Introduction
Atherosclerosis is the most common cause of
ischaemic heart disease and stroke; it is closely associated with
the occurrence of acute cardiovascular events, which have a huge
impact on human life and health, accounting for 20% of total
mortalities worldwide (1). It is
widely known that during the early stages of atherosclerosis,
monocytes enter the endothelial layer and differentiate into
macrophages under the action of adhesion molecules and cytokines
(2–4). For example, in one previous study,
autopsies of patients with atherosclerosis discovered the
accumulation of a large number of macrophages, lymphocytes and foam
cells in the coronary artery plaques (2). Macrophages are mainly divided into
two different subtypes: M1-type macrophages are mainly found in
unstable plaques, whereas M2-type macrophages are predominantly
found in stable plaque tissue (5,6).
During the development of the disease, various external factors
affect macrophages, and depending on the M1/M2 polarization status,
the corresponding inflammatory process is highly different
(5,7). Binesh et al (7) found that pro-inflammatory macrophages
(M1-type) were expressed in atherogenic diet-induced aorta, while
anti-inflammatory macrophages (M2-type) were expressed in
diosgenin-treated aorta. Therefore, improving the condition of the
disease by regulating the polarization of M1-type macrophages may
be one mechanism to control immune regulation in
atherosclerosis.
It has previously been reported that angiotensin II
(AngII) can induce macrophages to differentiate towards the
M1-type, produce inflammatory cytokines and resist pathogen
invasion, whilst also causing damage to the body (8). Increased levels of AngII have been
demonstrated to induce the secretion of anti-inflammatory
cytokines, and activate NF-κB, adhesion molecules, chemokines,
growth factors and oxidative stress (9). NF-κB serves a well-known role in the
regulation of immune responses and inflammation, and the NF-κB
transcription factor family is comprised of crucial regulatory
factors for immune development, immune responses, inflammation and
cancer (10). A recent study
reported that an increase in the mRNA expression levels of
AngII-induced pro-inflammatory cytokines, IL-1β and tumour necrosis
factor (TNF)-α, could be blocked by inhibiting the NF-κB pathway
(11).
Connexins (Cxs) are one of the basic components of
gap junction channels and hemichannels, and are composed of six
four-span proteins called connecting proteins (12). The two hemichannel connectors from
adjacent cells can be gathered together to form a gap junction that
crosses the membrane channel, thus promoting cell-cell
communication by transferring a ~1-kDa signalling molecule from one
cell to the adjacent cell (13).
Connective family molecules, such as Cx30, Cx32, Cx37, Cx40 and
Cx43, of which Cx43 is the most common, are also widely expressed
in immune cells; these proteins make up the gap junction channels
and hemichannels involved in a variety of immunomodulation
processes (14). It was previously
demonstrated that AngII increased the expression levels of Cx43 in
the vascular smooth muscle of hidden veins in a dose- and
time-dependent manner, whereas the p38 and ERK MAPK inhibitors,
SB203580 and PD98059, inhibited this effect (15). In addition, reduced conductivity of
Cx43 channels using Gap26 in a lipopolysaccharide (LPS)-induced
inflammatory mouse model effectively reduced the adhesion of
neutrophils and endothelial cells, significantly reduced residual
bacteria in the abdominal cavity of mice, and significantly
improved survival in infected mice (16). However, the mechanisms underlying
AngII-induced macrophage polarization have not been determined. The
present study aimed to investigate whether Cx43 and NF-κB are
involved in AngII-induced macrophage polarization processes by
studying the effect of Cx43/NF-κB signalling pathways on the
polarization of AngII-induced RAW264.7 macrophages to the
M1-type.
Materials and methods
Cell culture and experimental
grouping
RAW264.7 macrophages were purchased from the
Shanghai Institutes for Biological Sciences, Chinese Academy of
Sciences. Cells were plated into a culture flask at a density of
3×105/ml, and were cultured in a CO2
thermostatic incubator at 37°C and 5% CO2 in DMEM
(Gibco; Thermo Fisher Scientific, Inc.) supplemented with 10% FBS
(Gibco; Thermo Fisher Scientific, Inc.). Cells were passaged upon
reaching 70–80% confluence.
RAW264.7 macrophages were treated with
10−9, 10−8, 10−7, 10−6
and 10−5 mol/l AngII (APExBIO Technology LLC) for 0, 3,
6, 12, 24 and 48 h to determine the optimal treatment conditions.
RAW264.7 macrophages were cultured and treated in different ways:
i) Control group, macrophages cultured in DMEM/FBS without any
treatment; ii) AngII group, in which cells were treated with
10−6 mol/l AngII (APExBIO Technology LLC) for 12 h at
37°C (17,18); iii) Gap26 group, in which cells
were pre-treated with 10−4 mol/l Gap26 (APExBIO
Technology LLC) at 37°C for 45 min prior to 10−6 mol/l
AngII treatment for 12 h (19,20);
iv) Cx43 inhibitor Gap19 group, in which cells were pre-treated
with 2×10−4 mol/l Gap19 (APExBIO Technology LLC) at 37°C
for 45 min prior to treatment with 10−6 mol/l AngII for
12 h (20); v) DMSO group, in
which cells were treated with 10−6 mol/l DMSO (Beijing
Solarbio Science & Technology Co., Ltd.) for 12 h at 37°C; and
vi) NF-κB signaling pathway inhibitor BAY117082 group, in which
cells were pre-treated with 10−5 mol/l BAY117082 (cat.
no. ab141228; Abcam) for 1 h prior to treatment with
10−6 mol/l AngII for 12 h (21,22).
Cell viability assay
Cells (8×103/well) were seeded into a
96-well plate and treated with increasing concentrations
(10−9, 10−8, 10−7, 10−6
or 10−5 mol/l) of AngII for 12 h at 37°C. Cell viability
was subsequently measured using a Cell Counting Kit-8 (CCK-8) assay
(Dojindo Molecular Technologies), according to the manufacturer's
protocol.
Flow cytometry
The frequency of CD86 (Invitrogen; Thermo Fisher
Scientific, Inc.) expression was detected using flow cytometry. The
following were used as blocking (4°C for 15–30 min) reagents:
MouseBD Fc Block (BD Biosciences) and BSA (5%; Sigma-Aldrich; Merck
KGaA). Before use, the serum preparations were heat inactivated
(56°C for 45 min) and sterile filtered (0.2-µm filter); the
negative cells and CD86 were individually labelled as a control to
analyze the data. Cells (1×106/well) were cultured for
24 h, digested with 0.25% trypsin, centrifuged at 603 × g for 5 min
at 37°C and subsequently retained as a pellet; filter paper was
used to absorb as much of the liquid left in the tube as possible.
Each pellet was resuspended in 1 ml PBS, centrifuged at 603 × g at
37°C for 5 min and retained as a pellet. The pellets were
resuspended in 200 µl PBS in Eppendorf tubes and 2 µl phycoerythrin
(PE)-CD86 antibody (1:100, cat. no. 85-12-0862; Invitrogen; Thermo
Fisher Scientific, Inc.) was added to each Eppendorf tube and
incubated at 37°C in the dark for 30 min. Subsequently, PBS was
added to each tube and cells were centrifuged at 603 × g for 5 min
at 37°C. The supernatant was discarded and the pellets were
resuspended in 200 µl PBS and mixed. A formulated fixative Fixation
Concentrate (cat. no. 00-5521-00; 1%; BD Biosciences) was added to
each tube at room temperature for 20 min prior to incubation with a
PE-CD206 antibody (1:100, cat. no. 85-25-2061; Invitrogen; Thermo
Fisher Scientific, Inc.) at 37°C in the dark for 30 min.
