Introduction
Rheumatoid arthritis (RA) is a chronic systemic
disease accompanied by inflammatory synovitis that is mainly
characterized by symmetrical distribution of invasive joint
inflammation of the hand and foot (1,2). In
addition, RA exhibits increased interstitial inflammatory cell
infiltration and bone tissue destruction, resulting in joint
deformity and loss of function (3). Immune function is considered to be
the main aspect associated with RA; RA is characterized by the
induction of innate immune disorders, including immune
complex-mediated complement activation, osteoclast and chondrocyte
activation and cytokine network dysregulation, which develop
semi-autonomous features that contribute to disease progression
(4,5). However, the exact mechanism of RA
development remains elusive and further investigation is
required.
General, surgical and pharmaceutical therapies are
widely applied in RA treatment (6). The most commonly used pharmacological
RA drugs include the administration of non-steroidal
anti-inflammatory drugs, immunosuppressants, botanicals and
biological agents (7). Rituximab
(RTX), a chimeric monoclonal antibody against the CD20 ligand of B
lymphocytes, has been reported to exhibit therapeutic activity in
the clinical treatment of RA (8);
however, its therapeutic mechanism needs to be further
investigated. Although several drugs alleviate pain in patients
with RA, their efficacy is limited (9), therefore the development of novel and
effective drugs for RA is required.
The present study aimed to further elucidate the
pathogenesis of RA and identify potential drugs for RA treatment.
The expression profiles of normal, RA control and RTX-treated
tissues were analyzed. A series of immune-related genes, including
leukocyte immunoglobulin-like receptor subfamily B member 1
(LILRB1), were detected by screening the differentially expressed
genes (DEGs). The results revealed that LILRB1 was associated with
RA pathogenesis. LILRB1, an inhibitory receptor broadly expressed
in leukocytes, has been demonstrated to regulate immune responses
by binding to MHC class I molecules on antigen-presenting cells
(10). Finally, Traditional
Chinese Medicine (TCM) libraries were molecularly screened for this
key functional gene in order to identify potential therapeutic
drugs.
Materials and methods
Download of expression profile chip
data and DEGs analysis
The screening of DEGs (11,12)
in the synovial tissues of normal patients without RA and patients
with RA (GSE55235) (13) was
performed using the Gene Expression Omnibus (GEO) database
(14) and differential gene
analysis. In addition, DEG screening in RA and RTX-treated patients
(GSE24742) (15) was assessed
using the GEO database and R, version 3.6.2. Data quality was
determined by calculating residual sign, residuals, weight,
relative log expression, normalized unscaled standard errors and
RNA degradation. Finally, the differences in RNA expression
profiles between groups were analyzed using the pheatmap and limma
R packages (16,17). |Log2 fold-change (FC)|≥1
and P<0.05 were set as the cutoff criteria for DEGs.
Gene Ontology (GO) and Kyoto
Encyclopedia of Genes and Genomes (KEGG) analyses (18,19)
The functions of DEGs were analyzed using the ClueGO
plug-in application in Cytoscape 3.6.1 (https://cytoscape.org). In addition, KEGG pathway
enrichment analysis (20) was
carried out using ClueGO (21) and
visualized using CluePedia (22).
P<0.05 was set as the cutoff value.
Gene set enrichment analysis
(GSEA)
GSEA analysis was performed using the GSEA software
(23). In brief, the method
consisted of the following steps. First, the gene data, including
expression profiles and class distinctions, were listed. Given a
defined set of genes, the goal of GSEA was to determine whether the
members of the gene set were found at the top or bottom of the
list. Subsequently, an enrichment score was calculated in order to
identify the degree of the over-represented genes. Finally, a
weighted enrichment statistics was carried out and the number of
random permutations was set to 1,000 times.
Pathway analysis
DEGs were subjected to signal pathway enrichment
analysis using the ConsensusPathDB database (http://cpdb.molgen.mpg.de) (24,25).
The files containing the measured genetic data were uploaded to the
database and subsequently pathway enrichment analysis was carried
out using the gene set analysis function.
Protein-protein interaction (PPI)
analysis
The PPI network was analyzed using STRING software
(https://string-db.org) (26). The organism selected was ‘Homo
sapiens’. The interacting protein complexes that functionally
influence the physiological processes of a disease and the PPI
enrichment analysis reflected the interaction between DEGs.
TCM database and molecular docking
simulation of LILRB1
A total of 32,364 compounds were obtained from the
TCM database (http://tcm.cmu.edu.tw) (27). The conformational energy of small
molecules was minimized using the Maestro software (version 11.8,
Schrödinger, LLC) by adding hydrogen atoms and removing counter
ions and salts (28,29). The crystal structure of LILRB1 was
downloaded from Protein Data Bank (structure no. 1UGN; http://www.rcsb.org) (30) and the protein structure was refined
by removing crystalline water and ions. In addition, energy
minimization was performed on the LILRB1 protein structure. TCM
docking and the selection of candidate compounds was performed
using the virtual screening workflow model of Schrodinger and Glide
XP (extra precision), respectively (31–33).
