Introduction
China has a high incidence of diabetes mellitus,
with the number of diabetic patients exceeding 90 million (1). At present, ~300 million individuals
have been diagnosed with diabetes mellitus worldwide (2). Large-scale clinical studies have
reported that patients with diabetes have a significantly higher
risk of heart injury and cardiac insufficiency compared with those
without diabetes (3–7). Diabetic cardiomyopathy is defined as
a heart injury, which is independent of other diseases and is
caused by diabetes mellitus itself (8), and manifests as left ventricular
diastolic dysfunction at the early stage of onset and systolic
dysfunction at the later stage. If not treated properly, diabetic
cardiomyopathy can develop into heart failure, arrhythmia,
cardiogenic shock and even sudden death in severe cases (9).
There are multiple clinical strategies to prevent
and treat diabetic cardiomyopathy. However, the effect of reducing
the incidence and mortality of cardiovascular complications
requires improvement in patients with diabetes. Therefore, the
study of diabetic cardiomyopathy requires extensive investigation.
It is currently hypothesized that several mechanisms are involved
in the pathogenesis of diabetic cardiomyopathy, including changes
in myocardial energy metabolism and calcium signaling (10–13).
As an important secondary messenger, calcium plays
an important role in the excitation-contraction coupling of the
heart and regulates the expression of cardiac-related genes. The
role of Ca2+ signaling and its dependent signal
transduction pathway in cardiac hypertrophy and myocardial
apoptosis has been widely recognized by researchers.
Ca2+ is ubiquitous in cells and plays an important role
not only in the electrical activity of the heart but also as a
direct activator of myofilament contraction. Calcium enters the
cardiomyocytes through voltage-gated calcium channels which
selectively permeate calcium ions into the cell when there is a
change from a high voltage to a low voltage inside of the cell.
Voltage-gated calcium ion channels are subdivided into six
fundamental types according to their electrophysiology and
sensitivity to certain drugs and toxins. These voltage-gated
calcium ion channels are named L-, T-, N-, P-, Q- and R-channels:
L-channels have a long activation and high conductance, and are
mainly located in skeletal, cardiac and vascular muscle and are
involved in its contraction; T-channels have a transient opening
and are involved in calcium ion entry when the membrane is
depolarized; N-channels are involved in neurotransmitter release;
P/Q-channels are mainly localized in the nerve terminal of cells of
the cerebellum and are involved in neurotransmitter release; and
R-channels are mainly localized in cell bodies and are involved in
Ca2+-dependent action potentials (14).
In the myocardium, the L-type calcium channel is the
main pathway for calcium to enter the cell (15).
The role of the Ca2+-calcineurin
(CaN)-nuclear factor of activated T cells 3 (NFAT3) signaling
pathway in cardiac development has gained interest in recent years,
and several classes of antihypertensive drugs, including
angiotensin-converting enzyme inhibitors, angiotensin II receptor
blockers and calcium channel blockers (CCBs), have been
demonstrated to reverse hypertrophy in humans (16,17).
CCBs were introduced for the treatment of hypertension in the
1980s. Their use was subsequently expanded to disorders such as
angina pectoris, paroxysmal supraventricular tachycardia,
hypertrophic cardiomyopathy, coronary spasm and cerebral vasospasm
(18). Evidence from experimental
studies indicate that CCBs nifedipine, nisoldipine and amlodipine
additionally lead to regression of interstitial and perivascular
myocardial fibrosis, which may contribute to the improvement of
diastolic function and coronary reserve (19).
Norvasc is the brand name of amlodipine besylate.
Amlodipine does not increase cardiovascular morbidity or mortality
in patients with severe heart failure (20) and multiple studies have reported
that amlodipine can reduce cardiac remodeling in spontaneously
hypertensive rats and myocardial infarction in rats (21–23).
Therefore, Norvasc was selected for the current study to determine
whether it could inhibit hypertrophy of H9C2 cardiomyocytes induced
by high glucose.
The aim of the present study was to investigate high
glucose-induced hypertrophy of H9C2 cells and its possible
pathogenesis, from the perspective of the calcium signaling
pathway, to provide a theoretical basis for the identification of
potential targets for clinical prevention and treatment of this
disease.
Materials and methods
Materials
Cell culture reagents: FBS (cat. no. SH30087.01;
Hyclone; GE Healthcare Life Sciences); DMEM-high glucose culture
medium (50 mM; cat. no. 11965-092; Gibco; Thermo Fisher Scientific,
Inc.); DMEM-low glucose culture medium (5 mM; cat. no. 10567-014;
Gibco; Thermo Fisher Scientific, Inc.). H9C2 cells were rat
embryonic cardiomyocytes purchased from the Cell Resource Center of
Shanghai Academy of Life Sciences, Chinese Academy of Sciences;
Norvasc was obtained from Sigma-Aldrich (Merck KGaA); ELISA assay
kit of CaN (cat. no. E-EL-R0134c) was purchased from Elabscience
Biotechnology Co., Ltd.; Fluo-3 AM (calcium ion fluorescent probe;
cat. no. S1056) was from Beyotime Institute of Biotechnology; DNase
I (RNase-free) was purchased from Beijing Transgen Biotech Co.,
Ltd.; reverse transcription kit and real-time PCR kit were obtained
from Vazyme; CnAβ, nuclear factor of activated T cells 3 (NFAT3), β
type myosin heavy chain (β-MHC) and b-actin primary antibodies
(cat. nos. ab3673, ab66781, ab207926 and ab8227, respectively) were
all purchased from Abcam.
Cell culture
Following thawing, cells
(1×104−1×105/ml) were cultured in DMEM
containing 10% FBS in a 5% CO2 incubator, with saturated
humidity at 37°C.
Experimental grouping
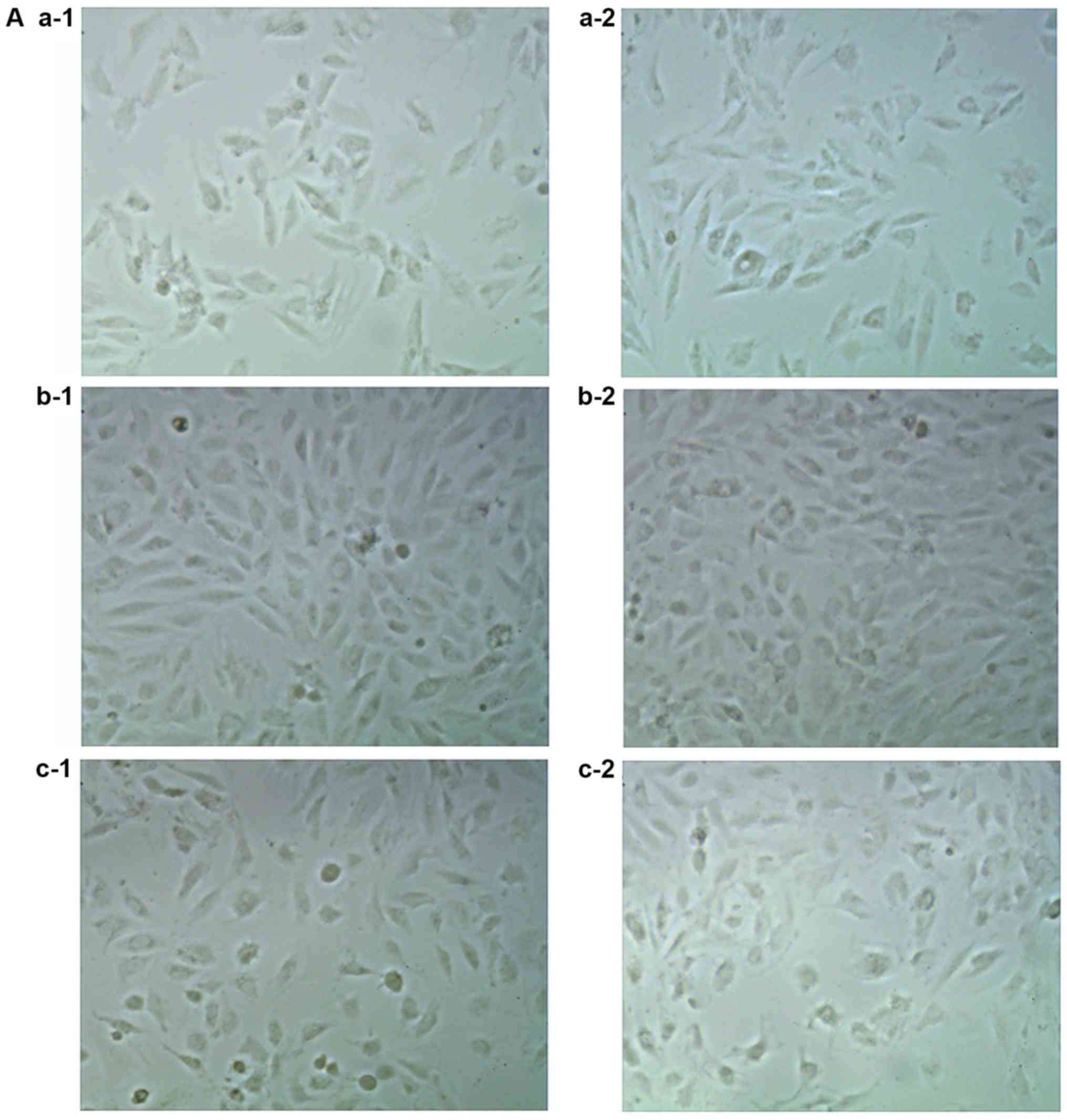
The experimental groups were as follows: i) A1, 48-h
culture group treated with 5 mM glucose; ii) B1, 48-h culture group
treated with 50 mM glucose; iii) C1, 48-h culture group treated
with 50 mM glucose + 25 nmol/l Norvasc; iv) A2, 72-h culture group
treated with 5 mM glucose; v) B2, 72-h culture group treated with
50 mM glucose; vi) C2, 72-h culture group treated with 50 mM
glucose + 25 nmol/l Norvasc. All cell treatments used DMEM
containing 10% FBS.
Determination of cell surface
area
Cell morphology were observed using an optical
microscope, and the cell surface area was measured by Image-Pro
Plus 6.1 software (Media Cybernetics, Inc.). A total of 3 fields of
view were observed at magnification ×20.
Determination of intracellular calcium
([Ca2+]i) activity
Cell culture media was removed and cells
(1×104−1×105/ml) were washed with PBS. Fluo-3
(2 µM), diluted in serum-free media was added for 1.5 h at
37°C, 5% CO2. Fluo-3 was removed, followed by two PBS
washes and the addition of cell culture media. Fluorescence
microscopy (magnification, ×20) was performed to detect
[Ca2+]i.