Subsequently, cells were resuspended in 1 ml 1X permeabilization
reagent (cat. no. 00-8333-56; BD Biosciences) for 30 min at 25°C
and centrifuged at 603 × g for 5 min at 37°C. The supernatant was
discarded and filter paper was used to absorb the liquid left in
the tube. Following the suspension of cell pellets in 200 µl PBS,
analysis of CD206 and CD86 expression levels was performed using
flow cytometry (FACSort; BD Biosciencnes) with BD CellQuest
Prosoftware (version 2.0, system OS2; Becton, Dickinson and
Company). To ensure the accuracy of the experimental results, all
samples were analysed within 3 h to avoid fluorescence changes,
which could affect the experimental results. The number of detected
cells per tube was 10,000-20,000.
Western blotting
The protein expression levels of CD86, CD206,
inducible nitric oxide synthase (iNOS), Cx43, p65 and
phosphorylated (p)-p65 were analysed using western blotting. Total
protein was extracted from cells using RIPA lysis buffer (cat. no.
R0020; Beijing Solarbio Science and Technology Co., Ltd.). Total
protein was quantified using a bicinchoninic acid protein assay kit
and equal quantities of protein (15 µg/lane) were separated by
SDS-PAGE (Beijing Solarbio Science & Technology Co., Ltd.) on
10% gels. The separated proteins were subsequently transferred onto
a PVDF membrane (EMD Millipore) and blocked with 5% skim milk in
TBS-0.2% Tween-20 (TBST; Sangon Biotech, Co., Ltd.) for 2 h at room
temperature. The membranes were incubated with the following
primary mouse and rabbit antibodies at 4°C overnight: Anti-CD86
(1:1,000; cat. no. ab53004; Abcam), anti-Cx43 (1:1,000; cat. no.
ab11370; Abcam), anti-p65 (1:1,000; cat. no. ab16502; Abcam),
anti-p-p65 (1:1,000; cat. no. ab86299; Abcam), anti-iNOS (1:500;
cat. no. ab15323; Abcam), anti-CD206 (1:500; cat. no. ab64693;
Abcam) and anti-β-actin (1:1,000; cat. no. TA-09; Beijing Fir
Jinqiao Biotechnology Co., Ltd.). Following primary antibody
incubation, the membrane was washed three times with TBST (10
min/wash) and the secondary antibody (1:10,000; horseradish
peroxidase-conjugated goat anti-rabbit; cat. no. ZB-2306; Beijing
Zhongshan Jinqiao Biotechnology Co. Ltd.) was subsequently
incubated with the PVDF membrane at room temperature for 2 h. The
membrane was washed three times with TBST (10 min/wash) and protein
bands were visualized on X-ray film using an ECL reagent (GE
Healthcare Life Sciences; Thermo Fisher Scientific, Inc.). Protein
expression was semi-quantified using ImageJ software (version
1.8.0; National Institutes of Health).
Reverse transcription-quantitative PCR
(RT-qPCR)
Total RNA was extracted from RAW264.7 macrophages
using TRIzol® reagent (Invitrogen; Thermo Fisher
Scientific, Inc.). Total RNA (0.5 µg) was RT using a Revert Aid
First Strand cDNA Synthesis kit (cat. no. K1622; Thermo Fisher
Scientific, Inc.) at 42°C for 60 min and 70°C for 5 min. Total RNA
(adjusted to 500 ng, 2.5 µl), Oligo dT PCR primer (50 µM, 1 µl), 5X
PrimeScript buffer (4 µl), RiboLock RNase Inhibitor (20 U/µl, 1
µl), 10 mM dNTP Mix (2 µl), RevertAidM-MuLV RT (200 U/µl, 1 µl) and
RNase free distilled H2O (8.5 µl). The synthesized cDNA
was amplified by RT-qPCR using SYBR (Applied Biosystems; Thermo
Fisher Scientific, Inc.). Each reaction mixture for PCR (total
volume 20 µl) consisted of cDNA (adjusted to 500 ng, 1 µl), forward
and reverse PCR primers (10 µM, 0.5 µl each), SYBR (10X, 10 µl) and
distilled H2O (8 µl). The themorcycling conditions were
as follows: Initital denaturation at 95°C for 2 min for one cycle,
followed by 40 cycles of denaturation at 95°C for 5 sec and
annealing and extension at 60°C for 31 sec. The relative expression
levels of target genes were normalized to rRNA (Applied Biosystems;
Thermo Fisher Scientific, Inc.) and were calculated using the
2−ΔΔCq method (23).
qPCR with SYBR Green detection was subsequently performed using an
Eppendorf Mastercycler ep realplex (Eppendorf). The gene primers
(Shanghai GenePharma Co., Ltd.) used were as follows: CD86, forward
5′-ACGGAGTCAATGAAGATTTCCT-3′, reverse 5′-GATTCGGCTTCTTGTGACATAC-3′;
iNOS, forward 5′-GTTTACCATGAGGCTGAAATCC-3′ reverse
5′-CCTCTTGTCTTTGACCCAGTAG-3′, TNF-α, forward,
5′-ATGTCTCAGCCTCTTCTCATTC-3′, reverse 5′-GCTTGTCACTCGAATTTTGAGA-3′;
IL-1β, forward 5′-TCGCAGCAGCACATCAACAAGAG-3′, reverse,
5′-TGCTCATGTCCTCATCCTGGAAGG-3′; IL-6, forward
5′-CTCCCAACAGACCTGTCTATAC-3′, reverse,
5′-CCATTGCACAACTCTTTTCTCA-3′; and β-actin, forward
5′-CACGATGGAGGGGCCGGACTCATC-3′ and reverse
5′-TAAAGACCTCTATGCCAACACAGT-3′.
Immunofluorescence
A total of 3×105/ml RAW264.7 macrophages
were seeded into 6-well plates at 37°C for 2–3 days with sterile
coverslips to make cell slides. Then, slides were washed with PBS
(Beijing Solarbio Science & Technology Co., Ltd.) 2–3 times and
then fixed with 4% paraformaldehyde for 10–20 min at room
temperature. The fixative was washed off three times with PBS (2–3
min/wash). Slides were permeabilized with 0.2% Triton X-100 for 5
min at room temperature, washed three times with PBS for 2–3 min
each time and subsequently blocked with 5% BSA (Sigma-Aldrich;
Merck KGaA) blocking solution at 37°C for 30 min. Sections were
then probed with various primary antibodies [anti-Cx43 polyclonal
antibody (1:100; cat. no. ab11370; Abcam), anti-CD86 polyclonal
antibody (1:100; cat. no. ab53004; Abcam) and anti-iNOS polyclonal
antibody (1:100; cat. no. ab15323; Abcam)], whilst as a negative
control, samples were incubated overnight at 4°C with the primary
antibody and rewarmed to 37°C for 30 min before the primary
antibody was discarded. Samples were washed three times with PBS (3
min/wash) prior to incubation with the fluorescent secondary
antibody (1:50; Goat anti-rabbit secondary antibodies; cat. no.
ZDR-5209; Beijing Zhongshan Jinqiao Biotechnology Co., Ltd.) at
37°C for 1 h. Following the incubation, the secondary antibody
solution was discarded and samples were washed three times with PBS
(5 min/wash). Slides were subsequently incubated with propidium
iodide (1:1,000; Beijing Solarbio Science & Technology Co.,
Ltd.) in the dark at room temperature for ~1 min prior to being
washed three times with PBS (5 min/wash). An anti-fluorescence
attenuating sealer was then added to the slides and stained slides
were observed using a Zeiss LSM 510 META laser confocal microscope
(magnification, ×630; Carl Zeiss AG).