Results
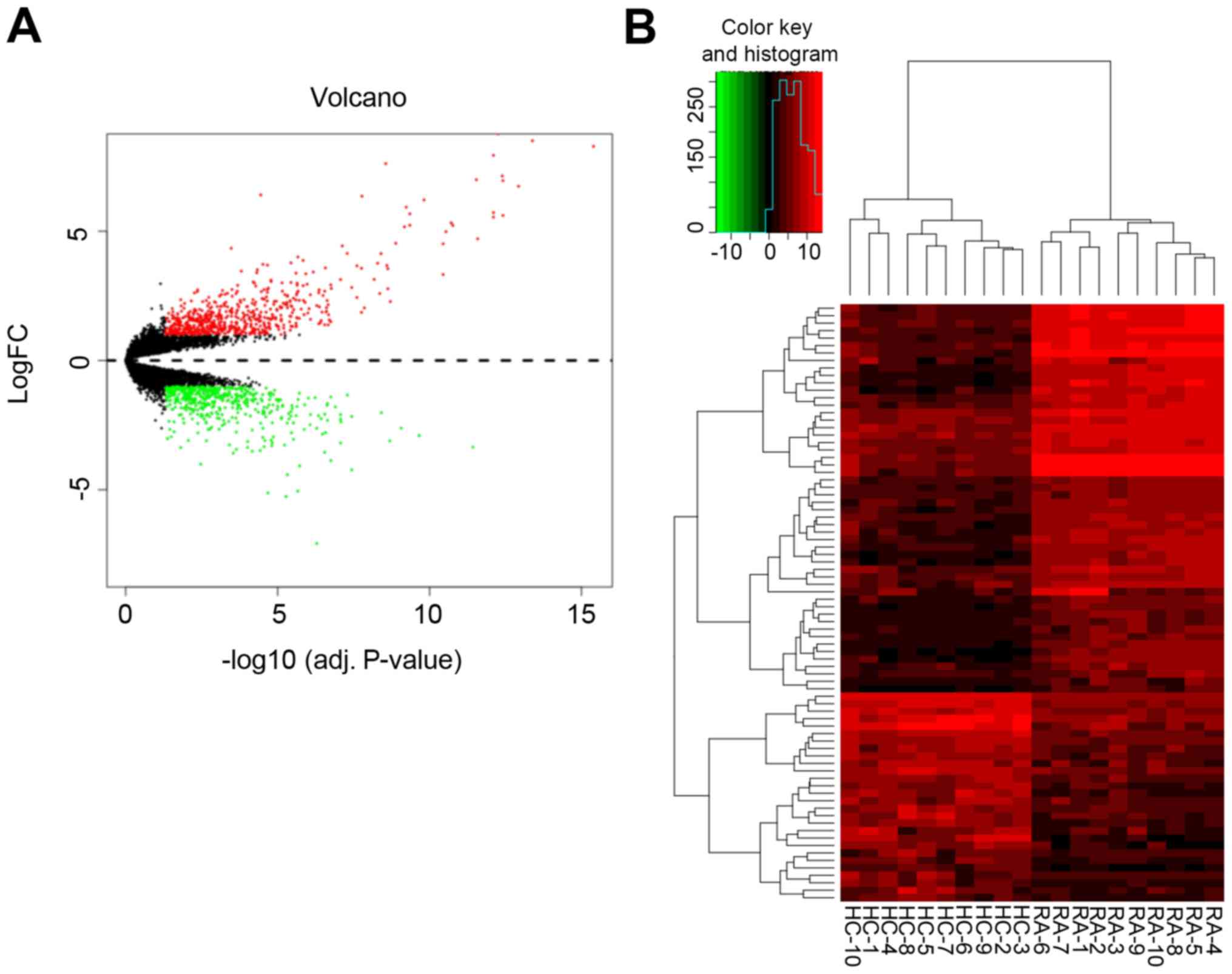
Identification of DEGs in RA
The gene expression profiles of the synovial tissues
of patients with RA from GSE55235 were obtained from the GEO
database. The microarray data from GSE55235 included synovial
tissues from 10 healthy and 10 RA joints. A total of 1,150 DEGs
were extracted from the expression profile data set, including 508
downregulated and 642 upregulated genes. |Log2FC|≥1 and
P<0.05 were set as the cutoff criteria for DEGs. The
distribution of DEGs is presented in a volcano plot (Fig. 1A). In addition, hierarchical
cluster analysis was performed in order to obtain an overview of
the expression profiles of normal and RA tissues (Fig. 1B). Finally, all heat maps
demonstrated different adjustment directions and significant
separation between normal and RA samples.
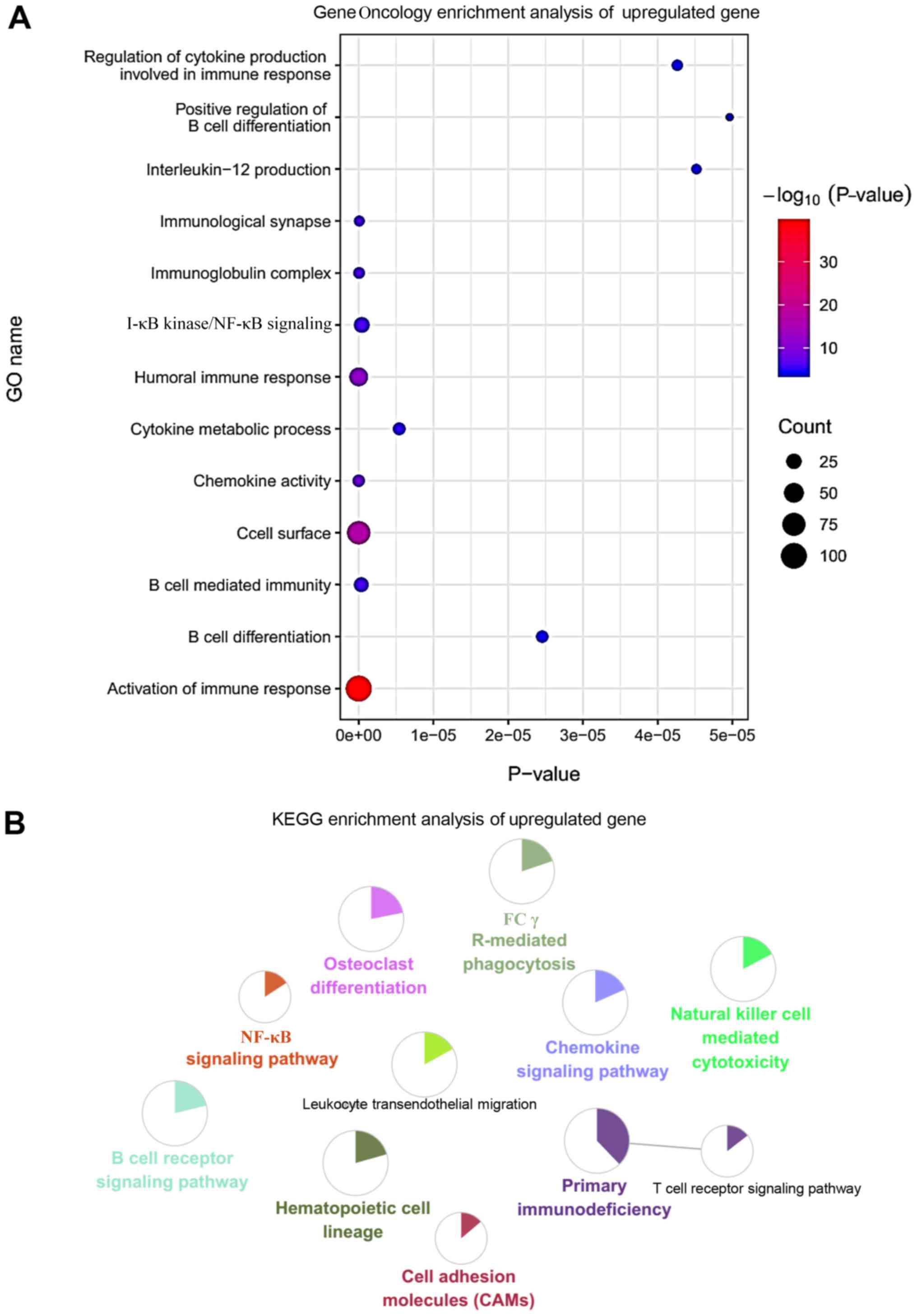
GO and KEGG analyses of DEGs in
RA
GO and KEGG enrichment analyses of DEGs were
performed using the ClueGO plug-in in Cytoscape software, and
subsequently the upregulated and downregulated genes were analyzed.
