Introduction
Diabetes is a disease that often manifests as
hyperglycemia and is caused both by genetic and environmental
factors. Diabetes is classified into type I and type II diabetes.
Type I diabetes is an autoimmune disease mediated by T lymphocytes,
characterized by inflammation and selective damage to pancreatic
β-cells, resulting in severe insulin deficiency (1,2).
Type II diabetes is characterized by chronic hyperglycemia and
insulin resistance caused by reduced sensitivity of liver, muscle
and adipose tissue to insulin (3).
An abnormal pancreatic β-cell count is a key factor
in the pathogenesis of type I and type II diabetes (4). Indeed, the advanced stage of type I
diabetes presents severe loss and elimination of pancreatic β-cells
(5). Moreover, previous studies
reported that the pancreatic β-cell count was significantly reduced
in patients with type II diabetes (6,7).
Thus, the regulation of pancreatic β-cell proliferation, apoptosis
and regeneration affects the progression of diabetes (8,9).
Apoptosis and proliferation of pancreatic β-cells
are essential to the maintenance of the blood glucose balance
(10,11). In type I diabetes, infiltrating
immune cells produce several cytokines, such as interleukin-1β,
interferon-γ and tumor necrosis factor-α. Pro-inflammatory
cytokines activate signaling cascades, such as NF-κB signaling,
that result in increased production of nitric oxide, causing
endoplasmic reticulum stress-induced pancreatic β-cell apoptosis
(12,13). Furthermore, increased cytokine
levels can also activate the JNK/p38 and STAT1 signaling pathways,
which promote mitochondria-dependent cell apoptosis by
downregulating Bcl-2 expression, which is essential for cytochrome
c release, ultimately inducing β-cell apoptosis (14–16).
Streptozotocin (STZ) is often used to establish
hyperglycemia in animal models (17). STZ enters pancreatic β-cells
through the glucose transporter GLUT2 and induces the production of
reactive oxygen species (ROS), which, in turn, damages DNA and
leads to pancreatic β-cell apoptosis (17,18).
Previous studies reported that STZ treatment significantly
increased the levels of intracellular ROS, Bax and
cleaved-caspase-9 and −3 in MIN6 cells, thereby enhancing apoptosis
(19–21). Thus, STZ stimulates β-cell
apoptosis in vitro.
Peroxiredoxins (Prxs) are crucial in maintaining the
balance of the redox system by maintaining intracellular
H2O2 homeostasis via their peroxidase
activity to decrease ROS redox reactions in cells. Prx I regulates
antioxidant and oxidation-sensitive signal transduction (22). A previous study reported that the
expression of Prx family members was altered under various
pathological conditions, such as cancer and diabetes (23). In response to cytokine stimulation,
both Prx I and Prx II expression levels are upregulated in
pancreatic β-cells, which suggests a possible involvement of Prx I
and Prx II in the progression of type I diabetes. Furthermore, it
has been reported that Prx I is more sensitive to cytokine
stimulation than Prx II (24).
Patients with type II diabetes present higher plasma levels of Prx
I, compared with healthy individuals, which was reported as an
indicator of glycemic control in type II diabetes (25). Pancreatic β-cell apoptosis and
function decline have been implicated not only in type I and type
II diabetes, but also in pancreatic transplantation (26).
In the present study, the role of Prx I in the
progression of type I diabetes was examined. The potential
regulatory function of Prx I in STZ-induced pancreatic β-cell
apoptosis was investigated both in vivo and in vitro,
using Prx I knockout mice and MIN6 pancreatic cells.
Materials and methods
Animals
Male, 8-week-old wild-type, Prx I−/− and
Prx II−/− mice (129/SvJ strain; n=6 in each group; Korea
Research Institute of Bioscience and Biotechnology) weighing 22–25
g were used in the present study. All mice were maintained in a
pathogen-free facility at 20–22°C, with 50–60% humidity and a
12-h-dark/light cycle, and had free access to food and water. Mice
received 50 mg/kg STZ by intraperitoneal injection (17,21,27,28)
once daily for five consecutive days to establish a type I diabetes
mice model. Mice were sacrificed by cervical dislocation under deep
anesthesia and the pancreas were sampled on day 7 (17,18,21,29).
All experiments were approved by The Institutional Animal Care and
Use Committee.
Cell culture
MIN6 cells were obtained from Shanghai Bogoo
Biological Technology Co., Ltd., and were maintained in Dulbecco's
modified Eagle medium (Gibco; Thermo Fisher Scientific, Inc.)
supplemented with 10% heat inactivated FBS (Beijing Solarbio
Science & Technology Co., Ltd.), 2 mM L-glutamine, 100 U/ml
penicillin and streptomycin (Beijing Solarbio Science &
Technology Co., Ltd.) at 37°C in a 5% CO2 humidified
incubator. MIN6 cells were seeded in a 6-well plate at a density of
3×105 cells/well and cultured to 80–90% confluence.
Cells were stimulated with 5 µM STZ (Biosharp Life Sciences) for
another 24 h at 37°C.
Establishment of Prx I-knockdown
stable cell line
For the construction of lentivirus packaging Prx I
short hairpin (sh)RNA, control scrambled shRNA
(5′-TTCTCCGAACGTGTCACGTTTC-3′) and shRNAs specifically targeting
Prx I (5′-CAGTGATAGAGCCGAT-3′) were synthesized and inserted into
the pGLV3/H1/GFP+Puro lentiviral vector (Shanghai GenePharma Co.,
Ltd.) (26). Prior to viral
infection, MIN6 cells were seeded in 6-wells plate at the density
of 3×105 cells/well and cultured to 70–80% confluence.
