| Biology
process |
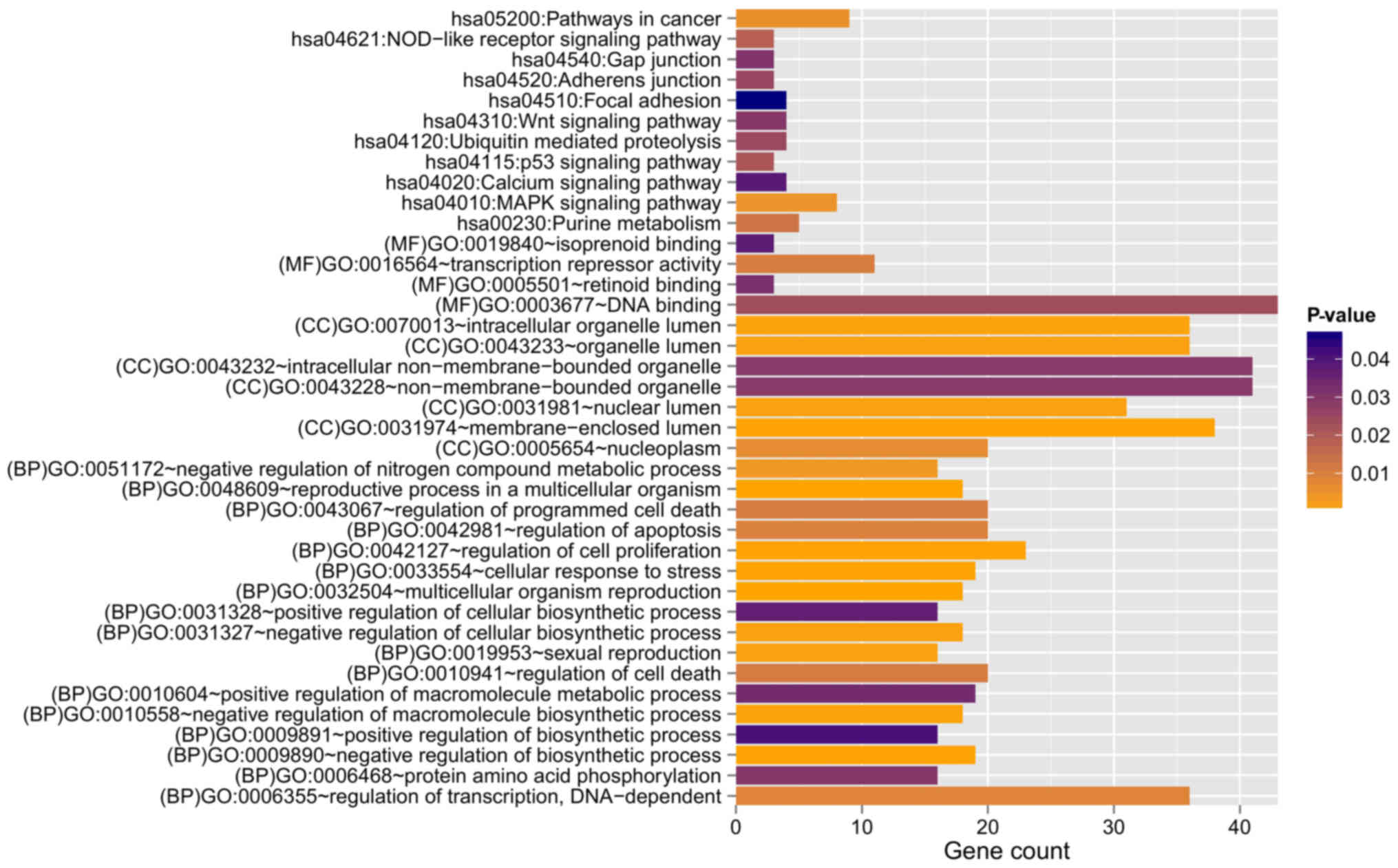
GO:0048609~reproductive process in a
multicellular organism | 18 | 2.390×10-4 | B4GALT1, RBP4,
HMGB2, AGFG1, ERBB2, ADCYAP1R1, FIGLA, ZBTB16, ELL3, BOLL, LEP,
INHBA, FANCD2, ZMIZ1, WIPF3, ADAMTS1, SPIN4, PNMA1 |
|
|
GO:0032504~multicellular organism
reproduction | 18 | 2.390×10-4 | B4GALT1, RBP4,
HMGB2, AGFG1, ERBB2, ADCYAP1R1, FIGLA, ZBTB16, ELL3, BOLL, LEP,
INHBA, FANCD2, ZMIZ1, WIPF3, ADAMTS1, SPIN4, PNMA1 |
|
| GO:0033554~cellular
response to stress | 19 | 4.810×10-4 | HMGB2, UBE2A,
DERL1, GEN1, PML, MBD4, RAD9A, SIRT7, TP73, TRIB1, SCAP, PRPF19,
CDKN1A, MRPS9, FANCD2, NSMCE1, TDP1, SUPT16H, GADD45A |
|
| GO:0009890~negative
regulation of biosynthetic process | 19 | 5.560×10-4 | HMGB2, CTBP1,
CDX4, SIX3, HDAC10, ZNF24, PML, ZHX3, RAD9A, SNW1, SIRT7, DMAP1,
ZBTB16, SCAP, INHBA, HHEX, HDAC3, OVOL2, IGFBP5 |
|
|
GO:0042127~regulation of cell
proliferation | 23 | 6.630×10-4 | B4GALT1, RBP4,
UBE2A, CTBP1, PTPRM, ERBB2, PML, ZBTB16, SSR1, TRIB1, LAMA1, HHEX,
S1PR3, TNFRSF9, CDKN1A, OSR2, OVOL2, ZMIZ1, PLA2G1B, ADAMTS1,
IL12B, F2R, IGFBP5 |
|
| GO:0010558~negative
regulation of macromolecule biosynthetic process | 18 | 8.940×10-4 | HMGB2, CTBP1,
CDX4, SIX3, HDAC10, ZNF24, PML, ZHX3, RAD9A, SNW1, SIRT7, DMAP1,
ZBTB16, INHBA, HHEX, HDAC3, OVOL2, IGFBP5 |
|
| GO:0019953~sexual
reproduction | 16 | 1.069×10-3 | B4GALT1, RBP4,
HMGB2, AGFG1, ADCYAP1R1, FIGLA, ZBTB16, ELL3, BOLL, LEP, FANCD2,
ZMIZ1, WIPF3, ADAMTS1, SPIN4, PNMA1 |
|
| GO:0031327~negative
regulation of cellular biosynthetic process | 18 | 1.178×10-3 | HMGB2, CTBP1,
