Introduction
Diabetes increases sensitivity to
ischemia-reperfusion injury, and may be associated with increased
levels of oxidative stress during hyperglycemia (1). The increased oxidative stress in
diabetes is primarily caused by an imbalance between the generation
and elimination of oxidative stress. Reactive oxygen species (ROS)
are associated with the endogenous antioxidant defense system
(2). In addition, due to elevated
levels of blood glucose, osmotic dehydration is observed in the
mouse model of diabetes, which leads to increased permeability of
the intestinal cells, an impaired normal mucosal barrier of the
small intestine and increased intestinal damage (3).
Intestinal ischemia reperfusion (IIR) injury is a
clinical challenge associated with high morbidity and mortality,
which has important roles during numerous pathological processes,
including neonatal necrotizing enterocolitis (4), acute mesenteric ischemia (5), intestinal torsion (6), bowel transplantation (7), trauma (8), shock (8,9), and
cardiopulmonary insufficiency (10). Ischemic injury is primarily caused
by interrupted blood flow triggered by hypoxia, which leads to the
death of intestinal mucosa cells and severe mucosal damage
(11). IIR occurs when blood flow
is restored, causing further damage to the viable mucosal cells and
resulting in the release of various inflammatory mediators, which
potentially leads to systemic inflammatory response syndrome and
ultimately to multiple organ failure (12). The pathophysiological mechanism
underlying IIR has yet to be fully elucidated; however,
considerable evidence has indicated that IIR-induced injury is
associated with the appearance of inflammatory mediators (including
ROS, cytokines and bacterial endotoxins), impaired mitochondrial
function and apoptosis (13–16).
Previous studies have reported that diabetes is
associated with increased susceptibility to cardiac
ischemia-reperfusion injury (1,17),
which may be associated with increased levels of underlying
oxidative stress that are secondary to hyperglycemia (18,19).
The increase in oxidative stress that occurs during diabetes is
primarily due to the imbalance between the production of reactive
oxygen species (ROS) and the removal of the endogenous antioxidant
defense system. The degree of imbalance may affect the severity of
ischemia-reperfusion injury under diabetic conditions (20). During diabetes, mitochondria are
the major intracellular sources of ROS, and ~4% of the oxygen
consumed by mitochondria is converted into ROS (21). Mitochondria are the main targets of
oxidative damage and damaged mitochondria can further increase ROS
production via ROS-induced release of ROS (22), which may lead to leakage of lethal
factors such as interleukin (IL)-1β and IL-6 (23), triggering a vicious cycle which
induces myocardial cell death (24). Diabetes is at least partly
considered to be a mitochondrial disease, since the overproduction
of superoxide in the mitochondrial electron transport chain is
involved in the mechanism underlying hyperglycemia-induced injury
(25,26). A previous study on diabetic
ischemic reperfusion of the myocardium revealed that decreased
levels of mitochondrial autophagy promote mitochondrial aggregation
in the myocardium, aggravate mitochondrial disturbances in
myocardial tissue, damage myocardial function and increase
mortality in a mouse model of diabetes (27). The discovery of autophagy during
ischemia-reperfusion provides a novel therapeutic target for
ischemic heart disease; however, only a few studies have reported
the effects of mitochondrial autophagy during diabetic IIR.
Therefore, the present study aimed to assess the effect of diabetes
on intestinal pathological alterations in a mouse model of type 1
diabetes mellitis with IIR.
Materials and methods
Animals
The present study was approved by the Animal Care
Committee of Wuhan University and protocols were conducted in
accordance with the National Institutes of Health Guidelines for
the Care and Use of Experimental Animals (28). A total of 40 healthy wild-type SPF
male C57BL/6J mice (age, 6–8 weeks; weight, 20–25 g) were purchased
from Beijing Weitong Lihua Experimental Animal Technology Co., Ltd.
All animals were housed in individual cages (n=2/3 per cage) in a
climate-controlled room (23±1°C; relative humidity, 60±5%) with
12-h light/dark cycles and free access to food and water for one
week. All mice were fasted with free access to water for 12 h prior
to the experiments.
Diabetes model
All mice were fasted with free access to water for
12 h prior to the experiments. After a 72-h fast, the diabetic
group were administrated streptozocin (220 mg/kg; Sigma-Aldrich;
Merck KGaA) intraperitoneally. After 72 h, the blood glucose level
in the tail blood was tested. Symptoms of polydipsia, polyphagia
and diabetes indicated successful induction of the mouse model of
diabetes. During the experimental period (~2 months), blood was
collected every week to monitor blood glucose levels. If the blood
glucose level was ≥16.7 mmol/l for three consecutive weeks, the
mouse model of diabetes was considered to have been successfully
established. The non-diabetic group received an equal volume of
saline (200 mg/kg).
IIR model
The IIR model was established according to the
protocol previously described by Meng et al (29). Mice were anesthetized by the
intraperitoneal injection of pentobarbital sodium solution (1%; 40
mg/kg) and were subsequently fixed in the supine position. The fur
in the abdominal region was shaved and the area was disinfected. A
midline incision was made and the roots of the superior mesenteric
artery (SMA) were clamped using micro-artery clamps. Subsequently,
vascular contractions were reduced, the intestinal wall became pale
and the mesentery was observed. After the arterial blood flow was
blocked, the wound was closed. After 45 min, the arterial clamp was
released to restore blood supply, modeling IIR. The Sham group and
DS groups were subjected to the same procedure; however, the
vessels were not clamped. Mice were sacrificed 2 h
post-ischemia-reperfusion clamping. The blood and intestine tissues
were collected and processed for subsequent biochemical analysis.
The small intestine tissue was isolated, washed with saline, fixed
with 4% paraformaldehyde at 4°C for 24–48 h and frozen in liquid
nitrogen.
Experimental grouping
Following surgical preparation, the mice were
randomly divided into four groups (n=10 per group): i) Control
group (Sham group); ii) normal IIR group (IIR group); iii) diabetic
non-ischemic reperfusion group (DS group); and iv) diabetic
ischemia-reperfusion group (DIIR group).
Histopathological assessment of the
intestines
The fresh ischemic area of the intestine was washed
three times in cold phosphate buffer (pH 7.4), fixed in 4%
formaldehyde (pH 7.4) at 4°C for 24–48 h, paraffin-embedded and
sectioned at a thickness of 5 µm. Sections were then stained in
Harris hematoxylin solution for 5–20 min and with eosin solution
(95% ethanol solution) for 3–30 sec at room temperature. Stained
sections were observed using an Olympus BX50 (Olympus Corporation)
light microscope at ×200 magnification. The HPIAS-1000 medical
color image analysis system (Olympus Corporation) was used to
collect images and Image Pro Plus software (version 6.0; Media
Cybernetics, Inc.) was used for analysis. For each specimen, two
sections were observed (10 fields of view), intestinal pathological
alterations were identified and Chiu's pathology scores were
calculated (25). Higher scores
indicated more severe damage. The Chiu grading system consists of 5
subdivisions according to alterations to the villus and gland of
the intestinal mucosa: Grade 0, normal mucosa; grade 1, development
of subepithelial Gruenhagen's space at the tip of the villus; grade
2, extension of the space with moderate epithelial lifting; grade
3, massive epithelial lifting with a few denuded villi; grade 4,
denuded villi with exposed capillaries; and grade 5, disintegration
of the lamina propria, ulceration and hemorrhage.
