Introduction
Although improved cardio-pulmonary resuscitation
(CPR) strategies have increased the return of spontaneous
circulation (ROSC) rate in cardiac arrest (CA) patients, both the
discharge survival rate and long-term prognosis of these patients
remain poor (1). Global cerebral
ischemia-reperfusion (GCIR) injury has been considered as one of
the most important contributions to poor prognosis (2). However, the exact mechanisms leading
to exacerbated post-GCIR remodeling and poor outcomes in ROSC
patients are incompletely understood, and effective therapeutic
interventions are limited (3).
Emerging evidence suggests that both apoptosis and
autophagy contribute to GCIR-induced neuronal death resulting from
CA (4). Numerous studies have
demonstrated that oxidative stress occurs during focal cerebral
ischemia- reperfusion promoting protein oxidation, which could
impair neuron protein synthesis and metabolism resulting in
inevitable neuron apoptosis (5–7).
However, definitive proof of the cause-and-effect relationship
between oxidative stress and neuron apoptosis following
CA-CPR-induced GCIR has remained elusive.
Autophagy is an evolutionarily conserved
stress-response mechanism, which often occurs in
ischemia-reperfusion (IR)-induced oxidative stress (8). Several studies indicated that
switching apoptosis to autophagy protects neurons exposed to focal
cerebral ischemia or GCIR induced by vertebral artery occlusion
(9,10). However, whether the crosstalk
between autophagy and apoptosis affects GCIR injury and improves
the survival and neurological outcomes of ROSC patients remains to
be elucidated, and the mechanisms are unclear (11).
Adiponectin (APN), also known as GBP-28, apM1,
AdipoQ and Acrp30, is a novel adipokine that maintains insulin
responsiveness, stimulates mitochondrial biogenesis, inhibits
inflammatory response and modulates autophagy and apoptosis in
different disease models (12,13).
A clinical study has revealed that the level of APN is
independently associated with mortality and low plasma APN is
related to an increased risk of 5-year mortality following the
first-ever ischemic stroke in humans (14). The preliminary experiment of the
present study demonstrated that APN relieved GCIR by impairing
apoptosis in brain tissues of mice suffering from CA-CPR. It has
been shown that, APN markedly decreased neuronal injury and infarct
size by inhibiting neuronal apoptosis and alleviating oxidative
stress in neurons subjected to middle cerebral artery occlusion
(MACO)-induced focal cerebral ischemia (FCI) (15). However, few previous studies had
clarified the definitive effects of APN on neuronal apoptosis and
autophagy following GCIR induced by CA-CPR, and the underlying
mechanisms of these effects remain poorly understood.
The present study reported that APN relieved the
brain injury via an adiponectin receptor 1 (AdipoR1)/AMP-activated
protein kinase (AMPK)-dependent mechanism in a GCIR mice model
induced by CA-CPR.
Materials and methods
Animals and reagents
In total, 60 adult male C57BL/6 (Chengdu Da Shuo
Laboratory Animal Co., Ltd., Chengdu, Sichuan) mice, at 8 weeks of
age (body weight, 30–35 g), were bred according to guidelines in
compliance with the current international laws and policies
(16). All animal studies were
approved by the Institutional Animal Care and Use Committee at West
China Hospital, Sichuan University (no. 2017071A). The mice were
housed in a conventional facility under adequate temperature (23°C)
and humidity (60%) conditions with a 12-h light/dark cycle and
could freely access food and tap water.
Recombinant mouse APN was provided by Professor
Xinliang Ma of the Thomas Jefferson University (Philadelphia, USA).
Antibodies against cleaved caspase-3 (cat. no. 9661T, Rabbit,
1:1,000), Bcl-2 (cat. no. 3498T, rabbit, 1:1,000), light chain 3
(LC3) II (cat. no. 3868T, rabbit, 1:1,000), Beclin-1 (cat. no.
3495T, rabbit, 1:1,000), AMPK (cat. no. 4182; rabbit; 1:1,000),
anti-β-actin (cat. no. 3700; mouse; 1:1,000) and p-AMPK (cat. no.
4186, rabbit, 1:1,000) were purchased from Cell Signaling
Technology, Inc. APN receptor 1 (rabbit, 1:1,000) was purchased
from Santa Cruz Biotechnology, Inc. Malondialdehyde (MDA; cat. no.
A003-1) and superoxide dismutase (SOD; cat. no. A001-1-1) detection
kits were purchased from Nanjing Jiancheng Bioengineering
Institute. A mouse APN/Acrp30 Quantikine ELISA kit (MRP300) was
purchased from R&D Systems, Inc. IgG horseradish peroxidase
(HRP)-conjugated secondary antibodies were purchased from Beyotime
Institute of Biotechnology (cat. no. A0208; 1:10,000) or Applygen
Technologies, Inc. (1:100,000; cat. no. C1710).
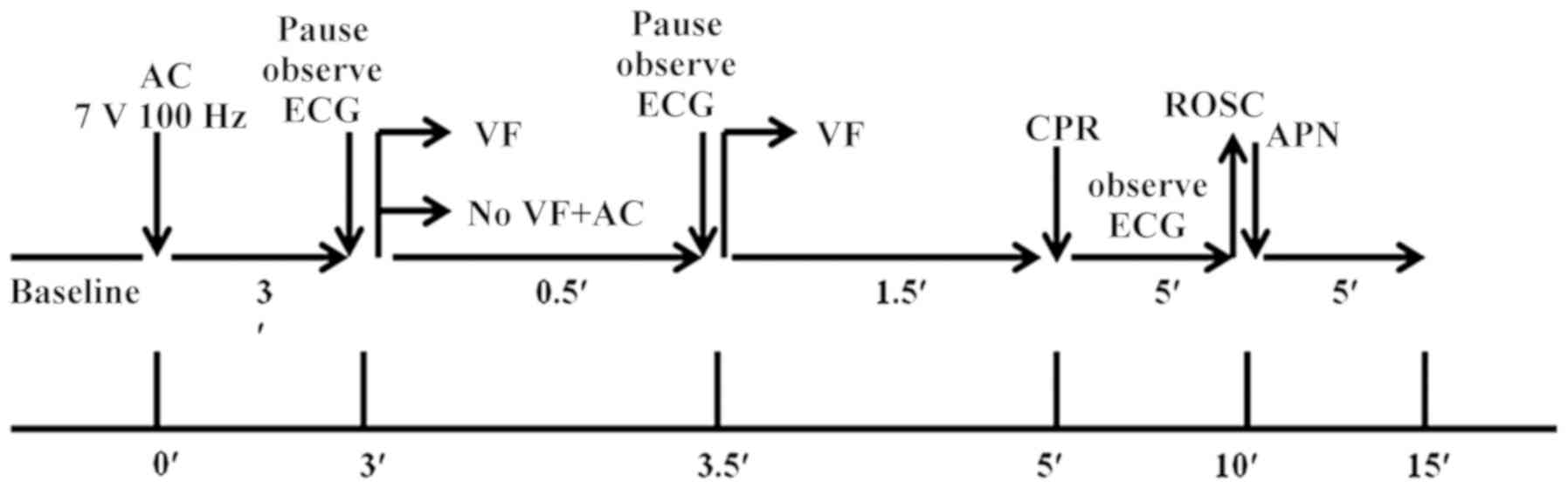
Experimental protocol
Mice were randomized into various treatment groups
using random number table: i) Sham group (n=20) was subjected to
invasive operation and injected with 0.9% saline. The mice
experience circulatory arrest for 5 min, 40 successful resuscitated
mice were randomly divided into the PCAS and APN groups, ii) PCAS
group (n=20), which was subjected to cardiac arrest/cardiopulmonary
resuscitation (CA/CPR) and injected with 0.9% saline; and iii) APN
group (n=20), which was subjected to CA/CPR and injected with APN
(0.17 mg/kg) as soon as the resuscitation was successful (15,17,18).