Determination of cellular CaN enzyme
activity
Enzyme activity of CaN was measured using a rat CaN
ELISA assay kit, according to the manufacturer's instructions.
Detection of CaN, NFAT3 and β-MHC mRNA
expression in myocardial cells by reverse
transcription-quantitative (RT-q)PCR
The extraction of RNA was achieved using
TRIzol® (Invitrogen; Thermo Fisher Scientific, Inc.).
Next, the reaction mixture was prepared with RNase-free DNase І,
followed by digestion for 30 min at 37°C and inactivation for 10
min at 65°C to remove DNA. Reverse transcription was conducted with
the addition of template RNA and primer mixtures at 42°C for 1 h,
then resting on ice for 2 min. This was followed by the addition of
cDNA (10X) to the primer reaction mixture. The RNA concentration
was measured with NanoDrop™ 1000 (Thermo Fisher Scientific, Inc.).
The cycling conditions for qPCR were as follows: 95°C for 30 sec,
40 cycles at 95°C for 10 sec followed by 60°C for 34 sec. The
melting curve was drawn within the temperature range of 60–95°C.
The primer sequences were: Rat-GAPDH forward (F),
5′-CATCAACGACCCCTTCATTG-3′ and reverse (R),
5′-GAAGATGGTGATGGGTTTCC-3′; rat-CaN F, 5′-ATGTTGCCTAGTGGAGTGTT-3′
and R, 5′-GGAGAGTATCCTCGTATTGCTT-3′; NFAT3 F,
5′-CCACAAGGCATTGAGACACAT−3′ and R, 5′-TCACCAGCAGCAGCAGCAG-3′; and
rat-β-MHC F, 5′-AATGAACACCGGAGCAAGG-3′; and R,
5′-CGGGTCAGCTGAGAGATAAGAGC-3′. mRNA levels were quantified by
relative quantification and analyzed using the 2−ΔΔCq
method (24).
Detection of CaN, NFAT3 and β-MHC
protein expression in myocardial cells by western blotting
Culture plates of H9C2 cells
(1×104−1×105/ml) were washed with pre-cooled
PBS and treated with pre-cooled lysis buffer (CWBio) on ice for 30
min. The lysis products were centrifuged for 20 min at 8,300 × g at
4°C to collect the supernatant. Furthermore, 2 µl supernatant was
used to determine the protein concentration by using a BCA protein
quantitative kit. Protein samples (50 µg/well) were separated by
SDS-PAGE on a 10% gel. Electrophoresis was performed for 20 min at
a constant voltage of 100 V, and for 40 min at a constant voltage
of 140 V. Subsequently, the proteins were transferred to a PVDF
membrane, followed by three TBS washes. Following the removal of
TBS, the membranes were blocked using TBS with Tween-20 (TBST)
buffer (20 mM Tris-HCl, 150 mM NaCl and 0.1% Tween-20) containing
5% non-fat milk for 2 h at room temperature. The membrane was then
incubated with TBST buffer (20 mM Tris-HCl, 150 mM NaCl and 0.1%
Tween-20) containing 5% non-fat milk with the following primary
antibodies, polyclonal rabbit anti-human CaN (1:1,000), polyclonal
rabbit anti-human NFAT3 (1:1,000), polyclonal rabbit anti-human
β-MHC (1:1,000) and polyclonal rabbit anti-human β-actin, overnight
at 4°C. Following three washes with 1X PBS, the PVDF membrane was
incubated with a horseradish peroxide-conjugated anti-rabbit
secondary antibodies (1:10,000; cat. no. 111-035-003; Jackson
ImmunoResearch Laboratories, Inc.) at room temperature for 2 h. The
PVDF membrane was washed five times with PBS (15 min each), and
developed using an ECL kit (Beijing ComWin Biotech Co., Ltd.).
Statistical analysis
GraphPad Prism 7 (GraphPad Software, Inc.) was used
for statistical analysis. Data were analyzed with two-way ANOVA
adjusted for multiple comparisons. Multiple comparisons between the
groups was performed using Bonferroni tests. The bilateral
inspection level for α was 0.05, and P<0.05 was considered to
indicate a statistically significant difference.
Results
Cell swelling analysis
The cell size in B1 and B2 groups was increased
compared with cells in A1 and A2 groups after 48 and 72 h of
culture under different conditions. Cell size was decreased
following the addition of Norvasc compared with those without the
addition of Norvasc under the same conditions. There was no
significant difference between the C1 and C2 and A1 and A2 groups,
respectively. Furthermore, there was no significant difference in
cell size between the A1 and A2 groups, B1 and B2 groups, and C1
and C2 groups, respectively. The statistical analysis chart is
presented in Fig. 1.
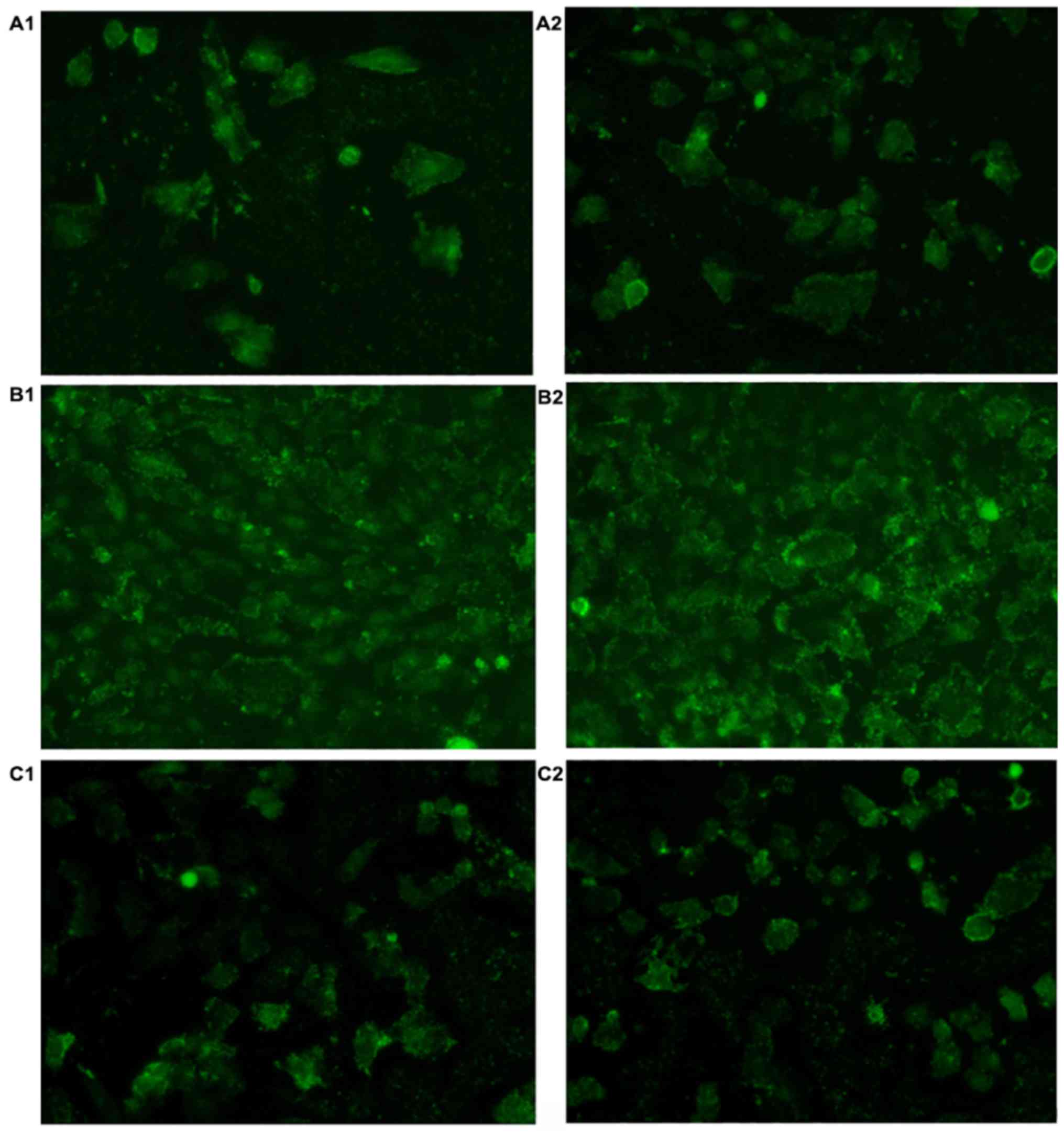
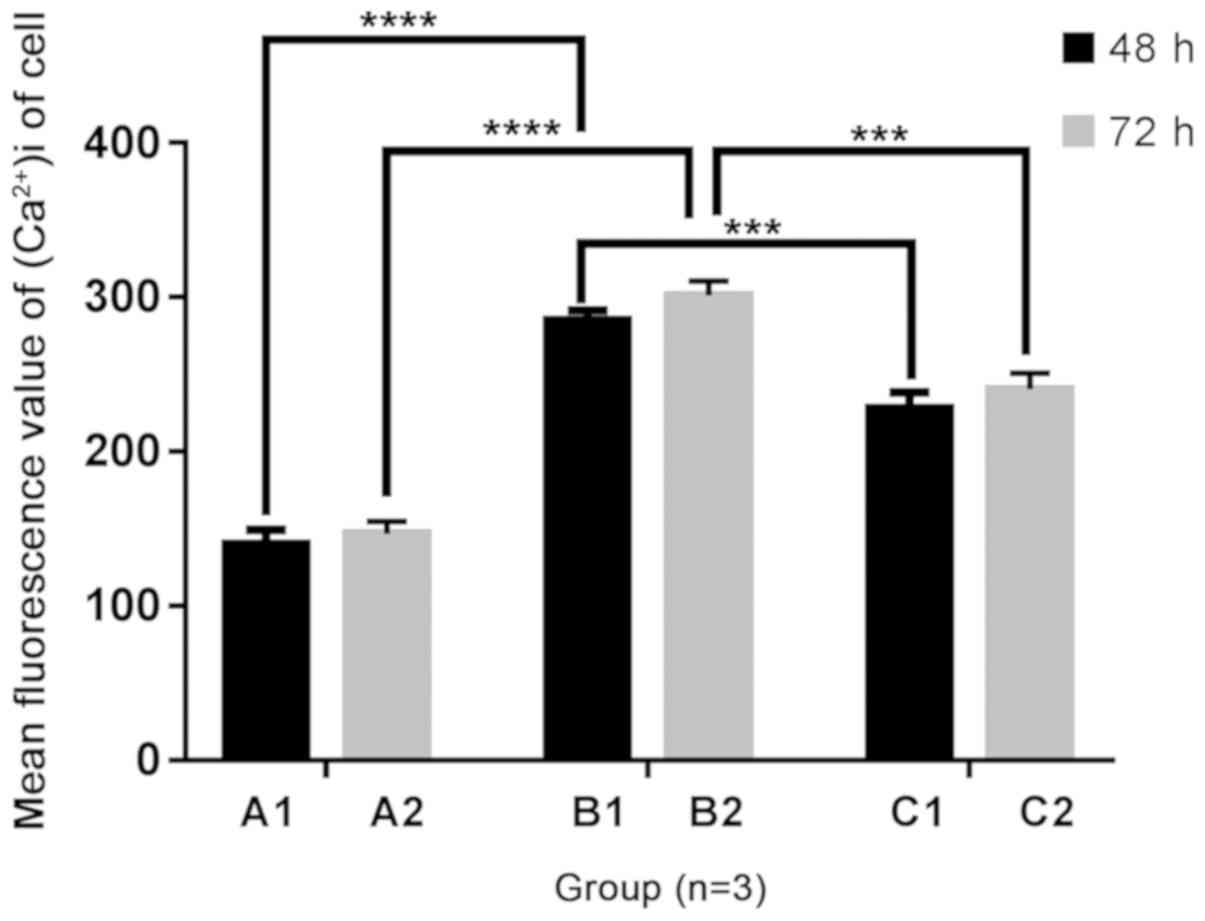
Determination of [Ca2+]i
activity in each group
Following 48 and 72 h of culture, the fluorescent
values of [Ca2+]i activity in B1, B2, C1 and C2 groups
were increased compared with the A1 and A2 groups, respectively.