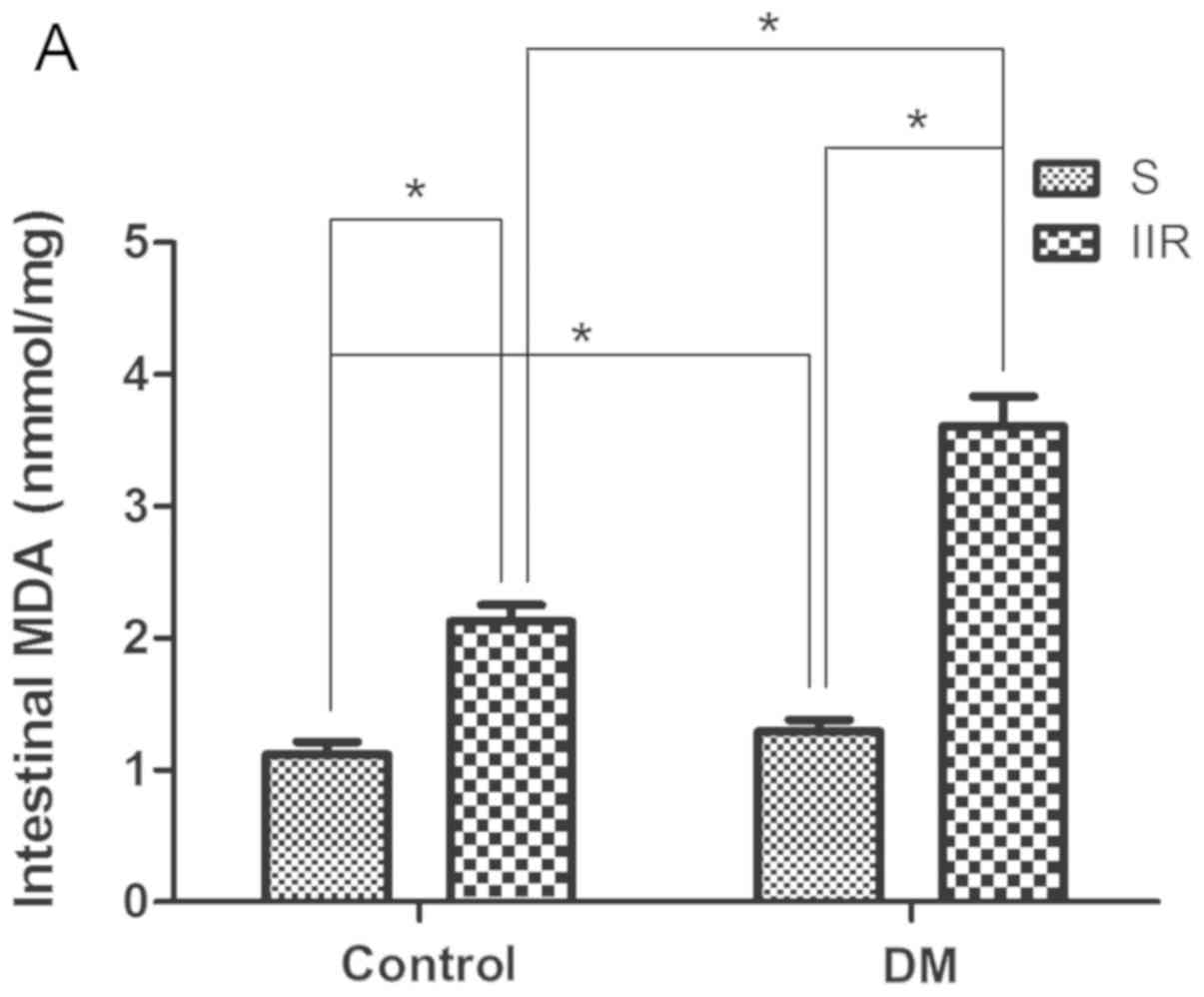
Measurement of malondialdehyde
(MDA)
Following homogenization of the tissue in normal
saline on ice or centrifugation at 13,000 × g for 10 min at 4°C of
the blood, the MDA levels of the supernatants were determined using
an MDA assay kit (cat. no. A001-3-2; Nanjing Jiancheng
Bioengineering Institute) according to the manufacturer's protocol
and the thiobarbituric acid colorimetric method, as previously
described (30). MDA concentration
is expressed as nmol/mg protein.
Activity of superoxide dismutase (SOD)
and glutathione peroxidase (GSH-Px) in intestinal tissues
SOD activity was measured using the SOD assay kit
(Nanjing Jiancheng Bioengineering Institute) according to the
manufacturer's protocol and calculated according to the following
method. Briefly, epinephrine undergoes autoxidation rapidly at pH
10.0 to produce adrenochrome, a pink-colored product that can be
detected at a wavelength of 480 nm using a UV-vis spectrophotometer
in the kinetic mode (31). The
amount of enzyme required to produce 50% inhibition was defined as
one unit of enzyme activity. SOD activity is expressed as units/mg
protein.
GSH-Px activity was measured using the GSH-Px assay
kit (Nanjing Jiancheng Bioengineering Institute), according to the
manufacturer's protocol. The reaction was initiated by the addition
of H2O2. A series of enzymatic reactions were
activated by GSH-Px in the homogenate that subsequently led to the
conversion of reduced glutathione (GSH) into oxidized glutathione
(GSSG). Alterations to the absorbance at a wavelength of 340 nm
during the conversion of GSH to GSSG were recorded using a
spectrophotometer. Enzyme activity was calculated according to a
formula provided by the manufacturer. GSH-Px activity is expressed
as nmol/mg protein.