Every five mice of each group were invasively monitored for an
additional of 0.5, 3, 6 and 12 h of reperfusion and were euthanized
to harvest the brain tissue and serum for further measurements.
CA/CPR model
CA/CPR was used to induce global cerebral ischemia
in vivo. Mice were anesthetized, which was maintained with
2% isoflurane, and mechanically ventilated to maintain respiration
using a rodent ventilator (RWD, Shen Zhen, China). CA/CPR was
induced as previously described (19). A cannula (Fine Science) was
inserted into the right femoral artery to measure arterial blood
pressure (Columbus Instruments). A PE 10 catheter was placed into
the right jugular vein for drug administration. An intubation tube
was connected to a ventilator (RWD Life Science) and mice were
ventilated with a tidal volume of 150 µl and a respiratory rate of
100 bpm. Following all the preparatory steps, mice were allowed to
stabilize for 10 min, during which blood pressure and temperature
were recorded. Cardiac arrest was induced by transesophageal
alternating current (AC) current stimulation (7 V, 100 Hz) for 180
sec. A pause in AC stimulation was then initiated for 1–2 s to
observe the change of electrocardiogram (ECG). As soon as the
ventricular fibrillation (VF) reverted spontaneously together with
an increase of blood pressure, additional 30-sec burst stimulation
was performed until either the VF reappeared and persisted, or
until asystole or pulseless electrical activity (PEA) occurred.
After 2 min with no intervention, cardiopulmonary resuscitation was
initiated via an injection of epinephrine (EPI, 0.04 mg/kg, 37°C;
Welman Pharmaceutical Co., Ltd.) into the jugular vein catheter
followed by manual chest compressions and ventilation with 100%
oxygen with a tidal volume of 4 ml/kg and a respiratory rate of 100
bpm. Manual chest compression at a rate of 200 compressions per
minute with equal compression-relaxation duration was always
performed by the same investigator who was blinded to the
hemodynamic monitor tracings and guided only by audio tones.
Compression depth was approximately 1/3 of the anteroposterior
chest diameter at maximal compression. ROSC was defined as the
return of ventricular rhythm with a mean aortic pressure of ≥20
mmHg for a minimum of 5 min (20).
When there was a failure to restore spontaneous circulation after
10 min, resuscitation efforts were discontinued. The flow chart of
the experimental protocol is given in Fig. 1.
The procedure described above was similar for the
CA/CPR and sham animals, except that the control animals were
injected with isotonic saline instead of epinephrine and did not
experience chest compressions.
Tissue processing
The brain tissue was harvested, followed by
post-fixing in the 4% paraformaldehyde overnight at room
temperature or homogenizing with lysis buffer (cat. no. P0013B;
Beyotime Institute of Biotechnology). The homogenized supernatants
and serums were stored in −80°C for western blot analysis and
biochemistry indexes.
Histology
Tissue sections were stained with hematoxylin and
eosin (H&E), following standard protocols (21). Briefly, the mice were sacrificed
and intracardially perfused with 1% PBS, followed by 4%
paraformaldehyde overnight at room temperature and were
paraffin-embedded. Then, the tissues were cut at the thickness of 5
µm, and stained using the hematoxylin and eosin (HE) via standard
processes at room temperature for 4–10 min. The images were
captured using the upright light microscope, according to the
previous study described (21).
TUNEL assay
TUNEL staining was used to evaluate the apoptosis of
neurons. The brain tissues were fixed with 4% paraformaldehyde for
12 h at room temperature, paraffin- embedded and cut into 5-µm
sections for the TUNEL staining by using the in situ
Apoptosis Detection kit (Roche Diagnostics). Cxylene was used to
de-paraffinize the paraffin-embedded brain tissues for 20 min, and
ethanol (75, 85, 95 and 100% for 3 min) series were used to
rehydrate the brain tissues. The brain tissues were incubated with
the proteinase K (at final concentration of 20 µg/ml in 10 mM
Tris/HCl) at 37°C for 30 min. The endogenous peroxidase activity
was blocked with 0.3% H2O2 in methanol for 10
min at room temperature. The cerebral cortex slices were
permeabilized by using 0.1% sodium citrate and 0.1% Triton-X-100
for 5 min. Then, the slices were washed three times with PBS for 10
min, and were incubated using TUNEL reaction mixture at 37°C for 60
min. The slices were incubated using a convertor-POD in humidity
chamber for 30 min at 37°C. The slices were washed with PBS again
for three times, and the color was developed by using a DAB
substrate solution for 15 min at room temperature. Finally, the
slices were observed using the light microscopy, and cells with an
apoptotic morphology and TUNEL-staining positive cells were
identified as apoptotic cells (21,22).
Immunofluorescence staining
Immunofluorescence staining was performed according
to a previous study (23).
Briefly, paraffin sections made in the preparation of HE staining,
were deparaffinized, rehydrated and pretreated with an antigen
unmasking solution (Vector Laboratories, Inc.) for 8 min followed
by blocking with peroxidase blocking reagent (Dako; Agilent
Technologies, Inc.) and 3% goat serum (ABC-Elite kit; Vector
Laboratories, Inc.). The sections were then incubated overnight
with the polyclonal goat anti-rabbit cleaved-caspase 3 and LC3II
antibody at 4°C, followed by a biotinylated anti-goat secondary
antibody (1:100,000; cat. no. C1711; Applygen Technologies, Inc.)
at 4°C for 30 min. The secondary antibody solution was discarded
and DAPI solution (1 µg/ml) was applied directly on slides for 5
min at room temperature. After washing slides three times each for
10 min in 0.05% TBS-Tween 20, the slides were incubated with
filtered Sudan Black B solution directly for 30 sec in the dark at
room temperature. A small amount of fluorescent mounting medium
(50–100 µl; cat. no. BL701A; Biosharp) and a cover slip was placed
over the specimen, avoiding bubbles. Then, clear nail polish was
used to seal the sides of the cover glass to the slide. A negative
control section was always included in which the primary antibody
was substituted by the corresponding isotype control.
Western blot analysis
Brain tissue was homogenized with lysis buffer. The
homogenates were centrifuged at 1,000 × g for 10 min at 4°C. The
protein concentration of the supernatant was measured using the
Bradford method (5000001, Bio-Rad Laboratories, Inc.). Equal
amounts of proteins at 20 µg derived from each sample were
separated on 10% SDS-PAGE gels (Thermo Fisher Scientific,
NPO335BOX) and electroblotted onto polyvinylidene fluoride
membranes (EMD Millipore, IPVH00010). After blocking by 5% milk for
30 min at room temperature, membranes were incubated overnight at
4°C with antibodies against target proteins. The membranes were
probed with HRP-conjugated secondary antibodies at room temperature
for 1 h. Immunoreactive protein bands were visualized by using an
enhanced chemiluminescence detection system (GE Healthcare Life
Sciences) according to the manufacturer's protocol. Densitometry
data were analyzed with ImageJ 1. 8. 0 (National Institutes of
Health).
SOD and MDA analyses
The tissue level of MDA and SOD contents was
analyzed following the manufacturer's instructions (Nanjing
Jiancheng Bioengineering Institute), as previously described
(24,25). The bicinchoninic acid method was
used to test tissue protein concentration. The content of MDA was
calculated as nmol/mg protein and the activity of SOD was expressed
as U/mg protein.