Lentiviral particles were produced by transfecting 293T cells
(30-40% confluence) with the 0.5 µg/ml expression plasmids and 1.5
µg/ml packaging vectors (pGag/Pol, pRev; pVSV-G; Shanghai
GenePharma Co., Ltd.) using the RNAi-mate transfection reagent
(Shanghai GenePharma Co., Ltd.), following the manufacturer's
protocol. After 72-h transfection, the supernatant containing
lentiviral particles was collected and centrifuged at 1,500 × g for
4 min at 4°C, then filtered with 0.45-µm cellulose acetate filters
to eliminate cell debris. Lentiviral supernatants were then
ultra-centrifuged at 48,400 × g for 2 h at 4°C. Viral stock
containing shPrx I at a multiplicity of infection of 100 was added
along with 5 µg/ml of polybrene (Shanghai GenePharma Co., Ltd.) to
the MIN6 cells. After 24 h, cells were inoculated in fresh culture
media. Transduced MIN6 cells were incubated in 10% FBS DMEM
containing 2 µg/ml puromycin for selection at 37°C in a 5%
CO2 humidified incubator. After two rounds of selection,
transduced MIN6 cells were incubated in 10% FBS DMEM without
puromycin for 24 h and transduction efficiency was confirmed by
flow cytometry and western blot analysis.
FGF2 treatment
Transduced MIN6 cells were cultured
(3×105 cells/well) in a 6-well plate. At 80–90%
confluence, cells were treated with 20 ng/ml FGF2 (GenScript) for 2
h at 37°C, followed by 5 µM STZ treatment for 24 h at 37°C.
Flow cytometry
MIN6 cells transduced with shRNA vectors expressing
GFP were cultured in DMEM containing 10% FBS and 2 µg/ml puromycin
for selection. After selection, 1×106 cells were
harvested and added to individual tubes. Data were acquired on a
CytoFLEX flow cytometer and GFP expression was analyzed using the
CytExpertsoftware (version 1.2.10.0, both from Beckman Coulter,
Inc.).
Annexin V staining
MIN6 cells were cultured for 24 h in 6-well plates
at a concentration of 4×105 cells/well. Cells were then
treated with 5 mM STZ for the indicated times (0, 12 or 24 h) at
37°C in a 5% CO2 humidified incubator. After treatment,
cells were washed with PBS, then incubated with 1 µl Annexin
V-phycoerythrin in 199 µl cell binding buffer for 15 min at room
temperature. After staining, the cells were washed twice with PBS,
then analyzed under a DM2500 fluorescence microscope (Leica
Microsystems GmbH; magnification, ×100).
Western blot analysis
Total protein was extracted using ice-cold cell
extraction buffer [20 mM HEPES (pH 7.0), 50 mM NaCl, 10% Triton
X-100, 10% glycerol, 1 mM β-ME, protease inhibitor cocktail tablet]
for 30 min. Lysates were centrifuged at 13,200 × g at 4°C for 20
min. Total protein (2 µg/µl) was quantified using the Bradford
assay. Proteins (2 µg/µl) were separated via 12% SDS-PAGE and
transferred to a nitrocellulose filter membrane (EMD Millipore).
After incubation in 5% skimmed milk blocking solution at room
temperature for 1 h, the membrane was probed overnight at 4°C using
primary antibodies. The blots were then incubated with anti-Prx I
(1:2,000; cat. no. sc-7381), Prx II (1:2,000; cat. no. sc-515429),
Bad (1:2,000; cat. no. sc-8044), Bcl-2 (1:2,000; cat. no. sc-7382),
pro-caspase-3 (1:2,000; cat. no. sc-373730), AKT (1:2,000; cat. no.
sc-8312), phosphorylated (p)-AKT (1:2,000; cat. no. sc-7985-R),
glycogen synthase kinase (GSK)-3β (1:2,000; cat. no. sc-377213),
p-GSK3β (1:2,000; cat. no. sc-81496), β-catenin (1:2,000; cat. no.
sc-7963), p-β-catenin (1:2,000; cat. no. sc-57533), inhibitor of
nuclear factor-κB α (IκB-α; 1:2,000; cat. no. sc-1643), p-p65
(1:2,000; cat. no. sc-166748) and β-actin (1:2,000; cat. no.
sc-47778) antibodies. The membranes were washed five times with
TBST (0.2% Tween-20) and incubated for 1 h at room temperature with
horseradish peroxidase-conjugated secondary antibodies (1:5,000;
cat. nos. sc-2004 and sc-2005). All antibodies were purchased from
Santa Cruz Biotechnology, Inc. After washing with TBST, the blots
were developed using a chemiluminescence detection system (GE
Healthcare). Band intensities were quantified using the ImageJ
software. (version 1.52a; National Institutes of Health).
Hematoxylin and eosin (H&E)
staining
Pancreatic tissues were fixed in 4% paraformaldehyde
overnight at room temperature, then embedded in paraffin. Tissues
were cut into 5-µm sections for H&E staining. Following which,
samples were maintained in xylene solution for 20 min, 100% alcohol
for 1.5 min, 90% alcohol for 1 min, 80% alcohol for 1 min, then 50%
alcohol for 1 min. The sections were stained with hematoxylin
solution for 5 min at room temperature, then treated by
acid-alcohol and 0.8% ammonia for 2 sec. The sections were then
stained with eosin solution for 20 sec, 95% alcohol for 1 min, 100%
alcohol for 1 min, xylene for 1 min, then sealed in neutral gum.
The sections were examined under a DM2500 fluorescence microscope
(Leica Microsystems GmbH; magnification, ×200).
Statistical analysis
Statistical analysis was carried out using two-way
ANOVA to analyze the changes across time and differences between
groups, followed by Tukey's post hoc test (α=0.05). P<0.05 was
considered to indicate a statistically significant difference.
Statistical analyses were conducted using SPSS software (version
25; IBM Corp). Experiments were performed in duplicate and were
repeated at least three times.