ELISA
The levels of TNF-α, IL-1β and IL-6 in RAW264.7
macrophages (1×106/well) supernatant (Repeated
freeze-thaw and high-speed centrifugation (603 × g at 4°C for 5
min) to obtain cell supernatant) were analysed using ELISA kits
(TNF-α, cat. no. PMTA00B; IL-1β, cat. no. DY401; IL-6, cat. no.
PM6000B; R&D Systems, Inc.), according to the manufacturer's
protocols. The absorbance was measured at 450 nm. All absorbance
values were within the linear range of the standard curve and the
final result was expressed in pg/ml.
Statistical analysis
Data are expressed as the mean ± SEM from ≥6
independent experiments. Statistical analyses were performed SPSS
24.0 (IBM Corp.). Statistical differences between >2 groups were
assessed using one-way ANOVA followed by Tukey's post hoc test,
whereas statistical differences between two groups were determined
using unpaired Student's t-test. P<0.05 was considered to
indicate a statistically significant difference.
Results
AngII can mediate the polarization of
RAW264.7 macrophages to the M1-type
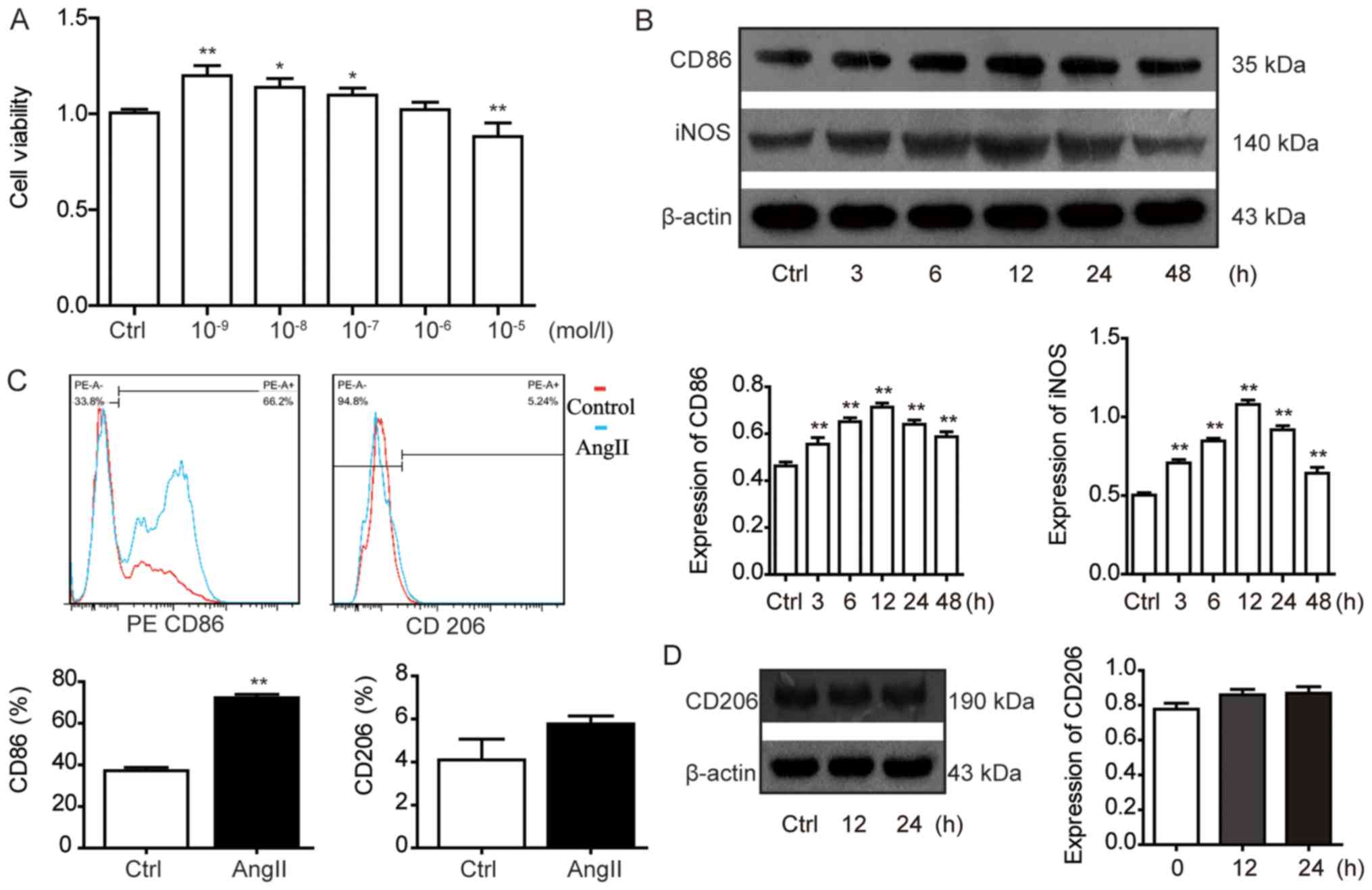
To investigate the effect of different
concentrations (0, 10−9, 10−8,
10−7, 10−6 and 10−5 mol/l) of
AngII on the cell viability of RAW264.7 macrophages, a CCK-8 assay
was used. It was observed that 10−6 mol/l AngII had no
significant effect on cell viability compared with the control
group; therefore, AngII at a concentration of 10−6 mol/l
was selected for use in further experiments (Fig. 1A). Flow cytometric analysis was
used to detect the expression levels of the M1-type macrophage
marker CD86 and the M2-type macrophage marker CD206 in RAW264.7
macrophages. The levels of CD86 expression were significantly
increased following AngII treatment compared with the control group
(Fig. 1C). There were no
significant differences observed in the expression levels of the M2
marker CD206 in the AngII group compared with the control group
(Fig. 1C). Furthermore, the
protein expression levels of CD86 and iNOS at 3, 6, 12, 24 and 48 h
were significantly increased in the AngII group compared with the
control group (Fig. 1B). Moreover,
this trend in protein expression levels of the M1-type macrophage
markers CD86 and iNOS occurred in a time-dependent manner; the
protein expression levels of CD86 and iNOS peaked following 12 h of
AngII treatment. However, the protein expression levels of the M2
macrophage marker CD206 were not significantly increased in the
AngII group compared with the control group after 12 and 24 h.
Therefore, in the subsequent experiments, a treatment intervention
time of 12 h was used (Fig.
1D).
