Introduction
Advances in the treatment of patients with acute
myocardial infarction, through coronary reperfusion, have led to a
decrease in hospital mortality rates. However, the injury generated
by the reestablishment of blood flow, known as reperfusion injury
(RI), still represents a great challenge (1,2).
There is strong evidence that reactive oxygen species (ROS) and
reactive nitrogen species (RNS), produced during the period of
ischemia, trigger an increased production of these reactive species
at the time of reperfusion (3–5).
As a way to increase myocardial tolerance to
ischemia, approaches such as pre-conditioning and post-conditioning
have been proposed; among these are oxidative stress: Hypoxic,
ischemic and hyperoxic (3,6). Hyperbaric therapy (HBO) has also been
used in different clinical conditions in which ischemia plays a
considerable role in tissue injury (7). The Undersea and Hyperbaric Medical
Society (North Palm Beach, FL, USA) (8) defines HBO as an intervention in which
an individual breathes nearly 100% oxygen, intermittently, inside a
hyperbaric chamber that is pressurized to greater than sea level
pressure (1 atmosphere absolute, or ATA). The benefits of 100%
oxygen therapy administered at pressures of 2.5 ATA result from the
increase in the volume of O2 dissolved in plasma from
0.3% at 1 ATA to more than 3.5% at 2.5 ATA, resulting in a 3- to
4-fold increase in the diffusion distance of O2 in
tissues (9–11). Thus, improvement in tissue oxygen
delivery provided by HBO therapy may limit the evolution of
necrosis during the event of myocardial ischemia.
Paradoxically, the increase in O2,
offered by HBO, can intensify oxidative stress due to ROS elevation
(12). Notwithstanding, a growing
body of evidence suggests that cell signaling mediated by increased
production of ROS results in increased activation of antioxidant
enzymes, increasing the tolerance of at-risk tissue as has been
described with cardiac cells (4,5,13–17).
In our previous study in a rat model, we reported
that application of HBO, after induction of acute myocardial
infarction (MI), promoted a decrease in the size of the infarct and
an increase in rat survival rate (18). In their study of HBO therapy, also
in a rat model, Guadalupe et al (19) reported higher values of the total
antioxidant response and 3-nitrotyrosine in the zone of tissue
damage of the left heart, compared to animals not treated with HBO.
We identified three other studies in which HBO was used for
post-conditioning. Kuhn et al (20) used a rat model of coronary
occlusion, using systemic embolization, reporting a decrease in the
mortality rate of rats treated with HBO, compared to no treatment.
Using a dog model, Mogelson et al (21) reported on the benefits of
therapeutic HBO in improving the outcomes of cardiac infarction.
Thomas et al (22) compared
the outcomes of HBO to rTPA therapy in a dog model of cardiac
infarct, concluding that all forms of treatment decrease the
severity of injury, with combined HBO therapy and recombinant
tissue plasminogen activator (rTPA) treatment providing maximal
recovery.
In humans, Yogaratnam et al (23) demonstrated that preconditioning of
patients with coronary heart disease using HBO, prior to on-pump
cardiac surgery, improved ventricular ejection and reduced
myocardial injury. Shandling et al (24), Stavitsky et al (25), Dekleva et al (26) and Vlahović et al (27) reported the benefits of HBO
treatment on cardiac function in patients with thrombolysis who
sustained MI. Zhdanov and Sokolov (28) reported that HBO therapy, combined
with conventional therapy for MI, effectively liquidated hypoxia
and improved the contractile and pumping function of the heart.
However, two recent studies (7,12)
indicated that the treatment outcomes of HBO therapy for the
treatment of patients with MI remains to be fully defined.
The objective of the present study was to
investigate the changes in the redox system associated with HBO
therapy maintained during the first hour after coronary occlusion
in an MI rat model. We analyzed the influence of HBO at the end of
the first hour after coronary occlusion, considering that, in the
rat, this period is sufficient to cause necrosis of the entire risk
area (29,30).
Materials and methods
Animals
Male Wistar rats weighing 250–330 g (11–12 weeks of
age) from the Central Animal Facilities of our institution were
used. The animals were housed under a 12-h light/dark cycle, at
22–23°C and 54–55% humidity. Rats were fed a pellet rodent diet
(Nuvilab CR1, manufactured by Nuvital, Curitiba, Brazil), ad
libitum, and had free access to water. Surviving male rats
(n=105) were randomly assigned to one of three groups: Sham (SH;
n=26), Myocardial infarction (MI; n=45) and submitted to Hyperbaric
therapy (HBO; n=34). Animals in the SH and MI groups were
maintained under oxygenation at ambient pressure for the same time
as the treated group.
All procedures were performed according to the
principles of ethics of animal experimentation adopted by the
Brazilian College of Animal Experimentation (COBEA-www.cobea.org.br), in conformity with the Guide for
the Care and Use of Laboratory Animals published by the US National
Institutes of Health (NIH publication no. 85-23, revised 1996). The
protocol of the present study was approved by the Research Ethics
Committee (CEP 0891/09) of the Federal University of São Paulo.
The rats were anesthetized with halothane, intubated
and ventilated with a ventilator for rodents (model 683; Harvard
Apparatus Co., South Natik, MA, USA). The rats were randomized
between the Sham (SH), myocardial infarction (MI) and hyperbaric
therapy (HBO) groups. MI was produced in the animals of the MI and
HBO groups by ligation of the anterior descending coronary artery,
as described previously (31,32).
Sham rats underwent a similar procedure, without coronary
ligation.
All experiments were initiated at the same time
(13:00) in order to avoid influence of the different biological
rhythms.
Hyperbaric therapy
Immediately after recovery from anesthesia
(approximately 2 min), animals from the HBO group were exposed to
100% O2 under a pressure of 2.5 atmospheres absolute
(ATA) for 60 min. within a hyperbaric chamber for small rodents, as
previously described (18). The
gas in the chamber was continuously vented to avoid CO2
retention (<0.1%) and the temperature was maintained in the
range of 25–27°C. Compression and decompression were performed at
the rate of 0.2 ATA/min (20 kPa/min) and the pressure inside the
chamber was stable over a period of 12 to 15 min.
Animals in the SH and MI groups were maintained
under oxygenation at ambient pressure for the same time as the
treated group. At the end of the protocol, the surviving rats
(SH=26, MI=45, and HBO=34). were sacrificed under halothane
anesthesia and each heart was removed and sectioned transversely at
the middle of the left ventricle (LV). The basal half was used to
measure the infarct size, with triphenyltetrazolium (TTZ),
according to a previously described protocol (18). The apical half was prepared in gel
(OCT-Sakura, Japan) and frozen with liquid nitrogen for cryostat
sectioning. The samples were stored at −80°C for further analysis
by microscopy.