Results
STZ treatment downregulates Prx I
expression and upregulates the expression of apoptosis-related
proteins in MIN6 cells
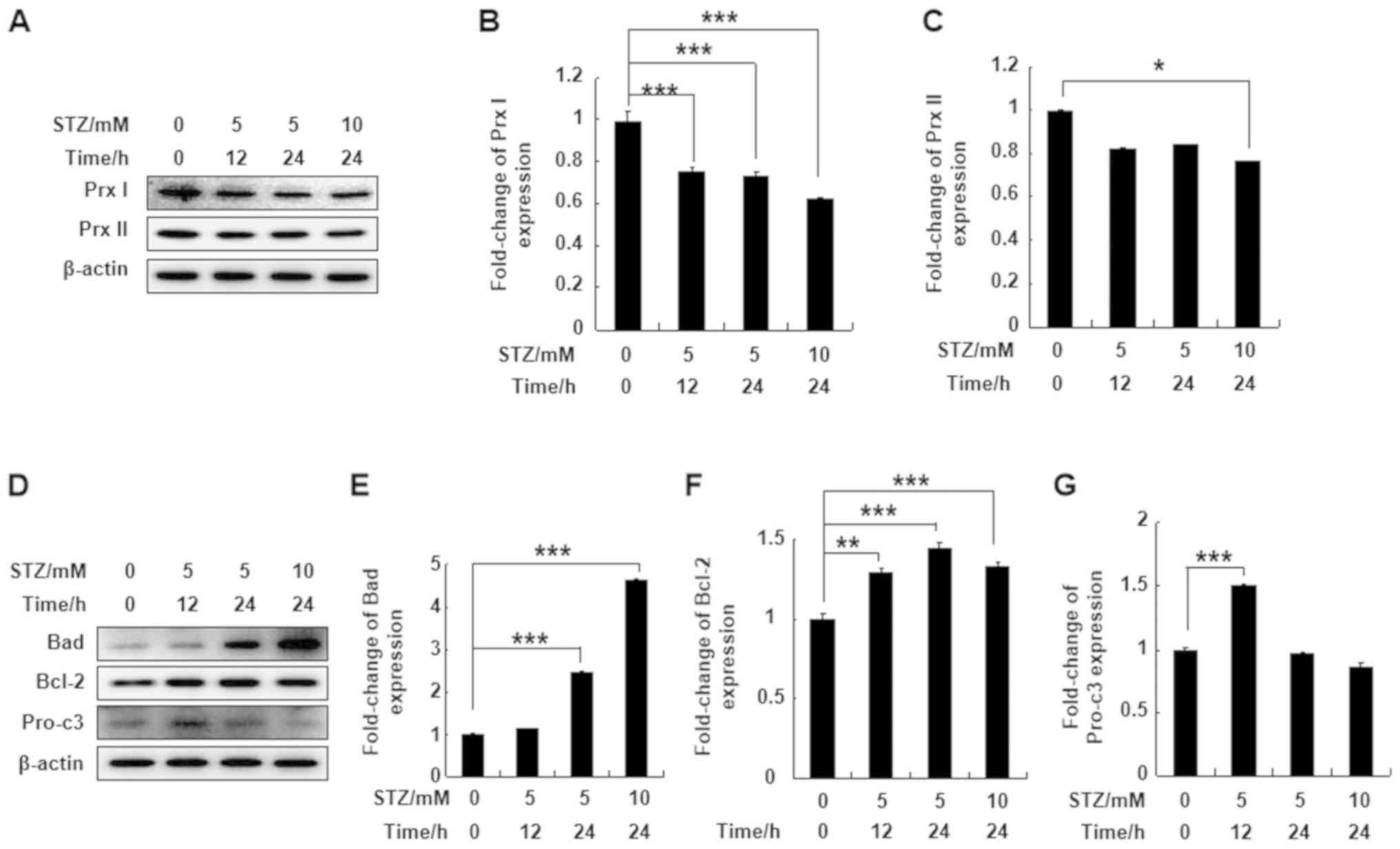
To understand the effect of STZ treatment on Prx I
and Prx II protein expression levels, MIN6 pancreatic cells were
treated with 5 or 10 mM STZ for 12 or 24 h. STZ significantly
downregulated expression levels of Prx I after 12 or 24 h of
treatment. However, Prx II protein expression in MIN6 cells was not
affected by STZ, except at a higher concentration and following
longer incubation times (10 mM for 24 h; Fig. 1A-C). Bad and Bcl-2 protein
expression levels were significantly upregulated at all
concentrations and time points compared with the control group.
Pro-caspase-3 protein expression was also upregulated following
incubation with 5 mM STZ for 12 h compared with the control group
(Fig. 1D-G). The results indicated
that Prx I was more sensitive to STZ treatment compared with Prx
II.
Prx I knockdown increases STZ-induced
β-cell death in vivo and in vitro
To understand the effect of Prx I and Prx II
deletion on acute pancreatic damage in vivo, wild-type, Prx
I−/− and Prx II−/− mice were injected with 50
mg/kg STZ according to the aforementioned protocol. After treatment
the mice were sacrificed, and pancreatic damage was examined with
H&E staining. Degeneration and shrinkage of pancreatic islet
tissue was observed in Prx I knockout mice after STZ treatment
compared with the wild- and Prx II knockout mice, suggesting that
Prx I is essential for protecting the pancreas islands against STZ
stimulation (Fig. 2A).