ELISA analysis
The plasma level of APN was determined by using an
ELISA kit (MRP300) from R&D Systems, Inc. according to the
manufacturer's instructions.
Statistical analysis
Data are presented as the means ± standard errors of
the mean. All statistical analyses were performed using IBM SPSS
Statistics 19.0 (IBM Corp.). Differences of the means among the
groups were statistically analyzed with one-way or two-way analysis
of variance followed by the Tukey post hoc test for pairwise
comparisons (details in the Figure legends). For all statistical
tests, P<0.05 was considered to indicate a statistically
significant difference.
Results
Physiological parameters
The HRs of the groups i, ii and iii pre-CA were
444.60±33.37, 466.0±37.34 and 471.8±18.15 bpm, respectively
(P>0.05). MAPs of the 3 groups were 83.00±5.09, 82.40±3.91 and
82.26±4.44 mmHg, respectively (P>0.05; Table I). In addition, the chest
compression manner affected the HRs and MAPs without significant
differences among the 3 groups at 30 min after ROSC (Table II).
 | Table I.Comparison of vital signs in each
group (means ± standard errors of the mean). |
Table I.
Comparison of vital signs in each
group (means ± standard errors of the mean).
| Vital sign | Sham | PCAS group | APN group | P-value |
|---|
| HR (bpm) | 444.60±33.37 | 466.0±37.34 | 471.8±18.15 | 0.36 |
| MAP (mmHg) | 83.00±5.09 | 82.40±3.91 | 82.26±4.44 | 0.28 |
| Table II.Comparison of HR and MAP for the
three group after 30-min reperfusion (means ± standard errors of
the mean). |
Table II.
Comparison of HR and MAP for the
three group after 30-min reperfusion (means ± standard errors of
the mean).
| Vital sign | Sham | PCAS group | APN group | P-value |
|---|
| HR (bpm) | 489.60±25.17 | 466.50±33.34 | 473.80±18.15 | 0.48 |
| MAP (mmHg) | 88.00±3.09 | 70.40±5.91 | 73.26±3.44 | 0.33 |
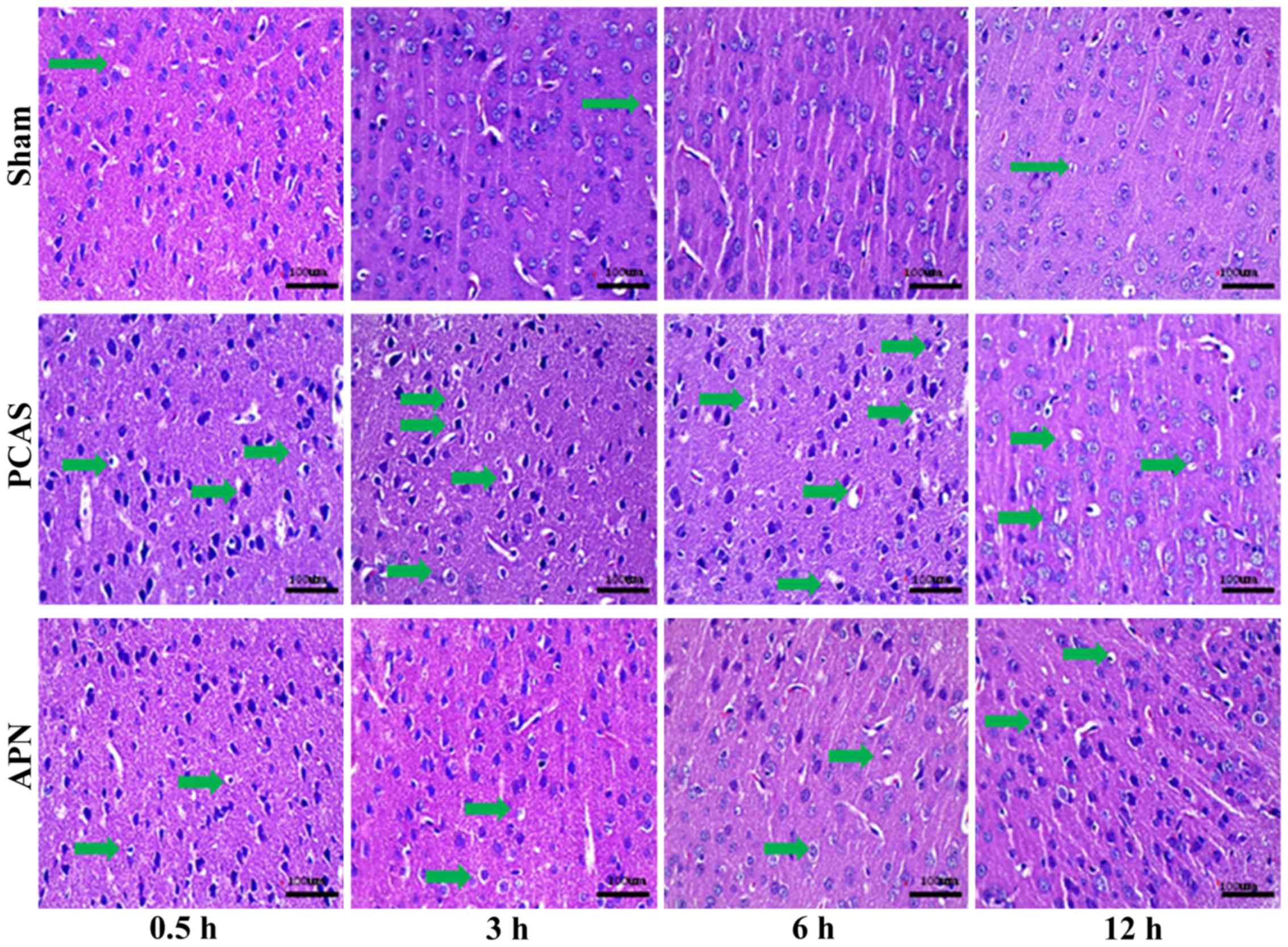
APN relieves GCIRI
H&E staining of the cerebral cortex indicated
that the PCAS and APN groups appeared to experience neuronal death
at 0.5 h of reperfusion and neuronal activity apoptosis occurred at
3 and 6 h of reperfusion. Parts of the blood vessels and the
parenchyma mesh shape changed (microscopic liquefactive necrosis)
in the PCAS group, whereas the severity was significantly reduced
in the APN group (Fig. 2).
APN inhibits the GCIR-induced
apoptosis in brain tissues and modulated the activation of AMPK
induced by GCIR
To further determine whether GCIR induces apoptosis
in the brain tissues, DAB-mediated TUNEL and immunofluorescence
staining analysis were performed. The results demonstrated that APN
relieved the apoptosis after 0.5 to 12 h of reperfusion (Fig. 3A and B).