CDX4, SIX3, HDAC10, ZNF24, PML, ZHX3, RAD9A, SNW1, SIRT7, DMAP1,
ZBTB16, INHBA, HHEX, HDAC3, OVOL2, IGFBP5 |
|
| GO:0051172~negative
regulation of nitrogen compound metabolic process | 16 | 3.578×10-3 | HMGB2, CTBP1,
CDX4, SIX3, ZNF24, PML, HDAC10, ZHX3, RAD9A, SNW1, SIRT7, DMAP1,
ZBTB16, HHEX, HDAC3, OVOL2 |
|
|
GO:0006355~regulation of transcription,
DNA-dependent | 36 | 8.581×10-3 | HMGB2, CDX4,
IRX2, ZNF557, ZBTB16, DMAP1, ZKSCAN2, OVOL2, PLA2G1B, POU4F1,
POU3F1, RFX8, CTBP1, RFX4, SIX3, ZNF24, HDAC10, ZHX3, DMRT2, SNW1,
SIRT7, ELL3, ZNF320, TP73, SCAP, RPS6KA5, ASCL2, INHBA, HHEX,
HDAC3, RNF4, ZMIZ1, ZNF460, MGA, UNCX, F2R |
|
|
GO:0042981~regulation of apoptosis | 20 | 9.553×10-3 | B4GALT1, ERBB2,
PML, MBD4, RAD9A, ZBTB16, TP73, MAP3K7, INHBA, TNFRSF9, HDAC3,
PLEKHG2, CDKN1A, HSP90B1, BNIP1, GSPT1, POU4F1, IL12B, TNFAIP3,
F2R |
|
|
GO:0043067~regulation of programmed cell
death | 20 | 1.054×10-2 | B4GALT1, ERBB2,
PML, MBD4, RAD9A, ZBTB16, TP73, MAP3K7, INHBA, TNFRSF9, HDAC3,
PLEKHG2, CDKN1A, HSP90B1, BNIP1, GSPT1, POU4F1, IL12B, TNFAIP3,
F2R |
|
|
GO:0010941~regulation of cell death | 20 | 1.094×10-2 | B4GALT1, ERBB2,
PML, MBD4, RAD9A, ZBTB16, TP73, MAP3K7, INHBA, TNFRSF9, HDAC3,
PLEKHG2, CDKN1A, HSP90B1, BNIP1, GSPT1, POU4F1, IL12B, TNFAIP3,
F2R |
|
| GO:0006468~protein
amino acid phosphorylation | 16 | 3.007×10-2 | CTBP1, ERBB2,
MKNK2, PML, ALK, TBCK, TRIB1, RPS6KA5, MAP3K7, RPS6KA6, ADCK1,
PLA2G1B, CAMK1, PRKACB, MYLK, F2R |
|
| GO:0010604~positive
regulation of macromolecule metabolic process | 19 | 3.329×10-2 | HMGB2, RFX4,
SIX3, PML, SIRT7, ELL3, BOLL, TP73, SCAP, HHEX, INHBA, OVOL2, RNF4,