The enriched upregulated genes were mainly associated with
‘interleukin-12 production’, ‘I-κB kinase/NF-κB signaling’,
‘regulation of cytokine production involved in immune response’,
‘positive regulation of B cell differentiation’, ‘cytokine
metabolic process’ and ‘activation of immune response’ (Fig. 2A). The results of the pathway
analysis showed that RA mainly affected the ‘Fcγ R-mediated
phagocytosis’, ‘natural killer cell-mediated cytotoxicity’, ‘B cell
receptor signaling pathway’, ‘NF-κB signaling pathway’ and
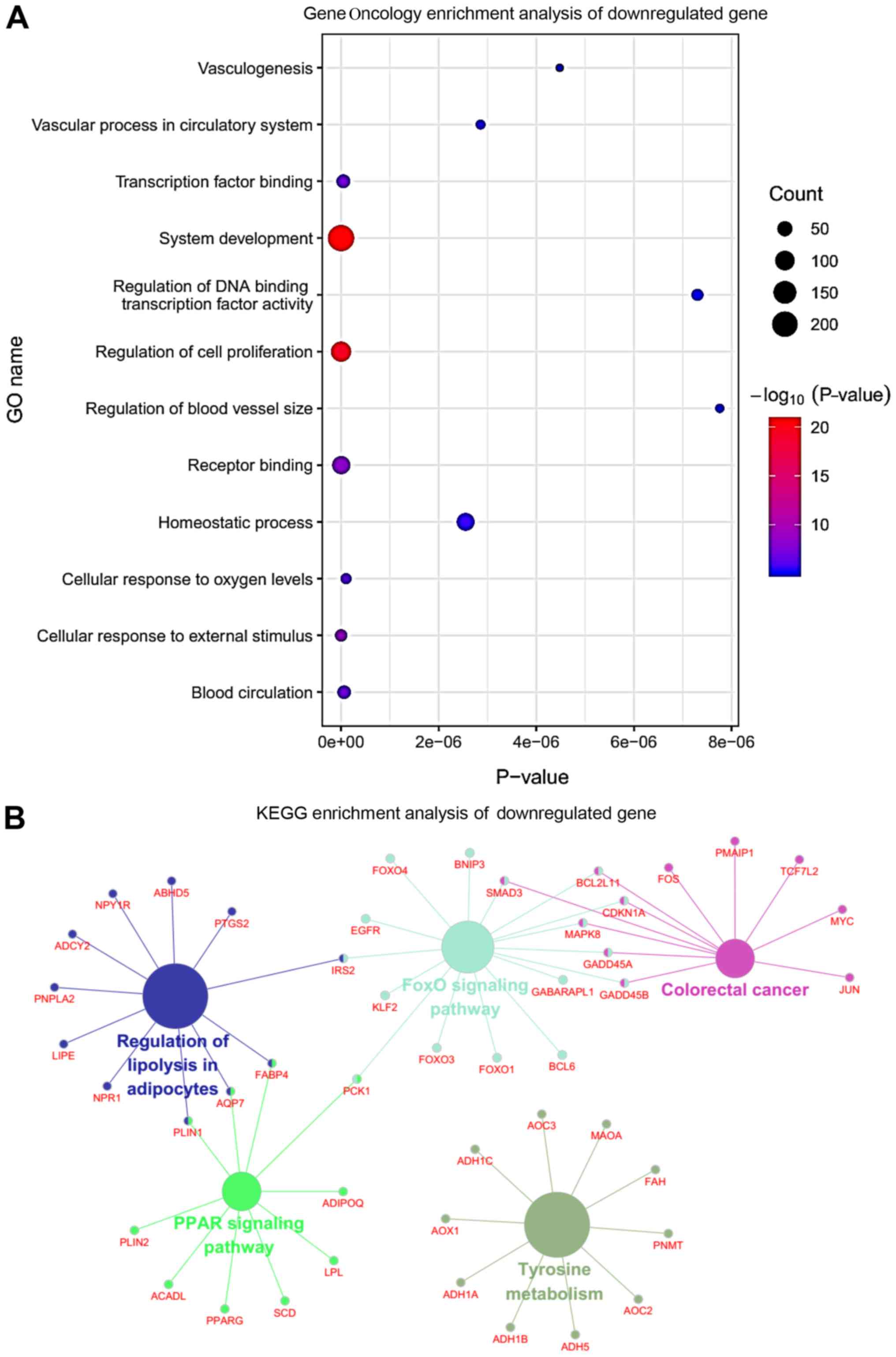
‘leukocyte transendothelial migration’ (Fig. 2B). By contrast, the downregulated
genes were mainly involved in ‘vasculogenesis’, ‘regulation of DNA
binding transcription factor activity’, ‘cellular response to
external stimulus’, ‘vascular process in circulatory system’,
‘blood circulation’ and ‘transcription factor binding’ (Fig. 3A). Finally, the pathway enrichment
analysis indicated that the downregulated DEGs were mainly enriched
in ‘tyrosine metabolism’, ‘FoxO signaling pathway’, ‘regulation of
lipolysis in adipocytes’ and ‘colorectal cancer’ (Fig. 3B).
