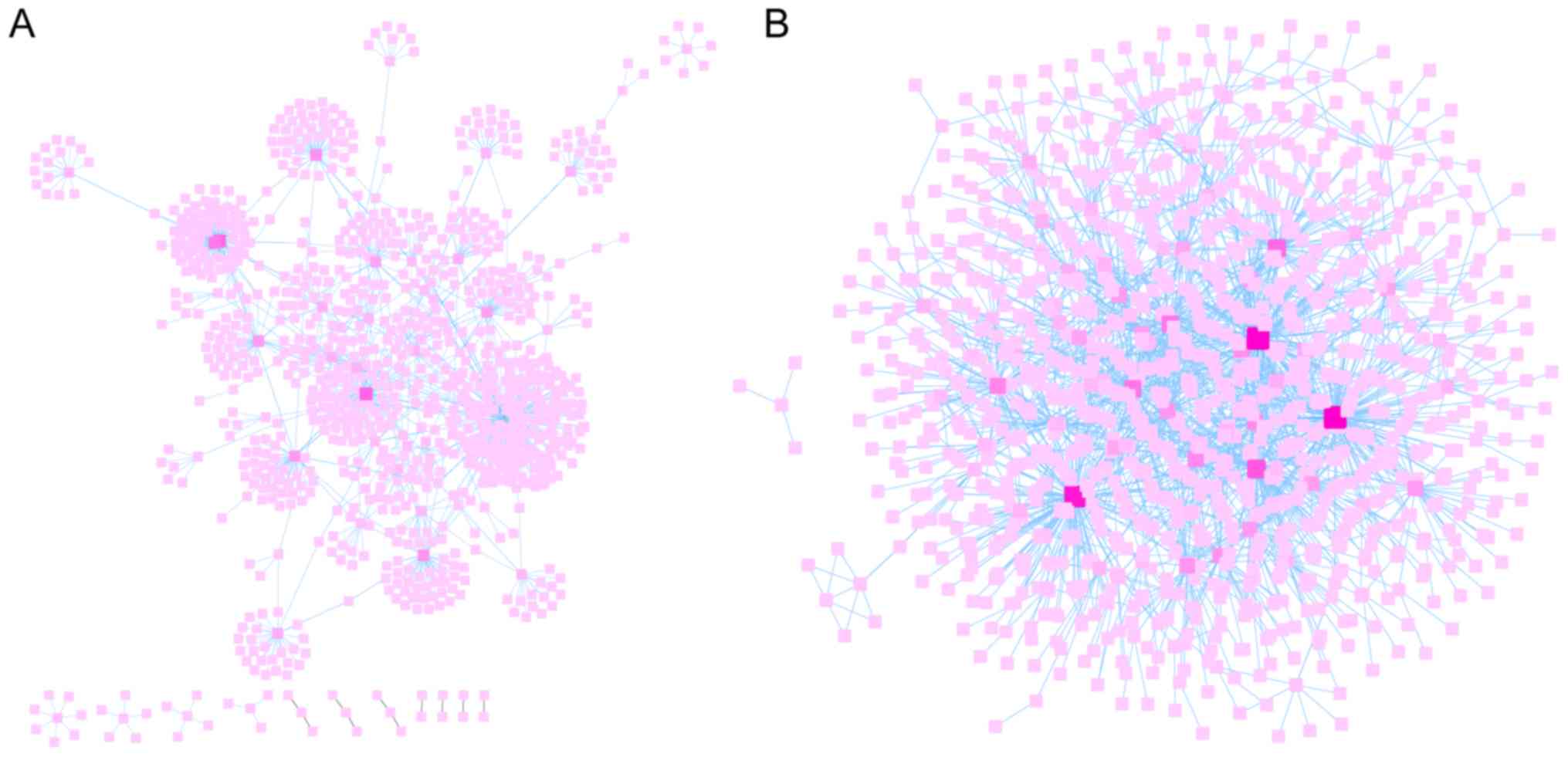
Selection of PPI network node
genes
FunRich was used to predict the relationships
between genes and proteins of the hypomethylation-high expression
and hypermethylation-low expression gene groups. Subsequently,
their interaction networks were visualized by Cytoscape v3.5.0
software as revealed Fig. 5A and
B. Cytoscape is a software focused on open source web
visualization and analysis. Its core is to provide the basic
functional layout and query network, and is based on the
combination of basic data into a visual network. Derived from
systems biology, Cytoscape is used to integrate biomolecular
interaction networks with high-throughput gene expression data and
other molecular state information. Its most powerful function is
for the analysis of large-scale PPIs, protein-DNA and genetic
interactions (21). The top ten
node genes in the hypomethylation-high expression and
hypermethylation-low expression gene groups are listed separately
in Tables II and III, based on the degree of distribution
of the nodes.
| Table II.Top ten node genes in the
hypomethylation-high expression genes. |
Table II.
Top ten node genes in the
hypomethylation-high expression genes.
| Name | Degree |
|---|
| ESR1 | 234 |
| NPM1 | 81 |
| NME2 | 77 |
| NME1-NME2 | 64 |
| UBQLN1 | 47 |
| CTBP1 | 45 |
| TRIM27 | 41 |
| CCT3 | 40 |
| ERBB3 | 35 |
| P4HB | 33 |
| Table III.Top ten node genes in the
hypermethylation-low expression genes. |
Table III.
Top ten node genes in the
hypermethylation-low expression genes.