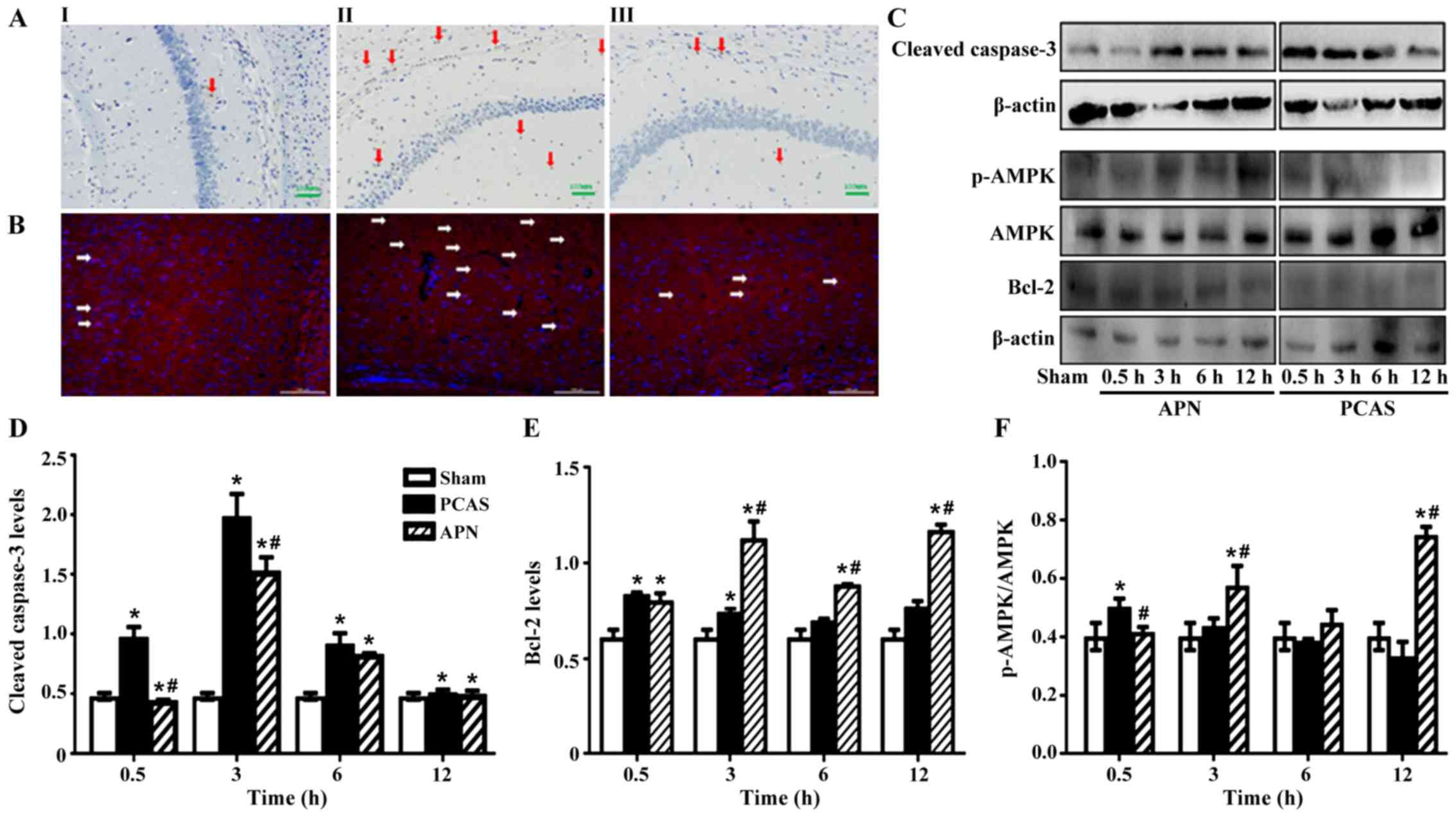 | Figure 3.Adiponectin inhibited GCIR-induced
apoptosis in brain tissues. (A) TUNEL-positive cells were detected
by HRP-conjugated probes with DAB staining in brain tissues of
different groups; I: Sham group, II: PCAS group, III: APN group,
arrows indicate apoptotic cells. (B) Expression of cleaved
casepase-3 in the cortex of different groups mice at 3 h of
reperfusion; I: Sham group, II: PCAS group, III: APN group, arrows
indicated the apoptotic cells. Nuclei were stained by DAPI (blue).
Apoptotic protein cleaved casepase-3, anti-apoptotic protein Bcl-2
and activation of AMPK were measured by western blotting in brain
tissues of different groups. (C) The grayscale values of (D)
casepase-3, (E) Bcl-2 and (F) activation of AMPK in different
groups. Data are presented as means ± standard errors of the mean
(n=5 per group). *P<0.05 vs. Sham group. #P<0.05
vs. PCAS group. GCIR, global cerebral ischemia-reperfusion; HRP,
horseradish peroxidase; DAB, 3,3′-diaminobenzidine; DAPI,
4′,6-diamidino-2-phenylindole; AMPK, AMP-activated protein kinase;
APN, adiponectin; PCAS, post cardiac arrest syndrome. |
GCIR significantly induced the expression of
apoptotic protein cleaved caspase-3 in the brain tissues at all
time points following reperfusion in the PCAS group compared to the
Sham group (P<0.05). Conversely, APN post-treatment
significantly inhibited the induction of cleaved caspase-3
expression by GCIR and nearly reversed it to the normal level
(P<0.05; Fig. 3C and D).
The anti-apoptotic protein, Bcl-2, increased
instantly following the initiation of GCIR, which seemed to be
significantly higher than the Sham group at 0.5 and 3 h of
reperfusion (Fig. 3E). APN
post-treatment increased the expression of Bcl-2 induced by GCIR
from 3 to 12 h (P<0.05; Fig. 3C and
E).
Chen et al (26) demonstrated that UVB radiation
impairs the autophagy response via the phosphorylation of AMPK at
the site of Ser182, as well as stimulating reactive oxidative
stress in keratinocytes. To determine the effects of GCIR on AMPK
activation in brain tissue, the p-AMPK (Ser182)/AMPK ratio was
measured. The results indicated that GCIR significantly activated
AMPK signaling with a significant elevation of p-AMPK (Ser182)/AMPK
ratio at early as 0.5 h of reperfusion (P<0.05). However, this
effect was later inhibited, especially at 6 and 12 h of reperfusion
(P>0.05). In contrast, APN post-treatment activated AMPK
signaling from 3 h of reperfusion (P<0.05; Fig. 3C and F).