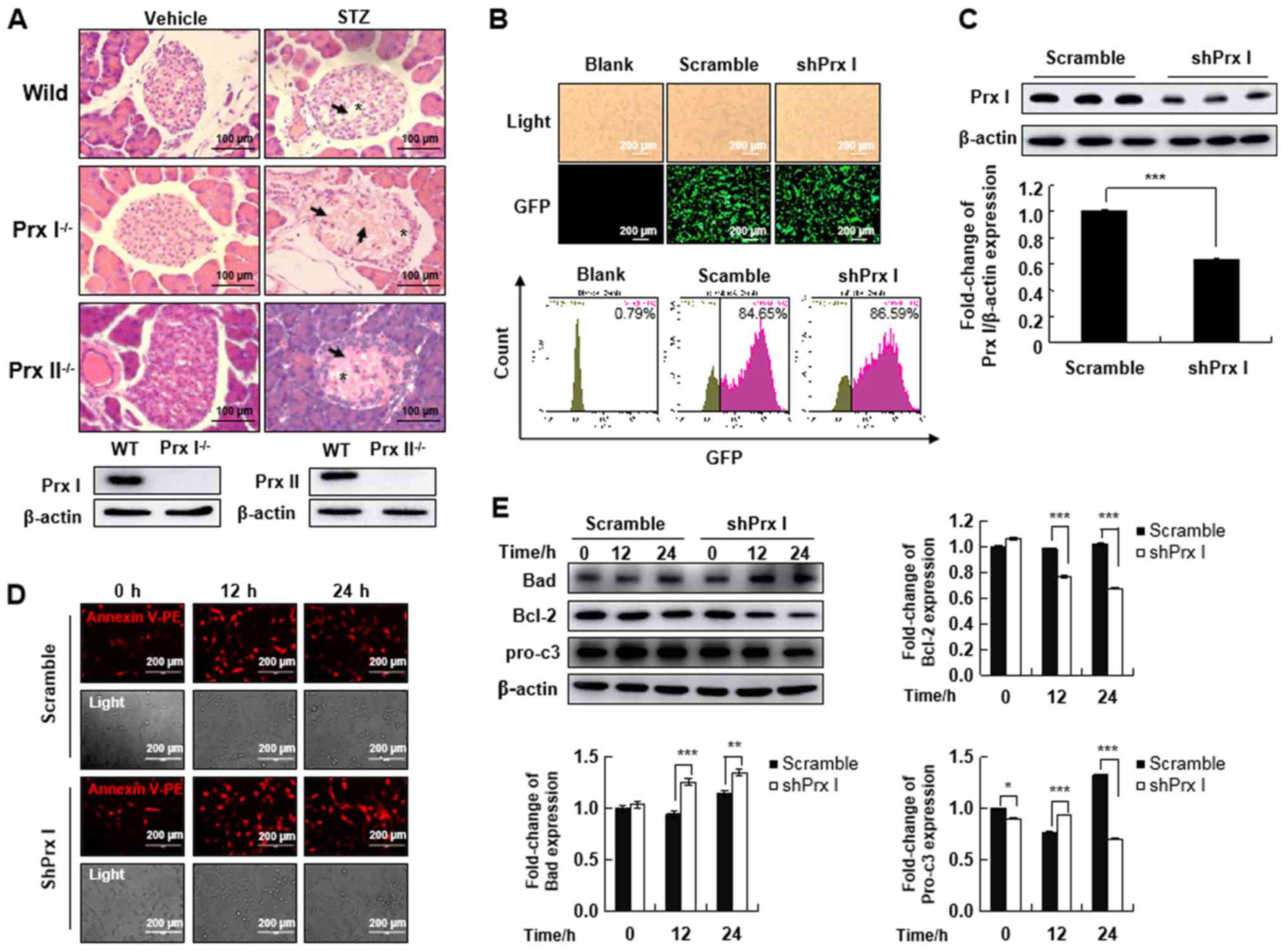 | Figure 2.Prx I knockdown increases apoptosis
of pancreatic β-cells. (A) Hematoxylin-eosin staining analysis of
pancreatic islets after STZ injection in wild-type, Prx
I−/− and Prx II−/− mice. STZ-treated mice
displayed degeneration and shrinkage of Islets of Langerhans,
illustrated by Islet amyloidosis with atrophied Islets β-cells
(arrows). *Indicates cytoplasmic vacuoles. Mouse genotype was
confirmed by western blotting. Scale bar, 100 µm. (B) GFP
expression was detected by immunofluorescence (upper panel) and
flow cytometry (lower panel) blank, scramble, and shPrx I MIN6 cell
lines. (C) Expression levels of Prx I in scramble and shPrx
I-transduced MIN6 cells. (D) Apoptosis was analyzed by Annexin V-PE
staining and observed using fluorescence microscopy after STZ
treatment in both scramble and shPrx I-transduced MIN6 cells. (E)
Bad, Bcl-2 and pro-c3 expression levels were evaluated by western
blot analysis in scramble and shPrx I-transduced MIN6 cells
following STZ treatment. Data are presented as the mean ± SD. n=6
in each group. *P<0.05, **P<0.01 and ***P<0.001. STZ,
streptozotocin; Prx, peroxiredoxin; pro-c3, pro-caspase-3; GFP,
green fluorescent protein; PE, phycoerythrin; WT, wild-type; shRNA;
short hairpin RNA. |
To understand the regulatory function of Prx I in
pancreatic β-cells, MIN6 pancreatic cells were transduced with Prx
I knockdown constructs. A scramble vector was used as a control.
Transduction efficiency was quantified by GFP expression using
fluorescence microscopy and flow cytometry (Fig. 2B). Based on GFP expression,
transduction efficiency was estimated to be 84.65 and 86.59% with
scramble and shPrx I, respectively (Fig. 2B). Transfection efficiency was
confirmed using western blotting, which showed that Prx I
expression was significantly reduced in shPrx I-transduced cells,
compared with cells transduced with the scramble control (Fig. 2C).
To investigate whether STZ-mediated cell death was
detectable following Prx I knockdown, scramble and shPrx I
transduced MIN6 cells were treated with 5 mM STZ for 12 or 24 h.
Examination of Annexin V staining by fluorescence microscopy
indicated that STZ-induced cell apoptosis was increased in the
shPrx I-transduced MIN6 cells, compared with scramble cells
(Fig. 2D). The expression of
apoptosis-related proteins was also examined following STZ
treatment (Fig. 2E). STZ
significantly upregulated the expression of the proapoptotic
protein Bad in shPrx I-transduced cells, compared with scramble
cells. However, expression of Bcl-2, which is an anti-apoptotic
protein, was significantly downregulated. The expression of
pro-caspase-3 was also significantly downregulated after 24-h STZ
treatment.
Prx I knockdown upregulates the
AKT/GSK3β/β-catenin and NF-κB signaling pathways
To understand the possible molecular mechanism
underlying the effects of Prx I on STZ-induced pancreatic β-cell
apoptosis, activation of the AKT/GSK3β/β-catenin and NF-κB
signaling pathways in shPrx I-transduced cells following 12 or 24-h
STZ treatment were assessed. The expression levels of p-AKT and
p-GSK3β were significantly reduced in shPrx I-transduced cells,
compared with scramble. By contrast, β-catenin phosphorylation was
significantly upregulated following Prx I knockdown and STZ
treatment (Fig. 3A-D).