| Name | Degree |
|---|
| CREBBP | 160 |
| SMAD3 | 159 |
| MCC | 142 |
| VIM | 92 |
| ATXN1 | 79 |
| CAV1 | 68 |
| FLNA | 68 |
| MAP3K14 | 55 |
| PRKCB | 53 |
| FHL2 | 52 |
Validation of the node genes
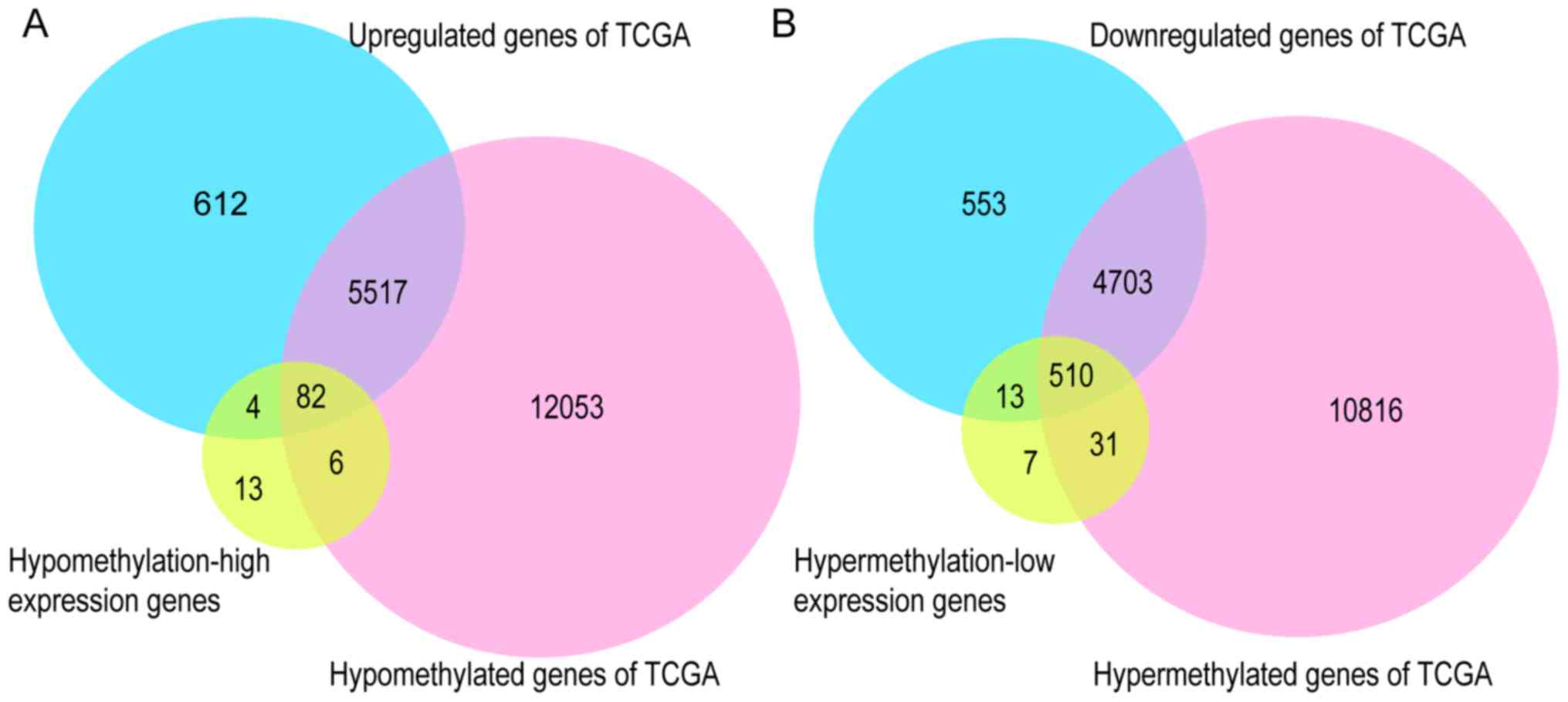
To further validate the present results, TCGA
database, the GEPIA tool, and the HPA database were employed. Venn
diagram analysis of the GEO database and TCGA database is presented
in Fig. 6, from which it can be
observed that most of the aberrantly methylated and expressed genes
in GEO were contained within TCGA dataset. The methylation and gene
expression status of the top ten node genes were similar between
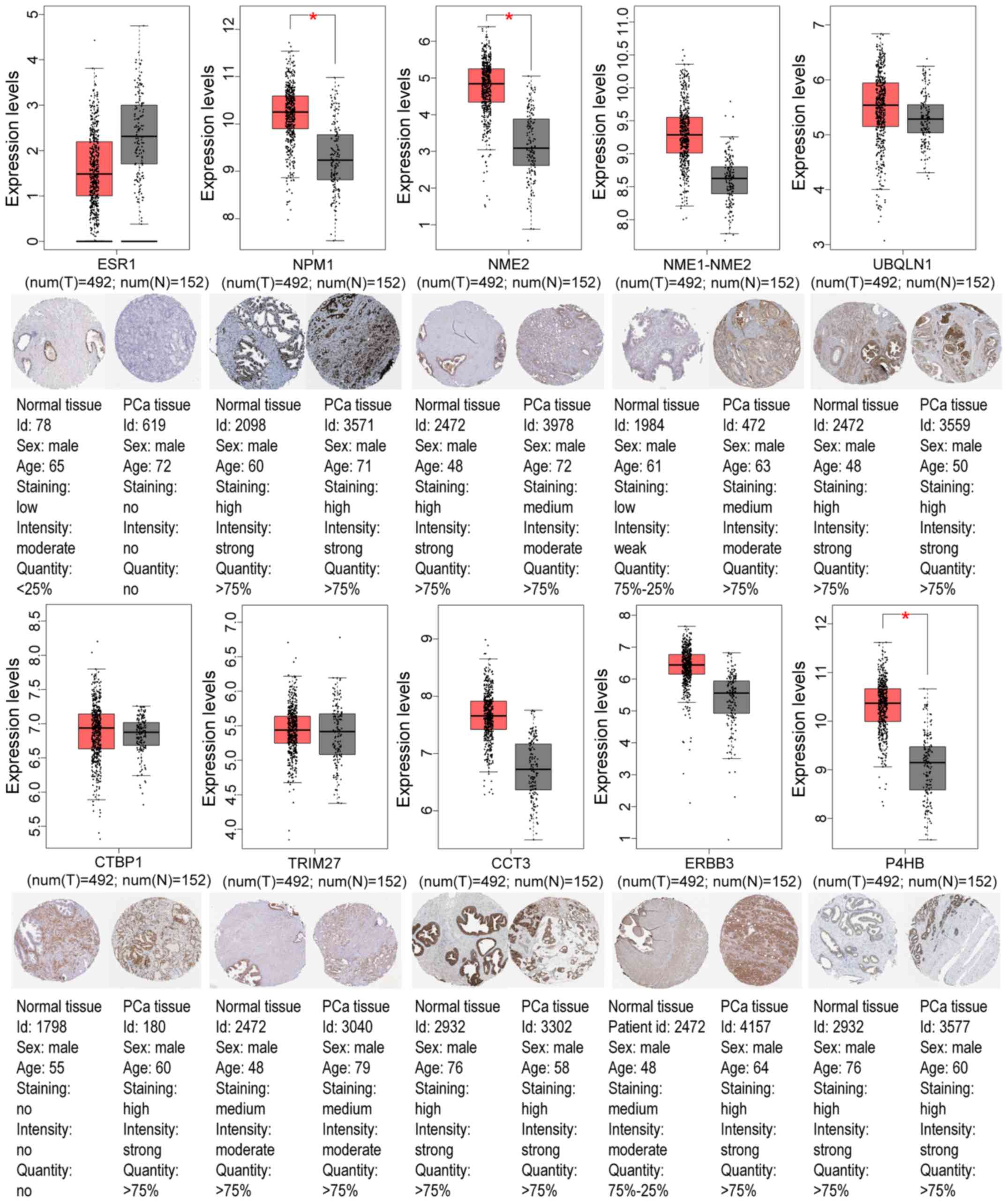
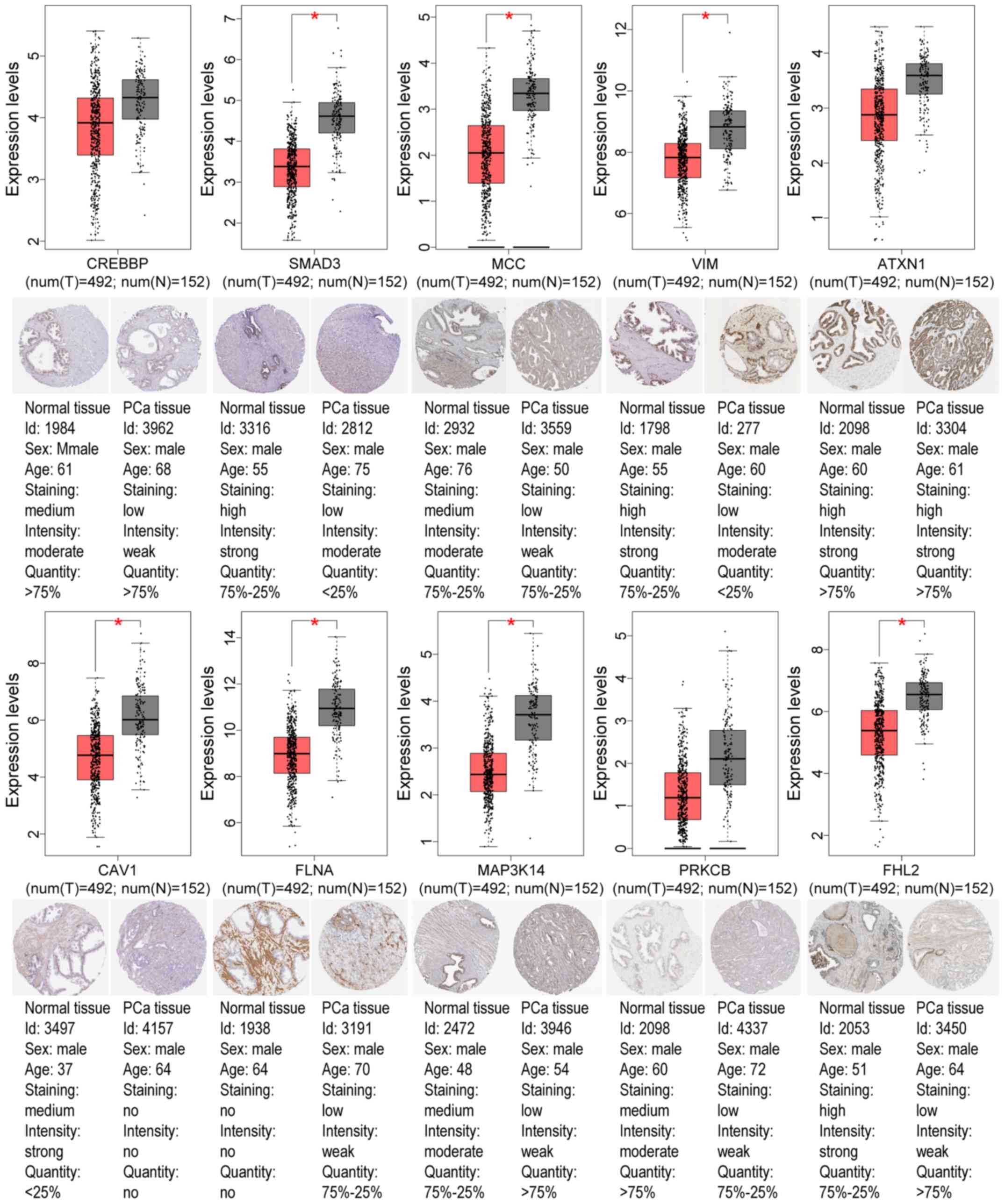
GEO and TCGA databases, as revealed in Tables IV and V. Using the GEPIA tool and HPA database,
the expression of node genes in tumor and normal prostate samples
were further verified and the immunohistochemical staining combined
with patient information, node gene expression, and the mRNA
levels, respectively, were obtained, as revealed in Figs. 7 and 8 and Tables
II and III. In Figs. 7 and 8, immunohistochemical images and data
were generated by the online GEPIA tool, log2(TPM+1) for
log-scale was used to express gene expression on the y-axis. Red
and gray were set as the colors of tumor and normal data sets
respectively. The size of jitter across the box was as 0.4. It was
revealed that the expression of node genes was generally consistent
with previous results.
| Table IV.Validation of the top ten
hypomethylation-high expression genes in TCGA database. |
Table IV.
Validation of the top ten
hypomethylation-high expression genes in TCGA database.
| Gene | Methylation
status | P-value | Expression
status | P-value |
|---|