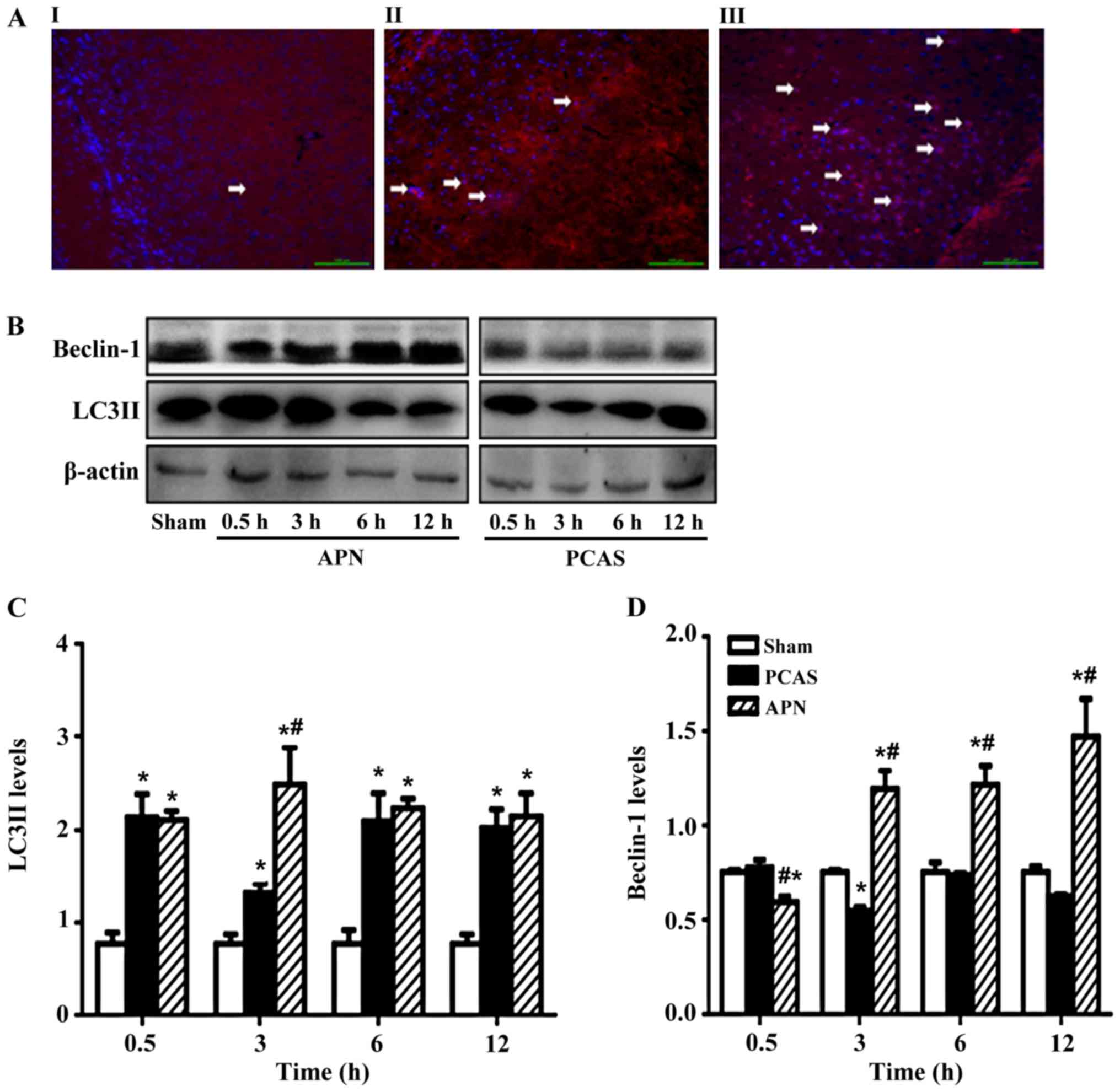
APN irrigates GCIR-induced autophagy
in brain tissues
LC3II was significantly increased in the
GCIR-induced brain tissues with or without APN treatment. APN
post-treatment increased the LC3II expression in the GCIR-induced
brain tissues, with the most significant increase occurring at 3 h
of reperfusion (P<0.05; Fig. 4A and
B).
In addition, GCIR increased the Beclin-1 after 0.5 h
of reperfusion, but inhibited it after 3 h of reperfusion
(P<0.05), with no more significant effects on Beclin-1 in the
following time points of reperfusion. By contrast, administration
of APN reversed the effect of GCIR on Beclin-1, except for the 0.5
h of reperfusion (Fig. 4C and
D).
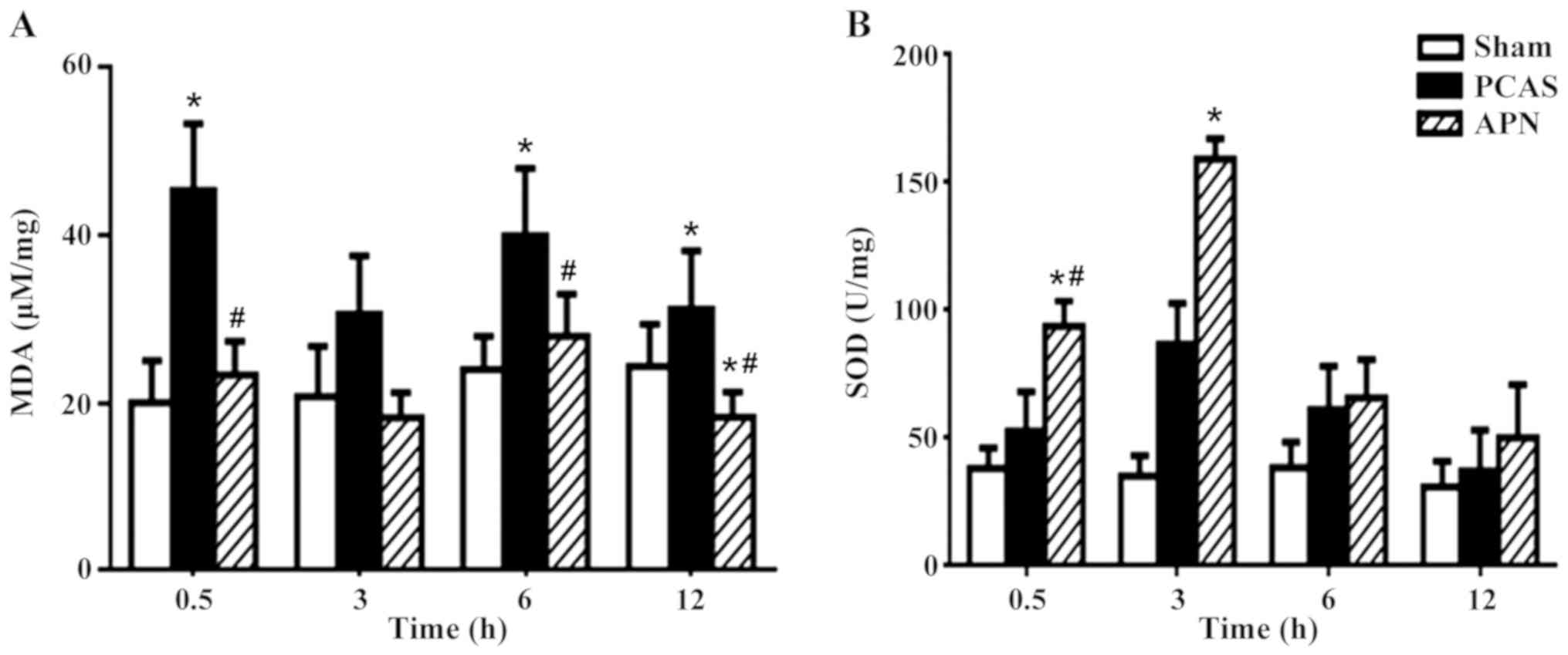
APN alleviates the GCIR-induced
unbalance of oxidative and anti-oxidative stress in brain
tissues
GCIR significantly induced the formation of MDA in
brain tissues after 0.5, 6 and 12 h of reperfusion (P<0.05),
which was significantly decreased by the post-treatment of APN at
0.5, 6 and 12 h of reperfusion (P<0.05; Fig. 5A).
The anti-oxidative SOD was stimulated by GCIR at all
time points of reperfusion (P<0.05), peaking at 3 h of
reperfusion. Post-treatment of APN further enhanced the stimulation
of GCIR on SOD formation as early as 0.5 h of reperfusion
(P<0.05), peaking at 3 h of reperfusion (P<0.05; Fig. 5B).