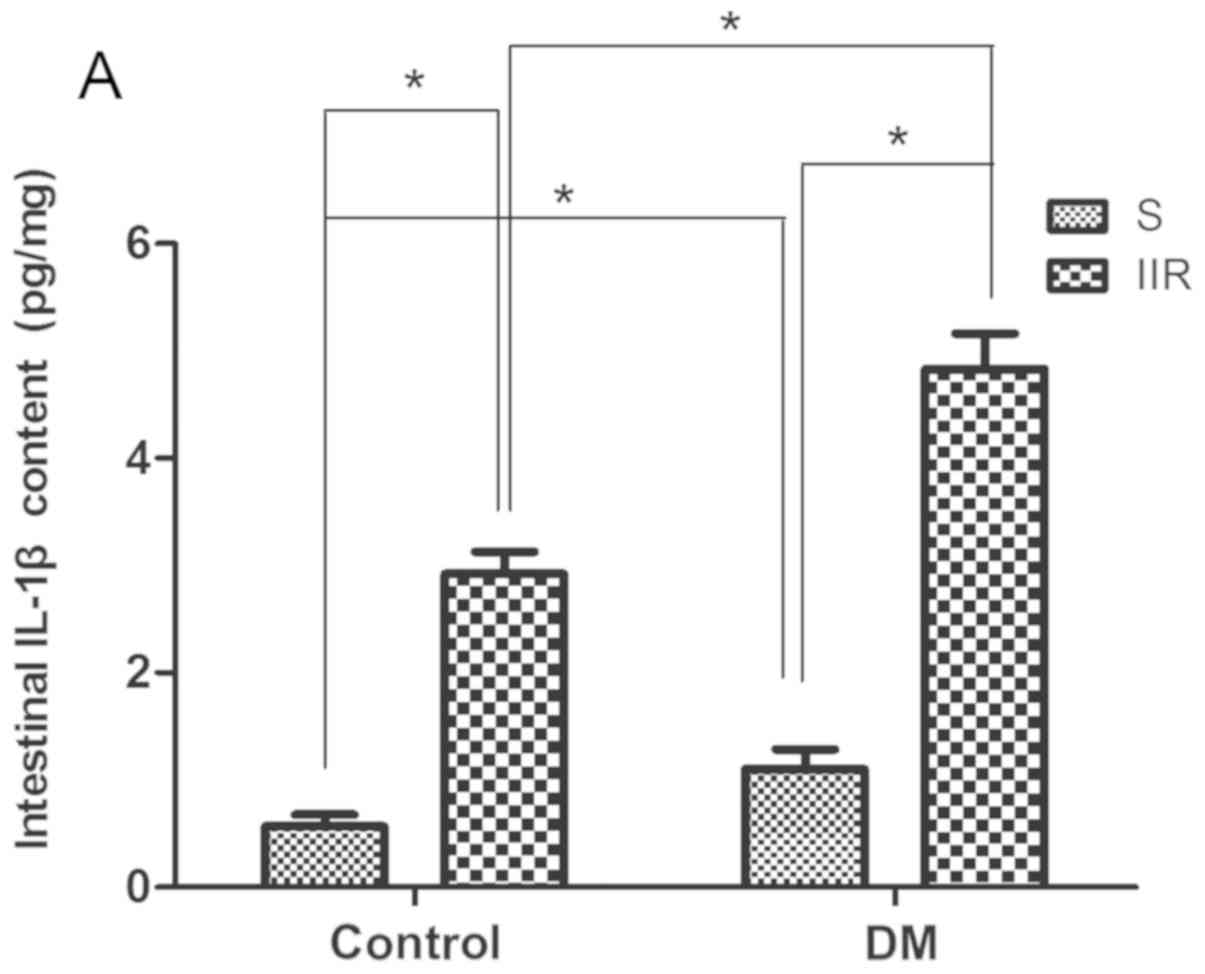
TNF-α, IL-6, IL-10 and IL-1β levels in
the blood plasma and intestine tissue
The inflammatory cytokine levels of TNF-α, IL-6 and
IL-10 in the blood plasma and intestinal tissues were quantified
using ELISA kits (32). The
following kits were used according to the manufacturer's protocol:
TNF-α ELISA kit (cat. no. H052; Nanjing Jiancheng Bioengineering
Institute), IL-1β ELISA kit (cat. no. H002; Nanjing Jiancheng
Bioengineering Institute) and IL-6 ELISA kit (cat. no. H007;
Nanjing Jiancheng Bioengineering Institute). Cytokine levels are
expressed as pg/ml.
Western blot analysis
The intestines were isolated and washed three times
with pre-chilled PBS at 4°C. Subsequently, total protein was
extracted from the intestinal tissues using RIPA lysate (cat. no.
MT006; Biolab Technology) on ice for 30 min. Cell suspensions were
lysed by sonification at 400w on crushed ice every 10 sec 15–20
times and then centrifuged at 4°C for 15 min at 12,000 × g. Total
protein was quantified using a bicinchoninic acid assay and
subsequently, 5X loading buffer (BioMart) was added to the samples
and boiled for 10 min. Equal amounts of protein (50 µg) were
separated via 12% SDS-PAGE at 100 V for 3 h and transferred onto
PVDF membranes. The membranes were blocked with 5% fat-free milk
powder for 1 h at room temperature. Subsequently, the membranes
were incubated overnight at 4°C in a shaker with rabbit anti-mice
polyclonal primary antibodies targeted against: Parkin (cat. no.
2132; 1:1,000; Cell Signaling Technology, Inc.), Pink1 (cat. no.
ab137361; 1:1,000; Abcam), LC3B (cat. no. ab48394; 1:1,000; Abcam)
and β-actin (cat. no. 4970; 1:2,000; Cell Signaling Technology,
Inc.). Subsequently, the membranes were washed three times with
TBST at room temperature for 10 min per wash. Following primary
incubation, the membranes were incubated with a LI-COR
IRDye800CW-conjugated goat anti-rabbit secondary antibody
(1:10,000; cat. no. 926-32211; LI-COR Biosciences) for 1 h at room
temperature. The membranes were washed three times with TBST at
room temperature. Protein bands were visualized and using the
ODYSSEY two-color infrared laser imaging system (LI-COR
Biosciences). Protein expression was quantified using Odyssey
software (version 3.0, LI-COR Biosciences) using β-actin as the
loading control.
Transmission electron microscopy
(TEM)
Intestinal tissue samples were pre-fixed with 4%
glutaraldehyde for 2 h at 4°C and subsequently fixed with 2% osmium
tetroxide for 2 h at 20°C. Following washing with PBS and
dehydration using a graded ethanol series, tissue samples were
embedded in epoxy-resin for 48 h at room temperature. Ultrathin
sections (60–80 nm) were cut using an Ultracut UCT ultramicrotome
(Leica Microsystems GmbH). Samples were stained with lead citrate
for 30 min at room temperature. The ultrastructure of the tissue
sections was observed using a H-7000 FA transmission electron
microscope (Hitachi, Ltd.) at ×2,000 magnification. Autophagosomes
or autophagolysosomes were identified by the characteristic
structure of a bilayer or multilayer smooth membrane that
completely surrounded the compressed mitochondria or membrane-bound
electron-dense material.
Statistical analysis
Data are expressed as the mean ± SD. Statistical
analyses were performed using GraphPad Prism software (version 5.0;
GraphPad Software Inc.). Statistical evaluation of the data was
performed by two-way ANOVA followed by Tukey's post hoc test.
P<0.05 was considered to indicate a statistically significant
difference.
Results
Diabetes increases the intestinal
vulnerability to IIR injury
To investigate the effects of diabetes on IIR
injury, H&E staining was performed and Chiu's pathology scores
were calculated for the four experimental groups of mice. The
intestinal mucosa of the Sham group was intact and the villi of the
small intestine were well-arranged (Fig. 1A). In the DS group, the intestinal
mucosa was thickened, the small intestine villous epithelial cells
were edematous, and a cystic space was observed under the apical
epithelium (Fig. 1A). In the IIR
group, the intestinal mucosa lost its normal structure, the villi
were broken and shed, the interstitium was broken, inflammatory
cells had infiltrated and the cellular components of the lamina
propria were increased (Fig. 1A).
In the DIIR group, destruction of the intestinal mucosa was even
more severe compared with the IIR group: The epithelial cell layer
was necrotic, and in the process of being shed; the innermost layer
was destroyed; and scattered bleeding and ulceration lesions were
observed (Fig. 1A). Chiu's
pathological scoring system indicated that the degree of intestinal
mucosal damage in the DS group was more extensive compared with the
Sham group, and the degree of intestinal mucosal injury in the DIIR
group was more extensive compared with the IIR group. The degree of
injury of the intestinal mucosa in the IIR and DIIR groups was more
extensive compared with the Sham and DS groups, respectively. The
aforementioned differences were statistically significant
(P<0.05; Fig. 1A and B).