| ESR1 |
Hypomethylation |
3.70×10−143 | Upregulated |
1.20×10−1 |
| NPM1 |
Hypomethylation |
5.51×10−119 | Upregulated |
3.12×10−15 |
| NME2 |
Hypomethylation |
2.76×10−119 | Upregulated |
1.71×10−5 |
| NME1-NME2 |
Hypomethylation |
6.35×10−132 | Upregulated |
6.43×10−9 |
| UBQLN1 |
Hypomethylation |
4.24×10−117 | Upregulated |
8.87×10−1 |
| CTBP1 |
Hypomethylation |
1.85×10−126 | Upregulated |
2.01×10−7 |
| TRIM27 |
Hypomethylation |
5.11×10−129 | Upregulated |
5.39×10−27 |
| CCT3 |
Hypomethylation |
1.92×10−129 | Upregulated |
2.40×10−18 |
| ERBB3 |
Hypomethylation |
1.46×10−121 | Upregulated |
2.89×10−18 |
| P4HB |
Hypomethylation |
2.12×10−119 | Upregulated |
4.29×10−18 |
| Table V.Validation of the top ten
hypermethylation-low expression genes in TCGA database. |
Table V.
Validation of the top ten
hypermethylation-low expression genes in TCGA database.
| Gene | Methylation
status | P-value | Expression
status | P-value |
|---|
| CREBBP |
Hypermethylation |
1.06×10−123 | Downregulated |
1.64×10−1 |
| SMAD3 |
Hypermethylation |
6.27×10−121 | Downregulated |
1.34×10−10 |
| MCC |
Hypermethylation |
2.39×10−126 | Downregulated |
2.13×10−20 |
| VIM |
Hypermethylation |
1.11×10−134 | Downregulated |
1.26×10−5 |
| ATXN1 |
Hypermethylation |
3.54×10−134 | Downregulated |
2.13×10−4 |
| CAV1 |
Hypermethylation |
1.31×10−133 | Downregulated |
7.54×10−26 |
| FLNA |
Hypermethylation |
8.64×10−126 | Downregulated |
1.12×10−15 |
| MAP3K14 |
Hypermethylation |
2.39×10−123 | Downregulated |
1.79×10−7 |
| PRKCB |
Hypermethylation |
1.24×10−133 | Downregulated |
6.56×10−20 |
| FHL2 |
Hypermethylation |
1.74×10−123 | Downregulated |
1.71×10−12 |
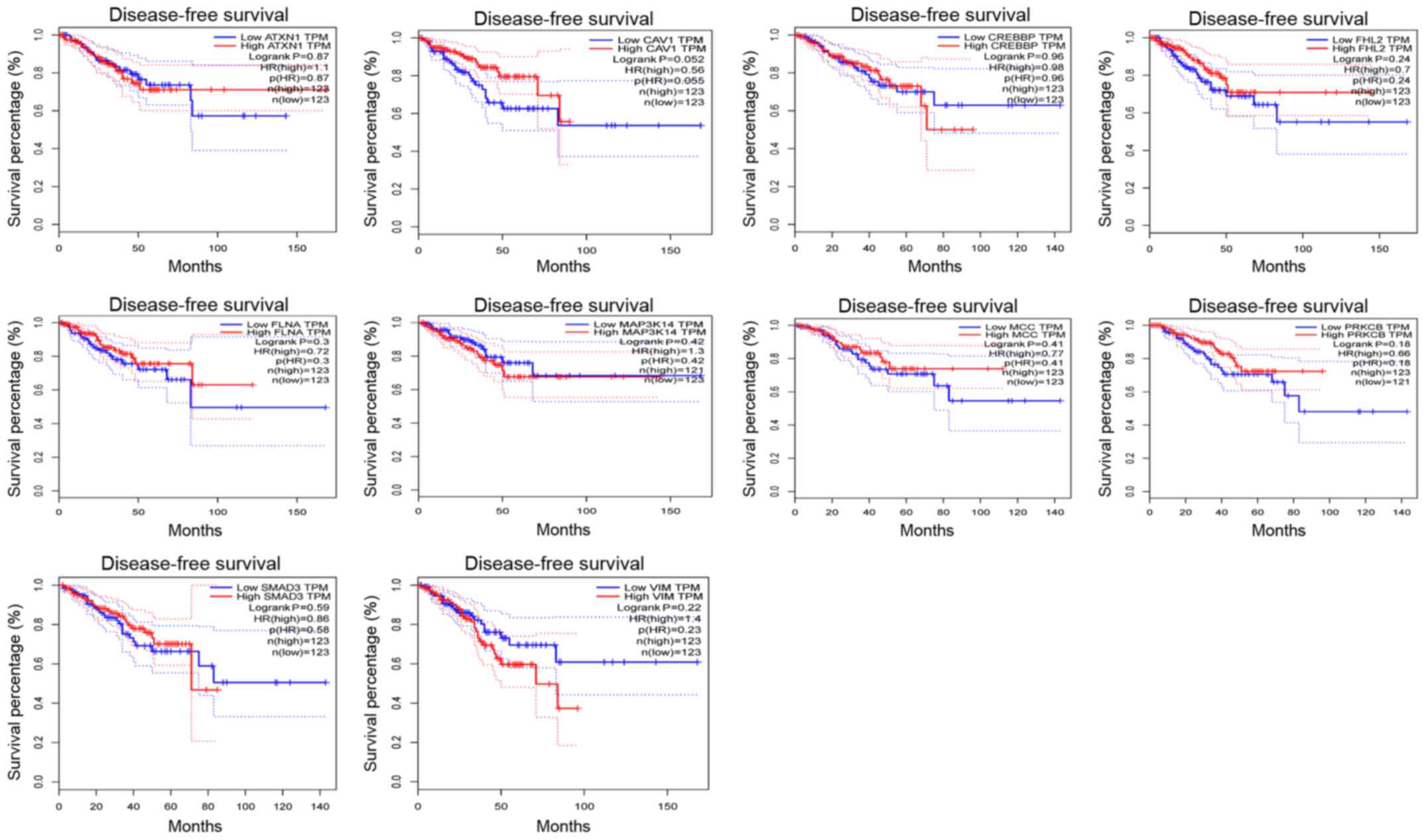
Survival analysis of the node
genes
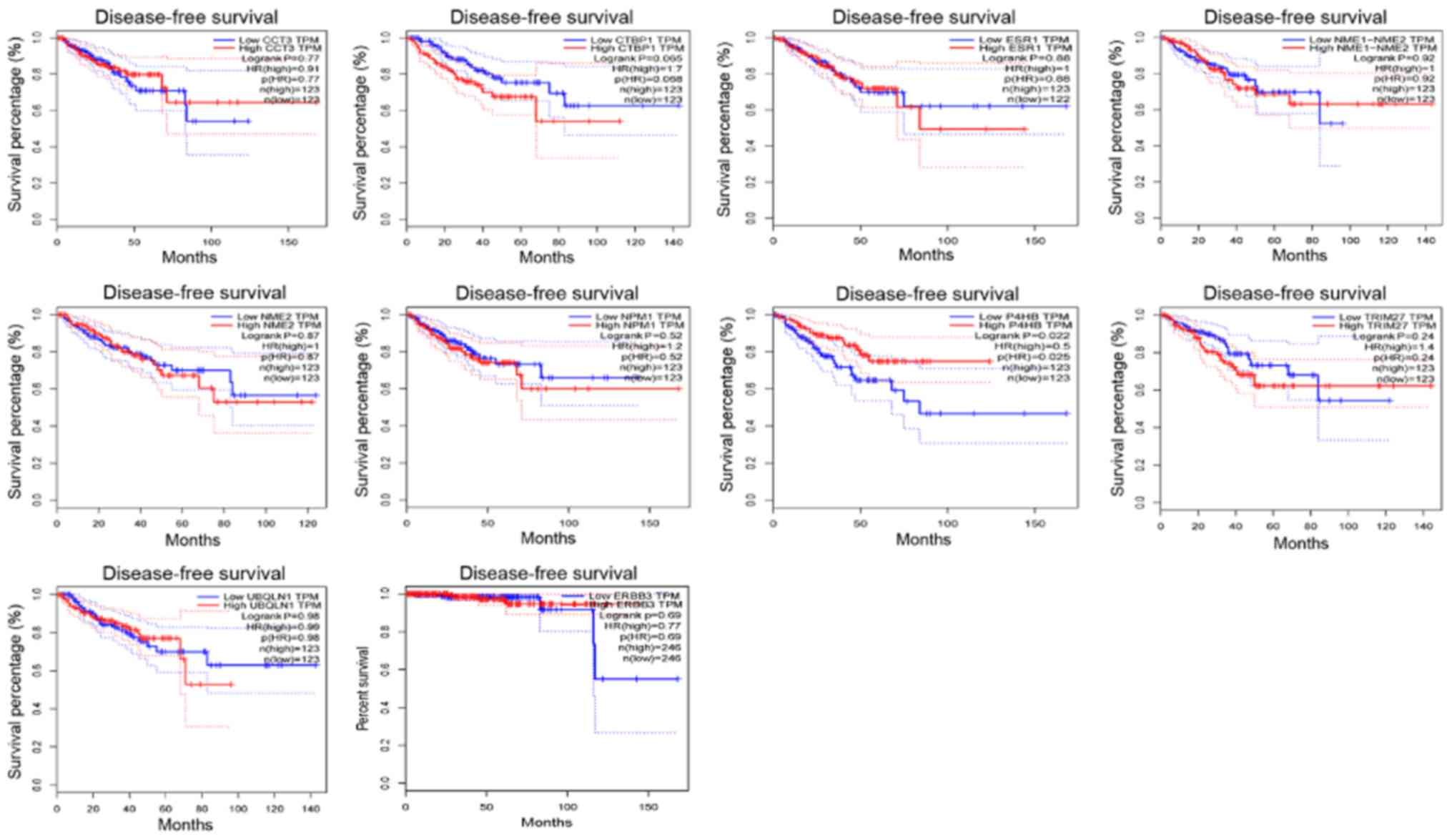