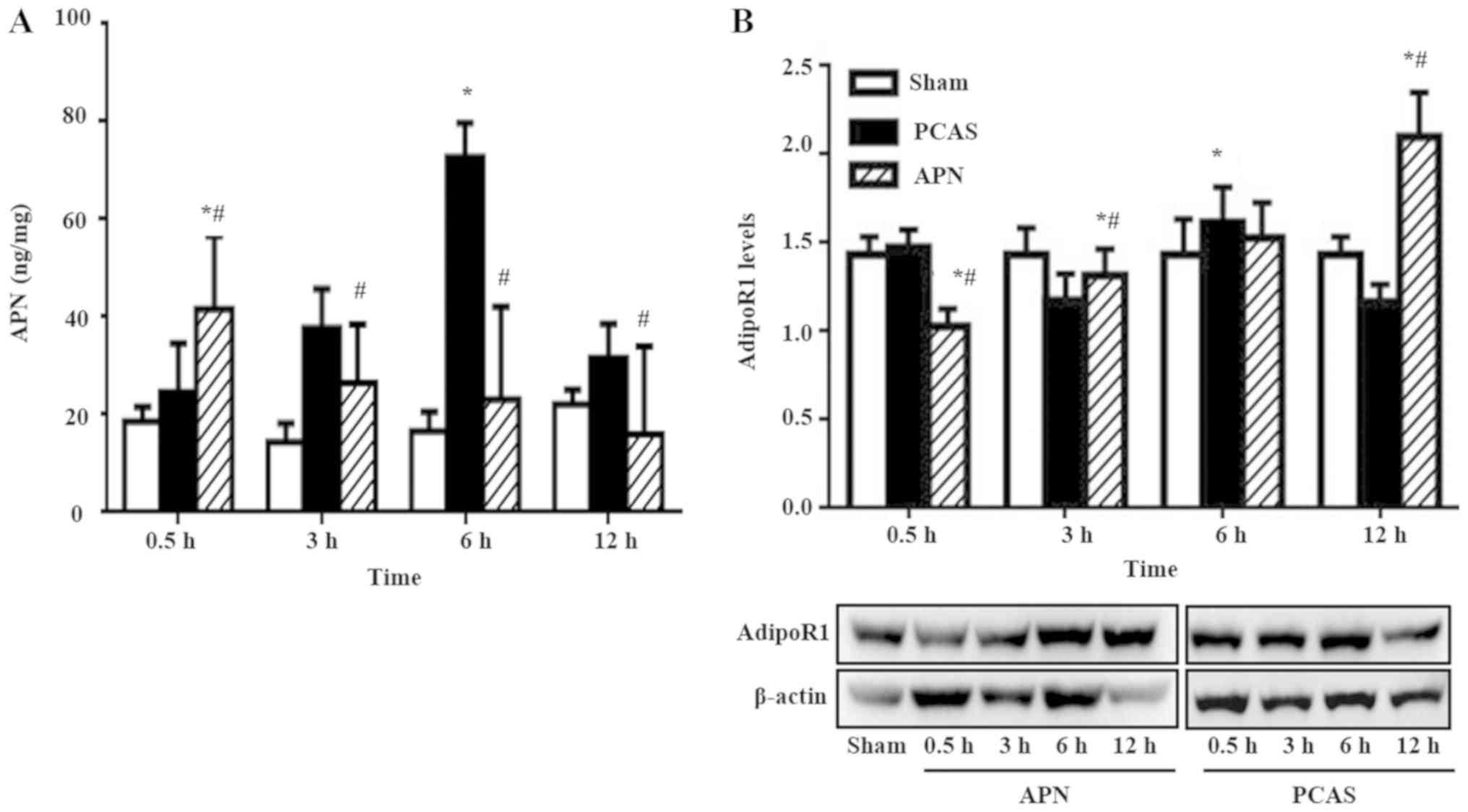
Variation of serum APN level and its
receptor expressed in different groups
GCIR significantly increased the serum APN level
following 3 and 6 h of reperfusion (P<0.05), peaking at 3 h. APN
post-treatment accelerated the increase of serum APN level induced
by GCIR, with its peak concentration appearing as early as 0.5 h of
reperfusion (P<0.05). However, this effect gradually
disappeared, because the serum APN level decreased to less than
that of the PCAS group from 3 to 12 h of reperfusion (Fig. 6A).
GCIR dramatically influenced the AdipoR1 expression
at different ROSC time points. For example, GCIR slightly increased
its expression at 0.5 h of reperfusion (P>0.05) and then was it
significantly inhibited at 3 h of reperfusion (P<0.05). However,
it significantly increased at 6 h of reperfusion, with levels
exceeding those of the Sham group. The injected exogenous APN
inhibited the AdipoR1 at the early stage of reperfusion (0.5 h;
P<0.05). However, this effect gradually disappeared since the
AdipoR1 expression was significantly higher than that in the PCAS
group at 3 and 12 h of reperfusion (P<0.05; Fig. 6B).
Discussion
Numerous studies have demonstrated that apoptosis
was one of the cell death types in different nervous disease
models, such as subarachnoid hemorrhage (27), stroke (28) and craniocerebral trauma (29). Our previous findings showed that
neuronal apoptosis also occurred in mice with CA (30). The results of the present study
also demonstrated that the activation of the apoptotic protein
caspase 3 and expression of the anti-apoptotic protein Bcl-2 in the
PCAS group were increased at 0.5 h after ROSC. These results
confirmed that apoptosis is one of the main mechanisms in the mice
brain following GCIR injury induced by CA-CPR.
APN, an adipocyte-derived secretory protein, plays
an important role in metabolic (31), anti-inflammatory (32) and anti-oxidative activities
(33). A number of studies have
reported that APN also specialized in alleviating myocardial, renal
and pancreatic ischemic reperfusion injury by reducing apoptosis
(34–36). Our previous study indicated that
APN reduced the number of apoptotic activities from neurons
following GCIR induced by CA-CPR (30). The present study indicated that APN
inhibited the apoptosis by reducing the expression of cleaved
caspase-3 in the mice brain tissue following GCIR induced by CA-CPR
and promoted the expression of anti-apoptotic protein Bcl-2. These
results confirmed the protective effects of APN on GCIR injury
induced by CA-CPR by inhibiting apoptosis.
Physiologically, autophagy is a self-protective
mechanism of the cells, as it helps maintain the functions of cell
structures, which might be damaged by apoptosis (37). However, excessive autophagy also
harms the cells, which also results in tissue injury (38). Previous studies have reported that
both autophagy and apoptosis are involved in the pathology of
cerebral hemorrhage, ischemic stroke and craniocerebral trauma
(26–28). Liu et al (39) indicates that arousing autophagy
relieves the brain injury by decreasing the apoptotic and necrotic
cells. Data in the present study demonstrated that LC3II, a protein
subsequently recruited to the autophagosomal membranes when
autophagy occurred, promptly increased in the brain tissue from 0.5
to 12 h after ROSC (P<0.05). By contrast, Beclin-1, an essential
mediator of autophagosome formation and another marker the
activation of autophagy slightly increased at 0.5 h after ROSC, but
decreased to less than that of the sham group from 3 to 12 h
following reperfusion. These results indicated that GCIR evoked
autophagy. In addition, the different expression trends of LC3II
and Beclin-1 may result from the interaction between LC3II and
Beclin-1 (35). A previous study
has demonstrated that activation of LC3I to LC3II inhibits Beclin-1
dissociation from the Beclin-1/Bcl-2 complex, further reducing the
polymerization of phosphatidyl inositol kinase (Vps34/PI3K) and
finally resulting in cell autophagy (39). This could be the reason LC3II was
detected earlier than Beclin-1 in the present study, a finding that
was in accordance with that of Guo et al (40).
The present study indicated that APN increased the
expression of LC3II and Beclin-1 in the brain tissue simultaneously
with inhibition of caspase-3 activation. To the best of the
authors' knowledge, the present study is the first to demonstrate
that APN protected the neuron from GCIR injury-induced excessive
apoptosis following CA-CPR by promoting autophagy, although Yue
et al (41) reported that
APN increases the expression of LC3II and Beclin-1 in the skeletal
muscle of diabetic mice to reduce apoptotic cells in a diabetic
mouse model. The mechanisms of how APN regulates the crosstalk
between apoptosis and autophagy in brain tissues injured by GCIR
following CA-CPR remain to be elucidated. However, some studies
have demonstrated that APN reduces neuronal apoptosis by reducing
reactive oxidative stress in an ischemic stroke model (15,42,43).