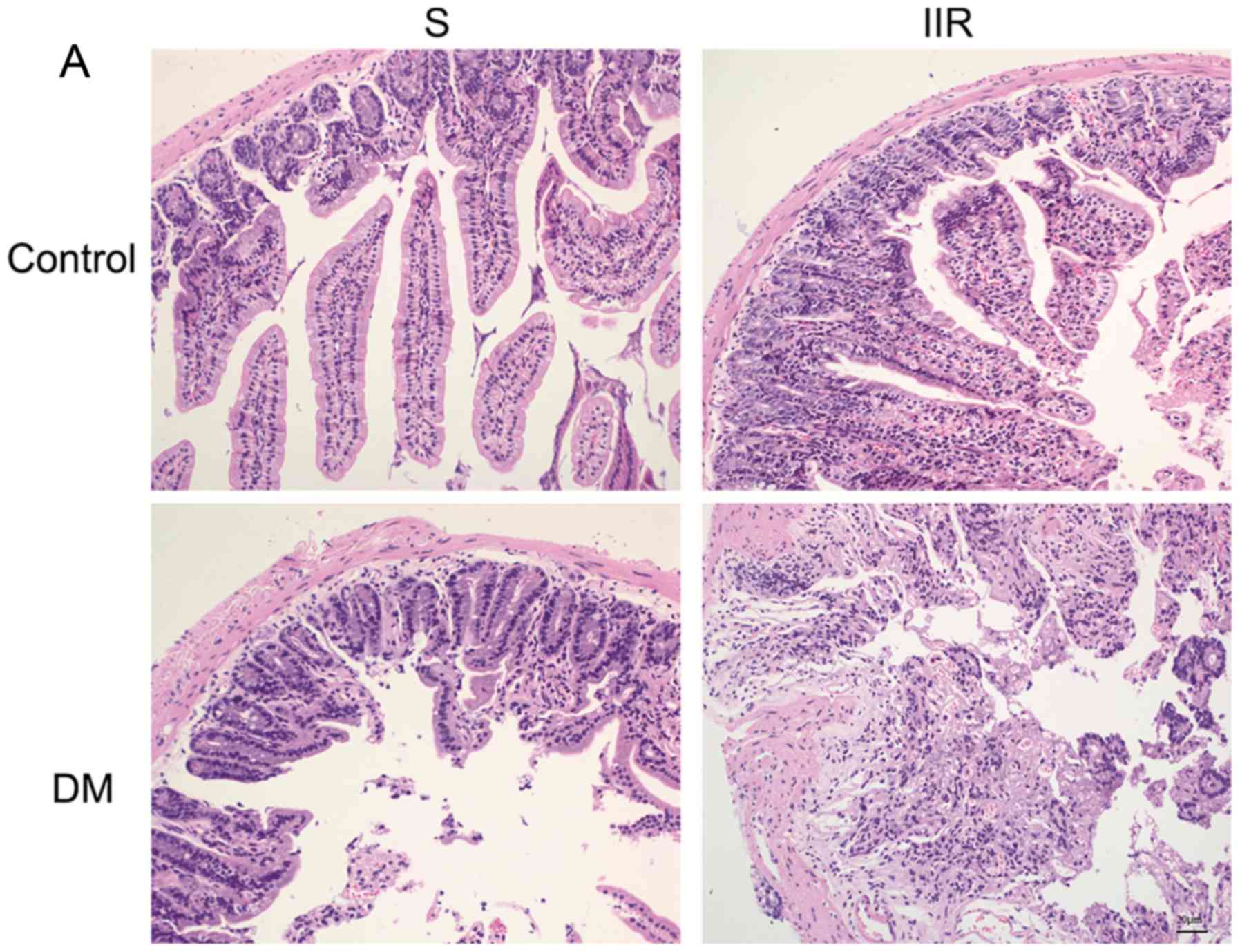 | Figure 1.Diabetes increases the intestinal
vulnerability to IIR injury. (A) Hematoxylin and eosin staining of
the intestinal tissue sections of the four groups (magnification,
×200). The Sham group displayed intact intestinal tissues with
well-arranged villi of the small intestine. The DS group displayed
thickened intestinal mucosa, edematous small intestine villous
epithelial cells and a cystic space under the apical epithelium.
The IIR group displayed a loss of normal intestinal mucosa
structure, broken and shed villi, broken interstitium, inflammatory
cell infiltration and increased cellular components of the lamina
propria. The DIIR group displayed a necrotic epithelial cell layer,
which was in the process of being shed, a destroyed innermost
layer, and scattered bleeding and ulceration lesions. (B) The
severity of intestinal injury was determined according to Chiu's
scoring system. *P<0.05, as indicated. IIR, intestinal ischaemia
reperfusion; DS, diabetic sham; DIIR, diabetes with IIR; S, Sham;
DM, diabetic mice. |
Diabetes increases the levels of the
inflammatory cytokines, IL-1β, IL-6 and TNF-α, in the blood plasma
and intestinal tissue
The levels of IL-1β, IL-6 and TNF-α in the blood
plasma and intestinal tissues of each group were measured. The
results suggested that IL-1β, IL-6 and TNF-α in the blood plasma
and intestinal tissues were significantly increased in the IIR
group compared with the Sham group (P<0.05; Fig. 2A-F). In addition, the IL-1β, IL-6
and TNF-α levels were significantly increased in the DIIR group
compared with the DS group (P<0.05; Fig. 2A-F).
Diabetes influences oxidative stress
during IIR
The levels of MDA content, as well as SOD and GSH-Px
activity, in the blood plasma and intestinal tissues of the
different groups were observed. The results suggested that the DS
and DIIR groups showed significantly increased MDA levels compared
with the Sham and IIR groups, respectively (P<0.05; Fig. 3A and B). By contrast, the DS and
DIIR groups showed significantly reduced levels of SOD and GSH-Px
activity compared with the Sham and IIR groups, respectively
(P<0.05; Fig. 3C and D).
Mitochondrial autophagy is
significantly increased following IIR in the mouse model of
diabetes
Subsequently, TEM was performed to examine the
intestinal tissue specimens of the different groups. The DS group
displayed mitochondrial swelling, structural integrity and
increased numbers of autophagosomes compared with the Sham group
(Fig. 4A and B). In the IIR group,
mitochondrial swelling was evident, the iliac crest had ruptured,
matrix density was decreased, mitochondria were vacuolated, and the
number of autophagosomes was increased compared with that of the
sham group (Fig. 4A and B). In the
DIIR group, a large number of mitochondria were destroyed, the
mitochondrial structure was undefined and the number of
autophagosomes was significantly increased compared to those of the
sham group (Fig. 4A and B).