AngII induces M1-type polarization in
RAW264.7 macrophages through the NF-κB (p65) signalling
pathway
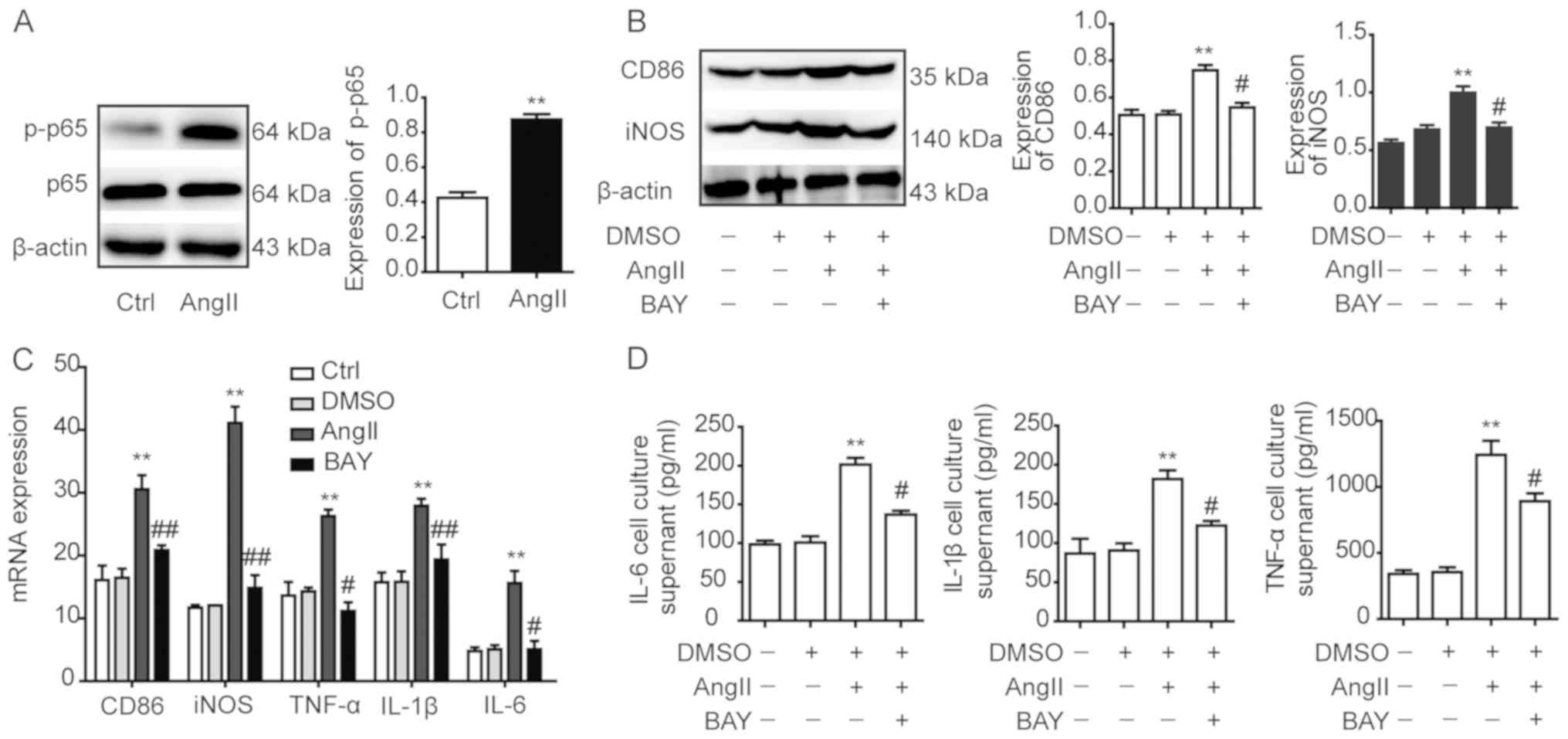
To investigate the role of NF-κB in AngII-induced
macrophage polarization, RAW264.7 macrophages were treated with
AngII for 12 h. Western blotting analysis observed that p-p65
expression levels in the AngII group were significantly increased
compared with the control group, which indicated that the NF-κB
(p65) signalling pathway may be activated by AngII (Fig. 2A). Subsequently, the RAW264.7
macrophages were pre-treated with the NF-κB (p65) signalling
pathway inhibitor BAY117082 prior to being incubated with AngII for
12 h. Western blotting revealed that the protein expression levels
of CD86 and iNOS in the DMSO group were not significantly different
compared with the control group (Fig.
2B); however, the expression levels of CD86 and iNOS in the
AngII group were significantly increased compared with the control
group. Notably, upon treatment with the NF-κB (p65) signalling
pathway inhibitor BAY117082, the protein expression levels of CD86
and iNOS in the BAY117082 group were significantly decreased
compared with the AngII group (Fig.
2B).
The results of RT-qPCR analysis demonstrated that
the mRNA expression levels of the M1-type markers iNOS, TNF-α,
IL-1β, IL-6 and CD86 in RAW264.7 macrophages DMSO group were not
significantly different compared with the control group (Fig. 2C). The results showed that DMSO had
no effect on cell mRNA expression levels. However, the M1-type
markers in the AngII group were significantly increased compared
with the control group, whereas they were significantly decreased
in the BAY117082 group compared with the AngII group (Fig. 2C). Furthermore, the levels of the
M1-type markers TNF-α, IL-1β and IL-6 were analysed in the
supernatants obtained from RAW264.7 macrophages using ELISA. The
expression levels in the AngII group were significantly increased
compared with the control group (Fig.
2D), whereas the expression levels in the BAY117082 group were
significantly decreased compared with the AngII group (Fig. 2D).
AngII increases Cx43 protein
expression levels in RAW264.7 macrophages
Following the treatment of RAW264.7 macrophages with
10−6 mol/l AngII for 3, 6, 12, 24 and 48 h, the
increases in the protein expression levels of Cx43 in macrophages
were time-dependent, with the protein expression levels reaching
their maximum at 12 h (Fig. 3A).
The localization of Cx43 in RAW264.7 macrophages was determined
using an immunofluorescence assay. The results discovered that Cx43
was expressed in RAW264.7 macrophages; compared with the control
group, the protein expression levels of Cx43 in macrophages was
consistent with the results obtained from western blotting at
different time points (Fig.
3B).
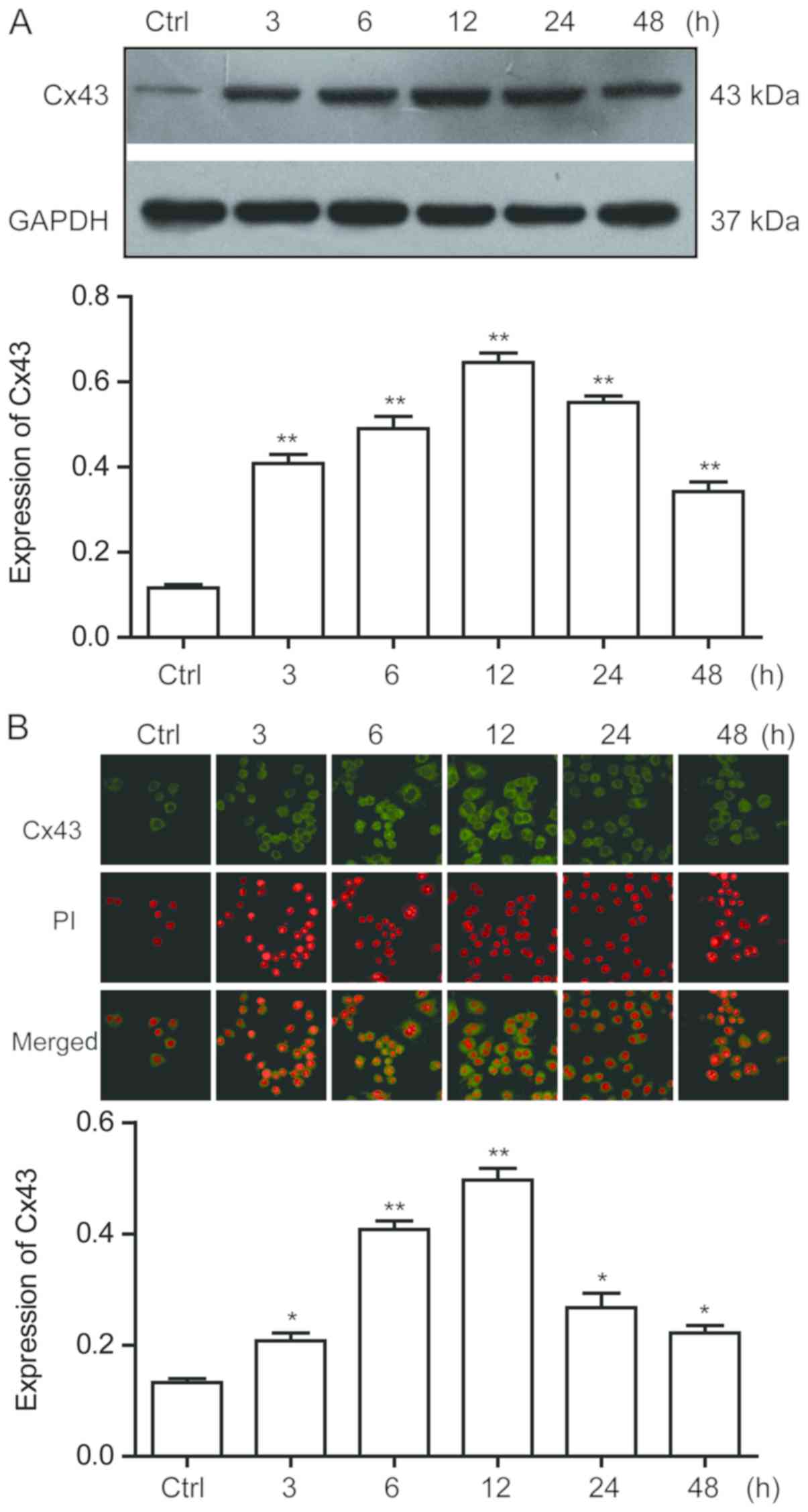 | Figure 3.AngII upregulates Cx43 protein
expression levels in RAW264.7 macrophages. (A) Western blotting was
used to detect the protein expression levels of Cx43 in RAW264.7
macrophages treated with 10−6 mol/l AngII at different
time points (3, 6, 12, 24 and 48 h). Semi-quantitative analysis of
Cx43 protein expression levels in macrophages was subsequently
performed. (B) Immunofluorescence assay was used to detect the
expression and localization of Cx43 protein in macrophages at
different time points (3, 6, 12, 24 and 48 h) following
10−6 mol/l AngII treatment and semi-quantitative
analysis of Cx43 protein levels in macrophages was conducted.