In addition, the Disease-Free Survival (RFS) method
of GEPIA online tool was used to analyze 20 aberrantly-methylated
genes. Group Cutoff was set as Quartile (Cutoff-High (%) was 75%
and Cutoff-Low (%) was 25%). The CI was 95%. Hypomethylated-high
expression genes (Fig. 9) and
hypermethylated-low expression genes (Fig. 10) were respectively represented in
red and blue colour. It was revealed that the survival analysis of
the P4HB gene exhibited a statistically significant difference in
survival between cancer and normal groups (log-rank P-value
0.022).
P4HB is a key protein of disulfide isomerase
(protein di-sulphideisomerase, PDI), which is also referred to as a
post-translational modifications collagen synthase, that is
involved in antioxidant and detoxification reactions. Its encoding
gene is on chromosome 17 q25 (22,23).
P4HB was revealed to be highly expressed in glioblastoma and liver
cancer (24,25). Notably, Xia et al (25) revealed that high expression of P4HB
in liver cancer tissues was associated with poor survival.
Discussion
In recent years, DNA methylation is a major part of
epigenetic modification and plays an important role in maintaining
chromosome stability and gene expression in mammals. It refers to
the process of transferring methyl groups to specific bases
catalyzed by DNA methyl transfer enzymes (DNMTs) using s-adenosine
methionine (SAM) as the methyl donor. Increasing studies (26) have revealed that aberrant DNA
methylation is intimately associated with tumorigenesis. It was
revealed that the overall methylation level of DNA in tumor cells
was lower than that in normal cells, however some specific gene CpG
islands were hypermethylated (27). DNA hypomethylation activates
proto-oncogenes and abnormal proliferation of cancer cells.