Western blot analysis
Proteins were extracted as previously described
(33). Homogenate protein samples
of 30 µg were subjected to SDS-PAGE on 12% polyacrylamide gel. The
separated proteins were transferred onto hydrophobic polyvinylidene
difluoride membranes (Hybond-P, Amersham Biosciences), and the
transfer efficiency was examined with 0.5% Ponceau S. The membranes
were soaked in a blocking buffer (5% nonfat dry milk and 0.1% Tween
20 in PBS, pH 7.5) for 1 h at room temperature and then incubated
overnight at 4°C with primary antibodies: Mouse monoclonal
anti-catalase (dilution 1:1,000, Sigma-Aldrich; Merck KGaA, catalog
number C0979), rabbit polyclonal anti-nitrotyrosine (dilution
1:2,000, EMD Millipore, catalog number 06-284), rabbit polyclonal
anti-superoxide dismutase 1 (dilution 1:2,000; Abcam, catalog
number ab16831), and rabbit polyclonal anti-peroxiredoxin (dilution
1:2,000, PrxSO2/3, Abcam, catalog number ab16830), which
recognizes sulfinic (-SO2) and sulfonic (SO3)
forms of Prx I to IV; mouse monoclonal anti-GAPDH (dilution
1:2,000, Abcam, catalog number ab8245). After incubation, the
membranes were washed three times and then incubated for 1 h at
room temperature with horseradish peroxidase-conjugated secondary
antibodies (dilution 1:5,000; Abcam, catalog number ab97051).
Detection was performed with chemiluminescence reagents (GE
Healthcare) and values for target protein were normalized to GAPDH.
Dot blot method for nitrotyrosine was analyzed as previously
described (34). Samples were
transferred onto a nitrocellulose membrane by vacuum for dot blot
fluorescence detection performed using Odyssey (LI-COR
Biosciences); intensities of dots were determined using Image J
1.43u software (available at http://imagej.nih.gov/ij/, developed by Wayne Rasband,
National Institutes of Health).
Determination of intracellular
glutathione concentration
Samples were homogenized in 100 mM phosphate buffer
(pH 7.0; 250 µl/10 mg tissue) and centrifuged (12,000 × g for 10
min, at 4°C). To the supernatant (200 µl), 5% of sulphosalicylic
acid (200 µl) was added to precipitate the proteins. Detect
X® kit (Arbor Assays, Ann Arbor, MI, USA) was then
utilized to quantify the total concentration of glutathione (GSH).
After incubating the mixture with ThioStar® (Detect X,
Arbor Assays) for 15 min, at room temperature, the fluorescence was
determined (excitation 390 nm, emission 510 nm), using a
spectrophotometer (U-2810; Hitachi), and the reduced GSH
concentration measured.
Subsequently, a reaction mixture was added to
convert all oxidized glutathione (GSSG) into free GSH, which then
reacted with the excess ThioStar® to yield the signal
related to the total GSH content. A standard curve was used to
calculate the total and reduced glutathione concentrations. The
oxidized glutathione concentration was obtained using the following
calculation: GSSG=(total GSH-reduced GSH)/2. Detection limits
ranged between 38 nM, for free GSH, and 42 nM, for the total GSH.
Results were normalized to the muscle fragment weight for
between-animal comparisons. All assays were performed in
triplicate.
In situ ROS generation
In situ microfluorotopography of
dihydroethidium (DHE) oxidation products was performed as
previously described (35), with 3
µmol/l final DHE concentration. Slides were analyzed by confocal
microscopy (Zeiss LSM510) with laser excitation at 488 nm and
emission at 610 nm. Controls, performed by incubating slides for 30
min with Peg-SOD (500 U/ml), indicated preferential detection of
superoxide with DHE. Quantitative analysis of fluorescence images
was performed with Leica Qwin Plus (Leica Microsystems Ltd.,
Switzerland) software.
Quantification of the by-products of
nitric oxide
Myocardial homogenates were prepared under liquid
N2. After centrifugation (13,400 × g for 20 min, at
4°C), 20 µl aliquots were injected into NOA (Nitric Oxide Analyzer
model 280; Sievers Instruments, USA), with VCl3 and HCl
(at 95°C) used as reductants, as previously described (36). Nitric oxide (•NO), nitrite
(NO2−) and nitrate
(NO3−) by-products were normalized for
protein concentration.
Statistical analysis
The data are expressed as mean ± SEM. The Student
t-test was used for comparisons of infarct sizes and the Chi-square
test was used to compare mortality. Two-way ANOVA was applied to
parametric data using Newman-Keuls to identify statistical
differences. Kruskal-Wallis was performed on non-parametric data,
associated with the Dunn's test to identify statistical
differences. The statistical program used was GraphPad Prism 6.0
(GraphPad Software Inc., San Diego, CA, USA). Differences with
P≤0.05 were considered significant.