Magnification, ×630. Data are presented as the mean ± SEM (n=6).
*P<0.05, **P<0.01 vs. Ctrl group. AngII, angiotensin II;
Ctrl, control; Cx43, connexin 43; PI, propidium iodide. |
AngII induces the polarization of
RAW264.7 macrophages to the M1-type, and polarization markers are
inhibited following pre-treatment of macrophages with the Cx43
blockers Gap26 and Gap19
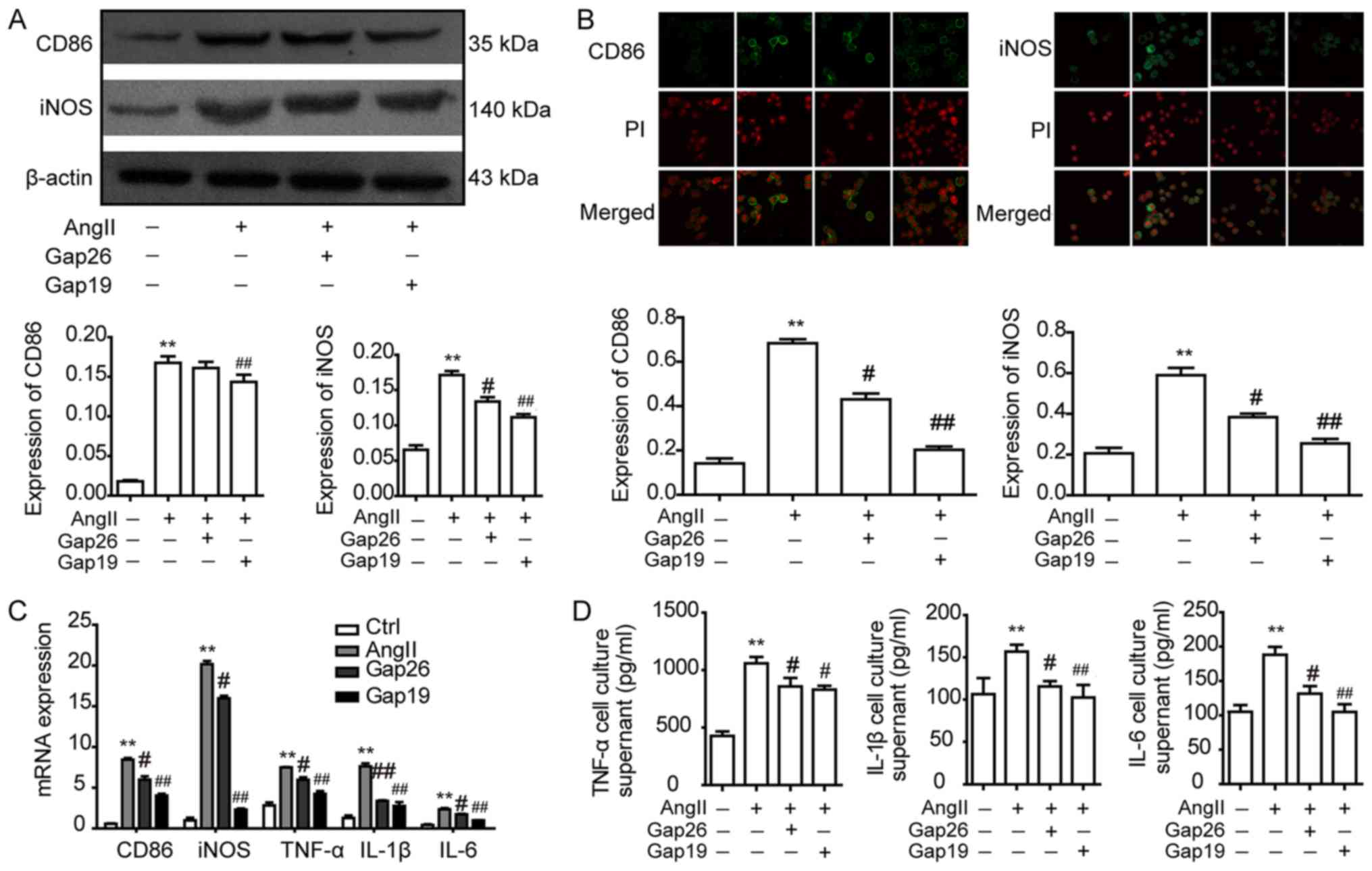
To investigate the role of Cx43 in macrophage
polarization, RAW264.7 macrophages were pre-treated with the Cx43
blockers Gap26 and Gap19 for 45 min prior to AngII treatment for 12
h. Western blotting observed that compared with the untreated
control group, the protein expression levels of the macrophage
M1-type markers, CD86 and iNOS, were significantly increased
following 12 h of AngII treatment (Fig. 4A). Notably, CD86 protein expression
was significantly decreased in the Gap19 group compared with the
AngII group, whereas iNOS protein expression levels were
significantly decreased in the Gap26 and Gap19 groups compared with
the AngII group (Fig. 4A). The
mRNA expression levels of M1-type markers were subsequently
investigated using RT-qPCR. The results demonstrated that the mRNA
expression levels of iNOS, TNF-α, IL-1β, IL-6 and CD86 in RAW264.7
macrophages were significantly increased following AngII treatment
compared with the control group; however, this effect was
significantly inhibited by pre-treatment with Gap26/Gap19 (Fig. 4C). The localization of the M1-type
markers CD86 and iNOS in RAW264.7 macrophages was analysed using
immunofluorescence. The results indicated that CD86 and iNOS were
expressed in RAW264.7 macrophages; compared with the control group,
the protein expression levels of M1-type markers CD86 and iNOS were
consistent with the results revealed with western blotting
(Fig. 4B). In addition, the levels
of M1-type cytokines TNF-α, IL-1β and IL-6 in the supernatants of
RAW264.7 macrophages were detected using ELISA; the levels of
TNF-α, IL-1β and IL-6 in the AngII group were significantly
increased compared with the control group; however, this increase
was significantly prevented following pre-treatment with
Gap26/Gap19 (Fig. 4D). These
results suggested that AngII may induce RAW264.7 macrophage
polarization to the M1-type by upregulating Cx43 expression.