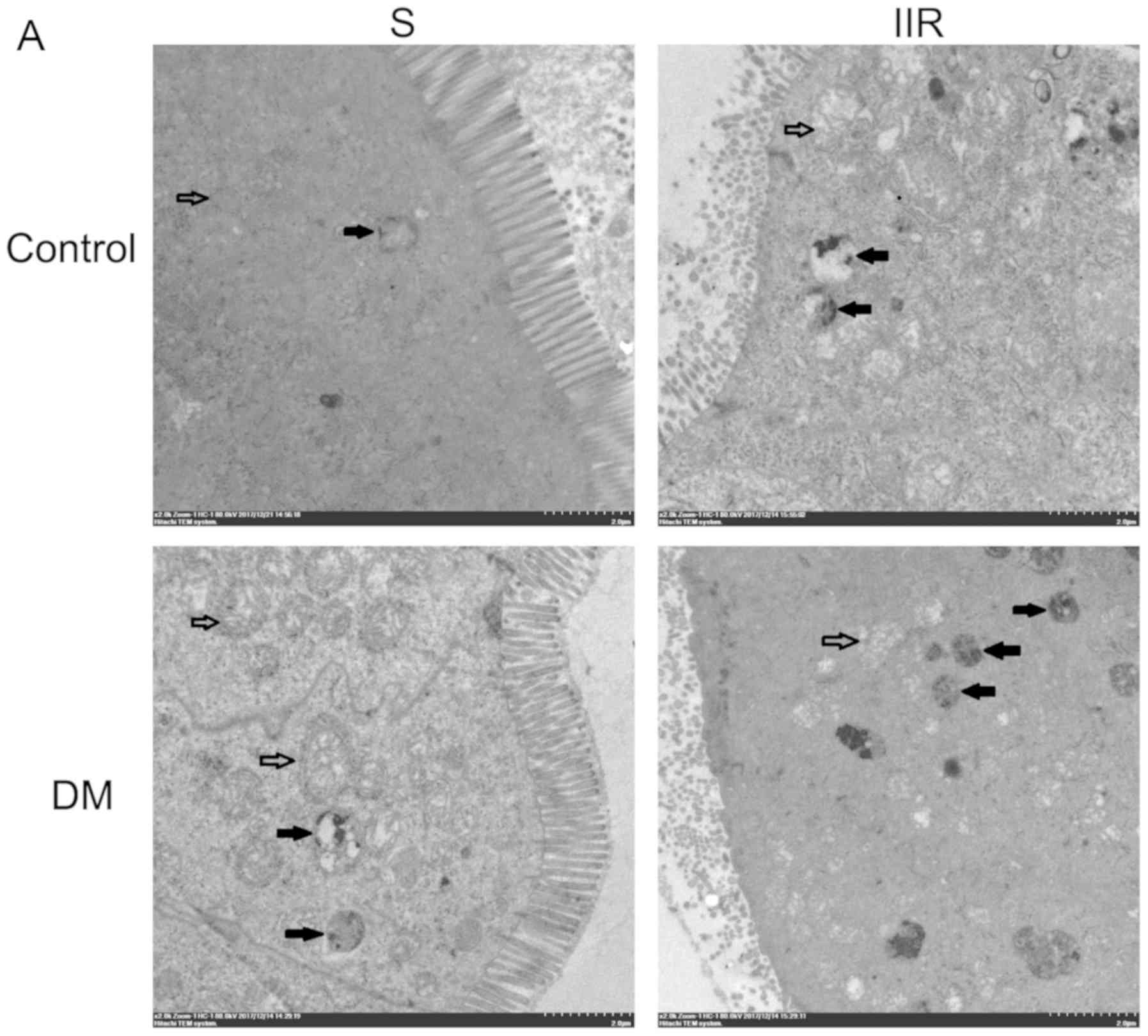 | Figure 4.Effects of diabetes on IIR-induced
intestinal. (A) Autophagosomes were observed under an electron
microscope at ×2,000 magnification. (Solid arrows indicate
mitochondria and hollow arrows indicate mitochondrial autophagy,
scale bar 2.0 µm). (B) Histogram shows the average number of
autophagosome structures per view (371 µm2) obtained by
examining at least 50 images per testing sample. Transmission
electron microscopy indicated that the diabetic group displayed
mitochondrial swelling, structural integrity and an increased
number of autophagosomes compared with the Sham group. The IIR
group displayed evident mitochondrial swelling, iliac crest
rupture, decreased matrix density, vacuolated mitochondria and an
increased number of autophagosomes compared with the Sham group.
The DIIR group displayed a large number of destroyed mitochondria,
undefined mitochondrial structure and an increased number of
autophagosomes compared with the IIR group. *P<0.05, as
indicated. IIR, intestinal ischaemia reperfusion; DIIR, diabetes
with IIR; S, Sham; DM, diabetic mice. |
Subsequently, the expression of the autophagy marker
LC3BII/I in intestinal tissues was detected by western blotting.
The levels of LC3BII/I were upregulated in the DS and DIIR groups
compared with the Sham and IIR groups, respectively. Treatment with
IIR significantly increased LC3BII/I levels in the IIR and DIIR
groups compared with the Sham and DS groups, respectively.
Additionally, the increase in LC3BII/I expression in the DIIR group
compared with the DS group was more obvious compared with the
increase in the IIR group compared with the Sham group (Fig. 5A and C).
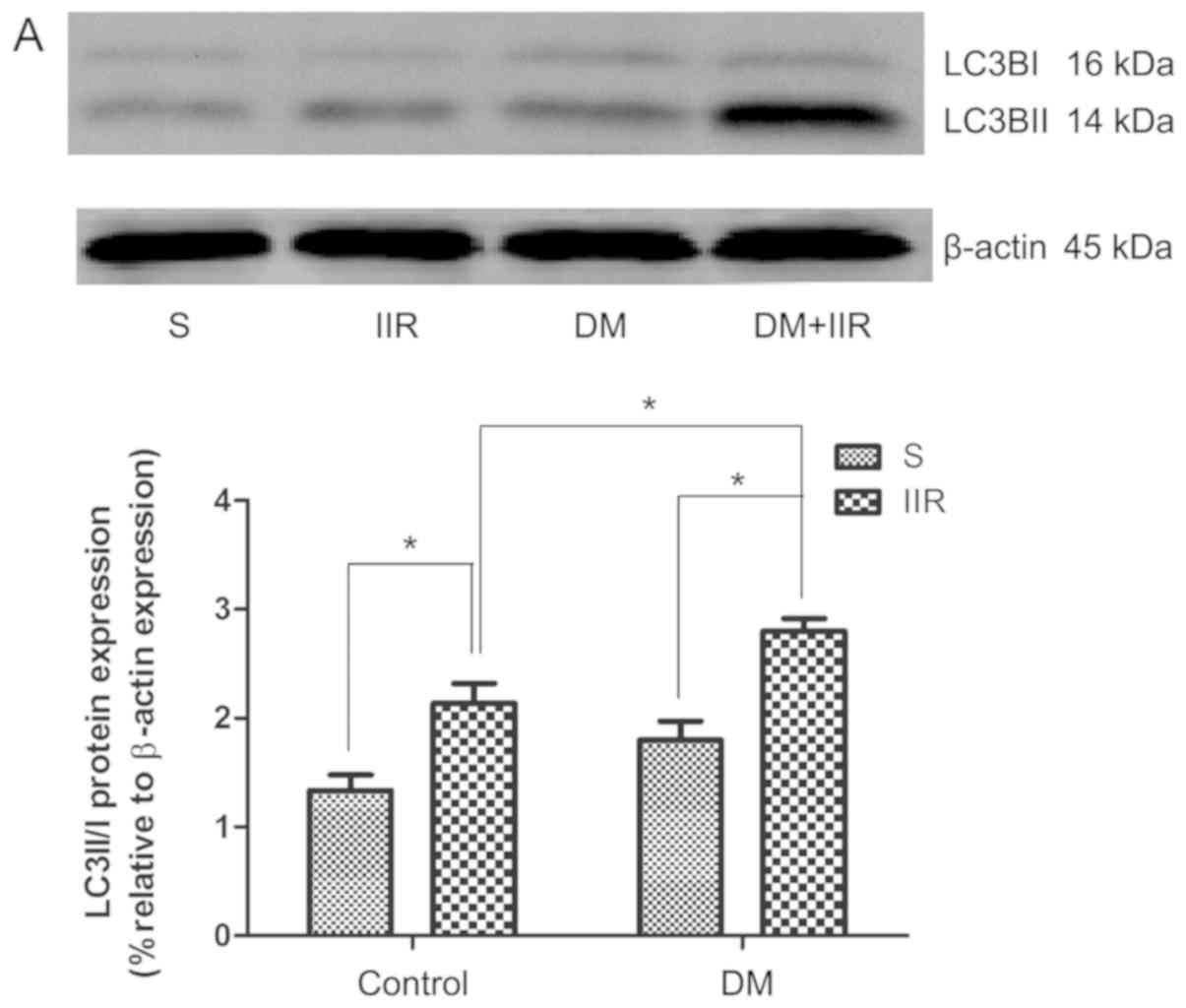 | Figure 5.Protein expression levels of
PINK1/Parkin and the ratio of LC3BII/I are upregulated following
IIR in the mouse model of diabetes. Western blotting was performed
to determine (A) the protein expression levels of PINK1 and Parkin,
and (B) the ratio of LC3BII/I expression. *P<0.05, as indicated.