Following the addition of Norvasc, the fluorescent value of
[Ca2+]i activity in each group was notably lower than
that without Norvasc under the same conditions. In addition, no
obvious difference was found in the fluorescent values of
intracellular [Ca2+]i activity between the A1 and A2
groups, B1 and B2 groups, and C1 and C2 groups, respectively.
Images of fluorescent staining are presented in Fig. 2. The statistical analysis chart is
presented in Fig. 3.
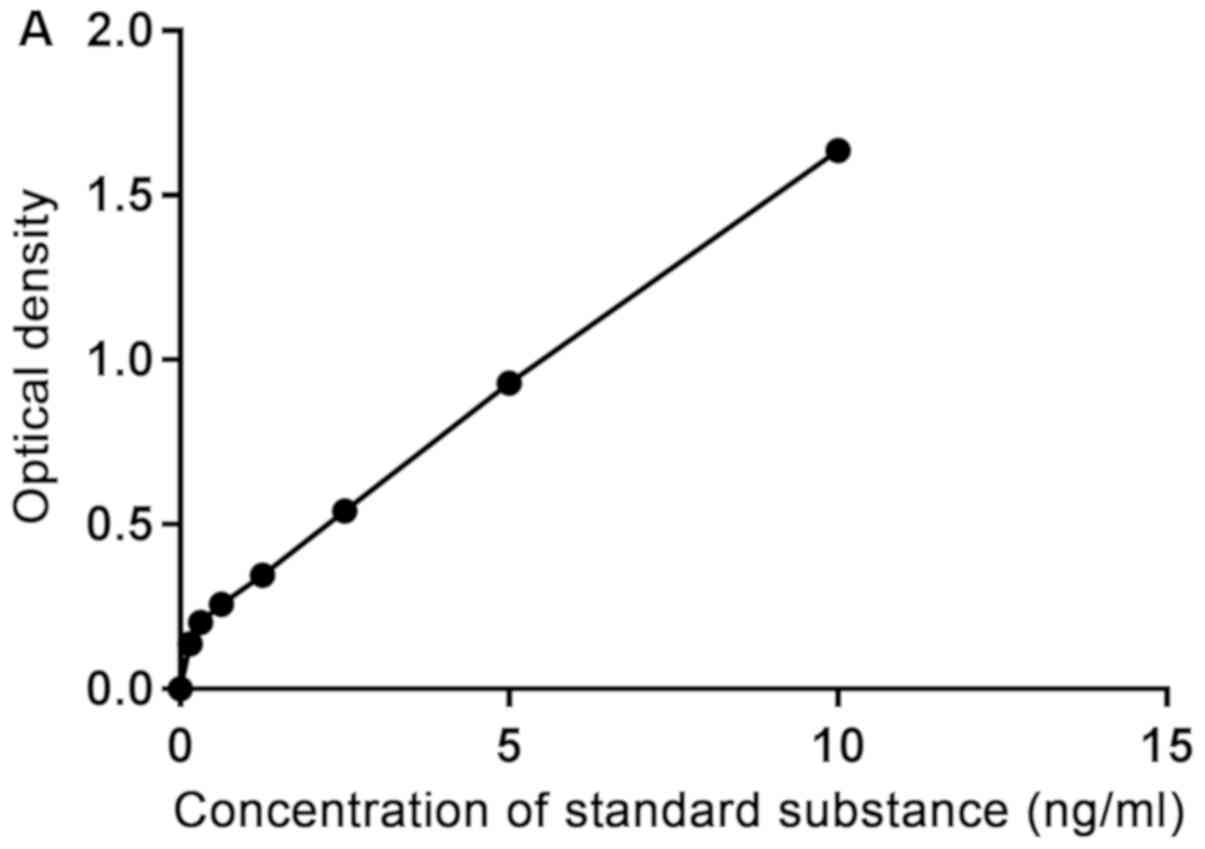
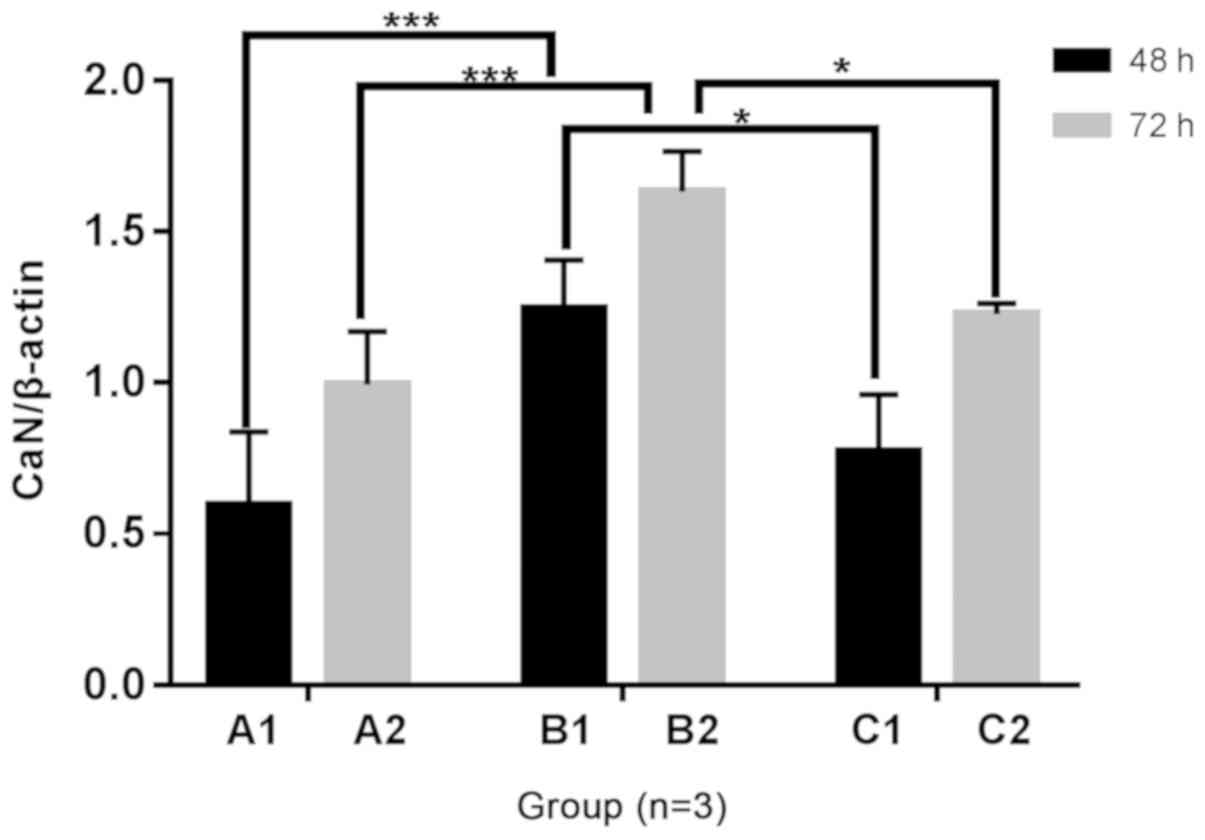
Cellular CaN enzyme activity
The CaN concentration was increased in the B1 and B2
groups compared with the A1 and A2 groups, following 48 and 72 h of
culture, respectively. With the addition of Norvasc, the
concentration of CaN in each group was significantly lower than
that without Norvasc. The concentration standard curve is presented
in Fig. 4A and the statistical
analysis chart is presented in Fig.
4B.