Results
Mortality and myocardial infarction
size
Immediate mortality was established when it occurred
in the period between the introduction of the animal into the HBO
chamber until the end of the decompression (~90 min). In the MI
group, 27 animals died during this period, whereas HBO therapy was
effective in improving post-infarction survival, with only 6
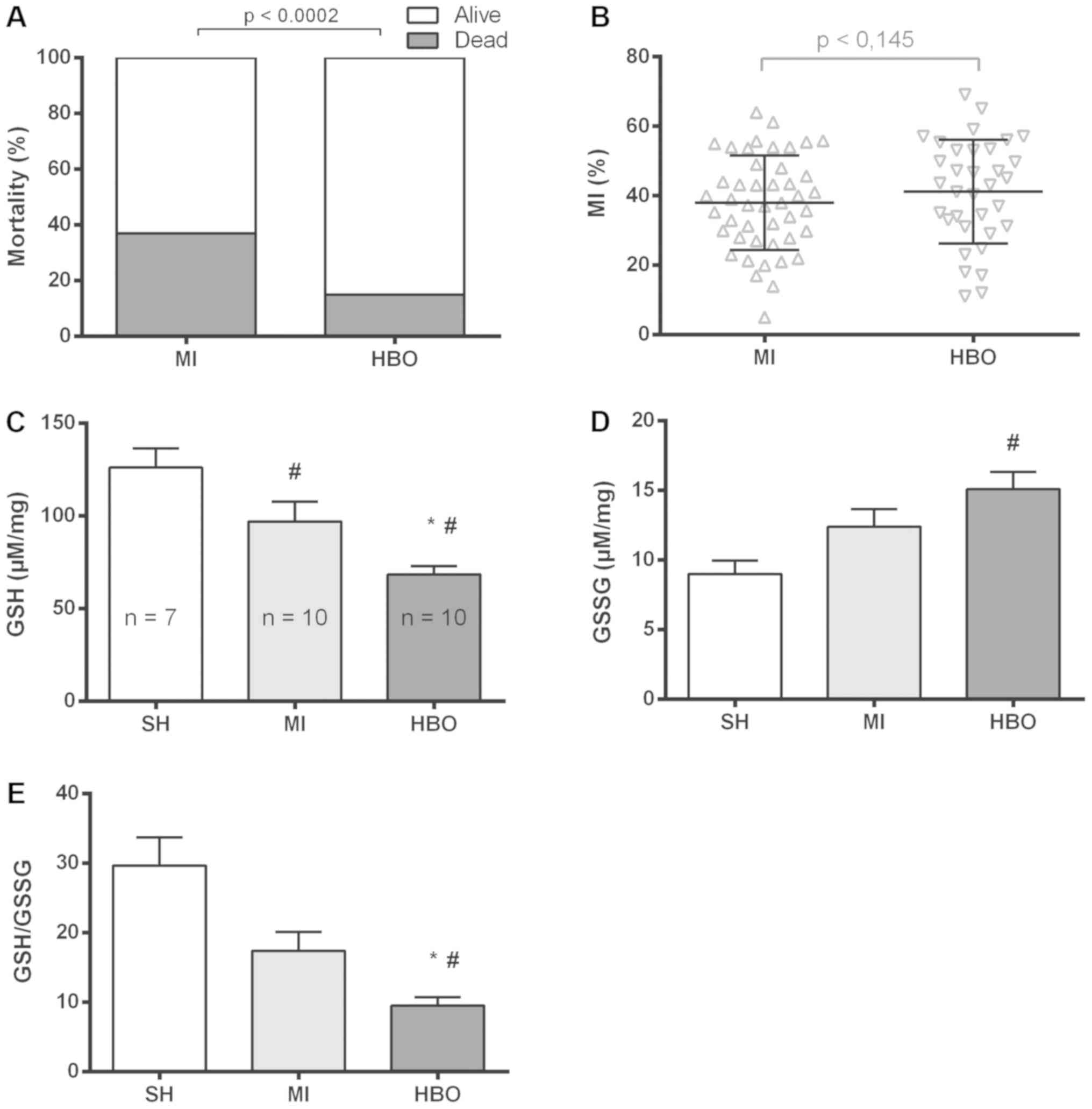
immediate deaths noted. Fig. 1
shows the percentage of mortality for all animals submitted to the
protocol, showing a clearly lower rate of mortality in the HBO
group (15%) than that in the MI group (37.5%). Additionally, no
significant difference in infarct size was observed (Fig. 1B) between the experimental groups
(MI 38±2.0% vs. HBO 43±2.5%).
Intracellular glutathione
concentration
In order to investigate whether redox processes are
involved in the protective effect induced by HBO therapy, we first
addressed the redox status in LV homogenates, by measuring the
amount of reduced and oxidized glutathione. The infarcted groups
showed (Fig. 1C) a reduction in
the GSH redox buffer (MI, 97±11 and HBO, 68±5 µM/mg) when compared
to the SH group (126±10 µM/mg). However, disulfide (Fig. 1D) was significantly increased only
in the HBO group (15±1 µg/mg) when compared to that in the SH (9±1
µM/mg) and MI (12±1 µM/mg) groups. The GSH/GSSG ratio showed that
the values in the HBO group (10±1) underwent a greater alteration
of this intracellular redox buffer, when compared to those of SH
(30±4) and MI (17±3) groups (Fig.
1E). There was no statistical significance in the comparison of
SH and MI groups (Fig. 1E).
Therefore, these results indicate that myocardial infarct decreases
the amount of reduced glutathione and HBO therapy accentuates such
a pro-oxidizing effect.
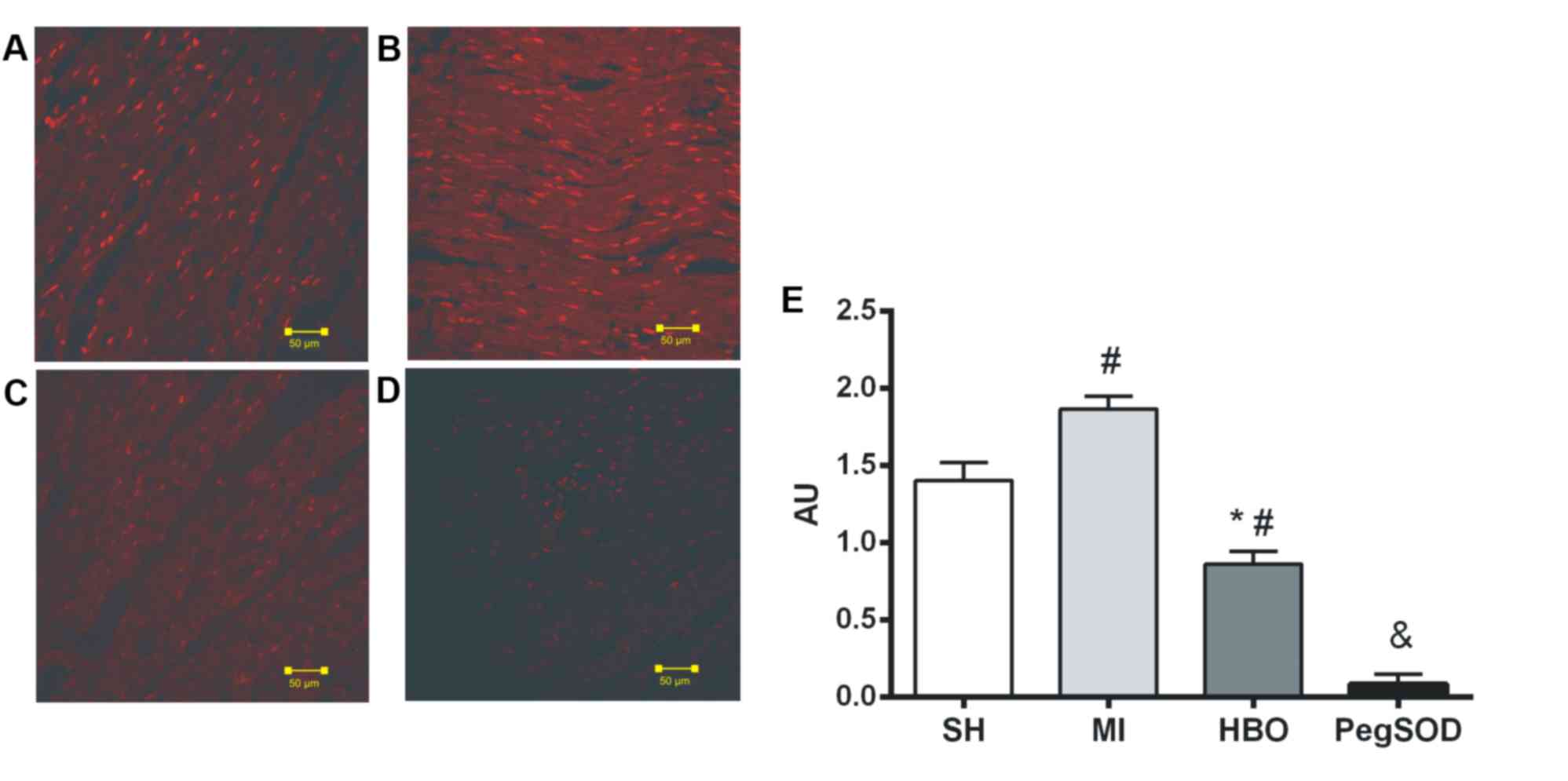
ROS generation
The higher oxidative environment promoted by MI may
be caused by increased oxidant generation or by reduced antioxidant
defense. First, we assessed the potential to generate ROS by
addressing the total amount of DHE-oxidation by-products by
microfluorotopography on slides of the myocardium in the SH group
(Fig. 2A), the MI group (Fig. 2B), the HBO group (Fig. 2C) and the MI group (Fig. 2D). The fluorescence of
DHE-oxidation products, measured in units/area, were significantly