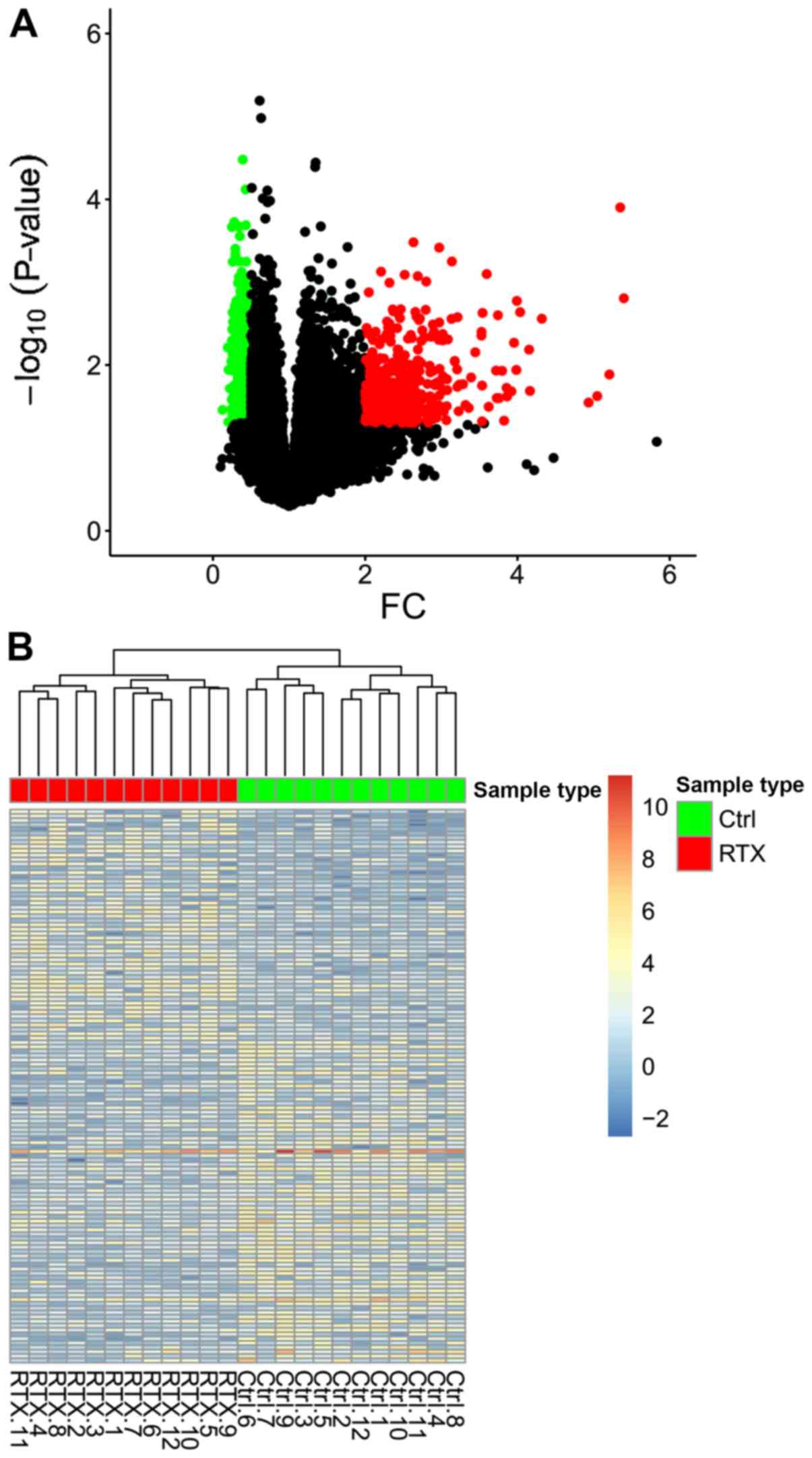
Identification of DEGs based on RTX
treatment data
A total of 54,675 genes were obtained from 12
control and 12 RTX-treated samples, and 941 DEGs (382 upregulated
and 559 downregulated) were identified. The volcano plot of DEGs is
presented in Fig. 4A. The red,
green and black dots indicate the upregulated, downregulated and
non-differentiated genes, respectively. Finally, the overview of
the expression profiles of DEGs before and after RTX treatment
(Fig. 4B), and the distribution of
genes between groups were revealed using a hierarchical cluster
analysis.
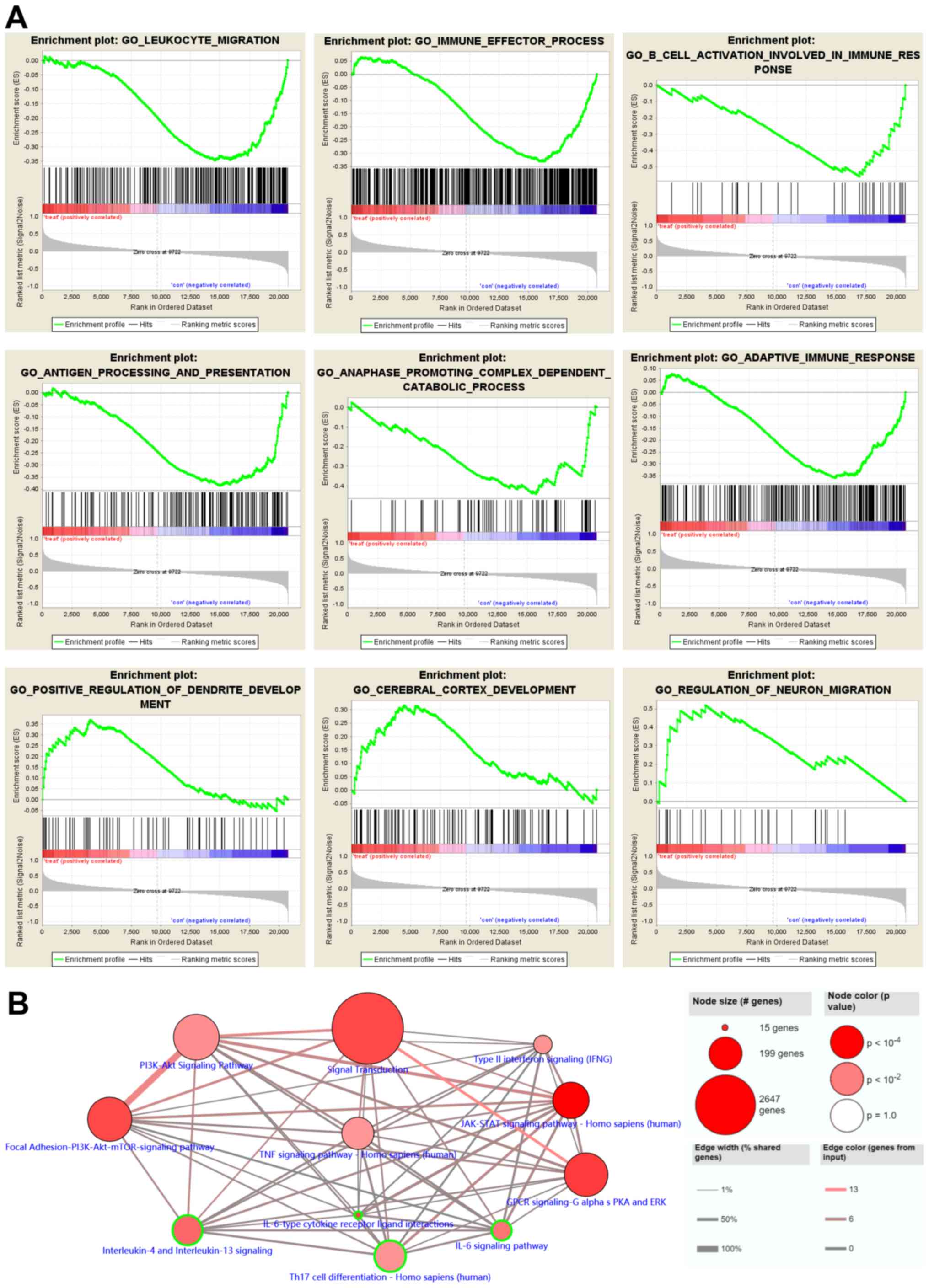
GSEA and pathway enrichment analyses
of DEGs after RTX treatment
GSEA was performed in order to further confirm the
selected DEGs from the GO functional enrichment analysis (Fig. 5A). GSEA revealed that RTX
upregulated the expression of genes associated with the ‘positive
regulation of dendritic development’ and the ‘regulation of neuron
migration’. By contrast, the ‘adaptive immune response’,
‘anaphase-promoting complex-dependent catabolic process’, ‘antigen
processing and presentation’, ‘B cell activation involved in immune
response’ and ‘immune effector process’ functions were suppressed.