CaN, NFAT3 and β-MHC mRNA expression
levels of H9C2 cells determined by RT-qPCR
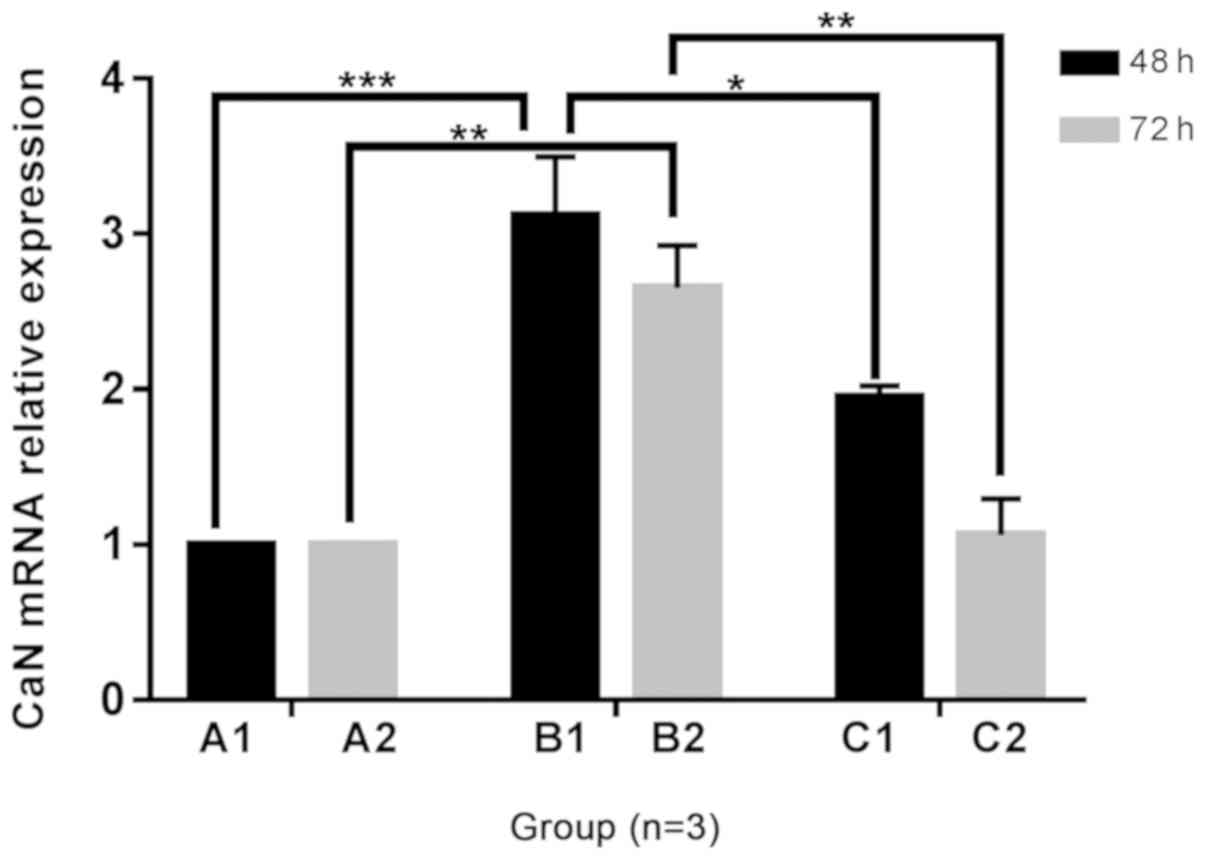
Changes in CaN mRNA expression in H9C2 cells
The mRNA expression of CaN in B1 and B2 groups was
increased compared with that in the A1 and A2 groups, following 48
and 72 h of culture, respectively. Furthermore, the mRNA expression
level of CaN was lower with the addition of Norvasc than that
without Norvasc. The mRNA expression of CaN in the C1 and C2 groups
had no significant difference between the A1 and A2 groups
respectively. The statistical analysis chart is presented in
Fig. 5.
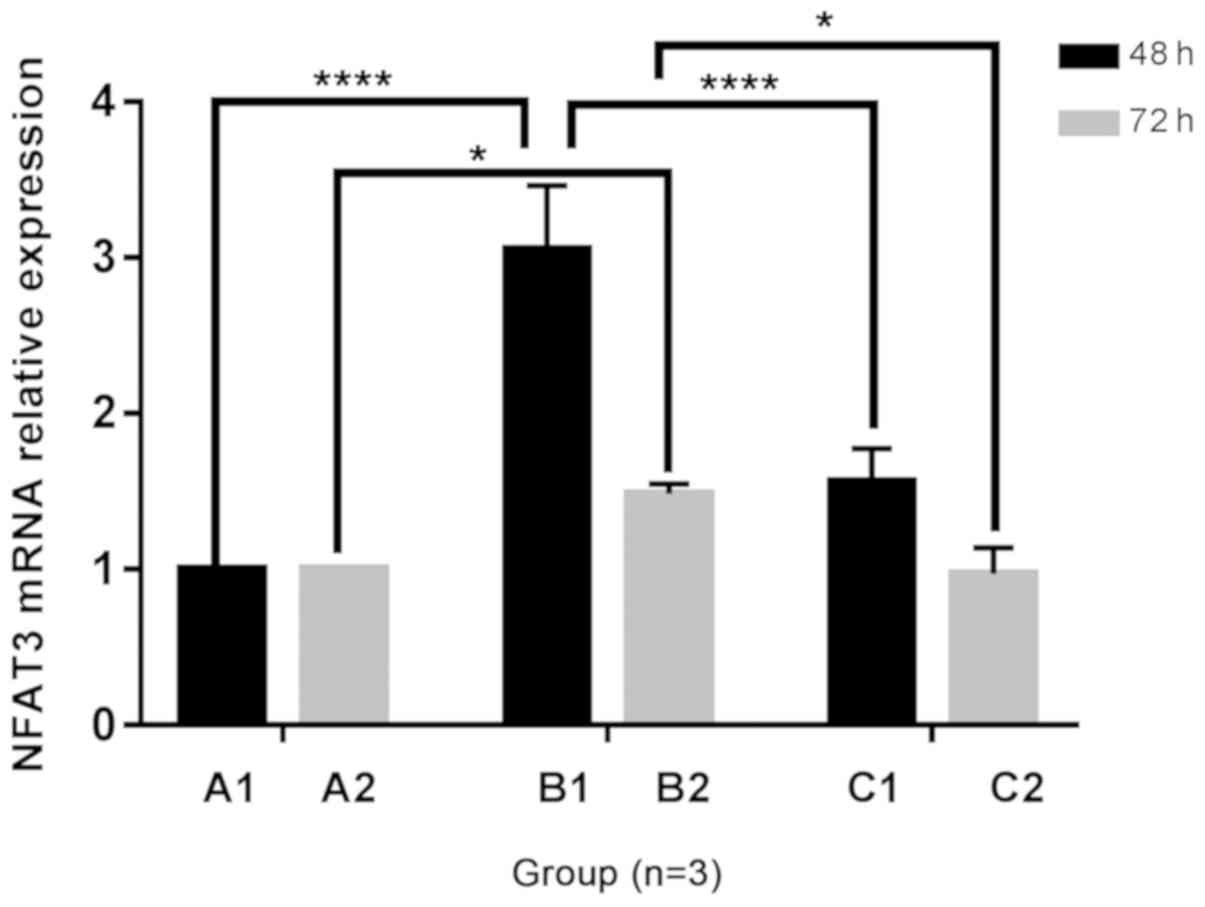
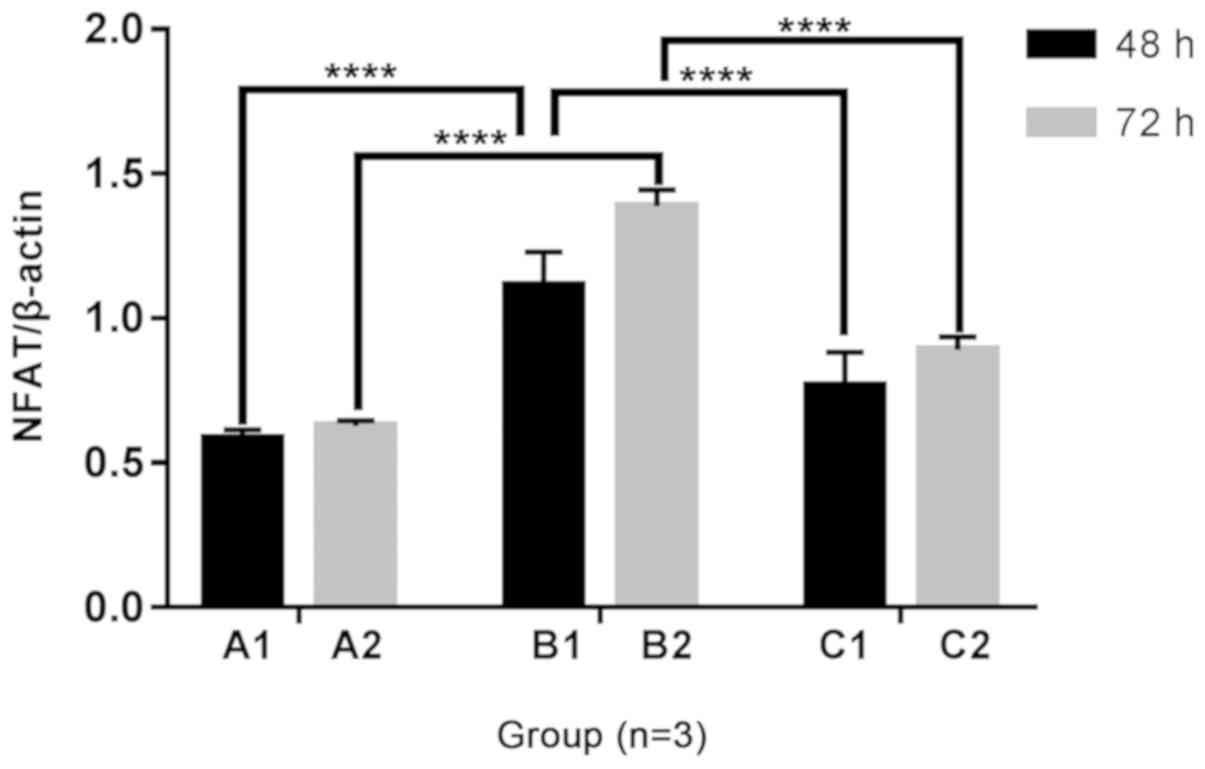
Changes in NFAT3 mRNA expression in H9C2
cells
Following 48 and 72 h of culture, the mRNA
expression of NFAT3 in the B1 and B2 groups was increased compared
with the A1 and A2 groups, and expression in the C1 group was
increased compared with the A1 group. The addition of Norvasc
resulted in decreased NFAT3 mRNA expression than that without
Norvasc. Additionally, no significant difference was observed in
the mRNA expression level of NFAT3 in the C2 groups compared with
that in the A2 groups. The statistical analysis chart is presented
in Fig. 6.