ZMIZ1, PLA2G1B, MLST8, POU3F1, IL12B, F2R |
|
| GO:0031328~positive
regulation of cellular biosynthetic process | 16 | 3.673×10-2 | HMGB2, RFX4,
SIX3, SIRT7, ELL3, TP73, BOLL, SCAP, HHEX, INHBA, OVOL2, RNF4,
ZMIZ1, PLA2G1B, IL12B, F2R |
|
| GO:0009891~positive
regulation of biosynthetic process | 16 | 4.086×10-2 | HMGB2, RFX4,
SIX3, SIRT7, ELL3, TP73, BOLL, SCAP, HHEX, INHBA, OVOL2, RNF4,
ZMIZ1, PLA2G1B, IL12B, F2R |
| Cellular
Component |
GO:0031974~membrane-enclosed lumen | 38 | 5.070×10-4 | PDP1, HMGB2,
PNMA2, POLR2J, PNPT1, FIGLA, PML, ZBTB16, DMAP1, POLR2D, PRPF19,
ALAS1, NUMA1, COX6B1, NPM3, POU3F1, ETFB, PDCD11, CTBP1, ZMYM3,
HDAC10, RAD9A, MBD4, SNW1, SIRT7, ELL3, RPS6KA5, NVL, CDKN1A,
HSP90B1, HDAC3, MRPS9, FANCD2, ZMIZ1, SUPT16H, MATR3, EP400,
PNMA1 |
|
|
GO:0070013~intracellular organelle
lumen | 36 | 9.530×10-4 | PDP1, HMGB2,
PNMA2, POLR2J, FIGLA, PML, ZBTB16, DMAP1, POLR2D, PRPF19, ALAS1,
NUMA1, NPM3, POU3F1, ETFB, PDCD11, CTBP1, ZMYM3, HDAC10, RAD9A,
MBD4, SNW1, SIRT7, ELL3, RPS6KA5, NVL, CDKN1A, HSP90B1, HDAC3,
MRPS9, FANCD2, ZMIZ1, SUPT16H, MATR3, EP400, PNMA1 |
|
| GO:0031981~nuclear
lumen | 31 | 1.079×10-3 | HMGB2, PNMA2,
POLR2J, FIGLA, PML, ZBTB16, DMAP1, POLR2D, PRPF19, NUMA1, NPM3,
POU3F1, PDCD11, CTBP1, ZMYM3, HDAC10, RAD9A, MBD4, SNW1, SIRT7,
ELL3, NVL, RPS6KA5, CDKN1A, HDAC3, FANCD2, ZMIZ1, SUPT16H, MATR3,
EP400, PNMA1 |
|
|
GO:0043233~organelle lumen | 36 | 1.430×10-3 | PDP1, HMGB2,
PNMA2, POLR2J, FIGLA, PML, ZBTB16, DMAP1, POLR2D, PRPF19, ALAS1,
NUMA1, NPM3, POU3F1, ETFB, PDCD11, CTBP1, ZMYM3, HDAC10, RAD9A,
MBD4, SNW1, SIRT7, ELL3, RPS6KA5, NVL, CDKN1A, HSP90B1, HDAC3,