Additionally, pathway analysis was performed, using the online
ConsensusPathDB database, in order to analyze the functional and
signaling pathway enrichment of the gene signatures (Fig. 5B).
The pathway enrichment analysis also revealed that
the downregulated DEGs were enriched in the ‘PI3K-Akt signaling
pathway’, ‘type II interferon signaling’, ‘Janus kinase/STAT
signaling pathway’, ‘interleukin-6 signaling pathway’, ‘T helper 17
cell differentiation’, and ‘interleukin-4 and interleukin-13
signaling’. The aforementioned results supported the conclusion
that RTX may treat RA via regulating body immunity.
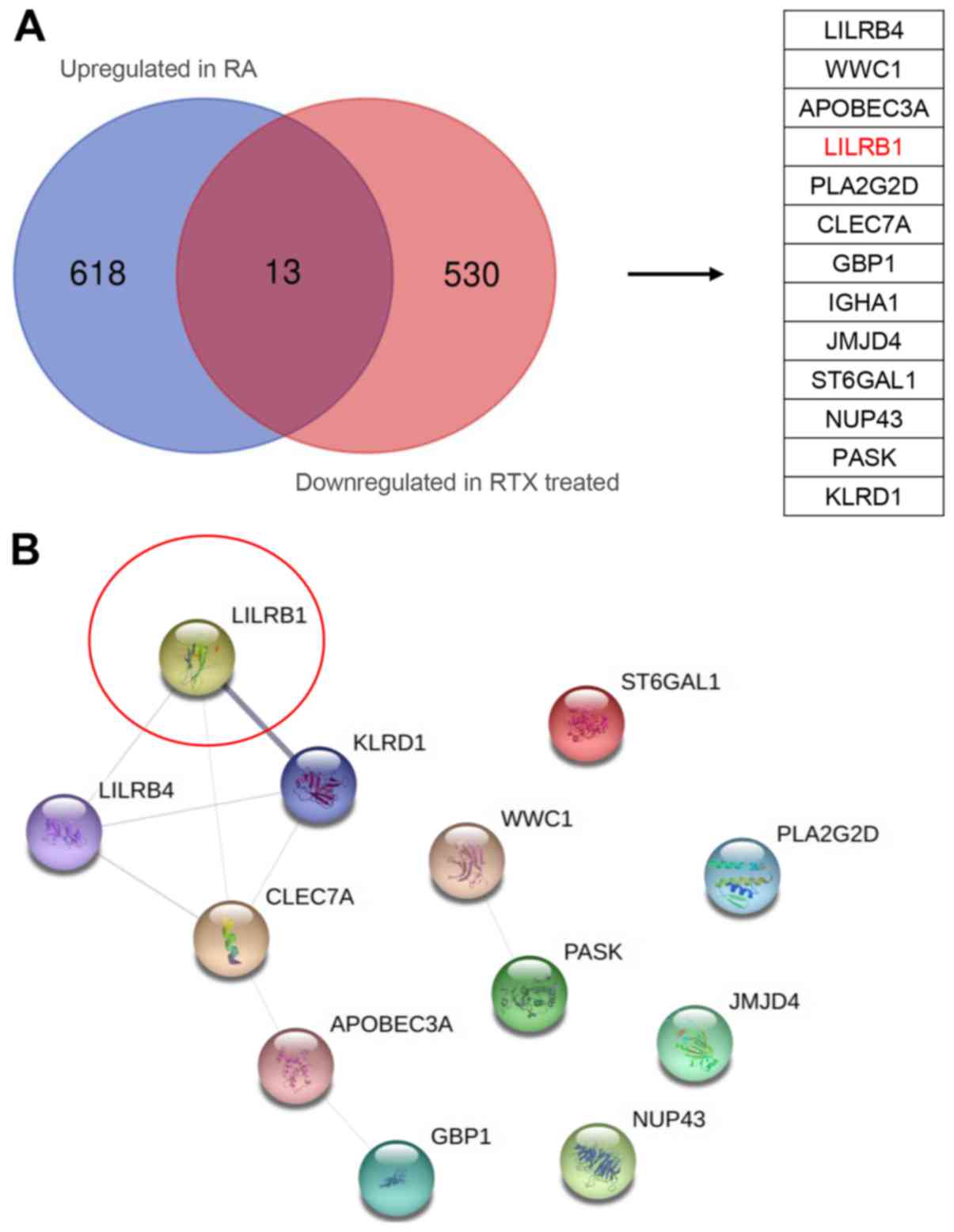
Identification of key candidate genes
using STRING protein interaction network
A Venn diagram analysis of the upregulated and
downregulated genes in RA and after RTX treatment groups,
respectively, was performed (34).
The analysis identified 13 key genes that were subsequently
analyzed using the STRING database (http://string-db.org) for PPI network analysis
(Fig. 6A). The results indicated
that LILRB1 exhibited the highest interactivity confidence
(Fig. 6B).