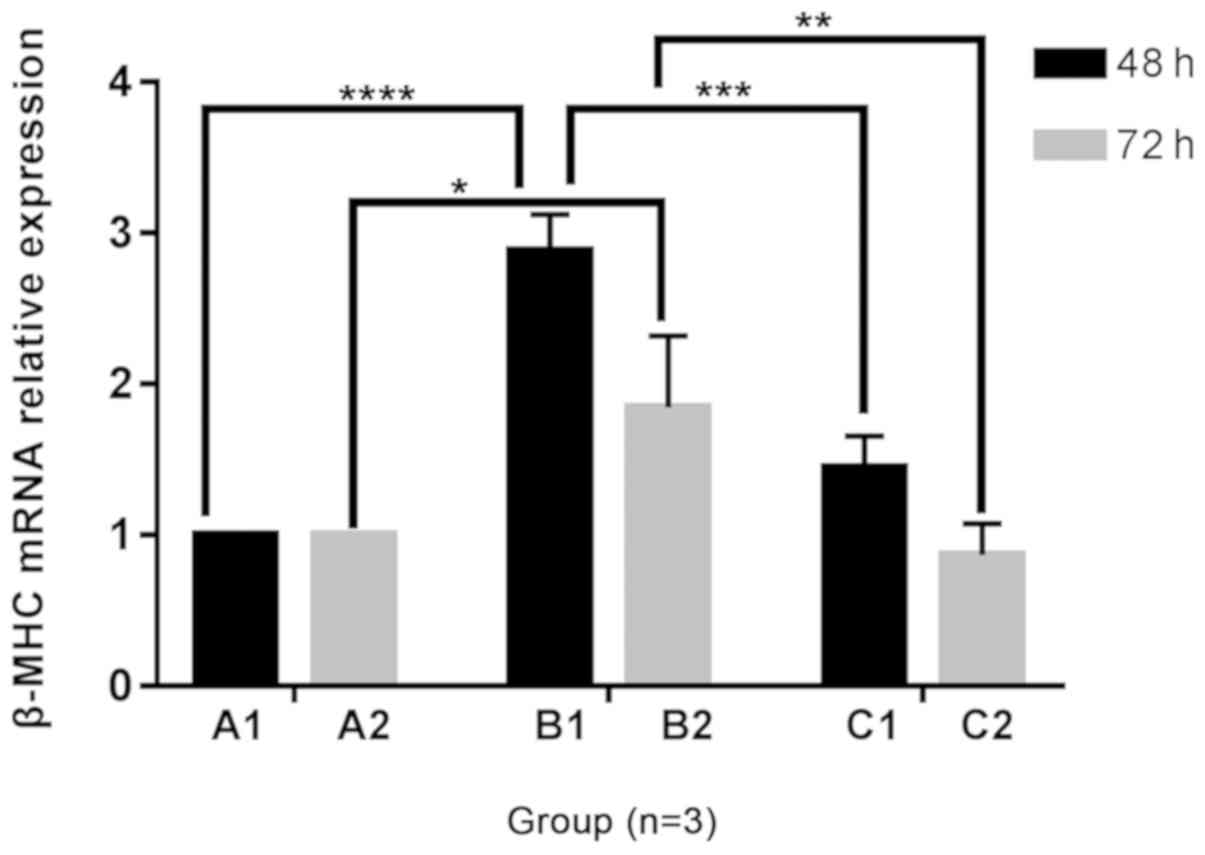
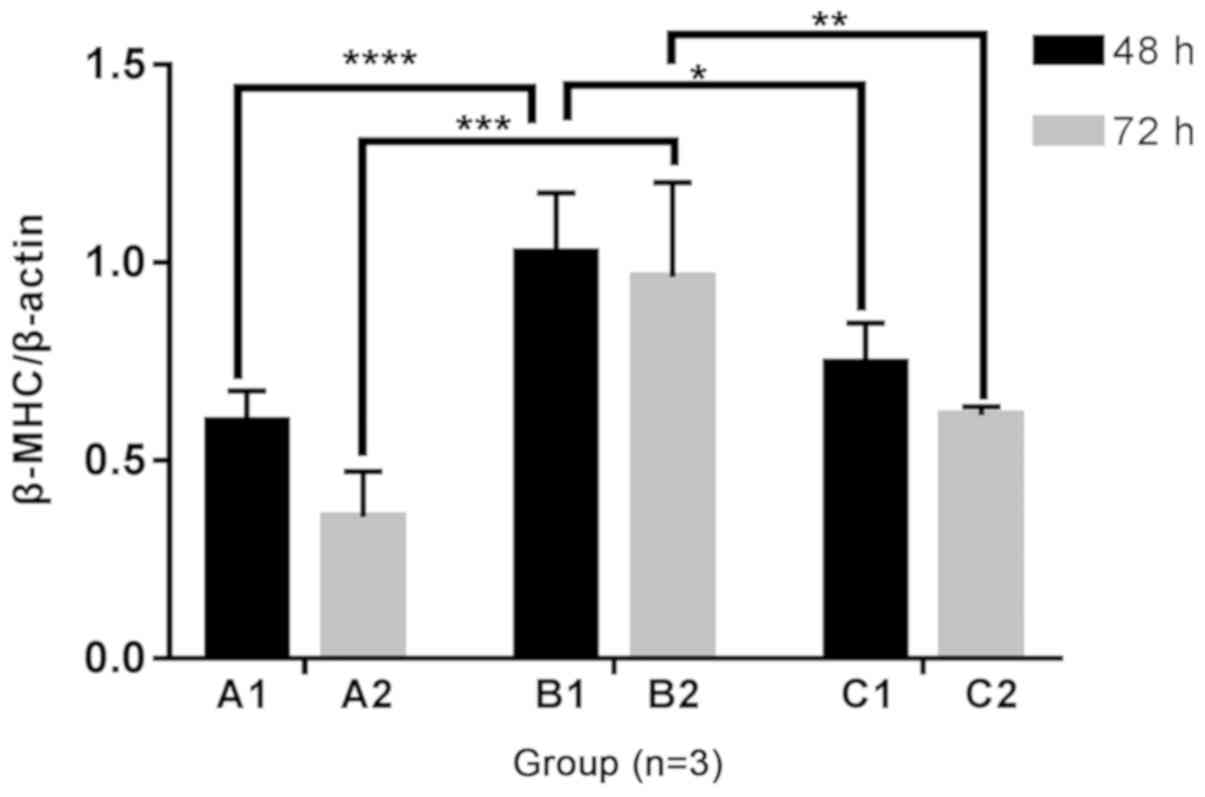
Changes in β-MHC mRNA expression in H9C2
cells
A trend towards increased β-MHC mRNA expression was
observed in B1 and B2 groups compared with the A1 and A2 groups
following 48 and 72 h of culture, under different culture
conditions. Following the addition of Norvasc, the mRNA expression
of β-MHC was lower than that without Norvasc under the same
conditions. The mRNA expression level of β-MHC in groups C1 and C2
were without any significant difference between groups A1 and A2,
respectively. Furthermore, there was no statistical difference
between the A1 and A2 groups or C1 and C2 groups. With the
prolongation of culture time, a trend towards decreased β-MHC mRNA
expression was observed in the B2 group compared with the B1 group.
The statistical analysis chart is shown in Fig. 7.
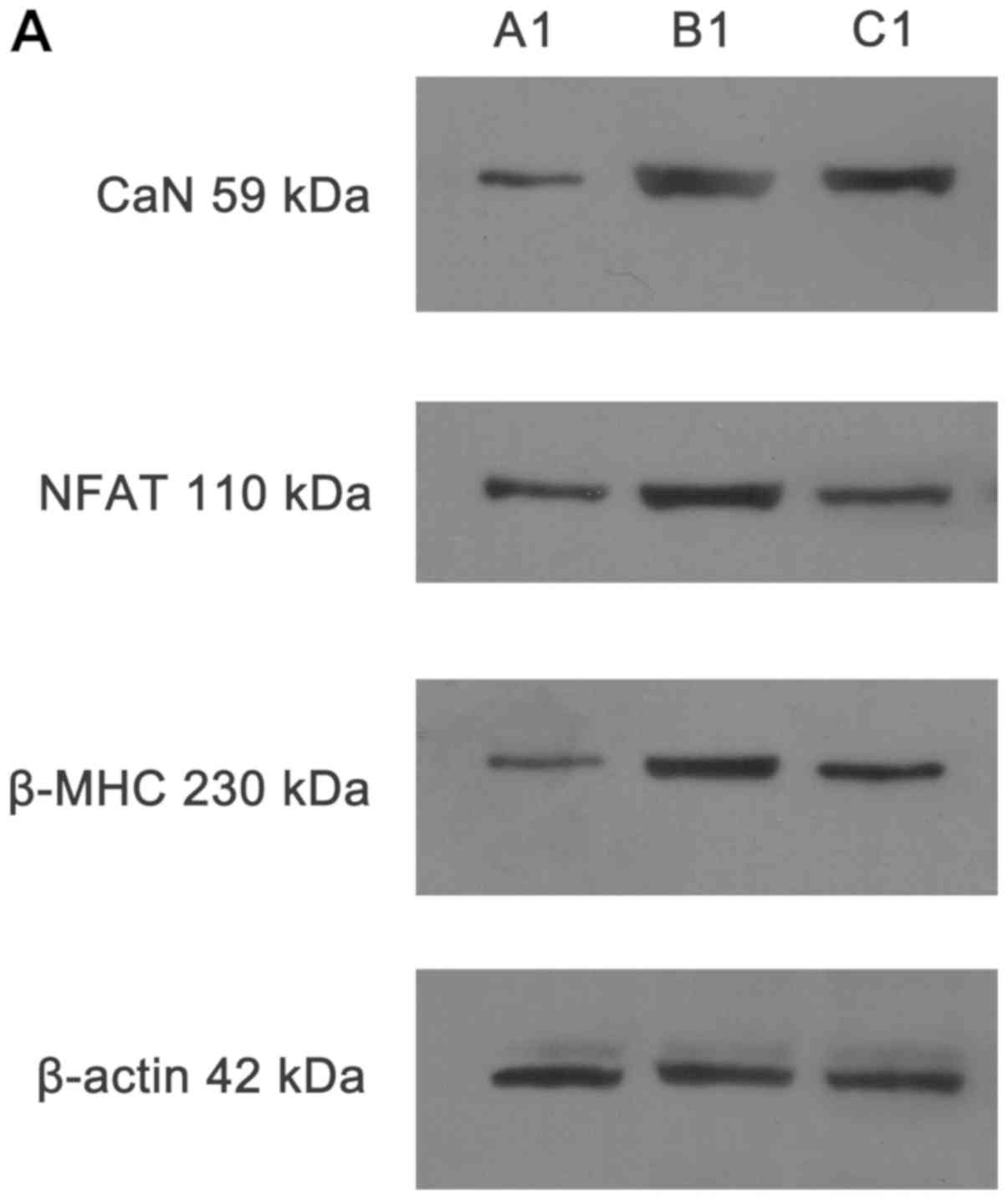
Detection of CaN, NFAT3 and β-MHC protein
expression in H9C2 cells by western blotting
Representative western blotting images are presented
in Fig. 8.