AngII activates the NF-κB (p65)
signalling pathway by upregulating Cx43 expression
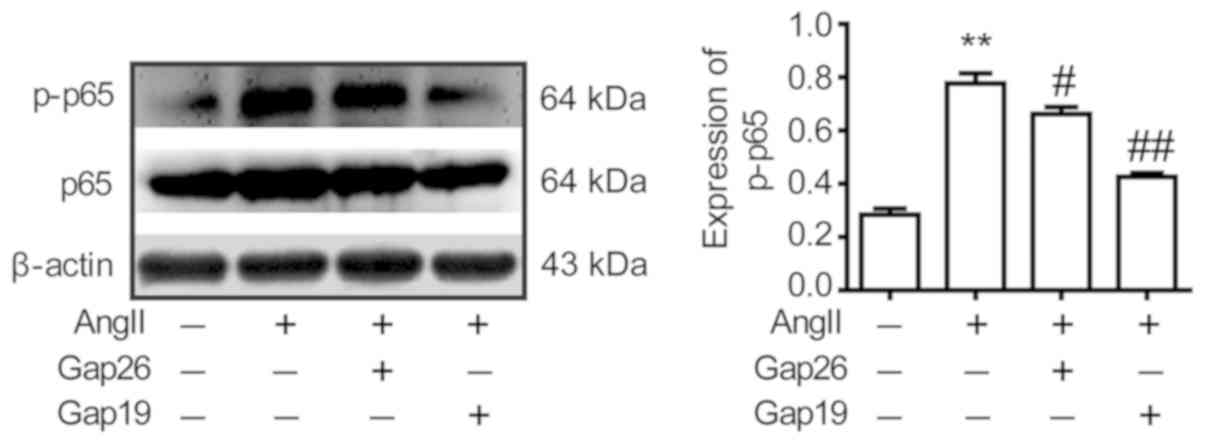
To investigate the relationship between Cx43 and the
NF-κB (p65) signalling pathway, RAW264.7 macrophages were
pre-treated with the Cx43 blockers Gap26 and Gap19 for 45 min prior
to AngII treatment for 12 h. It was revealed that the expression
levels of p-p65 in the Gap26 and Gap19 groups were significantly
decreased compared with the AngII group, indicating that AngII
activates the NF-κB (p65) signalling pathway by upregulating Cx43
expression (Fig. 5).
Discussion
Atherosclerosis is a multifactorial inflammatory
disease of the middle to large arterial wall (24), and is one of the leading causes of
death in industrialized countries, with incidence rates rapidly
increasing in developing countries (25). A large number of macrophages are
found to accumulate in atherosclerotic plaques, which subsequently
secrete various inflammatory cytokines, further aggravating the
accumulation of macrophages in blood vessels, and promoting the
continual change and development of atherosclerotic plaques
(26). Autopsies of patients with
atherosclerosis revealed that a large number of macrophages,
lymphocytes and foam cells are found in the coronary plaque, and
the ratio of macrophages and lymphocytes is largest in the rupture
plaque (25). Macrophages are
mainly divided into two different subtypes: M1 and M2, which are
predominantly located in unstable plaques and stable plaque
tissues, respectively (5,6). It has previously been reported that
iNOS, TNF-α, IL-1β, IL-6, IL-12 and CD86 are major markers of
M1-type macrophages (27). In
addition, AngII was found to stimulate RAW264.7 macrophages to not
only promote the production of pro-inflammatory factors, such as
TNF-α, IL-1β and IL-6, but also to increase the release of reactive
oxygen species (28). Therefore,
it was hypothesized that through inhibiting the polarization of
macrophages to the M1-type, it may be possible to stabilize
vascular plaques and reduce the formation of unstable plaques,
which would help identify mechanisms to prevent and treat
cardiovascular and cerebrovascular diseases caused by
atherosclerosis.
In the present study, RAW264.7 macrophages were used
to induce M1-type polarization and it was observed that
10−6 mol/l AngII treatment had no significant effect on
cell viability. This was consistent with previous findings by Li
et al (17) and Jiang et
al (29), who also used this
concentration of AngII in their studies. Therefore, this
concentration was used in subsequent experiments. It was revealed
that CD86 and iNOS expression levels in RAW264.7 macrophages were
significantly increased following treatment with AngII, whereas the
levels of the M2-type marker CD206 were not affected. The results
suggested that AngII may induce M1-type polarization in RAW264.7
macrophages.
It is well known that the NF-κB pathway serves an
important role in regulating immune and inflammatory processes, and
affects the expression of cytokines that mediate pro-inflammatory
responses and inflammation, apoptosis and proliferation (30). The NF-κB (p65) signalling pathway
is highly activated in the inflammatory sites with diverse enzymes,
which induces the transcription and production of inflammatory
cytokines, including IL-1β, IL-6 and TNF-α at the inflammatory
site; this pathway is known to serve an important role during the
acute phase of the immune response (31). It has previously been reported that
treatment with NF-кB (p65) inhibitors significantly reduced
mechanically induced inflammatory responses (32). In the present study, to investigate
the potential role of the NF-κB (p65) signalling pathway in
AngII-induced macrophage M1-type polarization, RAW264.7 macrophages
were pre-treated with inhibitors of the NF-κB (p65) signalling
pathway. Following treatment with NF-κB (p65) inhibitors, the
protein and mRNA expression levels of M1-type macrophage markers
were significantly reduced. These results suggested that
AngII-induced macrophage polarization to the M1-type may be
modulated by the NF-κB (p65) signalling pathway.
It has previously been demonstrated that gap
junction channels and hemichannels composed of Cxs are found in
almost all cells and tissues; however, their role in the immune
response and pathological conditions has only recently been studied
(33). Cx43 is one of the basic
components of gap junctions and hemichannels, which are opened by
changes in extracellular conditions (34). Cx43 is present in numerous types of
cells, such as smooth muscle cells, resident and circulating
leukocytes (neutrophils and dendritic cells), lymphocytes,
activated macrophages, mast cells and some endothelial cells
(35). Cx43 deficiency has been
reported to increase mortality in a mouse model of bacterial
peritonitis, and Cx43 is reportedly upregulated in macrophages
following LPS treatment (36).
Shen et al (36) suggested
that Cx43 expression in macrophages was associated with macrophage
migration, an important immune process in the host defence against
infection. In addition, Nie et al (37) demonstrated that the expression
levels of Cx43 in mouse dendritic cells were significantly
increased following AngII treatment. Immunofluorescence staining of
Cx43, CD86 and iNOS was used to investigate the effect of AngII on
macrophage polarization and it was discovered that Cx43 was
expressed in RAW264.7 macrophages. Moreover, some of these results
are consistent with the Shen et al (36) study. In addition, AngII treatment
increased the expression levels of CD86, iNOS and Cx43, which
indicated that Cx43 may be involved in the M1-type macrophage
polarization process.
In the present study, the pre-treatment of
macrophages with two specific blockers of Cx43, Gap26 (a gap
junction blocker) and Gap19 (a specific hemichannel blocker), was
observed to decrease the mRNA and protein expression levels of
M1-type macrophage polarization markers; Gap19 was more effective
compared with Gap26 in suppressing the M1-type polarization index.