MRPS9, FANCD2, ZMIZ1, SUPT16H, MATR3, EP400, PNMA1 |
|
|
GO:0005654~nucleoplasm | 20 | 6.379×10-3 | HMGB2, CTBP1,
POLR2J, HDAC10, PML, FIGLA, DMAP1, ZBTB16, ELL3, POLR2D, RPS6KA5,
PRPF19, HDAC3, NUMA1, CDKN1A, FANCD2, ZMIZ1, SUPT16H, POU3F1,
EP400 |
|
|
GO:0043232~intracellular
non-membrane-bounded organelle | 41 | 2.914×10-2 | HMGB2, PNMA2,
USP2, PML, ZBTB16, DMAP1, KIF2C, NUMA1, NPM3, SKA3, NDRG2, TRIP10,
CNKSR2, IPP, PDCD11, UBE2A, ZMYM3, RNF19A, PDE4D, SNW1, RAD9A,
MBD4, MID1IP1, SIRT7, PALLD, TNKS1BP1, NVL, HDAC3, TUBA8, MRPS9,
FANCD2, RPS4Y2, TPPP, SUPT16H, OPHN1, TUBA4A, ARL8B, TNFAIP3,
EP400, LCP1, PNMA1 |
|
|
GO:0043228~non-membrane-bounded
organelle | 41 | 2.914×10-2 | HMGB2, PNMA2,
USP2, PML, ZBTB16, DMAP1, KIF2C, NUMA1, NPM3, SKA3, NDRG2, TRIP10,
CNKSR2, IPP, PDCD11, UBE2A, ZMYM3, RNF19A, PDE4D, SNW1, RAD9A,
MBD4, MID1IP1, SIRT7, PALLD, TNKS1BP1, NVL, HDAC3, TUBA8, MRPS9,
FANCD2, RPS4Y2, TPPP, SUPT16H, OPHN1, TUBA4A, ARL8B, TNFAIP3,
EP400, LCP1, PNMA1 |
| Molecular
function | GO:0003677~DNA
binding | 43 | 2.360×10-2 | HMGB2, AGFG1,
CDX4, IRX2, POLR2J, ZNF557, FIGLA, PML, DMAP1, ZBTB16, ZKSCAN2,
APLP2, PRPF19, KIF2C, OVOL2, SERPINA3, POU4F1, POU3F1, BAHD1, RFX8,
CTBP1, RFX4, GEN1, SNAPC1, ZMYM3, SIX3, ZNF24, DMRT2, ZHX3, MBD4,
ZNF320, TP73, ASCL2, HHEX, HDAC3, RNF4, TDP1, ZNF460, MGA, TNFAIP3,
ZBTB1, EP400, UNCX |
|
|
GO:0016564~transcription repressor
activity | 11 | 1.021×10-2 | HHEX, CTBP1,
HMGB2, HDAC3, SIX3, PML, ZNF24, HDAC10, SNW1, ZBTB16,
DMAP1 |
|
| GO:0005501~retinoid
binding | 3 | 3.174×10-2 | RBP4, RBP7,
CYP26A1 |
|
|
GO:0019840~isoprenoid binding | 3 | 3.758×10-2 | RBP4, RBP7,
CYP26A1 |