Detection of CaN protein expression in H9C2 cells
by western blotting
The protein expression of CaN in B1 and B2 groups
was increased compared with the A1 and A2 groups, respectively,
following 48 and 72 h of culture under different culture
conditions. Protein expression of CaN was significantly decreased
in groups C1 and C2 compared with the B1 and B2 groups,
respectively, following the addition of Norvasc. There was no
significant difference between the C1 and C2 groups and A1 and A2
groups, respectively. The statistical analysis chart is presented
in Fig. 9.
Detection of NFAT3 protein expression in H9C2
cells by western blotting
The protein expression of NFAT3 was increased in the
B1 and B2 groups compared with that of the A1 and A2 groups,
respectively, following 48 and 72 h of culture under different
culture conditions. However, the protein expression of NFAT3 was
decreased with the addition of Norvasc compared to that without
Norvasc, under the same conditions. The NFAT3 protein expression
was also increased in the C1 and C2 groups compared with that in
the A1 and A2 groups. The statistical analysis chart is presented
in Fig. 10.
Detection of β-MHC protein expression in H9C2
cells by western blotting
The trend of increased protein expression of β-MHC
was observed in the B1 and B2 groups compared with that in the A1
and A2 groups, respectively, following 48 and 72 h of culture under
different culture conditions. The addition of Norvasc resulted in
decreased β-MHC protein expression compared with that without the
addition of Norvasc, under the same conditions. Additionally, the
expression of β-MHC protein in the C1 and C2 groups was not
significantly different with that in the A1 and A2 groups. There
was no significant difference in the protein expression of β-MHC in
group A and B and C with the prolongation of culture time. The
statistical analysis chart is presented in Fig. 11.
Discussion
The World Health Organization predicts that 300
million individuals worldwide will be diagnosed with diabetes
mellitus by 2025 (25). Patients
with diabetes are prone to cardiovascular diseases, and the
probability of myocardial infarction is 10 times higher than that
of non-diabetic patients (26,27).
First proposed by Rubler et al (28) in 1972, diabetic cardiomyopathy is a
type of heart disease that is independent of coronary heart
disease, valve disease and hypertensive heart disease, and is
mainly characterized by diastolic dysfunction in the early stage
and systolic dysfunction at the late stage. The pathogenesis is
complex, with the main pathological changes including myocardial
inflammation, metabolic disorders, myocardial cell apoptosis and
fibrosis (29,30). The duration of hyperglycemia serves
as an important indicator in determining the severity of heart
failure caused by diabetic cardiomyopathy (31). Therefore, hyperglycemia is studied
as an independent risk factor of myocardial injury caused by
diabetic cardiomyopathy (32,33).
High glucose will stimulate cardiac hypertrophy in a
variety of ways (34–37). In the present study, it was found
that high glucose could lead to an increase in the average
individual volume of H9C2 cells. Research investigating the
aberrant molecular processes that occur during cardiac hypertrophy
has used primary cardiomyocytes from neonatal rat hearts as the
standard experimental in vitro system. In addition, some
studies have made use of the H9C2 rat cardiomyoblast cell line
(38,39), as it was found by Watkins et
al (40) that the H9C2 cell
line and primary neonatal cardiomyocyte cells exhibit similar
hypertrophic responses in vitro. Therefore, the H9C2 cell
line was selected for the present study, which also has the
advantage of being an animal-free alternative. However, this is
also a limitation of the present study because primary neonatal
cardiomyocyte cells were not used. The role of the
Ca2+-CaN-NFAT3 signaling pathway in cardiac development
has become a research area of interest in recent years. The
activation of Ca2+-dependent CaN A subunit (CnA) has
been frequently observed in human heart hypertrophy and heart
failure (41,42). In mice, increased intracellular
calcium is known to activate CnA, which can bind and
dephosphorylate the NFAT transcription factor family of activated
T-nuclear factors. Subsequently, NFAT is transferred from the
cytoplasm to the nucleus, activating the specific expression of
various genes associated with cardiac hypertrophy, such as atrial
natriuretic factor, brain natriuretic peptide (BNP), and β-MHC in
the heart. This leads to increased nucleic acid synthesis of
myocardial cell protein and increased cardiac cell volume, and
finally cardiac hypertrophy (43,44).
Transgenic mice overexpressing a consistently active form of
CnA-specific cardiomyocytes, MHC-CnA, have been found to suffer
from cardiac hypertrophy at 18 days following birth, with varying
degrees of progression to heart failure and sudden death (45).
Several different studies have found that the
Ca2+-CaN-NFAT3 signaling pathway plays a role in cardiac
hypertrophy. Daskoulidou et al (46) found that increased Orai 1
expression could mediate an increase in the calcium current of the
store-operated calcium entry channel, in order to regulate calcium
in cardiac cells, which may be induced by hyperglycemia through the
activation of the CaN/NFATc3 signaling pathway. Somvanshi et
al (47) revealed that the
activation of the somatostatin receptor 2 (SSTR2) not only
inhibited the expression and activity of CaN phosphatase, but also
hindered the dephosphorylation of NFAT and nuclear translocation,
which provided evidence that SSTR2 could protect the heart by
regulating the Ca2+-associated signaling pathway,
leading to cardiac hypertrophy. The T-type calcium channel, Cav3.2
could be induced by Egr1 (early growth response 1), which is
released at the early stage of myocardial hypertrophy due to the
early pressure overload, to regulate cardiac hypertrophy through
the CaN phosphatase-NFAT signaling pathway (48). A previous study also revealed that
the activity of the CaN signaling pathway may be activated by the
bacteria Porphyromonas gingivalis, and further lead to
cardiomyocyte hypertrophy and cell death of cultured H9C2
cardiomyocytes (49). The
Ca2+-CaN-NFAT3 pathway is considered to be the crucial
mechanism in mediating the development of cardiac hypertrophy and
therefore, the role of the Ca2+-CaN-NFAT3 signaling
pathway in hyperglycemia-induced myocardial hypertrophy was
investigated in the present study. Following the determination of
[Ca2+]i and mRNA and protein expression levels of CaN,
NFAT and β-MHC, it was revealed that high glucose could induce an
increase in [Ca2+]i and CaN concentration. Furthermore,
high glucose increased the mRNA and protein levels of CaN, NFAT and
β-MHC, and in combination with the cell morphology data, suggest
the involvement of the Ca2+-CaN-NFAT signaling pathway
in hyperglycemia-induced H9C2 cells.
The activation of CaN mainly depends on the increase
in intracellular Ca2+, and its activity is regulated by
the change in intracellular Ca2+ concentration (50). The continuous maintenance of NFAT
in the nucleus requires a continuous and oscillating increase in
calcium (51) in order to maintain
the activated form of CaN phosphatase. This CaN-dependent signal is
sensitive to the inhibition of calcium channel blockers, such as
verapamil, or CaN inhibitors, such as cyclosporine A (CSA) and
tacrolimus (FK-506) (52–54). L-type Ca2+ channels are
widely distributed in the heart, smooth and skeletal muscles
(55,56). In the heart and smooth muscle
cells, the L-type Ca2+ channels are responsible for the
inward movement of Ca2+, thereby causing contraction. In
skeletal muscles, these channels act as voltage sensors in
excitation-contraction coupling. L-type calcium channels consist of
several subunits, namely, α1, α2, β, γ and δ (14). The α1 subunit consists of four
homologous structural domains, and contains transmembrane pores,
through which calcium ions can be obtained, as well as calcium
antagonist binding sites adjacent to the pores, which are connected
to each other and to the calcium channel via allosteric junctions
(57,58).
The L-type calcium channel is considered to be the
main source of calcium that can activate CaN-NFAT3 signal
transduction (59). Thus, L-type
calcium channel inhibitors have become a research area of interest
and an important therapeutic target for cardiac hypertrophy.
Cardiac remodeling, which involves structural and functional
changes at the molecular, cellular, tissue and whole-organ levels,
can be used to determine the clinical course of heart failure
(60). A number of studies have
reported that amlodipine can reduce cardiac remodeling in
spontaneously hypertensive rats and myocardial infarction in rats
(61,62). Valsartan monotherapy as well as
combination therapy with valsartan and either amlodipine or
cilnidipine similarly attenuated hypertension and left ventricular
hypertrophy (LVH) in hypertensive rats (22). Compared with amlodipine alone,
amlodipine combined with atorvastatin had a greater beneficial
effect on the myocardial hypertrophy. These benefits may be
associated with the cumulative effect of the drug on inhibiting
NADPH oxidase-mediated reactive oxygen species (63). Amlodipine has also been found to
improve cardiomyocyte hypertrophy by inhibiting the phosphorylation
of the epithelial growth factor receptor (64). Losartan, amlodipine, and
particularly fosinopril can inhibit myocardial cell apoptosis,
prevent myocardial fibrosis, and reverse cardiac hypertrophy; the
inhibition of the cardiac renin-angiotensin-aldosterone system may
be the mechanism of the cardioprotective effects of these three
drugs (65). Meo et al
(66) reported a decrease in
L-type calcium current in cardiomyocytes in a glycosuria mouse
model, induced by streptozotocin. In addition to the traditional
L-type calcium channel, it is generally considered that Orai
1-mediated calcium store-operated calcium channel also participates
in the calcium regulation of cardiomyocytes. In a diabetic model,
Orai 1 expression was increased in both cardiomyocytes and smooth
muscle cells (46,67). Amlodipine sulfonate is an L-type
calcium channel blocker, which can selectively inhibit the
transmembrane domain to inhibit calcium ions from entering
cardiomyocytes. Several studies have demonstrated that amlodipine
sulfonate could inhibit CaN-NFAT3 and thus inhibit cardiac
hypertrophy (68,69). Therefore, the L-type calcium
channel amlodipine bensulfonate, Norvasc, was selected to
investigate whether intracellular influx of Ca2+ was
observed in H9C2 cells cultured with high glucose, in order to
induce changes in intracellular calcium concentration and the
activation of CaN, thereby activating the signaling pathway, and
ultimately causing cardiomyocyte hypertrophy.
The main purpose of the present study was to
investigate whether Norvasc could induce the recovery of
cardiomyocyte hypertrophy and inhibit Ca2+-CaN-NFAT3
signaling. Following the addition of Norvasc, the activity of
[Ca2+]i, CaN concentration, CaN mRNA and protein
expression in the corresponding groups were all significantly
decreased, which indicated that Norvasc reduced CaN expression.
Thus, intracellular calcium could activate CaN and promote its
expression. The present study demonstrated that calcium channel
inhibitors could alleviate cell hypertrophy and are more effective
in inhibiting cell hypertrophy following prolonged action.