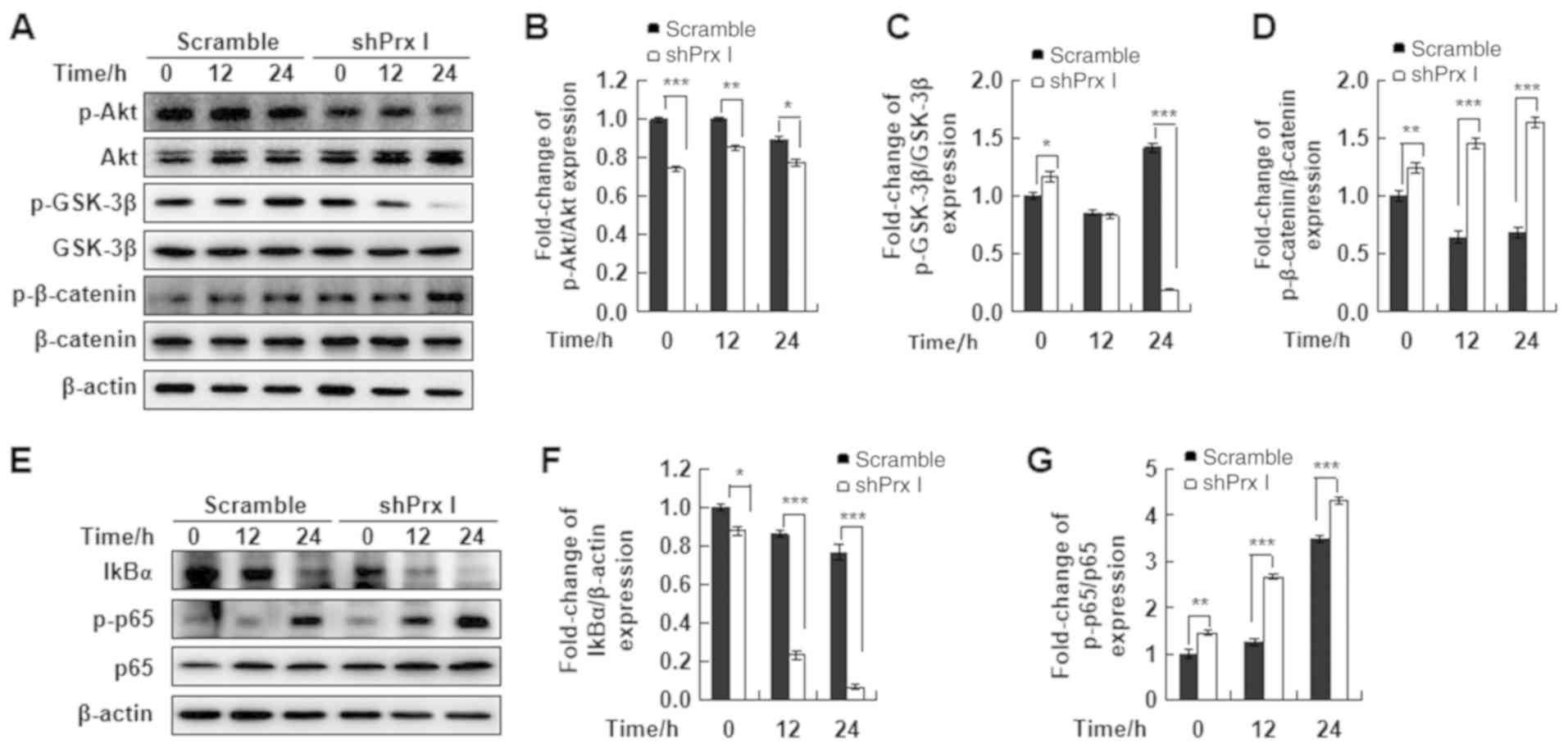 | Figure 3.Effect of Prx I knockdown on the
AKT/GSK-3β/β-catenin and NF-κB signaling pathways. (A) Western blot
analysis of p-AKT, AKT, p-GSK3β, p-β-catenin and β-catenin protein
expression in scramble and shPrx I-transduced MIN6 cells following
STZ stimulation. (B-D) Relative protein expression levels p-AKT,
p-GSK3β, and p-β-catenin. (E) Western blot analysis of IκBα and
p-p65 protein expression in scramble and shPrx I-transduced MIN6
cells following STZ stimulation. (F and G) Relative protein
expression levels of IκBα and p-p65. Data are presented as the mean
± SD. *P<0.05, **P<0.01 and ***P<0.001. STZ,
streptozotocin; Prx, peroxiredoxin; GSK-3β, glycogen synthase
kinase-3β; IκBα, inhibitor of nuclear factor-κB α; p,
phosphorylated; shRNA; short hairpin RNA. |
Moreover, to verify the effect of Prx I on
STZ-induced NF-κB signaling activation, scramble and Prx
I-knockdown MIN6 cells were treated with STZ for the indicated
times. STZ treatment significantly reduced the expression levels of
IκBα, which may suggest increased degradation of IκBα, compared
with scramble cells. In addition, p65 phosphorylation was
significantly increased in shPrx I-transduced cells, compared with
scramble (Fig. 3E-G).