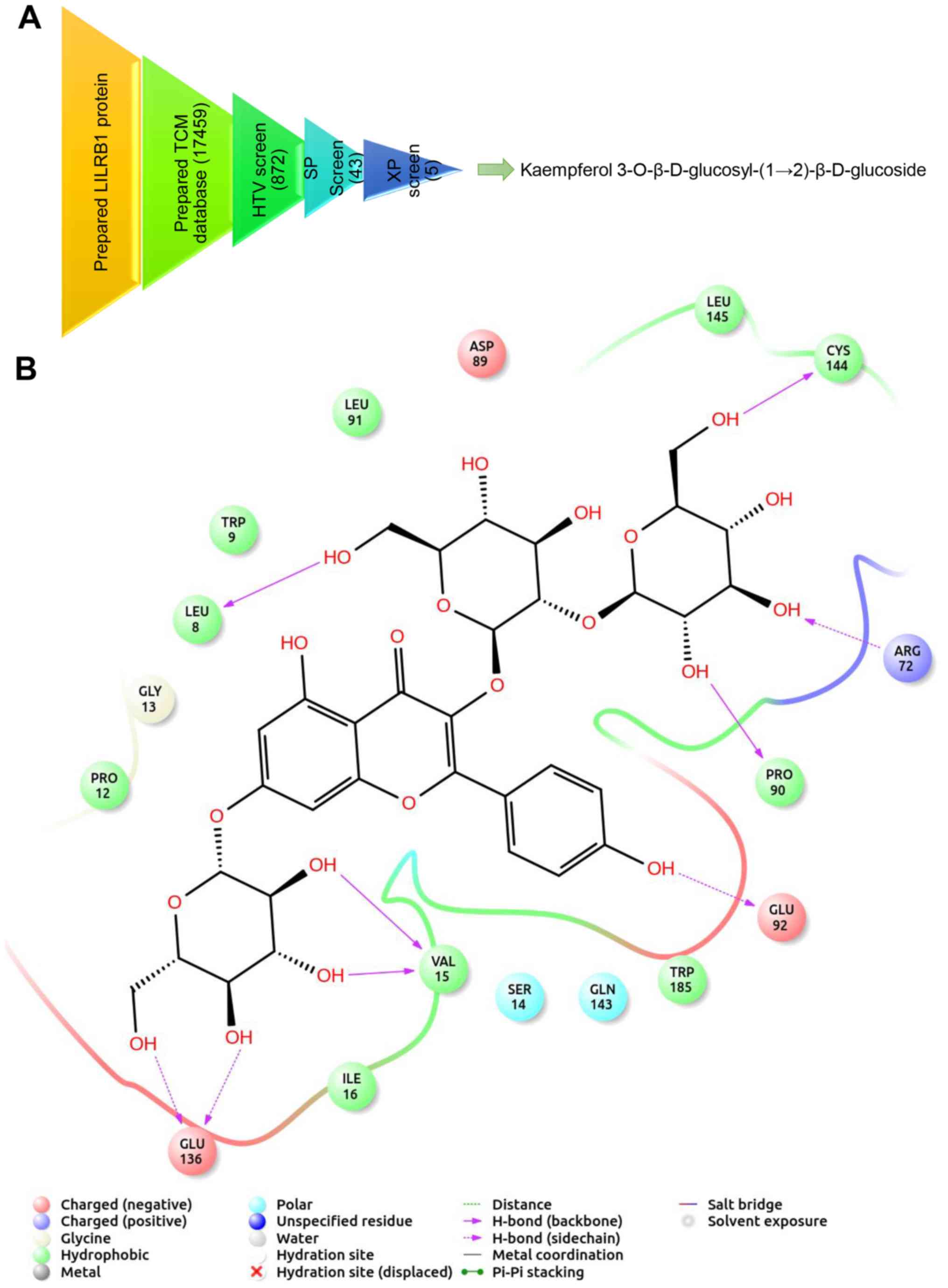
Screening of candidate compounds for
RA treatment
The PPI network analysis indicated that LILRB1 was a
key node gene associated with the mechanisms of RA pathogenesis.
Therefore, the LILRB1 gene was selected for virtual drug screening.
A set of molecular recognition strategies for TCM compounds
identification was designed via structure-based high-throughput
virtual screening. The filtering process is presented in Fig. 7A. Briefly, a total of 872 TCM
compounds demonstrating the highest docking score with LILRB1 were
screened using the high-throughput method, and 43 of them were
selected. Subsequently, 5 candidate TCM compounds were obtained via
precise docking. Among them, kaempferol
3-O-β-D-glucosyl-(1→2)-β-D-glucoside exhibited the highest binding
capacity. The interaction between LILRB1 and kaempferol
3-O-β-D-glucosyl-(1→2)-β-D-glucoside is shown in Fig. 7B. The key amino acid residues
Cys144, Arg72, Pro90, Val15 and Glu136 of the kaempferol
3-O-β-D-glucosyl-(1→2)-β-D-glucoside binding site interacted with
LILRB1 via hydrogen bonds (Fig.
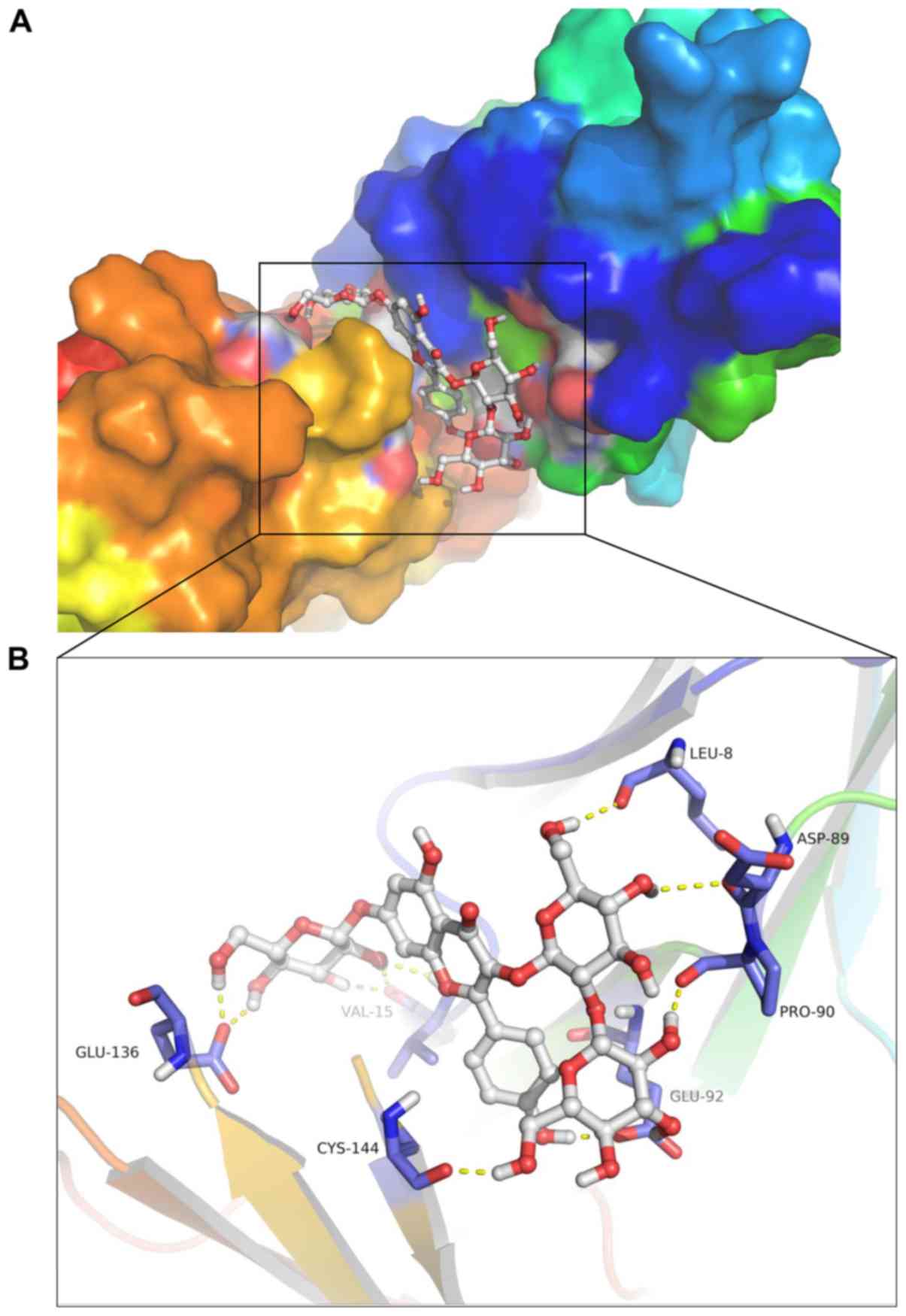
7B). The 3D conformation of the interaction and surface binding
of kaempferol 3-O-β-D-glucosyl-(1→2)-β-D-glucoside and LILRB1 with
other proteins were analyzed in order to identify additional
protein interactions (Fig. 8A and
B). The aforementioned results indicated that kaempferol
3-O-β-D-glucosyl-(1→2)-β-D-glucoside may decrease the biological
activity of LILRB1 by inhibiting its active center, thereby
exhibiting therapeutic effects on RA.