lower in the HBO group (0.86±0.08) compared to the MI group
(1.87±0.08) and the SH group (1.40±0.11) (Fig. 2E). Although we could not exclude
the potential interference by other peroxides (37), the pre-incubation with pegylated
SOD was able to inhibit the oxidation of DHE (0.09±0.06), showing
greater specificity of the technique to the superoxide anion. Thus,
hyperbaric therapy was able not only to reduce the production of
superoxide induced by infarction, but also to minimize myocardial
levels in relation to non-infarcted animals (Fig. 2E).
Results of western blot analysis of
SOD, catalase and peroxiredoxin
Due to the higher levels of superoxide promoted by
MI, combined with the protective effect induced by HBO, the
antioxidant capacity was investigated by assessing the expression
of SOD, whose values showed that HBO therapy significantly
increased enzyme expression in the HBO group (1.0±0.06 AU/µg)
relative to the MI (0.79±0.04 AU/µg) and SH (0.69±0.08 AU/µg)
groups, with no significant difference between the SH and MI groups
(Fig. 3A). In addition, expression
of catalase (CAT) was investigated due to the intriguing effect
observed in the HBO group concerning the higher levels of oxidized
glutathione. Similarly to that observed with the SOD enzyme, HBO
therapy significantly increased CAT enzyme expression in the HBO
group (0.97±0.06 AU/µg) in relation to the MI (0.73±0.07 AU/µg) and
SH (0.66±0.04 AU/µg) groups. There was no significant difference
between the SH and MI groups (Fig.
3B). Thus, HBO therapy was able to upregulate enzymatic
antioxidants. In order to better investigate the antioxidant
activity, the expression of peroxiredoxin (Prx) hyperoxidation was
assessed. Importantly, the expression of the sulfonylated form of
Prx (SO2/3) protein was lower in the HBO group
(0.65±0.05 AU/µg), when compared to MI (1.24±0.18 AU/µg) and SH
(1.45±0.26 AU/µg) values, corroborating with better redox control.
There was no significant difference between the MI group and the SH
group (Fig. 3C). Therefore, not
only the total expression of antioxidants was increased such as SOD
and CAT, but also the Prx hyperoxidation levels were decreased by
HBO therapy.
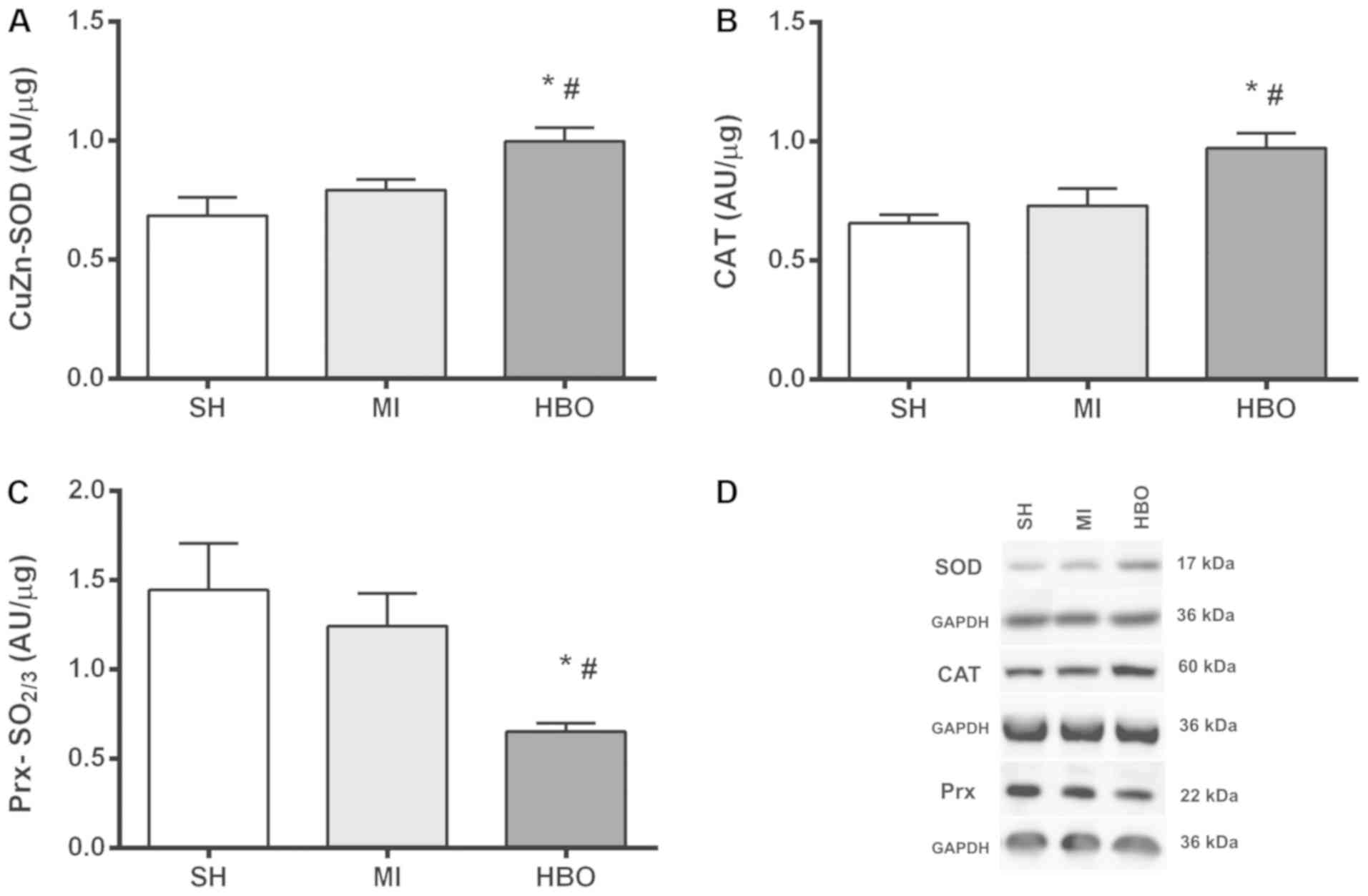 | Figure 3.Values (mean ± SEM) for (A) SOD
expression, (B) CAT expression and (C) Prx-SO2/3
expression in the SH, MI, and HBO groups. (D) Immunoblots.