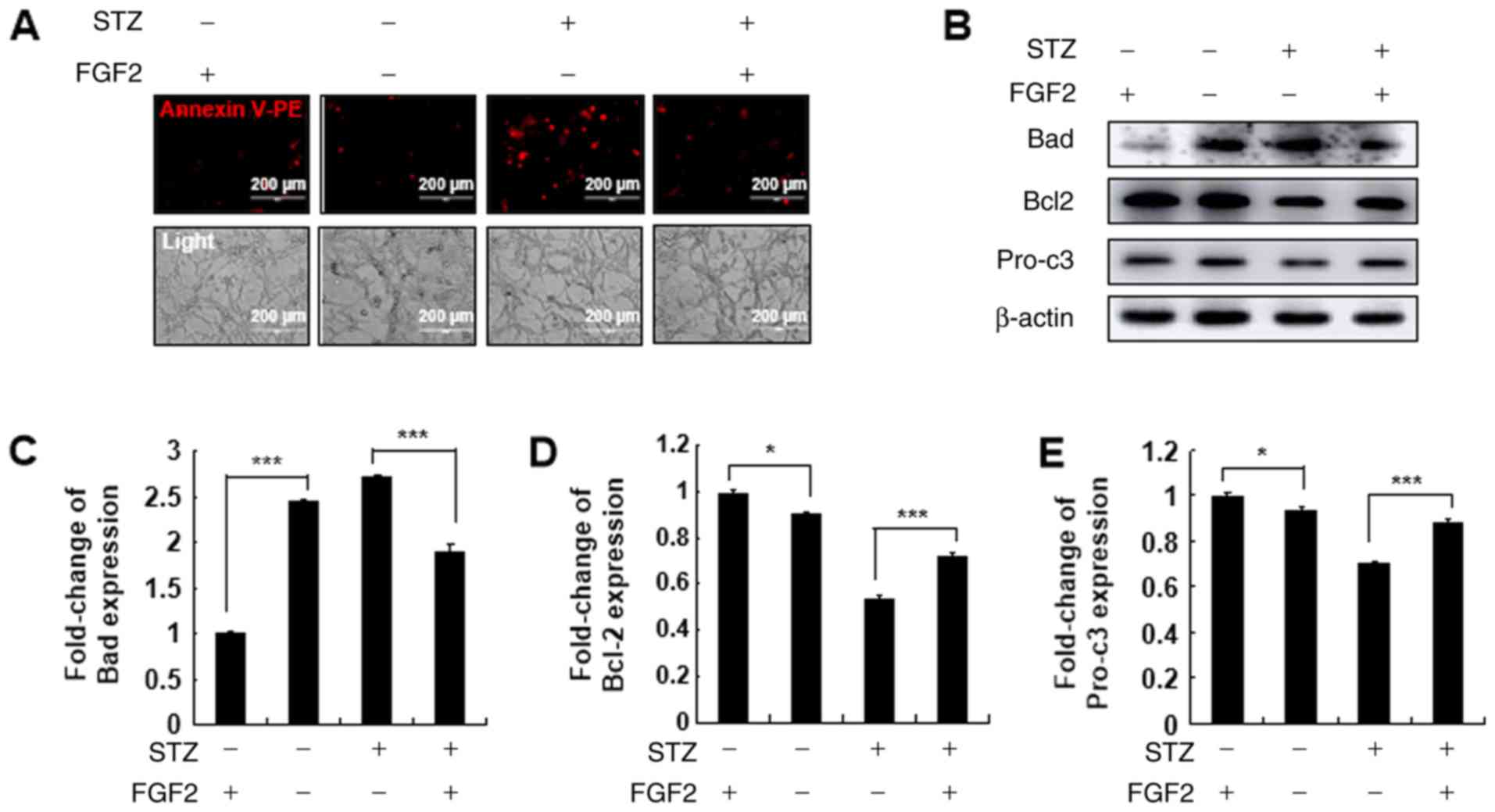
FGF2 treatment reverses STZ-induced
MIN6 cell apoptosis in Prx I-knockdown MIN6 cells
To the best of the authors' knowledge, FGF2 can
efficiently activate the AKT signaling pathways (30). Thus, to understand the regulatory
role of Prx I on the AKT signaling pathway following STZ
stimulation, shPrx I-transduced MIN6 cells were pre-treated with
FGF2 for 2 h, followed by STZ treatment for 24 h. Pre-treatment
with FGF2 reduced STZ-induced cellular apoptosis in shPrx
I-transduced cells, compared with scramble (Fig. 4A). Furthermore, FGF2 also reversed
the effects STZ, Bad expression decreased following treatment with
FGF2, whereas Bcl-2 and pro-caspase-3 expression increased compared
with STZ treatment alone (Fig.
4B-E).
Discussion
Streptozotocin was originally isolated from
Streptomyces achromogenes, and has been used as an
antibiotic and anticancer drug. Presently, STZ is more commonly
used in drug therapy for rare neuroendocrine tumors (31). STZ can induce diabetes in
experimental animals by intravenous or intraperitoneal injection
and is widely used in the study of diabetes (32). STZ is taken up by cells through the
GLUT2 glucose transporter, and has a selective destructive effect
on pancreatic β-cells (33). In
animal models, high doses of STZ leads to extensive destruction of
pancreatic β-cells, thereby increasing blood glucose levels and
urine output, thus replicating the characteristics of type I
diabetes (34). However, multiple
small doses of STZ in combination with a high-fat diet partly
destroy pancreatic cell function, and as peripheral tissues become
insensitive to insulin; the observed pathological changes are
closer to those of type II diabetes (29). Pancreatic β-cell injury is key in
the development and progression of diabetes, and damage to the
pancreatic β-cells results in reduced insulin secretion, thereby
accelerating the development of diabetes (35). STZ induces pancreatic β-cell
apoptosis mainly through ROS, reactive nitrogen species and DNA
damage (36). Moreover, pancreatic
β-cells are highly sensitive to ROS-mediated damage, which can
directly induce cell apoptosis (37). Additionally, ROS can also
indirectly damage the pancreatic β-cell via the signaling pathways
that affect insulin synthesis and secretion (38).
As STZ induces diabetes by increasing the production
of ROS and inhibiting the oxidative stress defense system (39), understanding the relationship
between antioxidant defense systems and ROS-induced pancreatic
β-cell injury would provide invaluable insight into the
pathogenesis of diabetes. In the present study, treatment with STZ
significantly downregulated Prx I, but not Prx II, and altered the
expression levels of apoptotic proteins, suggesting a possible
involvement of Prx I in STZ-induced β-cell apoptosis.
Peroxiredoxins represent a large and highly
conserved family of peroxidases, which reduce peroxides with a
conserved cysteine residue (40).