Bugyei-Twum et al (70)
also found that high glucose in vitro induced the activation
of Smad in H9C2 cells and promoted cardiac fibrosis and hypertrophy
through transcriptional coregulator p300, and Chen et al
(71) revealed that lercanidipine
may ameliorate cardiomyocyte hypertrophy, partially by blocking
Cn-NFAT3 and CaMKII-HDAC4 signaling. These findings are consistent
with the present study.
The signal transduction mechanism of myocardial
hypertrophy is complex. In addition to Ca2+-CaN-NFAT3
signaling pathway, there may be other signaling pathways, which may
have interactions with CaN-NFAT3. Thus, further studies are
required, including the use of specific inhibitors, such as CSA, to
identify other pathways. Liu et al (72) demonstrated that lipopolysaccharide
(LPS) treatment leads to myocardial hypertrophy via the
calcineurin/NFAT-3 signaling pathway in H9C2 cells. They further
provided a link between the LPS-induced inflammatory response and
the calcineurin/NFAT-3 signaling pathway that mediates the
development of cardiac hypertrophy. LPS treatment was found to
significantly promote the activation and nuclear translocation of
NFAT3 and mediated the development of cardiac hypertrophy as a
transcription factor. The cell size, actin fiber, atrial
natriuretic peptide and BNP levels were assessed following the use
of ERK1/2 inhibitors, p38 MAPK inhibitors, JNK 1/2 inhibitors, CaN
inhibitors and NF-KB inhibitors, which demonstrated that only the
CaN inhibitor could significantly inhibit NFAT3 nuclear
localization. The present study provides findings that can be used
to determine subsequent future studies.
In summary, the present experimental design and
results suggest that, the calcium channel inhibitor Norvasc may
inhibit hyperglycemia in H9C2 cells, which may result from the
activation of the Ca2+-CaN-NFAT3 signaling pathway by
high glucose levels.
Acknowledgements
The authors would like to thank Dr Shasha Han
(Department of Pediatrics, The First Clinical Medical College of
Jinan University, Guangzhou, China) for her technical support.
Funding
The present study was supported by NSFC (grant nos.
81741083, 81801492).
Availability of data and materials
The datasets used and/or analyzed are available from
the corresponding author on reasonable request
Authors' contributions
XX, LR, XT, FP and GL made substantial contributions
to conception and design of the study. XX, LR, XT, FP and CY
provided resources, contributed to research and validation of the
results, interpreted data and wrote the original draft preparation.
XX, LR, FP and GL reviewed and edited the manuscript. XX, LR and GL
supervised the study. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Chan JC, Malik V, Jia W, Kadowaki T,
Yajnik CS, Yoon KH and Hu FB: Diabetes in Asia: Epidemiology, risk
factors, and pathophysiology. JAMA. 301:2129–2140. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Guariguata L, Whiting DR, Hambleton I,
Beagley J, Linnenkamp U and Shaw JE: Global estimates of diabetes
prevalence for 2013 and projections for 2035. Diabetes Res Clin
Pract. 103:137–149. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Lamblin N, Fertin M, De Groote P and
Bauters C: Cardiac remodeling and heart failure after a first
anterior myocardial infarction in patients with diabetes mellitus.
J Cardiovasc Med (Hagerstown). 13:353–359. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Tochiya M, Makino H, Tamanaha T, Matsuo M,
Hishida A, Koezuka R, Ohata Y, Tomita T, Son C, Miyamoto Y, et al:
Effect of tofogliflozin on cardiac and vascular endothelial
function in patients with type 2 diabetes and heart diseases: A
pilot study. J Diabetes Investig. 11:400–404. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Sardu C, Barbieri M, Santamaria M,
Giordano V, Sacra C, Paolisso P, Spirito A, Marfella R, Paolisso G
and Rizzo MR: Multipolar pacing by cardiac resynchronization
therapy with a defibrillators treatment in type 2 diabetes mellitus
failing heart patients: Impact on responders rate, and clinical
outcomes. Cardiovasc Diabetol. 16:752017. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kannel WB and McGee DL: Diabetes and
cardiovascular disease: The Framingham study. JAMA. 241:2035–2038.
1979. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lind M, Bounias I, Olsson M,
Gudbjörnsdottir S, Svensson AM and Rosengren A: Glycaemic control
and incidence of heart failure in 20,985 patients with type 1
diabetes: An observational study. Lancet. 378:140–146. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Bayeva M, Sawicki KT and Ardehali H:
Taking diabetes to heart-deregulation of myocardial lipid
metabolism in diabetic cardiomyopathy. J Am Heart Assoc.
2:e4332013. View Article : Google Scholar
|
|
9
|
Xu Y, Wang L, He J, Bi Y, Li M, Wang T,
Wang L, Jiang Y, Dai M, Lu J, et al: Prevalence and control of
diabetes in Chinese adults. JAMA. 310:948–959. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kim HW, Ch YS, Lee HR, Park SY and Kim YH:
Diabetic alterations in cardiac sarcoplasmic reticulum
Ca2+-ATPase and phospholamban protein expression. Life
Sci. 70:367–379. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Cai L, Li W, Wang G, Guo L, Jiang Y and
Kang YJ: Hyperglycemia-induced apoptosis in mouse myocardium:
Mitochondrial cytochrome C-mediated caspase-3 activation pathway.
Diabetes. 51:1938–1948. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Way KJ, Isshiki K, Suzuma K, Yokota T,
Zvagelsky D, Schoen FJ, Sandusky GE, Pechous PA, Vlahos CJ,
Wakasaki H and King GL: Expression of connective tissue growth
factor is increased in injured myocardium associated with protein
kinase C beta2 activation and diabetes. Diabetes. 51:2709–2718.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Candido R, Forbes JM, Thomas MC, Thallas
V, Dean RG, Burns WC, Tikellis C, Ritchie RH, Twigg SM, Cooper ME
and Burrell LM: A breaker of advanced glycation end products
attenuates diabetes-induced myocardial structural changes. Circ
Res. 92:785–792. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Catterall WA: Structure and regulation of
voltage-gated Ca2+ channels. Annu Rev Cell Dev Biol.
16:521–555. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Dolphin AC: Voltage-gated calcium
channels: Their discovery, function and importance as drug targets.
Brain Neurosci Adv. 2:23982128187948052018. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Klingbeil AU, Schneider M, Martus P,
Messerli FH and Schmieder RE: A meta-analysis of the effects of
treatment on left ventricular mass in essential hypertension. Am J
Med. 115:41–46. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Fagard RH, Celis H, Thijs L and Wouters S:
Regression of left ventricular mass by antihypertensive treatment:
A meta-analysis of randomized comparative studies. Hypertension.
54:1084–1091. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Puscas L, Gilau L, Coltau M, Pasca R,
Domuta G, Baican M and Hecht A: Calcium channel blockers reduce
blood pressure in part by inhibiting vascular smooth muscle
carbonic anhydrase I. Cardiovasc Drug Ther. 14:523–528. 2000.
View Article : Google Scholar
|
|
19
|
Bangalore S, Fakheri R, Toklu B and
Messerli FH: Diabetes mellitus as a compelling indication for use
of renin angiotensin system blockers: Systematic review and
meta-analysis of randomized trials. BMJ. 352:i4382016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Packer M, O'Connor CM, Ghali JK, Pressler
ML, Carson PE, Belkin RN, Miller AB, Neuberg GW, Frid D, Wertheimer
JH, et al: Effect of amlodipine on morbidity and mortality in
severe chronic heart failure. Prospective randomized amlodipine
survival evaluation study group. N Engl J Med. 335:1107–1114. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Lu J, Hao J, Du H, Xiao B, Li Y, Yang X
and Cui W: Amlodipine and atorvastatin improved hypertensive
cardiac remodeling through regulation of MMPs/TIMPs in SHR RATS.
Cell Physiol Biochem. 39:47–60. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Nagasawa K, Takahashi K, Matsuura N,
Takatsu M, Hattori T, Watanabe S, Harada E, Niinuma K, Murohara T
and Nagata K: Comparative effects of valsartan in combination with
cilnidipine or amlodipine on cardiac remodeling and diastolic
dysfunction in Dahl salt-sensitive rats. Hypertens Res. 38:39–47.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Whittaker P, Zhang HP and Kloner RA:
Biphasic survival response to amlodipine after myocardial
infarction in rats: Association with cardiac vascular remodeling.
Cardiovasc Pathol. 2:85–93. 2000. View Article : Google Scholar
|
|
24
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta CT) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Verkhratsky A and Fernyhough P:
Mitochondrial malfunction and Ca2+ dyshomeostasis drive
neuronal pathology in diabetes. Cell Calcium. 1:112–122. 2008.
View Article : Google Scholar
|
|
26
|
Jouven X, Lemaître RN, Rea TD, Sotoodehnia
N, Empana JP and Siscovick DS: Diabetes, glucose level, and risk of
sudden cardiac death. Eur Heart J. 26:2142–2147. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Yeung EH, Pankow JS, Astor BC, Powe NR,
Saudek CD and Kao WH: Increased risk of type 2 diabetes from a
family history of coronary heart disease and type 2 diabetes.
Diabetes Care. 30:154–156. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Rubler S, Dlugash J, Yuceoglu YZ, Kumral
T, Branwood AW and Grishman A: New type of cardiomyopathy
associated with diabetic glomerulosclerosis. Am J Cardiol.
30:595–602. 1972. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Bergner DW and Goldberger JJ: Diabetes
mellitus and sudden cardiac death: What are the data? Cardiol J.
17:117–129. 2010.PubMed/NCBI
|
|
30
|
Aharinejad S, Andrukhova O, Lucas T,
Zuckermann A, Wieselthaler G, Wolner E and Grimm M: Programmed cell
death in idiopathic dilated cardiomyopathy is mediated by
suppression of the apoptosis inhibitor apollon. Ann Thorac Surg.
86:109–114. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Fang ZY, Schull-Meade R, Leano R, Mottram
PM, Prins JB and Marwick TH: Screening for heart disease in
diabetic subjects. Am Heart J. 149:349–354. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Boyer JK, Thanigaraj S, Schechtman KB and
Pérez JE: Prevalence of ventricular diastolic dysfunction in
asymptomatic, normotensive patients with diabetes mellitus. Am J
Cardiol. 93:870–875. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Boudina S and Abel ED: Diabetic
cardiomyopathy, causes and effects. Rev Endocr Metab Disord.