PINK1, phosphatase and tensin homolog-induced putative kinase;
LC3BII/I, light chain 3B II/I; S, Sham; IIR, intestinal ischaemia
reperfusion; DM, diabetic mice. (C) The difference in the ratio of
LC3BII/I expression between the two group (DIIR group compared with
the DS group vs. the IIR group compared with the Sham group).
#P<0.05, as indicated. Group D1, difference between
the IIR group and the Sham group; Group D2, difference between the
DIIR group and the DS group. |
Protein expression of PINK1/Parkin is
upregulated following IIR in the mouse model of diabetes
The protein expression levels of PINK1/Parkin were
investigated following treatment with IIR by western blotting. The
results demonstrated that PINK1/Parkin expression levels were
increased in the DIIR and IIR groups compared with the DS and Sham
groups, respectively (P<0.05; Fig.
5B). Furthermore, the increase in expression in the DIIR group
compared with the IIR group was more pronounced compared with the
increase in expression in the DS group compared with the Sham group
(Fig. 5B).
Discussion
In the present study, the results suggested that
diabetes enhanced intestinal damage following IIR, decreased
intestinal SOD and GSH-Px activity, and intensified the systemic
inflammatory response in mice. Of note, intestinal damage was
accompanied by an increase in mitochondrial autophagy in the
intestinal tissues, and diabetes further increased IIR-induced
increases in PINK1 and Parkin expression, as well as the ratio of
LC3BII/I.
Intestinal tissue is sensitive to inflammation due
to its anatomical structure, which possesses a large surface area
and strong permeability (33). Due
to high oxygen demand, the intestine is an ischemia-reperfusion
injury sensitive organ. The intestinal mucosal response to ischemia
can be divided into two phases: i) During hypoxia, hypoxia causes
death of intestinal mucosa cells and severe mucosal damage; and ii)
during reperfusion, re-oxidation further damages viable mucosal
cells (34). The primary indicator
of ischemic injury of the intestinal mucosa is degeneration of the
villous epithelium and desquamation of the epithelial layer. During
reperfusion, the villi and mucous membranes are severely damaged
due to the production of ROS (34). The main mechanism that triggers
ischemia-reperfusion damage is oxidative stress (35). During ischemia-reperfusion injury,
oxidative stress is followed by inflammatory cell activation,
production of systemic inflammatory mediators, increased bacterial
translocation, release of bacterial products including endotoxins,
activation of the systemic inflammatory response cascade and
multiple organ failure (36). In a
mouse model of diabetes, the mitochondria are the major source of
ROS in the cell, but the mitochondria are also the main target of
oxidative damage (37). Damaged
mitochondria further increase ROS production via ROS-induced ROS
release (22), resulting in
leakage of lethal factors such as IL-1β and IL-6, which triggers a
vicious cycle to induce cell death (23,24).
In addition, due to elevated blood glucose, osmotic dehydration is
often observed in the mouse model of diabetes, which leads to
increases in intestinal permeability, an impaired normal mucosal
barrier of the small intestine and increased intestinal damage
(3). Therefore, it was
hypothesized that diabetes aggravates inflammatory reactions and
oxidative stress, leading to catastrophic damage.
In the present study, the severity of pathological
intestinal tissue damage was evaluated by H&E staining combined
with Chiu's score analysis. The results suggested that the
intestinal mucosal structure, epithelial cell layer and lamina
propria were severely destroyed, and the Chiu's score was
significantly higher in the IIR groups compared with the
corresponding control groups. The results indicated that IIR injury
led to catastrophic damage to the intestinal tissue. The degree of
intestinal mucosal damage in the DS and DIIR groups was more
extensive compared with the Sham and IIR groups, respectively.
The mechanism that promotes the onset of IIR injury
is multifactorial, complex and highly integrated. A number of
biochemical pathways, including necrosis, apoptosis and autophagy,
interact with one another (38,39).
In the present study, inflammation was significantly increased by
upregulated expression levels of proinflammatory cytokines,
including IL-1β, IL-6 and TNF-α, in the DIIR group compared with
the DS group. Following IIR in diabetic and non-diabetic mice, the
level of MDA content increased, whereas the levels of SOD and
GPH-Px activity decreased, suggesting that oxidative stress levels
were significantly increased for all the experimental mice;
however, the effects were more pronounced for the diabetic groups
compared with the non-diabetic groups. The results indicated that
following IIR, diabetes further increased inflammation and
oxidative stress levels, and exacerbated intestinal tissue
damage.
Furthermore, previous studies have reported that
autophagy is closely related to inflammation and oxidative stress
(40,41). Zeng et al (42) demonstrated that the autophagy
agonist rapamycin may reduce apoptosis by inhibiting excessive
inflammatory responses and oxidative stress, thereby exerting a
protective effect on intestinal tissue. Autophagy serves dual roles
in a number of diseases via adaptive or maladaptive regulation.
Physiological autophagy serves as a protective mechanism to
maintain normal function; however, excessive autophagy contributes
to disease development (43). In
the present study, the western blotting results indicated that the
extent of autophagy was significantly increased in the DIIR group
compared with the IIR group. Furthermore, TEM experiments revealed
that the mitochondrial structure of the DS group was notably
damaged following IIR, and the basic outlines of the mitochondria
became less clearly defined. Compared with the non-diabetic groups,
mitochondrial destruction was more prevalent in the diabetic
groups. The results of the western blot analysis revealed that the
ratio of LC3BII/I was significantly higher in the diabetic groups
compared with the non-diabetic groups, and these differences were
more pronounced following IIR.