It has been reported that Prx VI-knockout mice have impaired
insulin signaling, which leads to reduced muscle glucose uptake
(41). Prx III protects pancreatic
β-cells from stress-related apoptosis (42). Moreover, Prx II also exhibits a
protective role in pancreatic β-cells under oxidative stress
(43). Collectively, these
previous studies indicate that members of the Prx family may play a
pivotal role in the development of diabetes. The results of present
study suggested that both Prx I−/− and Prx
II−/− mice pancreatic β-cells were destroyed following
STZ treatment. This result demonstrated that both Prx I and Prx II
may protect pancreatic β-cells from STZ-induced cell apoptosis.
Notably, Prx I was more sensitive to STZ stimulation. Sensitivity
of Prx I to other compounds has also been reported in related
studies. For instance, treatment of INS-1E β-cells with oleate and
palmitate resulted in a significant increase in Prx I expression
(44,45). In the present study, the protective
role of Prx I was also confirmed by silencing Prx I expression with
lentivirus vectors in the MIN6 cell line. Therefore, Prx I may play
a pivotal role in protecting pancreatic β-cells against STZ-induced
cell apoptosis.
The AKT signaling pathway is crucial in regulating
the proliferation and apoptosis of pancreatic β-cells. Activated
AKT can phosphorylate substrates for various biological processes,
thus regulating insulin-mediated glucose transport, protein and
glycogen synthesis, as well as cell proliferation, differentiation
and survival (46–48). It was previously demonstrated that
Akt2-deficient mice present mild glucose intolerance and insulin
resistance, and some of them developed diabetes (49). A previous study also reported that
AKT could promote cell survival (50). AKT-mediated phosphorylation of
GSK-3 and forkhead transcription factors (including forkhead box
O1) may also promote cell survival via downstream targets (51). For example, AKT-mediated
inactivation of GSK-3 reduces GSK-3-mediated phosphorylation of
β-catenin, leading to increased cell survival (52). In some cell types, β-catenin can be
transferred from the cytosol to the nucleus after phosphorylation
of GSK-3. GSK-3-mediated phosphorylation of β-catenin promotes its
degradation. In various cancer cells, decreased phosphorylation of
GSK3-β (ser9) leads to reduced Bcl-2/Bcl-xL expression and elevated
expression of Bax/Bad, which regulate apoptosis (40,53,54).
Phosphorylated AKT can inhibit the phosphorylation GSK3-β (ser21)
and GSK3-β (ser9), and inhibit their activity (54). The present study demonstrated that
the expression of p-AKT and p-GSK3-β (ser9) was significantly
decreased in Prx I-knockdown MIN6 cells after STZ treatment.
Notably, the addition of FGF2, which can upregulate the AKT
signaling pathway, significantly inhibited STZ-induced cell
apoptosis in Prx I-knockdown MIN6 cells, and reversed the effects
of STZ on apoptosis-related protein expression. These results
indicate that the AKT signaling pathway may be an important
signaling pathway that is stimulated by STZ in β-cell apoptosis,
and the function of Prx I on regulating STZ-induced cell death may
also occur via the same mechanism. Nevertheless, the possible
regulatory mechanism of Prx I on STZ-induced β-cell apoptosis
requires further study, and future studies should investigate the
regulatory mechanisms affecting the NF-κB signaling pathway,
mitochondrial function and cellular ROS-related pathways.
Furthermore, such study should extend to both type I and type II
diabetes pathogenesis.
In summary, deletion of Prx I increased STZ-induced
pancreatic β-cell apoptosis in MIN6 cells, and exacerbated
STZ-induced pancreatic damage in vivo. The increase in
apoptosis as a result of Prx I knockdown may occur via the
AKT/GSK/β-catenin and NF-κB signaling pathways. The present
findings provide a novel insight for treatment of pancreatic damage
caused by oxidative stress.
Acknowledgements
Not applicable.
Funding
The present study was supported by the Basic Science
Research Program through the National Research Foundation of Korea
funded by the Ministry of Education (grant no. 2020R1I1A2052417),
the Korean Research Institute of Bioscience and Biotechnology
Research Initiative Program (grant no. KGM5162021), the Research
Project of Heilongjiang Bayi Agricultural University (grant no.
XYB2013-17) and the Natural Science Foundation of Heilongjiang
Province of China (grant no. QC2015121).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
MHJ, GNS and YHJ constructed the disease model,
designed the study and wrote the manuscript. HNS, XZ and YQZ
performed the experiments. DSL, JSK, YDC and LYY performed carried
out data analysis. TK and YHH made substantial contributions to
conception and design. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
All procedures were approved by the Heilongjiang
Bayi Agricultural University Animal Care and Use Committee and
conformed to the National Institutes of Health Guide for the Care
and Use of Animals in Research.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Lincez PJ, Shanina I and Horwitz MS:
Reduced expression of the MDA5 gene IFIH1 prevents autoimmune
diabetes. Diabetes. 64:2184–2193. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Taborsky GJ Jr, Mei Q, Hackney DJ and
Mundinger TO: The search for the mechanism of early sympathetic
islet neuropathy in autoimmune diabetes. Diabetes Obes Metab. 16
(Suppl 1):S96–S101. 2014. View Article : Google Scholar
|
|
3
|
Yang ZH, Miyahara H and Hatanaka A:
Chronic administration of palmitoleic acid reduces insulin
resistance and hepatic lipid accumulation in KK-A y Mice with
genetic type 2 diabetes. Lipids Health Dis. 10:1202011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Guo T and Hebrok M: Stem cells to
pancreatic beta-cells: New sources for diabetes cell therapy.