Cxs are integral membrane proteins, and a hemichannel is formed by
six Cx monomers in the plasma membrane. Hemichannel interactions
facilitate the exchange of ions and small molecules, which forms
the basis of gap junction intercellular communication (38). Within an in vitro model of
ischaemia/reperfusion (I/R) injury, Yin et al (34) reported that the concentration of
anti-inflammatory cytokines in the oxygen-glucose
deprivation/reperfusion group is significantly decreased, while the
concentration of anti-inflammatory cytokines increases after Gap19
treatment; however, Gap26 treatment only increased IL-10
concentration. These results are consistent with the results
obtained in this study. Thus, it was hypothesized that the
hemichannel may serve a more significant role in the polarization
process compared with gap junction channels; however, the exact
mechanism that led to these results remains relatively unclear and
requires further investigations. Huang et al (39) studied the therapeutic effects of
two specific Cx43 inhibitors, Gap26 and Gap27, on the development
of allergic airway disease in mice. These studies suggest that
Gap26 also has protective effects against other diseases. Hawat
et al (40) demonstrated
that the Cx43 inhibitor Gap26 protected intact hearts from I/R
injury, either before or after ischaemia. The results from this
study are consistent with the results reported in the
aforementioned studies.
In a previous study, immunofluorescence staining of
pan-macrophages (CD68), pro-inflammatory M1 macrophages (CD86) and
remodelling M2 macrophages (CD206) was used to investigate the
effect of chitosan/hyaluronic acid (CS/HA) hydrogel on macrophage
polarization; the number of M1 pro-inflammatory cells was similar
among the three groups (the control group, Fibrin gel group and
CS/HA hydrogel group); however, the CS/HA hydrogel markedly
increased the number of M2-type cells (41). The present results are consistent
with this previous study (41),
suggesting that CD86 and CD206 are expressed in macrophages.
Another study investigated the effect of plumbagin (PL) on iNOS
expression in LPS-activated BV-2 microglial cells; it was
discovered that in the absence of LPS stimulation, iNOS was not
expressed in resting cells and PL-treated cells, whereas in the
presence of LPS, iNOS expression was detected; however, this effect
was attenuated in LPS and PL co-treated cells (42). Thus, it was concluded that AngII
may induce the polarization of RAW264.7 macrophages to the M1-type
through upregulating Cx43.
It has been reported that the activation of NF-κB
(p65) signalling is dependent on Cx43 (32). The phosphorylation of p65 is an
essential step in the NF-κB signalling cascade; and transfection
with small interfering RNA targeting Cx43 or treatment with a NF-кB
(p65) inhibitor was found to significantly reduce mechanically
induced inflammation (32). Chen
et al (32) reported that
the Cx43 hemichannel served an important role in the development of
ossification of the posterior longitudinal ligament through
mechanical signal transduction, which subsequently activated the
NF-κB (p65) signalling pathway in ligament fibroblasts and the
inflammatory response in longitudinal ligament ossification cells.
In the present study, the possible relationship between Cx43 and
the NF-кB (p65) signalling pathway was investigated in the cellular
model, as both Cx43 and NF-κB (p65) signalling pathways were
activated by AngII. Alonso et al (43) discovered that AngII could increase
the expression levels of Cx43 in A7r5 cells through activating the
NF-κB pathway. In addition, another study revealed that oxidized
low-density lipoprotein could increase the expression of Cx43 in
rat thoracic aortic vascular smooth muscle cells, and these effects
were prevented following pre-treatment with BAY117082 (44). Based on these studies, it was
hypothesized that AngII activation may upregulate Cx43 expression
through the NF-κB (p65) signalling pathway. In the present study,
it was further observed that when RAW264.7 macrophages were
pre-treated with Gap26 and Gap19, the protein expression levels of
p-p65 were decreased in macrophages, particularly following the
pre-treatment with Gap19. These results indicated that AngII
activation may activate the NF-кB (p65) signalling pathway through
upregulating Cx43 expression. In conclusion, the present study
discovered that AngII may activate the polarization of RAW264.7
macrophages to the M1-type through the Cx43/NF-κB (p65) signalling
pathway.
Acknowledgements
Not applicable.
Funding
This work was supported by grants from the National
Natural Science Foundation of China (grant nos. 81860286 and
81660271).
Availability of data and materials
The datasets used and/or analysed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
LWu, KC, LWa and KM performed study design. LWu, KC,
JiX and JuX performed data collection and statistical analysis. LZ,
XL, LL and JS performed data interpretation. LWu, KC, LWa and KM
performed manuscript preparation. LWu, KC, JiX and LZ performed
literature search. XL, LL, LWa and JS collected funds. All authors
read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Wang Y, Xie Y, Zhang A, Wang M, Fang Z and
Zhang J: Exosomes: An emerging factor in atherosclerosis. Biomed
Pharmacother. 115:1089512019. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Zhou M, Ren P, Zhang Y, Li S, Li M, Li P,
Shang J, Liu W and Liu H: Shen-Yuan-Dan capsule attenuates
atherosclerosis and foam cell formation by enhancing autophagy and
inhibiting the PI3K/Akt/mTORC1 signaling pathway. Front Pharmacol.
10:6032019. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Moore KJ, Sheedy FJ and Fisher EA:
Macrophages in atherosclerosis: A dynamic balance. Nat Rev Immunol.
13:709–721. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Groh L, Keating ST, Joosten LAB, Netea MG
and Riksen NP: Monocyte and macrophage immunometabolism in
atherosclerosis. Semin Immunopathol. 40:203–214. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Mohammadi S, Saghaeian-Jazi M, Sedighi S
and Memarian A: Sodium valproate modulates immune response by
alternative activation of monocyte-derived macrophages in systemic
lupus erythematosus. Clin Rheumatol. 37:719–727. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Stöger JL, Gijbels MJ, van der Velden S,
Manca M, van der Loos CM, Biessen EA, Daemen MJ, Lutgens E and de
Winther MP: Distribution of macrophage polarization markers in
human atherosclerosis. Atherosclerosis. 225:461–468. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Binesh A, Devaraj SN and Devaraj H:
Expression of chemokines in macrophage polarization and
downregulation of NF-κB in aorta allow macrophage polarization by
diosgenin in atherosclerosis. J Biochem Mol Toxicol. 34:e224222020.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zhou S, Lu H, Chen R, Tian Y, Jiang Y,
Zhang S, Ni D, Su Z and Shao X: Angiotensin II enhances the
acetylation and release of HMGB1 in RAW264.7 macrophage. Cell Biol
Int. 42:1160–1169. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Manucha W: Mitochondria and oxidative
stress participation in renal inflammatory process. Medicina (B
Aires). 74:254–258. 2014.(In Spanish). PubMed/NCBI
|
|
10
|
Mitchell S, Vargas J and Hoffmann A:
Signaling via the NF-κB system. Wiley Interdiscip Rev Syst Biol
Med. 8:227–241. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ding H, Zhou Y and Huang H: MiR-101a
ameliorates AngII-mediated hypertensive nephropathy by blockade of
TGFβ/Smad3 and NF-κB signalling in a mouse model of hypertension.
Clin Exp Pharmacol Physiol. 46:246–254. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Willebrords J, Crespo Yanguas S, Maes M,
Decrock E, Wang N, Leybaert L, Kwak BR, Green CR, Cogliati B and
Vinken M: Connexins and their channels in inflammation. Crit Rev
Biochem Mol Biol. 51:413–439. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Donahue HJ, Qu RW and Genetos DC: Joint
diseases: From connexins to gap junctions. Nat Rev Rheumatol.
14:42–51. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Glass AM, Snyder EG and Taffet SM:
Connexins and pannexins in the immune system and lymphatic organs.