Autophagy is a key mechanism for maintaining normal
cell function and stabilizing the internal environment for
self-protection (44). Autophagy
removes aging and damaged cells, and helps to eliminate pathogens,
indicating an important role during normal development and in
response to environmental stimuli (44). The mitochondrial kinase PINK1 and
the E3-ubiquitin ligase Parkin, two Parkinson's disease-related
proteins, function as the centers of mitochondrial quality control
(45). PINK1 identifies damaged
mitochondria and targets their degradation specifically via
mitophagy (25). Following PINK1
accumulation on defective mitochondria, Parkin translocation from
the cytosol is induced to mediate the clearance of damaged
mitochondria by mitochondrial autophagy (46).
Increasing evidence has demonstrated that the
activity of PINK1 is not restricted to mitophagy, but that
different subcellular and submitochondrial pools of PINK1 are
involved in distinct signaling cascades for the regulation of cell
metabolism and survival (47). At
the metabolic level, loss of PINK1 led to a significant inhibition
of glucose uptake by pancreatic β-cells and primary intact islets,
and was accompanied by increased insulin secretion and enhanced
glucose tolerance (48). On this
basis, the protein expression levels of PINK1 and Parkin were
investigated in the present study. The results suggested that the
expression of PINK1 and Parkin was significantly increased
following IIR, and the observed increase was more pronounced in the
diabetic groups compared with the non-diabetic groups. The results
of the present study demonstrated that diabetes exacerbated the
vulnerability to IIR, which was accompanied by alterations to
autophagy. PINK1/Parkin may serve an important regulatory role
during autophagy. Further studies are required to clarify the
mechanisms underlying increased susceptibility to
high-glucose-induced IIR by examining the effects of upregulation
or inhibition of autophagy.
At present, intestinal ischemia is impossible to
prevent and studies investigating IIR damage have been unsuccessful
in translating the results of basic research into effective
clinical prevention and treatment strategies. With the dramatic
increase in patients with diabetes, a higher probability of
undergoing surgery and suffering from IIR injury during the
perioperative period has been observed (49–51).
Further investigation into the pathological mechanisms underlying
diabetes in IIR may help to identify novel preventative and
therapeutic strategies, thereby improving survival.
To conclude, the present study indicated that
diabetes aggravated intestinal damage following IIR in mice, and
the increased vulnerability to IIR-induced intestinal damage due to
diabetes may be related to PINK1/Parkin-regulated mitochondrial
autophagy. However, the present study did not consider the
detection of ROS using DCFH-DA; therefore, future studies employing
this technique are required.
Acknowledgements
The authors would like to thank Dr Le Liu (Renmin
Hospital of Wuhan University) for providing technical support for
electron microscopy.
Funding
This study was supported by the National Natural
Science Foundation of China (grant nos. 81671948 and 81401574).
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
QTM and ZYX designed the study. ZZ, YYZ, RC and TS
performed the experiments. WL and HML provided vital reagents and
analytical tools, edited and revised the manuscript. ZZ, WL, YYZ
and QS performed the data analysis. ZZ, WL and HML wrote the
manuscript. ZZ, HML and QTM prepared the illustrations and
proofread the manuscript. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
The present study was approved by the Animal Care
Committee of Renmin Hospital of Wuhan University and protocols were
followed in accordance with the National Institutes of Health
guidelines for the care and use of experimental animals.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Miki T, Itoh T, Sunaga D and Miura T:
Effects of diabetes on myocardial infarct size and cardioprotection
by preconditioning and postconditioning. Cardiovasc Diabetol.
11:672012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Giacco F and Brownlee M: Oxidative stress
and diabetic complications. Circ Res. 107:1058–1070. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
D'Addio F and Fiorina P: Type 1 diabetes
and dysfunctional intestinal homeostasis. Trends Endocrinol Metab.
27:493–503. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Nankervis CA, Giannone PJ and Reber KM:
The neonatal intestinal vasculature: Contributing factors to
necrotizing enterocolitis. Semin Perinatol. 32:83–91. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Yasuhara H: Acute mesenteric ischemia: The
challenge of gastroenterology. Surg Today. 35:185–195. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Schwartz MZ: Novel therapies for the
management of short bowel syndrome in children. Pediatr Surg Int.
29:967–974. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Posma LA, Bleichrodt RP, Lomme RM, de Man
BM, van Goor H and Hendriks T: Early anastomotic repair in the rat
intestine is affected by transient preoperative mesenteric
ischemia. J Gastrointest Surg. 13:1099–1106. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Corcos O and Nuzzo A: Gastro-intestinal
vascular emergencies. Best Pract Res Clin Gastroenterol.
27:709–725. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Fishman JE, Sheth SU, Levy G, Alli V, Lu
Q, Xu D, Qin Y, Qin X and Deitch EA: Intraluminal nonbacterial
intestinal components control gut and lung injury following
trauma-hemorrhagic shock. Ann Surg. 260:1112–1120. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Sastry P, Hardman G, Page A, Parker R,
Goddard M, Large S and Jenkins DP: Mesenteric ischaemia following
cardiac surgery: The influence of intraoperative perfusion
parameters. Interact Cardiovasc Thorac Surg. 19:419–424. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Hart ML, Gorzolla IC, Schittenhelm J,
Dalton JH and Eltzschig HK: Retraction: Hypoxia-inducible
factor-1α-dependent protection from intestinal ischemia/reperfusion
injury involves Ecto-5′-nucleotidase (CD73) and the A2B adenosine
receptor. J Immunol. 199:19422017. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Puleo F, Arvanitakis M, Van Gossum A and
Preiser JC: Gut failure in the ICU. Semin Respir Crit Care Med.
32:626–638. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zhao W, Gan X, Su G, Wanling G, Li S, Hei
Z, Yang C and Wang H: The interaction between oxidative stress and
mast cell activation plays a role in acute lung injuries induced by
intestinal ischemia-reperfusion. J Surg Res. 187:542–552. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Daniel RA, Cardoso VK, Góis JE Jr, Parra
RS, Garcia SB, Rocha JJ and Féres O: Effect of hyperbaric oxygen
therapy on the intestinal ischemia reperfusion injury. Acta Cir
Bras. 26:463–469. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Liu L, Tan Q, Hu B, Wu H, Wang C, Liu R
and Tang C: Somatostatin Improved B cells mature in macaques during
intestinal ischemia-reperfusion. PLoS One. 10:e01336922015.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wu H, Deng YY, Liu L, Tan QH, Wang CH, Guo
MM, Xie YM and Tang CW: Intestinal ischemia-reperfusion of macaques
triggers a strong innate immune response. World J Gastroenterol.
20:15327–15334. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Whittington HJ, Babu GG, Mocanu MM, Yellon
DM and Hausenloy DJ: The diabetic heart: Too sweet for its own
good? Cardiol Res Pract. 2012:8456982012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ansley DM and Baohua W: Oxidative stress
and myocardial injury in the diabetic heart. J Pathol. 229:232–241.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Takayanagi R, Inoguchi T and Ohnaka K:
Clinical and experimental evidence for oxidative stress as an
exacerbating factor of diabetes mellitus. J Clin Biochem Nutr.
48:72–77. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhang P, Li T, Wu X, Nice EC, Huang C and
Zhang Y: Oxidative stress and diabetes: Antioxidative strategies.