Discussion
Immune dysregulation has been implicated in the
pathogenesis of RA through the anti-immunoglobulin G antibodies
(35). Although several mechanisms
regarding the role of immune regulation in RA pathology have been
proposed (36), the exact
mechanism should be further investigated. Therefore, in the present
study, the aim was to delineate the underlying molecular mechanisms
and identify the key regulatory genes involved in the developmental
process of RA.
As gene expression profiles may reflect disease
status (37), in the present study
the expression of DEGs was compared in normal, RA control and
RTX-treated tissues selected from the GO database. The DEGs
functional analysis indicated that RA was associated with the
induction of inflammation responses and immune activities. These
observations were consistent with the clinical characteristics of
the disease. However, following RTX treatment, inflammation and
immune-related signaling pathways were widely suppressed. These
results were consistently with the previously reported
inflammation- and immune-related characteristics of RA pathogenesis
(3). The upregulated and
downregulated genes in RA and RTX-treated groups, respectively,
were subjected to Venn analysis and subsequently 13 selected genes
were screened in order to identify the key regulatory genes.
Topological analysis was conducted to screen the node genes and
LILRB1, a receptor expressed on the membrane of immune cells
(10). The analysis demonstrated
that LILRB1 exhibited a high degree of connectivity. The results of
the present study indicated that LILRB1 may control inflammatory
responses and cytotoxicity by inducing a targeted immune response
and limiting autoreactivity. In addition, it was hypothesized that
upstream regulatory signaling pathways of the downregulated LILRB1
gene could serve as a target of RTX. However its pharmacological
mechanisms of action should be further investigated. Thus, the
results indicated that LILRB1 was a potential target of RA
treatment.
In order to clarify the target activity of LILRB1,
potential drugs that target LILRB1 were screened using the TCM
libraries. Kaempferol 3-O-β-D-glucosyl-(1→2)-β-D-glucoside showed
the strongest targeting activity. Kaempferol, a natural flavonol,
has been reported to act as an antioxidant by reducing oxidative
stress during the treatment of several diseases (38). However, the precise molecular
mechanisms of kaempferol need to be further studied. The results of
the present study revealed that kaempferol could bind to the active
pocket of LILRB1, suggesting inhibition of the immune response
signal transduction via the receptor. Thus, the activity of
kaempferol, a potential anti-RA drug, may be mediated via targeting
the immune-related receptor. Additional studies investigating the
clinical and molecular basis are required to further elucidate the
effect of LILRB1 in regulating the pathogenesis of RA and to
demonstrate the therapeutic value of kaempferol in RA
treatment.
In conclusion, this study may enhance the
understanding of RA development processes, and suggested that
kaempferol could be a potential novel and effective drug in RA
treatment in clinical practice.
Acknowledgements
Not applicable.
Funding
This work was supported by Tianjin Science and
Technology Commission (grant no. 16ZXMJSY00220).
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author upon reasonable
request.
Authors' contributions
CK conceived and designed the present study. YS and
WQ performed the bioinformatics analyses. CK and WQ wrote the
manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
RA
|
rheumatoid arthritis
|
|
LILRB1
|
leukocyte immunoglobulin-like receptor
subfamily B member 1
|
|
DEGs
|
differentially expressed genes
|
|
PPI
|
protein-protein interaction
|
|
GO
|
Gene Ontology
|
|
KEGG
|
Kyoto Encyclopedia of Genes and
Genomes
|
|
TCM
|
Traditional Chinese Medicine
|
References
|
1
|
Falconer J, Murphy AN, Young SP, Clark AR,
Tiziani S, Guma M and Buckley CD: Review: Synovial cell metabolism
and chronic inflammation in rheumatoid arthritis. Arthritis
Rheumatol. 70:984–999. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Nakajima A, Aoki Y, Sonobe M, Takahashi H,
Saito M, Terayama K and Nakagawa K: Radiographic progression of
large joint damage in patients with rheumatoid arthritis treated
with biological disease-modifying anti-rheumatic drugs. Mod
Rheumatol. 26:517–521. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Firestein GS and McInnes IB:
Immunopathogenesis of rheumatoid arthritis. Immunity. 46:183–196.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Arend WP and Firestein GS: Pre-rheumatoid
arthritis: Predisposition and transition to clinical synovitis. Nat
Rev Rheumatol. 8:573–586. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Firestein GS: Evolving concepts of
rheumatoid arthritis. Nature. 423:356–361. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Singh JA, Saag KG, Bridges SL Jr, Akl EA,
Bannuru RR, Sullivan MC, Vaysbrot E, McNaughton C, Osani M,
Shmerling RH, et al: 2015 American college of rheumatology
guideline for the treatment of rheumatoid arthritis. Arthritis
Rheumatol. 68:1–26. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
McInnes IB and Schett G: Pathogenetic
insights from the treatment of rheumatoid arthritis. Lancet.