In the present study, the data demonstrated that the APN group had
a significantly reduced MDA content and increased SOD level in the
brain tissue injured by GCIR following CA-CPR. These results
confirmed that APN reduced apoptosis and enhances autophagy by
relieving oxidative stress in the brain tissue injured by GCIR
following CA-CPR. The present study demonstrated that APN inhibited
the activation of AMPK (Ser182) in the brain tissue at the early
post-GCIR period (0.5 h) and then enhanced it at 3 and 12 h of
reperfusion, accompanied by decreased apoptosis and increased
autophagy. Although Chen et al (26) have demonstrated this hypothesis in
keratinocytes in cardiac and liver cells, no previous studies have
suggested that APN activated AMPK (Ser182) signaling pathway to
reduce ROS induced-apoptosis to autophagy in GCIR injury induced by
CA-CPR. Thus, it is concluded that APN may be involved in the
protection of neurons in the brain tissue of post-ROSC mice by
regulating the activation of AMPK (Ser182), although other studies
have reported the priority activation site being Thr172 (44,45).
To the best of the authors' knowledge, the present study has been
the first to reveal that APN promoted neuronal autophagy following
CA-CPR by activating AMPK and relieving oxidative stress as a
result of reducing apoptosis and reducing the brain tissue injury
following CA-CPR. In accordance with a previous study, the
expression trend of AdipoR1 in the present study was inverse to the
concentration of APN, confirming the phenomenon of decreased ligand
resulting in increased receptor expression reactively (46).
There are some limitations in the present study.
First, it is a short term study to explore the acute injury
following GCIR. Therefore, only histological and molecular
assessments were performed and no behavioral assessment was
conducted. In addition, APN knockout (APN-KO) mice and AMPK
inhibitor were not used; thus, further investigations on the
effects of APN on the regulation of the crosstalk between apoptosis
and autophagy are necessary.
In conclusion, the current study demonstrated that
APN administration shortly following ROSC inhibits GCIR injury by
reducing apoptosis and increasing autophagy through the activation
of the AdipoR1-AMPK signaling pathway, which is therefore important
from a translational point of view.
Acknowledgements
Not applicable.
Funding
This study was supported by the National Natural
Science Foundation of China (grant nos. 81772037 and 81471836to YC
and grant no. 81801883 to YRH), the 1•3•5 Project for Disciplines
of Excellence, West China Hospital, Sichuan University (grant no.
ZYJC18019) to YC and the Chengdu Science and Technology Project
(grant no. 2018SZ0390) to HFY.
Availability of data and materials
All data generated or analyzed during the present
study are included in this published article.
Authors' contributions
YH, BLi and ZWZ made contributions to acquisition of
data and article drafting. PY, YS, YCh, JZ and JW contributed to
analysis and interpretation. YCa, WH, XM, TAC and BLo made
substantial contributions to design of the study and revision. All
authors read and approved the final manuscript.
Ethics approval and consent to
participate
All animal studies were approved by the
Institutional Animal Care and Use Committee at West China Hospital,
Sichuan University (approval no. 2017071A).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Jerry N: Cardiac arrest and
cardiopulmonary resuscitation. Semin Neurol. 37:05–012. 2017.
View Article : Google Scholar
|
|
2
|
Wang JW, Qiu YR, Fu Y, Liu J, He ZJ and
Huang ZT: Transplantation with hypoxia-preconditioned mesenchymal
stem cells suppresses brain injury caused by cardiac arrest-induced
global cerebral ischemia in rats: Hypoxic stem cells rescue global
cerebral ischemia. J Neurosci Resh. 95:2059–2070. 2017. View Article : Google Scholar
|
|
3
|
Sanganalmath SK, Gopal P, Parker JR, Downs
RK, Parker JC Jr and Dawn BJ: Global cerebral ischemia due to
circulatory arrest: Insights into cellular pathophysiology and
diagnostic modalities. Mol Cell Biochem. 426:111–127. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Fakharnia F, Khodagholi F, Dargahi L and
Ahmadiani A: Prevention of cyclophilin d-mediated mPTP opening
using cyclosporine-A alleviates the elevation of necroptosis,
autophagy and apoptosis-related markers following global cerebral
ischemia-reperfusion. J Mol Neurosci. 61:52–60. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Cui D, Shang H, Zhang X, Jiang W and Jia
X: Cardiac arrest triggers hippocampal neuronal death through
autophagic and apoptotic pathways. Sci Rep. 6:276422016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Mirzaei H and Regnier F: Protein: Protein
aggregation induced by protein oxidation. J Chromatogr B Analyt
Technol Biomed Life Sci. 873:8–14. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Liu CL, Ge P, Zhang F and Hu BR:
Co-translational protein aggregation after transient cerebral
ischemia. Neuroscience. 134:1273–1284. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zhang YP, Zhang Y, Xiao ZB, Zhang YB,
Zhang J, Li ZQ and Zhu YB: CFTR prevents neuronal apoptosis
following cerebral ischemia reperfusion via regulating
mitochondrial oxidative stress. J Mol Med (Berl). 426:111–127.
2018.
|
|
9
|
Lavandero S, Chiong M, Rothermel BA and
Hill JA: Autophagy in cardiovascular biology. J Clin Invest.
125:55–64. 2015. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Rami A, Langhagen A and Steiger S: Focal
cerebral ischemia induces upregulation of Beclin 1 and
autophagy-like cell death. Neurobiol Dis. 29:132–141. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Yu B, Ruan M, Liang T, Huang SW, Yu Y,
Cheng HB and Shen XC: The synergic effect of tetramethylpyrazine
phosphate and borneol for protecting against ischemia injury in
cortex and hippocampus regions by modulating apoptosis and
autophagy. J Mol Neurosci. 63:70–83. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ryan F, Khodagholi F, Dargahi L,
Minai-Tehrani D and Ahmadiani A: Temporal pattern and crosstalk of
necroptosis markers with autophagy and apoptosis associated
proteins in ischemic hippocampus. Neurotox Res. 34:79–92. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Berg AH, Combs TP and Scherer PE:
ACRP30/adiponectin: An adipokine regulating glucose and lipid
metabolism. Trends Endocrinol Metab. 13:84–89. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Efstathiou SP, Tsioulos DI, Tsiakou AG,
Gratsias YE, Pefanis AV and Mountokalakis TD: Plasma adiponectin
levels and five-year survival after first-ever ischemic stroke.
Stroke. 36:1915–1919. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Li X, Guo H, Zhao L, Wang B, Liu H, Yue L,
Bai H, Jiang H, Gao L, Feng DY and Qu Y: Adiponectin attenuates
NADPH oxidase-mediated oxidative stress and neuronal damage induced
by cerebral ischemia-reperfusion injury. Biochim Biophys Acta Mol
Basis Dis. 1863:3265–3276. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
National Research Council (US), .
Committee Guide for the care and use of laboratory animals.
National Academies Press (US). 2011."ref-label" rowspan="1" colspan="1">
17
|
Yao R, Cao Y, He YR, Lau WB, Zeng Z and
Liang ZA: Adiponectin attenuates lung fibroblasts activation and
pulmonary fibrosis induced by paraquat. PLoS One. 10:e01251692015.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ding W, Cai Y, Wang W, Ji L, Dong Y and
Zhang X, Su M, Liu J, Lu G and Zhang X: Adiponectin protects the
kidney against chronic intermittent hypoxia-induced injury through
inhibiting endoplasmic reticulum stress. Sleep Breath.