*P<0.05 in relation to MI, #P<0.05 in relation to
SH. Groups: SH, Sham; MI, myocardial infarction; HBO, hyperbaric
therapy; SOD, superoxide dismutase; CAT, catalase; Prx,
peroxiredoxin. |
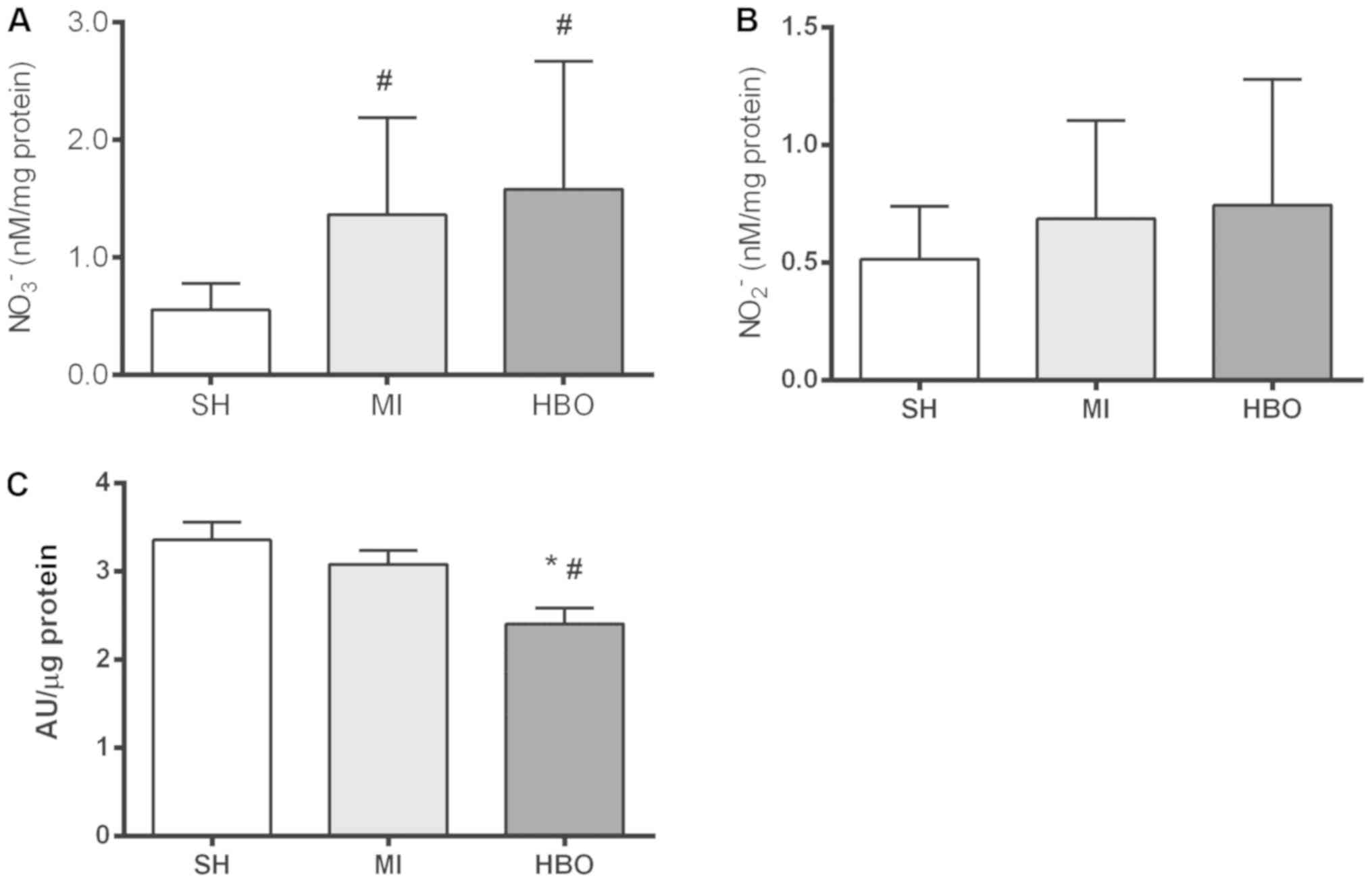
Nitric oxide by-products and
3-nitrotyrosine
Nitric oxide and its oxidation-derived by-products
show a dual role during normal and pathological conditions,
including MI (38,39). Coronary occlusion caused a
significant increase in nitrate (NO3−) (nM/mg
protein) in the MI (1.37±0.26) and HBO (1.58±0.41) groups in
relation to the SH group (0.56±0.08). No significant difference was
found between the two groups with MI (Fig. 4A), which represents a greater
availability of nitric oxide in an environment with increased
oxidative stress. No significant difference was found in levels of
nitrite (NO2−) (nM/mg protein) between the SH
(0.51±0.08), MI (0.69±0.13) and HBO (0.74±0.20) groups (Fig. 4B). In addition, the positive
labeling of 3-nitrotyrosine (AU/µg prot) by dot blot analysis
(Fig. 4C) showed a difference
between groups. A lower amount of nitrated protein was found in the
HBO group (2.40±0.18) compared to the MI (3.08±0.16) and SH
(3.36±0.20) groups. There was no significant difference between the
SH and MI groups. Therefore, these data suggest that MI induces
increased production of NO while HBO is effective to prevent such
NO-related modification in global target proteins, as reflected by
the lower levels of 3-nitrotyrosine.