Cell Mol Life Sci. 72:2899–2910. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Jia G, Mitra AK, Cheng G, Gangahar DM and
Agrawal DK: Angiotensin II and IGF-1 regulate connexin43 expression
via ERK and p38 signaling pathways in vascular smooth muscle cells
of coronary artery bypass conduits. J Surg Res. 142:137–142. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Sarieddine MZ, Scheckenbach KE, Foglia B,
Maass K, Garcia I, Kwak BR and Chanson M: Connexin43 modulates
neutrophil recruitment to the lung. J Cell Mol Med. 13:4560–4570.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Li M, Liu JT, Pang XM, Han CJ and Mao JJ:
Epigallocatechin-3-gallate inhibits angiotensin II and
interleukin-6-induced C-reactive protein production in macrophages.
Pharmacol Rep. 64:912–918. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Yang LX, Liu H, Guo RW, Ye J, Wang XM, Qi
F, Guo CM and Liang X: Angiotensin II induces EMMPRIN expression in
THP-1 macrophages via the NF-kappaB pathway. Regul Pept. 163:88–95.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Desplantez T, Verma V, Leybaert L, Evans
WH and Weingart R: Gap26, a connexin mimetic peptide, inhibits
currents carried by connexin43 hemichannels and gap junction
channels. Pharmacol Res. 65:546–552. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Li W, Bao G, Chen W, Qiang X, Zhu S, Wang
S, He M, Ma G, Ochani M, Al-Abed Y, et al: Connexin 43 hemichannel
as a novel mediator of sterile and infectious inflammatory
diseases. Sci Rep. 8:1662018. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Liang S, Chen Z, Jiang G, Zhou Y, Liu Q,
Su Q, Wei W, Du J and Wang H: Activation of GPER suppresses
migration and angiogenesis of triple negative breast cancer via
inhibition of NF-κB/IL-6 signals. Cancer Lett. 386:12–23. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Strickson S, Campbell DG, Emmerich CH,
Knebel A, Plater L, Ritorto MS, Shpiro N and Cohen P: The
anti-inflammatory drug BAY 11-7082 suppresses the MyD88-dependent
signalling network by targeting the ubiquitin system. Biochem J.
451:427–437. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Hansson GK, Robertson AK and
Söderberg-Nauclér C: Inflammation and atherosclerosis. Annu Rev
Pathol. 1:297–329. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Shao BZ, Han BZ, Zeng YX, Su DF and Liu C:
The roles of macrophage autophagy in atherosclerosis. Acta
Pharmacol Sin. 37:150–156. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Han X, Ni J, Wu Z, Wu J, Li B, Ye X, Dai
J, Chen C, Xue J, Wan R, et al: Myeloid-specific dopamine D2
receptor signaling controls inflammation in acute pancreatitis via
inhibiting M1 macrophage. Br J Pharmacol. Feb 14–2020.(Epub ahead
of print). View Article : Google Scholar
|
|
27
|
Tabas I and Bornfeldt KE: Macrophage
phenotype and function in different stages of atherosclerosis. Circ
Res. 118:653–667. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Guo F, Chen XL, Wang F, Liang X, Sun YX
and Wang YJ: Role of angiotensin II type 1 receptor in angiotensin
II-induced cytokine production in macrophages. J Interferon
Cytokine Res. 31:351–361. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Jiang Q, Pan D, Yang Y, Hu Y, Fang L,
Shang P, Xia Y and Li D: Luteolin regulates macrophage polarization
via the PI3K/Akt pathway to inhibit the apoptosis stimulated by
angiotensin II. Curr Pharm Biotechnol. 19:428–437. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kim KN, Ko SC, Ye BR, Kim MS, Kim J, Ko
EY, Cho SH, Kim D, Heo SJ and Jung WK:
5-Bromo-2-hydroxy-4-methyl-benzaldehyde inhibited LPS-induced
production of pro-inflammatory mediators through the inactivation
of ERK, p38, and NF-κB pathways in RAW 264.7 macrophages. Chem Biol
Interact. 258:108–114. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Gupta A, Niger C, Buo AM, Eidelman ER,
Chen RJ and Stains JP: Connexin43 enhances the expression of
osteoarthritis-associated genes in synovial fibroblasts in culture.
BMC Musculoskelet Disord. 15:4252014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Chen D, Chen Y, Li T, Shi L, Pan M and
Chen D: Role of cx43-mediated NFкB signaling pathway in
ossification of posterior longitudinal ligament: An in vivo and in
vitro study. Spine (Phila Pa 1976). 42:E1334–E1341. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Legein B, Temmerman L, Biessen EA and
Lutgens E: Inflammation and immune system interactions in
atherosclerosis. Cell Mol Life Sci. 70:3847–3869. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Yin X, Feng L, Ma D, Yin P, Wang X, Hou S,
Hao Y, Zhang J, Xin M and Feng J: Roles of astrocytic connexin-43,
hemichannels, and gap junctions in oxygen-glucose
deprivation/reperfusion injury induced neuroinflammation and the
possible regulatory mechanisms of salvianolic acid B and
carbenoxolone. J Neuroinflammation. 15:972018. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Pfenniger A, Chanson M and Kwak BR:
Connexins in atherosclerosis. Biochim Biophys Acta. 1828:157–166.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Shen C, Chen JH, Lee Y, Hassan MM, Kim SJ,
Choi EY, Hong ST, Park BH and Park JH: mTOR- and SGK-mediated
connexin 43 expression participates in
lipopolysaccharide-stimulated macrophage migration through the
iNOS/Src/FAK axis. J Immunol. 201:2986–2997. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Nie W, Yan H, Li S, Zhu W, Fan F and Zhu
J: Angiotensin II promotes atherogenesis through upregulating the
expression of connexin 43 in dendritic cells. Cell Mol Biol
(Noisy-le-grand). 61:96–101. 2015.PubMed/NCBI
|
|
38
|
Kim Y, Davidson JO, Green CR, Nicholson
LFB, O'Carroll SJ and Zhang J: Connexins and pannexins in cerebral
ischemia. Biochim Biophys Acta Biomembr. 1860:224–236. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Huang JQ, Chen XY, Huang F, Fan JM, Shi XW
and Ju YK: Effects of connexin 43 inhibition in an
ovalbumin-induced mouse model of asthma. Iran J Allergy Asthma
Immunol. 17:29–38. 2018.PubMed/NCBI
|
|
40
|
Hawat G, Benderdour M, Rousseau G and
Baroudi G: Connexin 43 mimetic peptide Gap26 confers protection to
intact heart against myocardial ischemia injury. Pflugers Arch.
460:583–592. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Deng Y, Ren J, Chen G, Li G, Wu X, Wang G,
Gu G and Li J: Injectable in situ cross-linking chitosan-hyaluronic
acid based hydrogels for abdominal tissue regeneration. Sci Rep.
7:26992017. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Messeha SS, Zarmouh NO, Mendonca P, Kolta
MG and Soliman KFA: The attenuating effects of plumbagin on
pro-inflammatory cytokine expression in LPS-activated BV-2
microglial cells. J Neuroimmunol. 313:129–137. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Alonso F, Krattinger N, Mazzolai L, Simon
A, Waeber G, Meda P and Haefliger JA: An angiotensin II- and
NF-kappaB-dependent mechanism increases connexin 43 in murine
arteries targeted by renin-dependent hypertension. Cardiovasc Res.
87:166–176. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Wang M, Wu Y, Yu Y, Fu Y, Yan H, Wang X,
Li T, Peng W and Luo D: Rutaecarpine prevented ox-LDL-induced VSMCs
dysfunction through inhibiting overexpression of connexin 43. Eur J
Pharmacol. 853:84–92. 2019. View Article : Google Scholar : PubMed/NCBI
|