20:1069–1074. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Chen MH, Liu TW, Xie L, Song FO, He T,
Zeng ZY and Mo SR: A simpler cardiac arrest model in rats. Am J
Emerg Med. 25:623–630. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Chen MH, Liu TW, Xie L, Song FQ and He T:
A comparison of transoesophageal cardiac pacing and epinephrine for
cardiopulmonary resuscitation. Am J Emerg Med. 24:545–552. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Song Y, Zhong M and Cai FC: Oxcarbazepine
causes neurocyte apoptosis and developing brain damage by
triggering Bax/Bcl-2 signaling pathway mediated caspase 3
activation in neonatal rats. Eur Rev Med Pharmacol Sci. 22:250–261.
2018.PubMed/NCBI
|
|
22
|
Bennett MW, O'connell J, O'sullivan GC,
Roche D, Brady C, Kelly J, Collins JK and Shanahan F: Expression of
Fas ligand by human gastric adenocarcinomas: A potential mechanism
of immune escape in stomach cancer. Gut. 44:156–162. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wolf AM, Wolf D, Avila MA, Moschen AR,
Berasain C, Enrich B, Rumpold H and Tilg H: Up-regulation of the
anti-inflammatory adipokine adiponectin in acute liver failure in
mice. J Hepatol. 44:537–543. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zhu X, Tang Z, Cong B, Du J, Wang C, Wang
L, Ni X and Lu J: Estrogens increase cystathionine-gamma-lyase
expression and decrease inflammation and oxidative stress in the
myocardium of ovariectomized rats. Menopause. 20:1084–1091. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Yang T, Mao YF, Liu SQ, Hou J, Cai ZY, Hu
JY, Ni X, Deng XM and Zhu XY: Protective effects of the free
radical scavenger edaravone on acute pancreatitis-associated lung
injury. Eur J Pharmacol. 630:152–157. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Chen X, Li L, Xu S, Bu W, Chen K, Li M and
Gu H: Ultraviolet B radiation down-regulates ULK1 and ATG7
expression and impairs the autophagy response in human
keratinocytes. J Photochem Photobiol B. 178:152–164. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Han Y, Zhang T, Su J, Zhao Y and Li X:
Apigenin attenuates oxidative stress and neuronal apoptosis in
early brain injury following subarachnoid hemorrhage. J Clin
Neurosci. 40:157–162. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Li Y, Mei Z, Liu S, Wang T, Li H, Li XX,
Han S, Yang Y, Li J and Xu ZD: Galanin protects from
caspase-8/12-initiated neuronal apoptosis in the ischemic mouse
brain via GalR1. Aging Dis. 8:85–100. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zhang M, Wu J, Ding H, Wu W and Xiao G:
Progesterone provides the pleiotropic neuroprotective effect on
traumatic brain injury through the Nrf2/ARE signaling pathway.
Neurocrit Care. 26:292–300. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Osellame LD, Blacker TS and Duchen MR:
Cellular and molecular mechanisms of mitochondrial function. Best
Pract Res Cl En. 26:711–723. 2012. View Article : Google Scholar
|
|
31
|
Mongardon N, Dumas F, Ricome S, Grimaldi
D, Hissem T, Pène F and Cariou A: Postcardiac arrest syndrome: From
immediate resuscitation to long-term outcome. Ann Intensive Care.
1:452011. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Denicola A and Radi R: Peroxynitrite and
drug-dependent toxicity. Toxicology. 208:273–288. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zhu Y, Yang GY, Ahlemeyer B, Pang L, Che
XM, Culmsee C, Klumpp S and Krieglstein J: Transforming growth
factor-β1 increases bad phosphorylation and protects neurons
against damage. Neurosci. 22:3898–3909. 2002. View Article : Google Scholar
|
|
34
|
Sun Y, Zhao D, Yang Y, Gao C, Zhang X, Ma
Z, Jiang S, Zhao L, Chen W, Ren KJ, et al: Adiponectin exerts
cardioprotection against ischemia/reperfusion injury partially via
calreticulin mediated anti-apoptotic and anti-oxidative actions.
Apoptosis. 22:108–117. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Yi W and Yang Q: Adiponectin improves
diabetic nephropathy by inhibiting necrotic apoptosis. AMS.
15:1321–1328. 2019.PubMed/NCBI
|
|
36
|
Wang Y, Li Y, Qiao J, Li N and Qiao S:
AMPK α1 mediates the protective effect of adiponectin against
insulin resistance in INS-1 pancreatic β cells. Cell Biochem Funct.
37:625–632. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Wiklund L, Martijn C, Miclescu A, Semenas
E, Rubertsson S and Sharma HS: Central nervous tissue damage after
hypoxia and reperfusion in conjunction with cardiac arrest and
cardiopulmonary resuscitation: Mechanisms of action and
possibilities for mitigation. Int Rev Neurobiol. 102:173–187. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Xu KY, Zweier JL and Becker LC: Hydroxyl
radical inhibits sarcoplasmic reticulum Ca(2+)-ATPase function by
direct attack on the ATP binding site. Circ Res. 80:76–81. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Liu Z, Gan L, Wu T, Feng F, Luo D, Gu H,
Liu S and Sun C: Adiponectin reduces ER stress-induced apoptosis
through PPARα transcriptional regulation of ATF2 in mouse adipose.
Cell Death Dis. 7:e24872016. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Guo F, Jiang T, Song W, Wei H, Wang F, Liu
L, Ma L, Yin H, Wang Q and Xiong L: Electroacupuncture attenuates
cerebral ischemia-reperfusion injury in diabetic mice through
adiponectin receptor 1-mediated phosphorylation of GSK-3β. Mol
Neurobiol. 51:685–695. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Yue L, Zhao L, Liu H, Li X, Wang B, Guo H,
Gao L, Feng D and Qu Y: Adiponectin protects against
glutamate-induced excitotoxicity via activating SIRT1-dependent
PGC-1 expression in HT22 hippocampal neurons. Oxid Med Cell Longev.
2016:29573542016. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Adrie C, Adib-Conquy M, Laurent I, Monchi
M, Vinsonneau C, Fitting C, Fraisse F, Dinh-Xuan AT, Carli P,
Spaulding C, et al: Successful cardiopulmonary resuscitation after
cardiac arrest as a ‘sepsis-Like’ syndrome. Circulation.
106:562–568. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Gairolla J, Kler R, Modi M and Khurana DJ:
Leptin and adiponectin: Pathophysiological role and possible
therapeutic target of inflammation in ischemic stroke. Rev
Neurosci. 28:295–306. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Duan J, Yin Y, Cui J, Yan J, Zhu Y, Guan
Y, Wei G, Weng Y, Wu X, Guo C, et al: Chikusetsu saponin IVa
ameliorates cerebral ischemia reperfusion injury in diabetic mice
via adiponectin-mediated AMPK/GSK-3beta pathway in vivo and in
vitro. Mol Neurobiol. 53:728–743. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Piao L, Yu C, Xu W, Inoue A, Shibata R, Li
X, Nan Y, Zhao G, Wang H, Meng X, et al: Adiponectin/AdiopR1 signal
inactivation contributes to impaired angiogenesis in mice of
advanced age. Int J Cardiol. 267:150–155. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Gamberi T, Modesti A, Magherini F, D'Souza
DM, Hawke T and Fiaschi T: Activation of autophagy by globular
adiponectin is required for muscle differentiation. Biochim Biophys
Acta. 1863:694–702. 2016. View Article : Google Scholar : PubMed/NCBI
|