However, hypermethylation of CpG islands in the promoter region can
inhibit gene expression and inactivate tumor suppressor genes, thus
promoting the occurrence and development of tumors. Therefore,
methylation is considered to be another mechanism of tumorigenesis
(28). DNA methylation is not only
involved in the regulation of the cell cycle, proliferation,
apoptosis and metastasis, but also in the regulation of drug
resistance and intracellular signal transduction pathway of tumor
cells (29).
At present, some progress has been made in genetics
and in the molecular pathogenesis of PCa, however effective
diagnosis and treatment of PCa still require further advances
(30). In the present study, the
gene expression and methylation datasets of PCa were analyzed using
a variety of online tools. A total of 105 hypomethylation-high
expression genes and 561 hypermethylation-low expression genes were
obtained.
GO annotations of the hypomethylation-high
expression gene group in PCa predominantly included translational
initiation, translation, SRP-dependent co-translational protein
targeting to the membrane, ribosome biogenesis, regulation of
oxidative stress-induced intrinsic apoptotic signaling pathways,
viral transcription, GTPase activator activity, structural
constituents of ribosomes, protein histidine kinase activity,
protein homodimerization activity, protein binding, and aldehyde
dehydrogenase [NAD(P)+] activity. Multiple translation initiation
factors were regularly magnified or diminished in tumors, promoting
proliferation, survival, angiogenesis, and metastasis (31,32).
Ribosome biogenesis is a process closely correlated to cell growth
and proliferation, which are upregulated in the vast majority of
cancers including PCa. This leads to downregulation of the
expression and activity of p53, and thus promotes tumorigenesis
(33,34). As a GTPase activator, DOCK4 can
promote intercellular adhesion by activating Rap GTPase (35).
According to the PPI network for the
hypomethylation-high expression gene group constructed by
Cytoscape, the node degree of each gene was obtained and the top 10
node genes were selected, including ESR1, NPM1, NME2, NME1-NME2,
UBQLN1, CTBP1, TRIM27, CCT3, ERBB3, and P4HB. Toy et al
(36) suggested that activation of
the ESR1 ligand binding domain mutations had different effects on
the efficacy of estrogen receptor antagonists. Nucleophosmin (NPM1)
is generally overexpressed, mutated, and rearranged in cancer and
has been revealed to be overexpressed in PCa (37,38).
In addition, NPM1 was revealed to participate in the progression,
invasion, and metastasis of tumors in high-grade serous ovarian
adenocarcinoma (39).
Underexpression of NME2 can inhibit the metastasis of lung cancer
and other studies concluded that hypomethylation and high
expression of NME2 can cause apoptosis of the mouse testes cells
(40,41). UBQLN1 was revealed to be
overexpressed in gastric cancer. Reduced UBQLN1 expression can
cause enhanced cell migration and invasion, actin cytoskeleton
rearrangements, and stimulation of the epithelial-mesenchymal
transition (EMT) (42,43). Scholars have revealed that CTBP1 is
carcinogenic in PCa and other adenomas, and overexpression of CTBP1
is thought to be involved in cell survival, proliferation,
migration, invasion, and EMT (44–46).
TRIM27 was initially considered to be part of a fusion gene RET
(rearranged during transfection), which is a proto-oncogene that is
upregulated in multitudinal cancers, including PCa (47,48).
CCT3 was significantly correlated with cancer cell proliferation
and was revealed to be upregulated in osteosarcoma and
hepatocellular carcinoma (49–51).
ERBB3 is the only gene identified whose impaired kinase domain may
be involved in the progression and invasion of PCa (52,53).
P4HB was overexpressed in colon cancer and downregulation of
expression may promote cancer cell apoptosis (54).
For the hypermethylation-low expression gene group
in this PCa study, the BP revealed from the GO annotations were
associated with cell adhesion, extracellular matrix organization,
response to hypoxia, muscle contraction, positive regulation of
cell migration, and regulation of phosphatidylinositol 3-kinase
signaling. Loss of cell adhesion is one of the critical steps in
tumor progression (55). Several
studies have demonstrated that the extracellular matrix, hypoxia,
cell migration, and the phosphatidylinositol 3-kinase signaling
pathway may play crucial roles in cancer metastasis (56–58).
Furthermore, the phosphatidylinositol 3-kinase signaling pathway is
involved in tumorigenesis and progression (58). The molecular functions have
primarily focused on actin, integrin, collagen, scaffold protein,
and actin filament binding, as well as signal transducer activity.
Previous studies have revealed that actin-binding proteins have
inhibitory roles in PCa cell growth and metastasis (59,60).
Integrins play a pivotal role in cell adhesion by connecting the
cytoskeleton and extracellular matrix, promoting tumor cell
proliferation, metastasis, and invasion (61,62).
In the present study, the top 10
hypermethylation-low expression node genes that were obtained from
the PPI network included CREBBP, SMAD3, MCC, VIM, ATXN1, CAV1,
FLNA, MAP3K14, PRKCB, and FHL2. There is evidence that CREBBP is
downregulated in PCa and is a tumor suppressor gene in small-cell
lung cancer and other neuroendocrine tumors (63,64).
Smad3/Sox5/Twist1 was reported to promote EMT and cancer
progression in PCa (65). MCC, a
candidate tumor suppressor gene, was revealed to be often silenced
by hypermethylation of its promoter in colorectal cancer (66). Several studies have revealed that
the EMT marker, VIM, was positively associated with bone metastasis
in PCa, and its promoter is often hypermethylated in cervical
cancer cells (67,68). It has been reported that
downregulation of ATXN1 induced EMT in cervical cancer, but its
role in PCa remains to be elucidated (69). Studies have revealed that CAV1 can
promote growth and metastasis of PCa, and the hypermethylation of
its promoter could make CAV1 a tumor suppressor gene in PCa
(70–72). The results of FLNA expression are
consistent with those of down-regulation in PCa, indicating that
FLNA may play a critical role as a negative regulator in PCa
(73). MAP3K14, also known as NIK,
is a regulatory component of the mitochondrial division machinery
and has a dominant effect on cancer cell invasion (74). PRKCB is a member of the PKC family,
and when its expression is downregulated, cell apoptosis and
suppression of tumorigenesis occur (75,76).
A previous study reported that FHL2 could be considered as a PCa
biomarker (77).
GEPIA is a TCGA database-based online tool that can
be used to study the prognostic effects of genes in various
cancers. Survival analysis was conducted on 20
aberrantly-methylated genes which exhibit potential as diagnostic
markers of PCa using GEPIA. Notably, the expression of P4HB was the
only one among the 20 genes that was significantly correlated with
survival. P4HB is found to be highly expressed in glioblastoma and
liver cancer, decreasing the apoptosis of tumor cells (24,25).
Xia et al (25) have
revealed that high expression of P4HB in liver cancer tissues was
associated with poor survival. However, there is also evidence
suggesting that the low expression of P4HB could lead to proteasome
inhibition-induced autophagy (78), since the boundary between apoptosis
and autophagy has always been controversial due to tumor
heterogeneity (79). Based on the
present results revealing that the patients with hypermethylation
and low expression of P4HB had poor prognosis, it was hypothesized
that hypermethylation of P4HB in prostate cancer would activate
autophagy of prostate cancer cells and promote the tumor
progression. Further experiments are required to verify the
hypothesis.