Discussion
In the present study, the mortality rate of the
animals in the hyperbaric oxygenation (HBO) group was significantly
lower than that of the animals in the myocardial infarction (MI)
group, indicating a favorable action of hyperbaric oxygenation in
survival. This result is in accordance with a recent review of the
literature published by Bennett et al (7). These authors related the reduced
mortality by HBO in MI to the limitation of infarcted muscle mass
and decreasing cardiac arrhythmias. As in our animals there was no
difference noted in MI size, the reduced mortality rate of the HBO
animals should be linked to the beneficial effect on the alteration
of infarct-induced redox processes, which are important for
maintaining a favorable environment for cell survival and heart
rhythm (40,41).
Our results indicate that therapy with 2.5 ATA of
HBO resulted in similar mean infarct sizes in the MI and HBO group.
In a previous study by our group (18), we reported that hyperbaric
treatment reduced MI size and a lower mortality rate was not
observed. The discrepancy between the present data with data of our
previous study may result from differences in the protocols
employed. In the present protocol, the control of temperature
(~25°C) and the longer handling time in our protocol, due to the
collection and preparation of samples taken after treatment with
HBO, are crucial factors in the enzymatic action and may be
responsible for the verified differences.
The lower level found in the GSH/GSSG ratio in the
HBO group suggests an increase in glutathione (GSH) use, indicating
a higher efficiency of the antioxidant mechanisms to protect
against oxidative stress present in these animals. The same
response was not observed in the untreated animals of the MI group.
The higher glutathione disulfide (GSSG) formation indicates the
increased levels of reactive oxygen species (ROS) formation induced
by MI.
Importantly, the improved buffering capacity of
antioxidants promoted by HBO therapy is supported by the higher
amount of superoxide dismutase (SOD) and catalase (CAT), which
resulted in reduced levels of oxidant generation and their
post-translational modifications, such as ROS and 3-nitrotyrosine,
respectively. Moreover, the lower levels of Prx-SO2/3
strengthened such a protective effect. Collectively, the
pro-survival effect of HBO, may be promoted by a hormetic-like
effect (40,42). The acute exposure to the higher
oxidation conditions was able to affect a major ROS buffering
system, triggering a global response that prevented the more
accentuated disruption of the redox processes. These factors
resulted in a more prepared system to counteract impairments of the
myocardial milieu.
The lower levels of ROS indicated by DHE oxidation
may have resulted from increased SOD and CAT expression. Although
the mechanisms by which HBO therapy induces an antioxidant response
remain unclear, it has been reported to involve a Nrf2-dependent
effect (43,44) triggered in a pro-oxidizing
environment (31). Indeed, we
observed that HBO decreased the GSH/GSSG ratio, confirming such
Nrf2-inducing conditions. Moreover, due to the fast induction of
SOD and CAT reported here, we cannot exclude potential regulation
by protein degradation systems, as both enzymes were reported to be
targeted by proteasome degradation (45,46).
Thus, further studies are necessary to better understand the
mechanism by which acute HBO induces an antioxidant response.
The transient sulfinic acid peroxiredoxin (Prx)
intermediate has been attributed to control redox signaling. Such
reversibility, which is consistent with signaling events, is lost
by its hyperoxidation to sulfinic and sulfonic irreversible forms
(47). Importantly, HBO decreased
the amount of Prx-SO2/3H providing additional evidence
that the normal redox signaling is favored by acute hyperoxic
treatment. It is thought that the intensity of Prx I/II
hyperoxidation modulates ROS, which regulates signal transduction
pathways for cell apoptosis, survival or repair of damaged proteins
(12,48). Therefore, accumulation of the
inactive form of Prx (Cyx-SO2/3H) enzyme may lead to an
even greater increase in cellular H2O2
levels, resulting in increased apoptosis, as shown in cells that do
not express sulfiredoxin (49).
Surprisingly, the non-infarcted group showed higher levels of
Prx-SO2/3H, which may reflect a temporal Prx
inactivation to sustain ROS-mediated regulation promoted by stress
(50), such as associated with
sham surgical intervention. Further studies may clarify the effect
of these results on apoptosis in the later phase of MI.
Regarding reactive nitrogen species, we observed
lower values of 3-nitrotyrosine in the animals of the HBO group in
relation to the SH and MI groups, indicating lower nitration of
proteins by peroxynitrite resulting from the interaction of NO with
•O2− (51).
NO production under physiological conditions occurs via the
L-arginine/NO synthase pathway, which is O2-dependent
(38). Thus, the hypoxia and/or
ischemia present in the MI end up compromising NO production by
this route. Nitrate and nitrite, which are NO oxidation products,
can be recycled in vivo to form NO again, representing an
important alternative source of NO under physiological and
pathological conditions (38). NO
and its by-products have been identified as important signaling
agents, regulating mitochondrial respiration and ROS production
(39,52). Since the nitrate levels in the MI
and HBO groups were not different, the most marked nitration in the
MI animals was probably due to the higher
•O2− levels, allowing a greater formation of
peroxynitrite. It is important to note that the higher level of
nitrate found in the HBO group, when associated with less
nitration, shows greater NO bioavailability in this group.
One limitation of the present study must be
mentioned. The aim of assessing the acute effects of HBO following
coronary occlusion restricted our interests solely to the immediate
effects of the treatment. The effects of HBO on the evolution of MI
in the long term, remains to be studied.
In conclusion, our data showed that HBO animals had
greater ability to control pro-oxidants and antioxidants during
myocardial ischemia. This action should be the reason for the
reduced mortality rate in the treated rats.
Acknowledgements
The authors would like to thank Mr. Victor Debbas
and Ms. Ana Lucia Garippo (Laboratory of Vascular Biology, Heart
Institute, University of São Paulo) for technical assistance.
Funding
Financial support was provided by CAPES, FAPESP
(grant no. 09/54225-8) and CNPq (grant no. 478740-5).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
MO, EA, LB and LS contributed to the design of the
study, acquisition of data, and result analyses. AS performed
statistical analyses and LT introduced a new research technique,
analyzed the data and revised the manuscript for important
intellectual content. JK, FL and PT raised grant funding,
contributed to the design of the study, and revised the manuscript.
All the authors read and approved the final manuscript. All authors
take responsibility for all aspects of the reliability and freedom
from bias of the data presented and their discussed
interpretation.