389:2328–2337. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lopez-Olivo MA, Amezaga Urruela M, McGahan
L, Pollono EN and Suarez-Almazor ME: Rituximab for rheumatoid
arthritis. Cochrane Database Syst Rev. 1:CD0073562015.PubMed/NCBI
|
|
9
|
Smolen JS and Aletaha D: Rheumatoid
arthritis therapy reappraisal: Strategies, opportunities and
challenges. Nat Rev Rheumatol. 11:276–289. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Young NT, Waller EC, Patel R, Roghanian A,
Austyn JM and Trowsdale J: The inhibitory receptor LILRB1 modulates
the differentiation and regulatory potential of human dendritic
cells. Blood. 111:3090–3096. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wang X, Xu Z, Ren X, Chen X, Wei J, Lin W,
Li Z, Ou C, Gong Z and Yan Y: Function of low ADARB1 expression in
lung adenocarcinoma. PLoS One. 14:e02222982019. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Yan Y, Xu Z, Hu X, Qian L, Li Z, Zhou Y,
Dai S, Zeng S and Gong Z: SNCA is a functionally low-expressed gene
in lung adenocarcinoma. Genes (Basel). 9:E162018. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Woetzel D, Huber R, Kupfer P, Pohlers D,
Pfaff M, Driesch D, Häupl T, Koczan D, Stiehl P, Guthke R and Kinne
RW: Identification of rheumatoid arthritis and osteoarthritis
patients by transcriptome-based rule set generation. Arthritis Res
Ther. 16:R842014. View
Article : Google Scholar : PubMed/NCBI
|
|
14
|
Edgar R, Domrachev M and Lash AE: Gene
Expression Omnibus: NCBI gene expression and hybridization array
data repository. Nucleic Acids Res. 30:207–210. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Lauwerys BR, Hernández-Lobato D, Gramme P,
Ducreux J, Dessy A, Focant I, Ambroise J, Bearzatto B, Nzeusseu
Toukap A, Van den Eynde BJ, et al: Heterogeneity of synovial
molecular patterns in patients with arthritis. PLoS One.
10:e01221042015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Perry M: Heatmaps: Flexible Heatmaps for
Functional Genomics and Sequence Features. R package version
1.12.0. 2020.10.18129/B9.bioc.heatmaps.
|
|
17
|
Ritchie ME, Phipson B, Wu D, Hu Y, Law CW,
Shi W and Smyth GK: limma powers differential expression analyses
for RNA-sequencing and microarray studies. Nucleic Acids Res.
43:e472015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ashburner M, Ball CA, Blake JA, Botstein
D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT,
et al: Gene ontology: Tool for the unification of biology. The gene
ontology consortium. Nat Genet. 25:25–29. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
The Gene Ontology Consortium, . The gene
ontology resource: 20 years and still GOing strong. Nucleic Acids
Res. 47:D330–D338. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kanehisa M, Sato Y, Furumichi M, Morishima
K and Tanabe M: New approach for understanding genome variations in
KEGG. Nucleic Acids Res. 47:D590–D595. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Bindea G, Mlecnik B, Hackl H, Charoentong
P, Tosolini M, Kirilovsky A, Fridman WH, Pagès F, Trajanoski Z and
Galon J: ClueGO: A Cytoscape plug-in to decipher functionally
grouped gene ontology and pathway annotation networks.
Bioinformatics. 25:1091–1093. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Bindea G, Galon J and Mlecnik B: CluePedia
Cytoscape plugin: Pathway insights using integrated experimental
and in silico data. Bioinformatics. 29:661–663. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Subramanian A, Kuehn H, Gould J, Tamayo P
and Mesirov JP: GSEA-P: A desktop application for gene set
enrichment analysis. Bioinformatics. 23:3251–3253. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Herwig R, Hardt C, Lienhard M and Kamburov
A: Analyzing and interpreting genome data at the network level with
ConsensusPathDB. Nat Protoc. 11:1889–1907. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kamburov A, Wierling C, Lehrach H and
Herwig R: ConsensusPathDB-a database for integrating human
functional interaction networks. Nucleic Acids Res. 37:D623–628.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Snel B, Lehmann G, Bork P and Huynen MA:
STRING: A web-server to retrieve and display the repeatedly
occurring neighbourhood of a gene. Nucleic Acids Res. 28:3442–3444.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Sanderson K: Databases aim to bridge the
East-West divide of drug discovery. Nat Med. 17:15312011.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Halgren TA, Murphy RB, Friesner RA, Beard
HS, Frye LL, Pollard WT and Banks JL: Glide: A new approach for
rapid, accurate docking and scoring. 2. Enrichment factors in
database screening. J Med Chem. 47:1750–1759. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Friesner RA, Banks JL, Murphy RB, Halgren
TA, Klicic JJ, Mainz DT, Repasky MP, Knoll EH, Shelley M, Perry JK,
et al: Glide: A new approach for rapid, accurate docking and
scoring. 1. Method and assessment of docking accuracy. J Med Chem.
47:1739–1749. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Nair SK and Burley SK: X-ray structures of
Myc-Max and Mad-Max recognizing DNA. Molecular bases of regulation
by proto-oncogenic transcription factors. Cell. 112:193–205. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Gupta S and Bajaj AV: Extra precision
glide docking, free energy calculation and molecular dynamics
studies of 1,2-diarylethane derivatives as potent urease
inhibitors. J Mol Model. 24:2612018. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Friesner RA, Murphy RB, Repasky MP, Frye
LL, Greenwood JR, Halgren TA, Sanschagrin PC and Mainz DT: Extra
precision glide: Docking and scoring incorporating a model of
hydrophobic enclosure for protein-ligand complexes. J Med Chem.
49:6177–6196. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zhong W, Liu P, Zhang Q, Li D and Lin J:
Structure-based QSAR, molecule design and bioassays of
protease-activated receptor 1 inhibitors. J Biomol Struct Dyn.
35:2853–2867. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Yan Y, Su W, Zeng S, Qian L, Chen X, Wei
J, Chen N, Gong Z and Xu Z: Effect and mechanism of tanshinone I on
the radiosensitivity of lung cancer cells. Mol Pharm. 15:4843–4853.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Janossy G, Panayi G, Duke O, Bofill M,
Poulter LW and Goldstein G: Rheumatoid arthritis: A disease of
T-lymphocyte/macrophage immunoregulation. Lancet. 2:839–842. 1981.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Catrina AI, Joshua V, Klareskog L and
Malmström V: Mechanisms involved in triggering rheumatoid
arthritis. Immunol Rev. 269:162–174. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Barrett T, Suzek TO, Troup DB, Wilhite SE,
Ngau WC, Ledoux P, Rudnev D, Lash AE, Fujibuchi W and Edgar R: NCBI
GEO: Mining millions of expression profiles-database and tools.
Nucleic Acids Res. 33:D562–566. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Kashyap D, Sharma A, Tuli HS, Sak K, Punia
S and Mukherjee TK: Kaempferol-A dietary anticancer molecule with
multiple mechanisms of action: Recent trends and advancements. J
Funct Foods. 30:203–219. 2017. View Article : Google Scholar : PubMed/NCBI
|