To validate the present results, TCGA database, the
GEPIA tool, and the HPA database were employed to determine node
gene methylation and gene expression status, and the results were
generally consistent with our previous results, revealing the
reliability of this study. However, there are still several
limitations in the present study, including the focus on data
mining and analysis without experimental confirmation. In addition,
the methylation status of the node genes was only verified in TCGA
database. Therefore, further experiments are required to verify our
results.
In conclusion, the present study identified 20
aberrantly methylated-differentially expressed genes that may be
used as biomarkers in PCa. The bioinformatics approach included the
analysis of both gene expression and gene methylation microarrays,
providing a novel and practical approach for the diagnosis,
treatment, and prognosis of PCa. However, based on the limitations
of the present study, additional molecular biologal experiments are
required to confirm these findings.
It is anticipated that further study on the
relationship between DNA methylation and histone codes and their
mutual regulatory mechanism could provide new breakthroughs in the
gene regulation of tumor genesis. Prostate cancer has high
morbidity and mortality, however its pathogenesis has not been
fully elucidated, and its therapeutic effect is not satisfactory.
The study of the relationship between DNA methylation and histone
codes is crucial to improve understanding of some signaling
pathways which widely participate in cell physiology and pathology,
making subtle adjustments to the complex signaling network, yet
there have been few studies conducted on its clinical application.
With the deeper understanding of the role of DNA methylation in
tumor and the increasing number of high-throughput data of DNA
methylation in tumors, bioinformatics technology has laid the
foundation for the early diagnosis of tumors, the search for
molecular markers for prognosis evaluation and new tumor
therapeutic targets.
In the present study, high throughput microarray
data of biological databases was mined by bioinformatics
technology, to the best of our knowledge for the first time, to
determine the aberrantly-methylated biomarkers of prostate cancer.
The strategy applied was the selection of the intersecting high
methylation and low expression genes as biological markers. The low
methylation and high expression genes were analyzed by the same
procedure. The markers were further enriched to display their
related signaling pathways. Thus, the carcinogenic mechanism of
prostate cancer could be clarified, to provide theories for
clinical research and treatment.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
ZQ conceived and designed the present study. LW and
BW collected, extracted and analyzed the data. LW and BW wrote the
manuscript. LW and ZQ reviewed the final manuscript. All authors
read and approved the final manuscript and agree to be accountable
for all aspects of the research in ensuring that the accuracy or
integrity of any part of the work are appropriately investigated
and resolved.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
PCa
|
prostate cancer
|
|
DEGs
|
differentially expressed genes
|
|
GEO
|
Gene Expression Omnibus
|
|
PPI
|
protein-protein interaction
|
|
TCGA
|
The Cancer Genome Atlas
|
|
GEPIA
|
Gene Expression Profiling Interactive
Analysis
|
|
HPA
|
Human Protein Atlas
|
|
NCBI
|
National Center for Biotechnology
Information
|
|
DMGs
|
differentially methylated genes
|
|
GO
|
Gene Ontology
|
|
DAVID
|
Database for Annotation,
Visualization, and Integrated Discovery
|
|
BP
|
biological processes
|
|
EMT
|
epithelial-mesenchymal transition
|
References
|
1
|
Bray F, Ferlay J, Soerjomataram I, Siegel
RL, Torre LA and Jemal A: Global cancer statistics 2018: GLOBOCAN
estimates of incidence and mortality worldwide for 36 cancers in
185 countries. CA Cancer J Clin. 68:394–424. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Jemal A, Bray F, Center MM, Ferlay J, Ward
E and Forman D: Global cancer statistics. CA Cancer J Clin.
61:69–90. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Feinberg AP, Ohlsson R and Henikoff S: The
epigenetic progenitor origin of human cancer. Nat Rev Genet.
7:21–33. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Nephew KP and Huang TH: Epigenetic gene
silencing in cancer initiation and progression. Cancer Lett.
190:125–133. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Nakao M: Epigenetics: Interaction of DNA
methylation and chromatin. Gene. 278:25–31. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Strahl BD and Allis CD: The language of
covalent histone modifications. Nature. 403:41–45. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
De Carvalho DD, Sharma S, You JS, Su SF,
Taberlay PC, Kelly TK, Yang X, Liang G and Jones PA: DNA
methylation screening identifies driver epigenetic events of cancer
cell survival. Cancer Cell. 21:655–667. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Jones PA and Baylin SB: The fundamental
role of epigenetic events in cancer. Nat Rev Genet. 3:415–428.
2002. View
Article : Google Scholar : PubMed/NCBI
|
|
9
|
Mundbjerg K, Chopra S, Alemozaffar M,
Duymich C, Lakshminarasimhan R, Nichols PW, Aron M, Siegmund KD,
Ukimura O, Aron M, et al: Identifying aggressive prostate cancer
foci using a DNA methylation classifier. Genome Biol. 18:32017.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Xia D, Wang D, Kim SH, Katoh H and DuBois
RN: Prostaglandin E2 promotes intestinal tumor growth via DNA
methylation. Nat Med. 18:224–226. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Iizuka N, Oka M, Yamada-Okabe H, Nishida
M, Maeda Y, Mori N, Takao T, Tamesa T, Tangoku A, Tabuchi H, et al:
Oligonucleotide microarray for prediction of early intrahepatic
recurrence of hepatocellular carcinoma after curative resection.
Lancet. 361:923–929. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kulasingam V and Diamandis EP: Strategies
for discovering novel cancer biomarkers through utilization of
emerging technologies. Nat Clin Pract Oncol. 5:588–599. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Verma M, Khoury MJ and Ioannidis JP:
Opportunities and challenges for selected emerging technologies in
cancer epidemiology: Mitochondrial, epigenomic, metabolomic, and
telomerase profiling. Cancer Epidemiol Biomarkers Prev. 22:189–200.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lu W and Ding Z: Identification of key
genes in prostate cancer gene expression profile by bioinformatics.
Andrologia. 51:e131692019. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ye Y, Li SL and Wang SY: Construction and
analysis of mRNA, miRNA, lncRNA, and TF regulatory networks reveal
the key genes associated with prostate cancer. PLoS One.
13:e01980552018. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
He Z, Tang F, Lu Z, Huang Y, Lei H, Li Z
and Zeng G: Analysis of differentially expressed genes, clinical
value and biological pathways in prostate cancer. Am J Transl Res.
10:1444–1456. 2018.PubMed/NCBI
|
|
17
|
Barrett T, Wilhite SE, Ledoux P,
Evangelista C, Kim IF, Tomashevsky M, Marshall KA, Phillippy KH,
Sherman PM, Holko M, et al: NCBI GEO: Archive for functional
genomics data sets-update. Nucleic Acids Res. 41 (Database
Issue):D991–D995. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Pathan M, Keerthikumar S, Ang CS, Gangoda
L, Quek CY, Williamson NA, Mouradov D, Sieber OM, Simpson RJ, Salim
A, et al: FunRich: An open access standalone functional enrichment
and interaction network analysis tool. Proteomics. 15:2597–2601.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Lin P, He RQ, Dang YW, Wen DY, Ma J, He Y,
Chen G and Yang H: An autophagy-related gene expression signature
for survival prediction in multiple cohorts of hepatocellular
carcinoma patients. Oncotarget. 9:17368–17395. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Morris JH, Wu A, Yamashita RA,
Marchler-Bauer A and Ferrin TE: cddApp: A Cytoscape app for
accessing the NCBI conserved domain database. Bioinformatics.