Ethics approval and consent to
participate
All procedures were performed according to the
principles of ethics of animal experimentation adopted by the
Brazilian College of Animal Experimentation (COBEA-www.cobea.org.br), in conformity with the Guide for
the Care and Use of Laboratory Animals published by the US National
Institutes of Health (NIH publication no. 85-23, revised 1996). Our
protocol was approved by the Research Ethics Committee (CEP
0891/09) of the Federal University of São Paulo.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
HBO
|
hyperbaric oxygenation
|
|
ROS, rROS
|
reactive oxygen species
|
|
I/R
|
reperfusion injury
|
|
AMI
|
acute myocardial infarction
|
|
ATA
|
atmospheres absolute
|
|
GSH
|
glutathione
|
|
GSSG
|
glutathione disulfide
|
|
DHE
|
dihydroethidium
|
|
NO
|
nitric oxide
|
|
•NO3-
|
nitrate
|
|
NO2-
|
nitrite
|
|
SOD
|
superoxide dismutase
|
|
CAT
|
catalase
|
|
Prx
|
peroxiredoxin
|
References
|
1
|
Braunwald E and Kloner RA: Myocardial
reperfusion: A double-edged sword? J Clin Invest. 76:1713–1719.
1985. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Bulluck H, Yellon DM and Hausenloy DJ:
Reducing myocardial infarct size: Challenges and future
opportunities. Heart. 102:341–348. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Facundo HT, Carreira RS, de Paula JG,
Santos CC, Ferranti R, Laurindo FR and Kowaltowski AJ: Ischemic
preconditioning requires increases in reactive oxygen release
independent of mitochondrial K+ channel activity. Free
Radic Biol Med. 40:469–479. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Granger DN and Kvietys PR: Reperfusion
injury and reactive oxygen species: The evolution of a concept.
Redox Biol. 6:524–551. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Zhang Y, Sano M, Shinmura K, Tamaki K,
Katsumata Y, Matsuhashi T, Morizane S, Ito H, Hishiki T, Endo J, et
al: 4-hydroxy-2-nonenal protects against cardiac
ischemia-reperfusion injury via the Nrf2-dependent pathway. J Mol
Cell Cardiol. 49:576–586. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Fröhlich GM, Meier P, White SK, Yellon DM
and Hausenloy DJ: Myocardial reperfusion injury: Looking beyond
primary PCI. Eur Heart J. 34:1714–1722. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Bennett MH, Lehm JP and Jepson N:
Hyperbaric oxygen therapy for acute coronary syndrome. Cochrane
Database Syst Rev. CD0048182015.PubMed/NCBI
|
|
8
|
Undersea and Hyperbaric Medical Society:
Indications for hyperbaric oxygen therapy. https://www.uhms.org/resources/hbo-indications.htmlMarch
25–2019
|
|
9
|
Sterling DL, Thornton JD, Swafford A,
Gottlieb SF, Bishop SP, Stanley AW and Downey JM: Hyperbaric oxygen
limits infarct size in ischemic rabbit myocardium in vivo.
Circulation. 88:1931–1936. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Tibbles PM and Edelsberg JS:
Hyperbaric-oxygen therapy. N Engl J Med. 334:1642–1648. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Whalen RE and Saltzman HA: Hyperbaric
oxygenation in the treatment of acute myocardial infarction. Prog
Cardiovasc Dis. 10:575–583. 1968. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Poff AM, Kernagis D and D'Agostino DP:
Hyperbaric Environment: Oxygen and Cellular damage versus
protection. Compr Physiol. 7:213–234. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Cabigas BP, Su J, Hutchins W, Shi Y,
Schaefer RB, Recinos RF, Nilakantan V, Kindwall E, Niezgoda JA and
Baker JE: Hyperoxic and hyperbaric-induced cardioprotection: Role
of nitric oxide synthase 3. Cardiovasc Res. 72:143–151. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Jones DP: Redefining oxidative stress.
Antioxid Redox Signal. 8:1865–1879. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kim CH, Choi H, Chun YS, Kim GT, Park JW
and Kim MS: Hyperbaric oxygenation pretreatment induces catalase
and reduces infarct size in ischemic rat myocardium. Pflugers Arch.
442:519–525. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Tavares AM, da Rosa Araujo AS, Llesuy S,
Khaper N, Rohde LE, Clausell N and Belló-Klein A: Early loss of
cardiac function in acute myocardial infarction is associated with
redox imbalance. Exp Clin Cardiol. 17:263–267. 2012.PubMed/NCBI
|
|
17
|
Thom SR: Hyperbaric oxygen: Its mechanisms
and efficacy. Plast Reconstr Surg. 127 (Suppl 1):131S–141S. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
dos Santos L, Serra AJ, Antônio EL, Hull
HF and Tucci PJ: Hyperbaric oxygenation applied immediately after
coronary occlusion reduces myocardial necrosis and acute mortality
in rats. Clin Exp Pharmacol Physiol. 36:594–598. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Guadalupe NBM, Castillo-Hernandez MDC,
Kormanovski A, Lara-Padilla E, Pimentel-Montejano VH, Olaf GV,
Lopez-Mayorga RM and Gustavo GB: Effect of hyperbaric oxygenation
in total antioxidant system, nitric oxide and 3 nitrotyrosine
levels in a rat model of acute myocardial infarct in the absence of
reperfusion. Int J Pharmacol. 11:834–839. 2015. View Article : Google Scholar
|
|
20
|
Kuhn LA, Kline HJ, Wang M, Yamaki T and
Jacobson JH II: Hemodynamic effects of hyperbaric oxygenation in
experimental acute myocardial infarction. Circ Res. 16:499–509.
1965. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Mogelson S, Davidson J, Sobel BE and
Roberts R: The effect of hyperbaric oxygen on infarct size in the
conscious animal. Eur J Cardiol. 12:135–146. 1981.PubMed/NCBI
|
|
22
|
Thomas MP, Brown LA, Sponseller DR,
Williamson SE, Diaz JA and Guyton DP: Myocardial infarct size
reduction by the synergistic effect of hyperbaric oxygen and
recombinant tissue plasminogen activator. Am Heart J. 120:791–800.
1990. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Yogaratnam JZ, Laden G, Guvendik L, Cowen
M, Cale A and Griffin S: Hyperbaric oxygen preconditioning improves
myocardial function, reduces length of intensive care stay, and
limits complications post coronary artery bypass graft surgery.
Cardiovasc Revasc Med. 11:8–19. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Shandling AH, Ellestad MH, Hart GB, Crump
R, Marlow D, Van Natta B, Messenger JC, Strauss M and Stavitsky Y:
Hyperbaric oxygen and thrombolysis in myocardial infarction: The
‘HOT MI’ pilot study. Am Heart J. 134:544–550. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Stavitsky Y, Shandling AH, Ellestad MH,
Hart GB, Van Natta B, Messenger JC, Strauss M, Dekleva MN,
Alexander JM, Mattice M and Clarke D: Hyperbaric oxygen and
thrombolysis in myocardial infarction: The ‘HOT MI’ randomized
multicenter study. Cardiology. 90:131–136. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Dekleva M, Neskovic A, Vlahovic A,
Putnikovic B, Beleslin B and Ostojic M: Adjunctive effect of
hyperbaric oxygen treatment after thrombolysis on left ventricular
function in patients with acute myocardial infarction. Am Heart J.