Front Med. Apr 4–2020.(Epub ahead of print). View Article : Google Scholar
|
|
21
|
Lenaz G, Bovina C, D'Aurelio M, Fato R,
Formiggini G, Genova ML, Giuliano G, Merlo Pich M, Paolucci U,
Parenti Castelli G and Ventura B: Role of mitochondria in oxidative
stress and aging. Ann NY Acad Sci. 959:199–213. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zorov DB, Juhaszova M and Sollott SJ:
Mitochondrial ROS-induced ROS release: An update and review.
Biochim Biophys Acta. 1757:509–517. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Tumurkhuu G, Koide N, Dagvadorj J,
Morikawa A, Hassan F, Islam S, Naiki Y, Mori I, Yoshida T and
Yokochi T: The mechanism of development of acute lung injury in
lethal endotoxic shock using alpha-galactosylceramide
sensitization. Clin Exp Immunol. 152:182–191. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Shen X, Zheng S, Metreveli NS and Epstein
PN: Protection of cardiac mitochondria by overexpression of MnSOD
reduces diabetic cardiomyopathy. Diabetes. 55:798–805. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Sack MN: Type 2 diabetes, mitochondrial
biology and the heart. J Mol Cell Cardiol. 46:842–849. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Brownlee M: Biochemistry and molecular
cell biology of diabetic complications. Nature. 414:813–820. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Wang S, Wang C, Yan F, Wang T, He Y, Li H,
Xia Z and Zhang Z: N-Acetylcysteine attenuates diabetic myocardial
ischemia reperfusion injury through inhibiting excessive autophagy.
Mediators Inflamm. 2017:92572912017. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
International C: Guide for the care and
use of laboratory animals. Publication. 327:963–965. 2011.
|
|
29
|
Meng QT, Cao C, Wu Y, Liu HM, Li W, Sun Q,
Chen R, Xiao YG, Tang LH, Jiang Y, et al: Ischemic
post-conditioning attenuates acute lung injury induced by
intestinal ischemia-reperfusion in mice: Role of Nrf2. Lab Invest.
96:1087–1104. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Yu C, Tan S, Zhou C, Zhu C, Kang X, Liu S,
Zhao S, Fan S, Yu Z, Peng A and Wang Z: Berberine reduces
uremia-associated intestinal mucosal barrier damage. Biol Pharm
Bull. 39:1787–1792. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Misra HP and Fridovich I: The role of
superoxide anion in the autoxidation of epinephrine and simple
assay for superoxide dismutase. J Biol Chem. 247:3170–3175.
1972.PubMed/NCBI
|
|
32
|
Crane JD, Devries MC, Safdar A, Hamadeh MJ
and Tarnopolsky MA: The effect of aging on human skeletal muscle
mitochondrial and intramyocellular lipid ultrastructure. J Gerontol
A Biol Sci Med Sci. 65:119–128. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Swank GM and Deitch EA: Role of the gut in
multiple organ failure: Bacterial translocation and permeability
changes. World J Surg. 20:411–417. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Gonzalez LM, Moeser AJ and Blikslager AT:
Animal models of ischemia-reperfusion-induced intestinal injury:
Progress and promise for translational research. Am J Physiol
Gastrointest Liver Physiol. 308:G63–G75. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Simos Y, Karkabounas S, Verginadis I,
Charalampidis P, Filiou D, Charalabopoulos K, Zioris I, Kalfakakou
V and Evangellou A: Intra-peritoneal application of catechins and
EGCG as in vivo inhibitors of ozone-induced oxidative stress.
Phytomedicine. 18:579–585. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Zu G, Guo J, Che N, Zhou T, Zhang X, Wang
G, Ji A and Tian X: Protective effects of ginsenoside Rg1 on
intestinal ischemia/reperfusion injury-induced oxidative stress and
apoptosis via activation of the Wnt/β-catenin pathway. Sci Rep.
6:384802016. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Nishikawa T, Edelstein D, Du XL, Yamagishi
S, Matsumura T, Kaneda Y, Yorek MA, Beebe D, Oates PJ, Hammes HP,
et al: Normalizing mitochondrial superoxide production blocks three
pathways of hyperglycaemic damage. Nature. 404:787–790. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Wen Z, Hou W, Wu W, Zhao Y, Dong X, Bai X,
Peng L and Song L: 6′-O-Galloylpaeoniflorin attenuates cerebral
ischemia reperfusion-induced neuroinflammation and oxidative stress
via PI3K/Akt/Nrf2 Activation. Oxid Med Cell Longev.
2018:86782672018. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Wu H, Ye M, Yang J and Ding J: Modulating
endoplasmic reticulum stress to alleviate myocardial ischemia and
reperfusion injury from basic research to clinical practice: A long
way to go. Int J Cardiol. 223:630–631. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Tsai YG, Wen YS, Wang JY, Yang KD, Sun HL,
Liou JH and Lin CY: Complement regulatory protein CD46 induces
autophagy against oxidative stress-mediated apoptosis in normal and
asthmatic airway epithelium. Sci Rep. 8:129732018. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Zhou M, Xu W, Wang J, Yan J, Shi Y, Zhang
C, Ge W, Wu J, Du P and Chen Y: Boosting mTOR-dependent autophagy
via upstream TLR4-MyD88-MAPK signalling and downstream NF-κB
pathway quenches intestinal inflammation and oxidative stress
injury. EBioMedicine. 35:345–360. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Zeng Z, Zhang YY, Tao S, et al: Protective
effects of rapamycin on intestinal injury after intestinal
ischemia-reperfusion in mouse. Med J Wuhan Univ. 40:192–196.
2019.
|
|
43
|
Jin S, Wei J, You L, Liu H and Qian W:
Autophagy regulation and its dual role in blood cancers: A novel
target for therapeutic development (Review). Oncol Rep.
39:2473–2481. 2018.PubMed/NCBI
|
|
44
|
Klionsky DJ and Emr SD: Autophagy as a
regulated pathway of cellular degradation. Science. 290:1717–1721.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Pickrell AM and Youle RJ: The Roles of
PINK1, Parkin, and mitochondrial fidelity in Parkinson's disease.
Neuron. 85:257–273. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Durcan TM and Fon EA: The three ‘P's of
mitophagy: PARKIN, PINK1, and post-translational modifications.
Genes Dev. 29:989–999. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Arena G and Valente EM: PINK1 in the
limelight: Multiple functions of an eclectic protein in human
health and disease. J Pathol. 241:251–263. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Deas E, Piipari K, Machhada A, Li A,
Gutierrez-del-Arroyo A, Withers DJ, Wood NW and Abramov AY: PINK1
deficiency in β-cells increases basal insulin secretion and
improves glucose tolerance in mice. Open Biol. 4:1400512014.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Rosenberg CS: Wound healing in the patient
with diabetes mellitus. Nurse Clin North Am. 25:247–253. 1990.
|
|
50
|
Scannel G, Waxman K, Vaziri ND, Zhang J,
Kaupke CJ, Jalali M and Hecht CC: Hypoxia-induced alterations of
neutrophil membrane receptors. J Surg Res. 59:141–145. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Thomaz Neto FJ, Koike MK, Abrahão Mde S,
Carillo Neto F, Pereira RK, Machado JL and Montero EF: Ischemic
preconditioning attenuates remote pulmonary inflammatory
infiltration of diabetic rats with an intestinal and hepatic
ischemia-reperfusion injury. Acta Cir Bras. 28:174–178. 2013.
View Article : Google Scholar : PubMed/NCBI
|