31:134–136. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Shannon P, Markiel A, Ozier O, Baliga NS,
Wang JT, Ramage D, Amin N, Schwikowski B and Ideker T: Cytoscape: A
software environment for integrated models of biomolecular
interaction networks. Genome Res. 13:2498–2504. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Galligan JJ and Petersen DR: The human
protein disulfide isomerase gene family. Hum Genomics. 6:62012.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Pajunen L, Jones TA, Goddard A, Sheer D,
Solomon E, Pihlajaniemi T and Kivirikko KI: Regional assignment of
the human gene coding for a multifunctional polypeptide (P4HB)
acting as the beta-subunit of prolyl 4-hydroxylase and the enzyme
protein disulfide isomerase to 17q25. Cytogenet Cell Genet.
56:165–168. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Goplen D, Wang J, Enger PØ, Tysnes BB,
Terzis AJ, Laerum OD and Bjerkvig R: Protein disulfide isomerase
expression is related to the invasive properties of malignant
glioma. Cancer Res. 66:9895–9902. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Xia W, Zhuang J, Wang G, Ni J, Wang J and
Ye Y: P4HB promotes HCC tumorigenesis through downregulation of
GRP78 and subsequent upregulation of epithelial-to-mesenchymal
transition. Oncotarget. 8:8512–8521. 2017.PubMed/NCBI
|
|
26
|
Li Y and Tollefsbol TO: Impact on DNA
methylation in cancer prevention and therapy by bioactive dietary
components. Curr Med Chem. 17:2141–2151. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Ehrlich M: DNA methylation in cancer: Too
much, but also too little. Oncogene. 21:5400–5413. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Davis CD and Uthus EO: DNA methylation,
cancer susceptibility, and nutrient interactions. Exp Biol Med
(Maywood). 229:988–995. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Toyota M, Itoh F and Imai K: DNA
methylation and gastrointestinal malignancies: Functional
consequences and clinical implications. J Gastroenterol.
35:727–734. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Bjurlin MA and Taneja SS: Prostate cancer.
Urol Clin North Am. 44:xv–xvi. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Ruggero D: Translational control in cancer
etiology. Cold Spring Harb Perspect Biol. 5(pii):
a0123362013.PubMed/NCBI
|
|
32
|
Chu J, Cargnello M, Topisirovic I and
Pelletier J: Translation initiation factors: Reprogramming protein
synthesis in cancer. Trends Cell Biol. 26:918–933. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Ray S, Johnston R, Campbell DC, Nugent S,
McDade SS, Waugh D and Panov KI: Androgens and estrogens stimulate
ribosome biogenesis in prostate and breast cancer cells in receptor
dependent manner. Gene. 526:46–53. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Derenzini M, Montanaro L and Trerè D:
Ribosome biogenesis and cancer. Acta Histochem. 119:190–197. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Yajnik V, Paulding C, Sordella R,
McClatchey AI, Saito M, Wahrer DC, Reynolds P, Bell DW, Lake R, van
den Heuvel S, et al: DOCK4, a GTPase activator, is disrupted during
tumorigenesis. Cell. 112:673–684. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Toy W, Weir H, Razavi P, Lawson M,
Goeppert AU, Mazzola AM, Smith A, Wilson J, Morrow C, Wong WL, et
al: Activating ESR1 mutations differentially affect the efficacy of
ER antagonists. Cancer Discov. 7:277–287. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Box JK, Paquet N, Adams MN, Boucher D,
Bolderson E, O'Byrne KJ and Richard DJ: Nucleophosmin: From
structure and function to disease development. BMC Mol Biol.
17:192016. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Destouches D, Sader M, Terry S, Marchand
C, Maillé P, Soyeux P, Carpentier G, Semprez F, Céraline J, Allory
Y, et al: Implication of NPM1 phosphorylation and preclinical
evaluation of the nucleoprotein antagonist N6L in prostate cancer.
Oncotarget. 7:69397–69411. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Fan X, Wen L, Li Y, Lou L, Liu W and Zhang
J: The expression profile and prognostic value of APE/Ref-1 and
NPM1 in high-grade serous ovarian adenocarcinoma. APMIS.
125:857–862. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Thakur RK, Yadav VK, Kumar A, Singh A, Pal
K, Hoeppner L, Saha D, Purohit G, Basundra R, Kar A, et al:
Non-metastatic 2 (NME2)-mediated suppression of lung cancer
metastasis involves transcriptional regulation of key cell adhesion
factor vinculin. Nucleic Acids Res. 42:11589–11600. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Gu Y, Xu W, Nie D, Zhang D, Dai J, Zhao X,
Zhang M, Wang Z, Chen Z and Qiao Z: Nicotine induces Nme2-mediated
apoptosis in mouse testes. Biochem Biophys Res Commun. 472:573–579.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Bao J, Jiang X, Zhu X, Dai G, Dou R, Liu
X, Sheng H, Liang Z and Yu H: Clinical significance of ubiquilin 1
in gastric cancer. Medicine. 97:e97012018. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Shah PP, Lockwood WW, Saurabh K, Kurlawala
Z, Shannon SP, Waigel S, Zacharias W and Beverly LJ: Ubiquilin1
represses migration and epithelial-to-mesenchymal transition of
human non-small cell lung cancer cells. Oncogene. 34:1709–1717.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Porretti J, Dalton GN, Massillo C, Scalise
GD, Farré PL, Elble R, Gerez EN, Accialini P, Cabanillas AM,
Gardner K, et al: CLCA2 epigenetic regulation by CTBP1, HDACs,
ZEB1, EP300 and miR-196b-5p impacts prostate cancer cell adhesion
and EMT in metabolic syndrome disease. Int J Cancer. 143:897–906.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Phelps RA, Chidester S, Dehghanizadeh S,
Phelps J, Sandoval IT, Rai K, Broadbent T, Sarkar S, Burt RW and
Jones DA: A two-step model for colon adenoma initiation and
progression caused by APC loss. Cell. 137:623–634. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Blevins MA, Huang M and Zhao R: The role
of CtBP1 in oncogenic processes and its potential as a therapeutic
target. Mol Cancer Ther. 16:981–990. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Ma Y, Wei Z, Bast RC Jr, Wang Z, Li Y, Gao
M, Liu Y and Wang X, Guo C, Zhang L and Wang X: Downregulation of
TRIM27 expression inhibits the proliferation of ovarian cancer
cells in vitro and in vivo. Lab Invest. 96:37–48. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Shaikhibrahim Z, Lindstrot A, Ochsenfahrt
J, Fuchs K and Wernert N: Epigenetics-related genes in prostate
cancer: Expression profile in prostate cancer tissues,
androgen-sensitive and -insensitive cell lines. Int J Mol Med.
31:21–25. 2013.PubMed/NCBI
|
|
49
|
Zhang Y, Wang Y, Wei Y, Wu J, Zhang P,
Shen S, Saiyin H, Wumaier R, Yang X, Wang C and Yu L: Molecular
chaperone CCT3 supports proper mitotic progression and cell
proliferation in hepatocellular carcinoma cells. Cancer Lett.
372:101–109. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Xiong Y, Wu S, Du Q, Wang A and Wang Z:
Integrated analysis of gene expression and genomic aberration data
in osteosarcoma (OS). Cancer Gene Ther. 22:524–529. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Cui X, Hu ZP, Li Z, Gao PJ and Zhu JY:
Overexpression of chaperonin containing TCP1, subunit 3 predicts
poor prognosis in hepatocellular carcinoma. World J Gastroenterol.