148:E142004. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Vlahović A, Nesković AN, Dekleva M,
Putniković B, Popović ZB, Otasević P and Ostojić M: Hyperbaric
oxygen treatment does not affect left ventricular chamber stiffness
after myocardial infarction treated with thrombolysis. Am Heart J.
148:e12004. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zhdanov GG and Sokolov IM: Tissue hypoxia
in acute myocardial infarction and possible approaches to its
correction. Anesteziol Reanimatol. 51–53. 2001.(In Russian).
PubMed/NCBI
|
|
29
|
Hedström E, Engblom H, Frogner F,
Aström-Olsson K, Öhlin H, Jovinge S and Arheden H: Infarct
evolution in man studied in patients with first-time coronary
occlusion in comparison to different species-implications for
assessment of myocardial salvage. J Cardiovasc Magn Reson.
11:382009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Vetterlein F, Schrader C, Volkmann R,
Neckel M, Ochs M, Schmidt G and Hellige G: Extent of damage in
ischemic, nonreperfused, and reperfused myocardium of anesthetized
rats. Am J Physiol Heart Circ Physiol. 285:H755–H765. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
dos Santos L, Mello AF, Antonio EL and
Tucci PJF: Determination of myocardial infarction size in rats by
echocardiography and tetrazolium staining: Correlation, agreements,
and simplifications. Braz J Med Biol Res. 41:199–201. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Kanashiro RM, Nozawa E, Murad N, Gerola
LR, Moisés VA and Tucci PJ: Myocardial infarction scar plication in
the rat: Cardiac mechanics in an animal model for surgical
procedures. Ann Thorac Surg. 73:1507–1513. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
de Melo BL, Vieira SS, Antônio EL, Dos
Santos LF, Portes LA, Feliciano RS, de Oliveira HÁ, Silva JÁ Jr, de
Carvalho PT, Tucci PJ and Serra AJ: Exercise training attenuates
right ventricular remodeling in rats with pulmonary arterial
stenosis. Front Physiol. 7:5412016. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Tanito M, Elliott MH, Kotake Y and
Anderson RE: Protein modifications by 4-hydroxynonenal and
4-hydroxyhexenal in light-exposed rat retina. Invest Ophthalmol Vis
Sci. 46:3859–3868. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Miller FJ Jr, Gutterman DD, Rios CD,
Heistad DD and Davidson BL: Superoxide production in vascular
smooth muscle contributes to oxidative stress and impaired
relaxation in atherosclerosis. Circ Res. 82:1298–1305. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Leite PF, Danilovic A, Moriel P, Dantas K,
Marklund S, Dantas AP and Laurindo FR: Sustained decrease in
superoxide dismutase activity underlies constrictive remodeling
after balloon injury in rabbits. Arterioscler Thromb Vasc Biol.
23:2197–2202. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Fernandes DC, Wosniak J Jr, Pescatore LA,
Bertoline MA, Liberman M, Laurindo FR and Santos CX: Analysis of
DHE-derived oxidation products by HPLC in the assessment of
superoxide production and NADPH oxidase activity in vascular
systems. Am J Physiol Cell Physiol. 292:C413–C422. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Lundberg JO, Weitzberg E and Gladwin MT:
The nitrate-nitrite-nitric oxide pathway in physiology and
therapeutics. Nat Rev Drug Discov. 7:156–167. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Omar SA, Webb AJ, Lundberg JO and
Weitzberg E: Therapeutic effects of inorganic nitrate and nitrite
in cardiovascular and metabolic diseases. J Intern Med.
279:315–336. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Godman CA, Joshi R, Giardina C, Perdrizet
G and Hightower LE: Hyperbaric oxygen treatment induces antioxidant
gene expression. Ann N Y Acad Sci. 1197:178–183. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Soyer OS, Salathé M and Bonhoeffer S:
Signal transduction networks: Topology, response and biochemical
processes. J Theor Biol. 238:416–425. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Ristow M and Schmeisser S: Extending life
span by increasing oxidative stress. Free Radic Biol Med.
51:327–336. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Bocci V and Valacchi G: Nrf2 activation as
target to implement therapeutic treatments. Front Chem. 3:42015.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Yin X, Wang X, Fan Z, Peng C, Ren Z, Huang
L, Liu Z and Zhao K: Hyperbaric oxygen preconditioning attenuates
myocardium ischemia-reperfusion injury through upregulation of heme
oxygenase 1 expression: PI3K/Akt/Nrf2 pathway involved. J
Cardiovasc Pharmacol Ther. 20:428–438. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Aquilano K, Rotilio G and Ciriolo MR:
Proteasome activation and nNOS down-regulation in neuroblastoma
cells expressing a Cu, Zn superoxide dismutase mutant involved in
familial ALS. J Neurochem. 85:1324–1335. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Cao C, Leng Y, Liu X, Yi Y, Li P and Kufe
D: Catalase is regulated by ubiquitination and proteosomal
degradation. Role of the c-Abl and Arg tyrosine kinases.
Biochemistry. 42:10348–10353. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Poole LB, Karplus PA and Claiborne A:
Protein sulfenic acids in redox signaling. Annu Rev Pharmacol
Toxicol. 44:325–347. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Buettner GR, Wagner BA and Rodgers VG:
Quantitative redox biology: An approach to understand the role of
reactive species in defining the cellular redox environment. Cell
Biochem Biophys. 67:477–483. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Baek JY, Han SH, Sung SH, Lee HE, Kim YM,
Noh YH, Bae SH, Rhee SG and Chang TS: Sulfiredoxin protein is
critical for redox balance and survival of cells exposed to low
steady-state levels of H2O2. J Biol Chem. 287:81–89. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Day AM, Brown JD, Taylor SR, Rand JD,
Morgan BA and Veal EA: Inactivation of a peroxiredoxin by hydrogen
peroxide is critical for thioredoxin-mediated repair of oxidized
proteins and cell survival. Mol Cell. 45:398–408. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Pacher P, Beckman JS and Liaudet L: Nitric
oxide and peroxynitrite in health and disease. Physiol Rev.
87:315–424. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Totzeck M, Hendgen-Cotta UB and Rassaf T:
Nitrite-Nitric oxide signaling and cardioprotection. Adv Exp Med
Biol. 982:335–346. 2017. View Article : Google Scholar : PubMed/NCBI
|