11:31–39. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Kanai M, Otsuka Y, Otsuka K, Sato M,
Nishimura T, Mori Y, Kawaguchi M, Hatano E, Kodama Y, Matsumoto S,
et al: A phase I study investigating the safety and
pharmacokinetics of highly bioavailable curcumin (Theracurmin) in
cancer patients. Cancer Chemother Pharmacol. 71:1521–1530. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Sasaki H, Sunagawa Y, Takahashi K,
Imaizumi A, Fukuda H, Hashimoto T, Wada H, Katanasaka Y, Kakeya H,
Fujita M, et al: Innovative preparation of curcumin for improved
oral bioavailability. Biol Pharm Bull. 34:660–665. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
van Heerebeek L, Hamdani N, Handoko ML,
Falcao-Pires I, Musters RJ, Kupreishvili K, Ijsselmuiden AJ,
Schalkwijk CG, Bronzwaer JG, Diamant M, et al: Diastolic stiffness
of the failing diabetic heart: Importance of fibrosis, advanced
glycation end products, and myocyte resting tension. Circulation.
117:43–51. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Gonzalez-Quesada C, Cavalera M, Biernacka
A, Kong P, Lee DW, Saxena A, Frunza O, Dobaczewski M, Shinde A and
Frangogiannis NG: Thrombospondin-1 induction in the diabetic
myocardium stabilizes the cardiac matrix in addition to promoting
vascular rarefaction through angiopoietin-2 upregulation. Circ Res.
113:1331–1344. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Li MD, Cheng WP, Shi MX, Ge TD, Zheng XL,
Wu DY, Hu XY, Luo JC, Li FL and Li H: Role of tRNA selenocysteine 1
associated protein 1 in the proliferation and apoptosis of
cardiomyocyte-like H9c2 cells. Mol Med Rep. 15:988–994. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Saito M, Sakiyama K, Shiota T and Ito M:
Isoproterenol produces a rapid increase in sialidase activity in
rat heart tissue and cardiomyocyte-derived H9c2 cells in culture.
FEBS Lett. 542:105–108. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Watkins SJ, Borthwick GM and Arthur HM:
The H9C2 cell line and primary neonatal cardiomyocyte cells show
similar hypertrophic responses in vitro. In Vitro
Cell Dev Biol Anim. 47:125–131. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Diedrichs H, Chi M, Boelck B, Mehlhorn U
and Schwinger RH: Increased regulatory activity of the
calcineurin/NFAT pathway in human heart failure. Eur J Heart Fail.
6:3–09. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Haq S, Choukroun G, Lim H, Tymitz KM, del
Monte F, Gwathmey J, Grazette L, Michael A, Hajjar R, Force T and
Molkentin JD: Differential activation of signal transduction
pathways in human hearts with hypertrophy versus advanced heart
failure. Circulation. 103:670–677. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Zhu SJ, Yang YJ, Yu LJ and Huang L:
CaN-NFAT3 signal pathway: A crucial hinge relates Ca2+
signal with cardiomyocyte hypertrophy. Zhonghua Nei Ke Za Zhi.
43:19–21. 2004.(In Chinese). PubMed/NCBI
|
|
44
|
Wilkins BJ, Dai YS, Bueno OF, Parsons SA,
Xu J, Plank DM, Jones F, Kimball TR and Molkentin JD:
Calcineurin/NFAT coupling participates in pathological, but not
physiological, cardiac hypertrophy. Circ Res. 94:110–118. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Molkentin JD, Lu JR, Antos CL, Markham B,
Richardson J, Robbins J, Grant SR and Olson EN: A
calcineurin-dependent transcriptional pathway for cardiac
hypertrophy. Cell. 93:215–228. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Daskoulidou N, Zeng B, Berglund LM, Jiang
H, Chen GL, Kotova O, Bhandari S, Ayoola J, Griffin S, Atkin SL, et
al: High glucose enhances store-operated calcium entry by
upregulating ORAI/STIM via calcineurin-NFAT signalling. J Mol Med
(Berl). 93:511–521. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Somvanshi RK, Zou S, Qiu X and Kumar U:
Somatostatin receptor-2 negatively regulates β-adrenergic receptor
mediated Ca(2+) dependent signaling pathways in H9c2
cells. Biochim Biophys Acta. 1843:735–745. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Hsu SC, Chang YT and Chen CC: Early growth
response 1 is an early signal inducing Cav3.2 T-type calcium
channels during cardiac hypertrophy. Cardiovasc Res. 100:222–230.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Lee SD, Kuo WW, Lin DY, Chen TH, Kuo WH,
Hsu HH, Chen JZ, Liu JY, Yeh YL and Huang CY: Role of calcineurin
in Porphyromonas gingivalis-induced myocardial cell hypertrophy and
apoptosis. J Biomed Sci. 13:251–260. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Strack S, Wadzinski BE and Ebner FF:
Localization of the calcium/calmodulin-dependent protein
phosphatase, calcineurin, in the hindbrain and spinal cord of the
rat. J Comp Neurol. 375:66–76. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Smedler E and Uhlén P: Frequency decoding
of calcium oscillations. Biochim Biophys Acta. 1840:964–969. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Fric J, Zelante T, Wong AY, Mertes A, Yu
HB and Ricciardi-Castagnoli P: NFAT control of innate immunity.
Blood. 120:1380–1389. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Sommerer C, Meuer S, Zeier M and Giese T:
Calcineurin inhibitors and NFAT-regulated gene expression. Clin
Chim Acta. 413:1379–1386. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Maguire O, Tornatore KM, O'loughlin KL,
Venuto RC and Minderman H: Nuclear translocation of nuclear factor
of activated T cells (NFAT) as a quantitative pharmacodynamic
parameter for tacrolimus. Cytometry A. 83:1096–1104. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Hess P: Calcium channels in vertebrate
cells. Annu Rev Neurosci. 13:337–356. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Rampe D and Triggle DJ: New synthetic
ligands for L-type voltage-gated calcium channels. Prog Drug Res.
40:191–238. 1993.PubMed/NCBI
|
|
57
|
Katz AM: Calcium channel diversity in the
cardiovascular system. J Am Coll Cardiol. 28:522–529. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Triggle DJ: Calcium-channel antagonists:
Mechanisms of action, vascular selectivities, and clinical
relevance. Cleve Clin J Med. 59:617–627. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Wild AR, Sinnen BL, Dittmer PJ, Kennedy
MJ, Sather WA and Dell'Acqua ML: Synapse-to-nucleus communication
through NFAT is mediated by L-type Ca2+ channel
Ca2+ spike propagation to the Soma. Cell Rep.
26:3537–3550.e4. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Cohn JN, Ferrari R and Sharpe N: Cardiac
remodeling-concepts and clinical implications: A consensus paper
from an international forum on cardiac remodeling. Behalf of an
international forum on cardiac remodeling. J Am Coll Cardiol.
35:569–582. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Yamazaki T, Komuro I, Zou Y, Kudoh S,
Shiojima I, Mizuno T, Hiroi Y, Nagai R and Yazaki Y: Efficient
inhibition of the development of cardiac remodeling by a
long-acting calcium antagonist amlodipine. Hypertension. 31:32–38.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Sandmann S, Claas R, Cleutjens JP, Daemen
MJ and Unger T: Calcium channel blockade limits cardiac remodeling
and improves cardiac function in myocardial infarction-induced
heart failure in rats. J Cardiovasc Pharmacol. 37:64–77. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Lu JC, Cui W, Zhang HL, Liu F, Han M, Liu
DM, Yin HN, Zhang K and Du J: Additive beneficial effects of
amlodipine and atorvastatin in reversing advanced cardiac
hypertrophy in elderly spontaneously hypertensive rats. Clin Exp
Pharmacol Physiol. 36:1110–1119. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Liao Y, Asakura M, Takashima S, Kato H,
Asano Y, Shintani Y, Minamino T, Tomoike H, Hori M and Kitakaze M:
Amlodipine ameliorates myocardial hypertrophy by inhibiting EGFR
phosphorylation. Biochem Biophys Res Commun. 327:1083–1087. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Yu GL, Liang XQ and Zheng JQ: Contrast of
losartan, fosinopril and amlodipine on cardiomyocyte apoptosis and
left ventricular remolding in hypertensive rats. Hunan Yi Ke Da Xue
Xue Bao. 26:405–408. 2001.(In Chinese). PubMed/NCBI
|
|
66
|
Meo M, Meste O, Signore S, Sorrentino A,
Cannata A, Zhou Y, Matsuda A, Luciani M, Kannappan R, Goichberg P,
et al: Reduction in Kv current enhances the temporal dispersion of
the action potential in diabetic myocytes: Insights from a novel
repolarization algorithm. J Am Heart Assoc. 5:e0030782016.
View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Sung HH, Kam SC, Lee JH, Chae MR, Hong C,
Ko M, Han DH, So I and Lee SW: Molecular and functional
characterization of ORAI and STIM in human corporeal smooth muscle
cells and effects of the transfer of their dominant-negative mutant
genes into diabetic rats. J Urol. 187:1903–1910. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Liu CZ, Tan JX, Wang Y, Huang YG and Huang
DL: L-type calcium channel blocker suppresses calcineurin signal
pathway and development of right ventricular hypertrophy. J Formos
Med Assoc. 104:798–803. 2005.PubMed/NCBI
|
|
69
|
Zou Y, Yamazaki T, Nakagawa K, Yamada H,
Iriguchi N, Toko H, Takano H, Akazawa H, Nagai R and Komuro I:
Continuous blockade of L-type Ca2+ channels suppresses
activation of calcineurin and development of cardiac hypertrophy in
spontaneously hypertensive rats. Hypertens Res. 25:117–124. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Bugyei-Twum A, Advani A, Advani SL, Zhang
Y, Thai K, Kelly DJ and Connelly KA: High glucose induces Smad
activation via the transcriptional coregulator p300 and contributes
to cardiac fibrosis and hypertrophy. Cardiovasc Diabetol.
13:892014. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Chen Y, Yuan J, Jiang G, Zhu J, Zou Y and
Lv Q: Lercanidipine attenuates angiotensin II-induced cardiomyocyte
hypertrophy by blocking calcineurin-NFAT3 and CaMKII-HDAC4
signaling. Mol Med Rep. 16:4545–4552. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Liu CJ, Cheng YC, Lee KW, Hsu HH, Chu CH,
Tsai FJ, Tsai CH, Chu CY, Liu JY, Kuo WW and Huang CY:
Lipopolysaccharide induces cellular hypertrophy through
calcineurin/NFAT-3 signaling pathway in H9c2 myocardiac cells. Mol
Cell Biochem. 313:167–178. 2008. View Article : Google Scholar : PubMed/NCBI
|