21:8588–8604. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Jaiswal BS, Kljavin NM, Stawiski EW, Chan
E, Parikh C, Durinck S, Chaudhuri S, Pujara K, Guillory J, Edgar
KA, et al: Oncogenic ERBB3 mutations in human cancers. Cancer Cell.
23:603–617. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Koumakpayi IH, Le Page C, Delvoye N, Saad
F and Mes-Masson AM: Macropinocytosis inhibitors and Arf6 regulate
ErbB3 nuclear localization in prostate cancer cells. Mol Carcinog.
50:901–912. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Zhou Y, Yang J, Zhang Q, Xu Q, Lu L, Wang
J and Xia W: P4HB knockdown induces human HT29 colon cancer cell
apoptosis through the generation of reactive oxygen species and
inactivation of STAT3 signaling. Mol Med Rep. 19:231–237.
2019.PubMed/NCBI
|
|
55
|
Le Bras GF, Taubenslag KJ and Andl CD: The
regulation of cell-cell adhesion during epithelial-mesenchymal
transition, motility and tumor progression. Cell Adh Migr.
6:365–373. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Gilkes DM, Semenza GL and Wirtz D: Hypoxia
and the extracellular matrix: Drivers of tumour metastasis. Nat Rev
Cancer. 14:430–439. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Jacquemet G, Hamidi H and Ivaska J:
Filopodia in cell adhesion, 3D migration and cancer cell invasion.
Curr Opin Cell Biol. 36:23–31. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Liu X, Xu Y, Zhou Q, Chen M, Zhang Y,
Liang H, Zhao J, Zhong W and Wang M: PI3K in cancer: Its structure,
activation modes and role in shaping tumor microenvironment. Future
Oncol. 14:665–674. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Haffner MC, Esopi DM, Chaux A, Gürel M,
Ghosh S, Vaghasia AM, Tsai H, Kim K, Castagna N, Lam H, et al: AIM1
is an actin-binding protein that suppresses cell migration and
micrometastatic dissemination. Nat Commun. 8:1422017. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Vanaja DK, Grossmann ME, Cheville JC, Gazi
MH, Gong A, Zhang JS, Ajtai K, Burghardt TP and Young CY: PDLIM4,
an actin binding protein, suppresses prostate cancer cell growth.
Cancer Invest. 27:264–272. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Ramovs V, Te Molder L and Sonnenberg A:
The opposing roles of laminin-binding integrins in cancer. Matrix
Biol. 57-58:213–243. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Desgrosellier JS and Cheresh DA: Integrins
in cancer: Biological implications and therapeutic opportunities.
Nat Rev Cancer. 10:9–22. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Jia D, Augert A, Kim DW, Eastwood E, Wu N,
Ibrahim AH, Kim KB, Dunn CT, Pillai SPS, Gazdar AF, et al: Crebbp
loss drives small cell lung cancer and increases sensitivity to
HDAC inhibition. Cancer Discov. 8:1422–1437. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Shaikhibrahim Z, Lindstrot A, Buettner R
and Wernert N: Analysis of laser-microdissected prostate cancer
tissues reveals potential tumor markers. Int J Mol Med. 28:605–611.
2011.PubMed/NCBI
|
|
65
|
Hu J, Tian J, Zhu S, Sun L, Yu J, Tian H,
Dong Q, Luo Q, Jiang N, Niu Y and Shang Z: Sox5 contributes to
prostate cancer metastasis and is a master regulator of
TGF-β-induced epithelial mesenchymal transition through controlling
Twist1 expression. Br J Cancer. 118:88–97. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Kohonen-Corish MR, Sigglekow ND, Susanto
J, Chapuis PH, Bokey EL, Dent OF, Chan C, Lin BP, Seng TJ, Laird
PW, et al: Promoter methylation of the mutated in colorectal cancer
gene is a frequent early event in colorectal cancer. Oncogene.
26:4435–4441. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Lang SH, Hyde C, Reid IN, Hitchcock IS,
Hart CA, Bryden AA, Villette JM, Stower MJ and Maitland NJ:
Enhanced expression of vimentin in motile prostate cell lines and
in poorly differentiated and metastatic prostate carcinoma.
Prostate. 52:253–263. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Jung S, Yi L, Kim J, Jeong D, Oh T, Kim
CH, Kim CJ, Shin J, An S and Lee MS: The role of vimentin as a
methylation biomarker for early diagnosis of cervical cancer. Mol
Cells. 31:405–411. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Kang AR, An HT, Ko J and Kang S: Ataxin-1
regulates epithelial-mesenchymal transition of cervical cancer
cells. Oncotarget. 8:18248–18259. 2017.PubMed/NCBI
|
|
70
|
Li L, Yang G, Ebara S, Satoh T, Nasu Y,
Timme TL, Ren C, Wang J, Tahir SA and Thompson TC: Caveolin-1
mediates testosterone-stimulated survival/clonal growth and
promotes metastatic activities in prostate cancer cells. Cancer
Res. 61:4386–4392. 2001.PubMed/NCBI
|
|
71
|
Tahir SA, Yang G, Goltsov AA, Watanabe M,
Tabata K, Addai J, Fattah el MA, Kadmon D and Thompson TC: Tumor
cell-secreted caveolin-1 has proangiogenic activities in prostate
cancer. Cancer Res. 68:731–739. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Cui J, Rohr LR, Swanson G, Speights VO,
Maxwell T and Brothman AR: Hypermethylation of the caveolin-1 gene
promoter in prostate cancer. Prostate. 46:249–256. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Sun GG, Lu YF, Zhang J and Hu WN: Filamin
A regulates MMP-9 expression and suppresses prostate cancer cell
migration and invasion. Tumour Biol. 35:3819–3826. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Jung JU, Ravi S, Lee DW, McFadden K,
Kamradt ML, Toussaint LG and Sitcheran R: NIK/MAP3K14 regulates
mitochondrial dynamics and trafficking to promote cell invasion.
Curr Biol. 26:3288–3302. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Hagiwara K, Ito H, Murate T, Miyata Y,
Ohashi H and Nagai H: PROX1 overexpression inhibits protein kinase
C beta II transcription through promoter DNA methylation. Genes
Chromosomes Cancer. 51:1024–1036. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Surdez D, Benetkiewicz M, Perrin V, Han
ZY, Pierron G, Ballet S, Lamoureux F, Rédini F, Decouvelaere AV,
Daudigeos-Dubus E, et al: Targeting the EWSR1-FLI1 oncogene-induced
protein kinase PKC-β abolishes ewing sarcoma growth. Cancer Res.
72:4494–4503. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Kahl P, Gullotti L, Heukamp LC, Wolf S,
Friedrichs N, Vorreuther R, Solleder G, Bastian PJ, Ellinger J,
Metzger E, et al: Androgen receptor coactivators lysine-specific
histone demethylase 1 and four and a half LIM domain protein 2
predict risk of prostate cancer recurrence. Cancer Res.
66:11341–11347. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Rui YN, Xu Z, Chen Z and Zhang S: The
GST-BHMT assay reveals a distinct mechanism underlying proteasome
inhibition-induced macroautophagy in mammalian cells. Autophagy.
11:812–832. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Ouyang L, Shi Z, Zhao S, Wang FT, Zhou TT,
Liu B and Bao JK: Programmed cell death pathways in cancer: A
review of apoptosis, autophagy and programmed necrosis. Cell
Prolif. 45:487–498. 2012. View Article : Google Scholar : PubMed/NCBI
|