Endocr Rev. 30:214–227. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ilonen J, Lempainen J and Veijola R: The
heterogeneous pathogenesis of type 1 diabetes mellitus. Nat Rev
Endocrinol. 15:635–650. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Spijker HS, Song H, Ellenbroek JH, Roefs
MM, Engelse MA, Bos E, Koster AJ, Rabelink TJ, Hansen BC, Clark A,
et al: Loss of β-cell identity occurs in type 2 diabetes and is
associated with islet amyloid deposits. Diabetes. 64:2928–2938.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Rhodes CJ: Type 2 diabetes-a matter of
beta-cell life and death? Science. 307:380–384. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Bouwens L and Rooman I: Regulation of
pancreatic beta-cell mass. Physiol Rev. 85:1255–1270. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
McKinnon C and Docherty K: Pancreatic
duodenal homeobox-1, PDX-1, a major regulator of beta cell identity
and function. Diabetologia. 44:1203–1214. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Porat S, Weinberg-Corem N,
Tornovsky-Babaey S, Schyr-Ben-Haroush R, Hija A, Stolovich-Rain M,
Dadon D, Granot Z, Ben-Hur V, White P, et al: Control of pancreatic
β cell regeneration by glucose metabolism. Cell Metab. 13:440–449.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Muruganathan U, Srinivasan S and
Vinothkumar V: Antidiabetogenic efficiency of menthol, improves
glucose homeostasis and attenuates pancreatic β-cell apoptosis in
streptozotocin-nicotinamide induced experimental rats through
ameliorating glucose metabolic enzymes. Biomed Pharmacother.
92:229–239. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Pukel C, Baquerizo H and Rabinovitch A:
Destruction of rat islet cell monolayers by cytokines: Synergistic
interactions of interferon-γ, tumor necrosis factor, lymphotoxin,
and interleukin 1. Diabetes. 37:133–136. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Campbell IL, Kay T, Oxbrow L and Harrison
L: Essential role for interferon-gamma and interleukin-6 in
autoimmune insulin-dependent diabetes in NOD/Wehi mice. J Clin
Invest. 87:739–742. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Gurzov EN and Eizirik DL: Bcl-2 proteins
in diabetes: Mitochondrial pathways of β-cell death and
dysfunction. Trends Cell Biol. 21:424–431. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Czabotar PE, Lessene G, Strasser A and
Adams JM: Control of apoptosis by the BCL-2 protein family:
Implications for physiology and therapy. Nat Rev Mol Cell Biol.
15:49–63. 2014. View
Article : Google Scholar : PubMed/NCBI
|
|
16
|
Taniguchi S, Kang L, Kimura T and Niki I:
Hydrogen sulphide protects mouse pancreatic beta-cells from cell
death induced by oxidative stress, but not by endoplasmic reticulum
stress. Br J Pharmacol. 162:1171–1178. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Lenzen S: The mechanisms of alloxan- and
streptozotocin- induced diabetes. Diabetologia. 51:216–226. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Green AD, Vasu S and Flatt PR:
Functionality and antidiabetic utility of β-and L-cell containing
pseudoislets. Exp Cell Res. 344:201–209. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhang B, Lu Y, Campbell-Thompson M,
Spencer T, Wasserfall C, Atkinson M and Song S: Alpha1-antitrypsin
protects beta-cells from apoptosis. Diabetes. 56:1316–1323. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhao Y, Zhang X, Chen J, Lin C, Shao R,
Yan C and Chen C: Hexarelin protects rodent pancreatic β-cells
function from cytotoxic effects of streptozotocin involving
mitochondrial signalling pathways in vivo and in vitro. PLoS One.
11:e01497302016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Elango B, Dornadula S, Paulmurugan R and
Ramkumar KM: Pterostilbene ameliorates streptozotocin-induced
diabetes through enhancing antioxidant signaling pathways mediated
by Nrf2. Chem Res Toxicol. 29:47–57. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Rhee SG and Woo HA: Multiple functions of
peroxiredoxins: Peroxidases, sensors and regulators of the
intracellular messenger H2O2, and protein
chaperones. Antioxid Redox Signal. 15:781–794. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Tang Z, Xia N, Yuan X, Zhu X, Xu G, Cui S,
Zhang T, Zhang W, Zhao Y, Wang S and Shi B: PRDX1 is involved in
palmitate induced insulin resistance via regulating the activity of
p38MAPK in HepG2 cells. Biochem Biophys Res Commun. 465:670–677.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Bast A, Wolf G, Oberbäumer I and Walther
R: Oxidative and nitrosative stress induces peroxiredoxins in
pancreatic beta cells. Diabetologia. 45:867–876. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Al-Masri AA, El Eter E, Tayel S and Zamil
H: Differential associations of circulating peroxiredoxins levels
with indicators of glycemic control in type 2 diabetes mellitus.
Eur Rev Med Pharmacol Sci. 18:710–716. 2014.PubMed/NCBI
|
|
26
|
Lee YJ, Song DS, Yoo JS, Hyung KE, Lee MJ,
Moon YH, Lee IH, Go BS, Park SY and Hwang KW: Protective functions
of peroxiredoxin-1 against cytokine-induced MIN6 pancreatic β-cell
line death. Can J Physiol Pharmacol. 91:1037–1043. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Song G, Ouyang G and Bao S: The activation
of Akt/PKB signaling pathway and cell survival. J Cell Mol Med.
9:59–71. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Pathak S, Dorfmueller HC, Borodkin VS and
van Aalten DMF: Chemical dissection of the link between
streptozotocin, O-GlcNAc, and pancreatic cell death. Chem Biol.
15:799–807. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Srinivasan K, Viswanad B, Asrat L, Kaul CL
and Ramarao P: Combination of high-fat diet-fed and low-dose
streptozotocin-treated rat: A model for type 2 diabetes and
pharmacological screening. Pharmacol Res. 52:313–320. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Mossahebi M, Quan M, Zhang JS and Li X:
FGF Signaling pathway: A key regulator of stem cell pluripotency.
Front Cell Dev Biol. 8:792020. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Turner NC, Strauss SJ, Sarker D, Gillmore
R, Kirkwood A, Hackshaw A, Papadopoulou A, Bell J, Kayani I,
Toumpanakis C, et al: Chemotherapy with 5-fluorouracil, cisplatin
and streptozocin for neuroendocrine tumours. Br J Cancer.
102:1106–1112. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Deeds MC, Anderson JM, Armstrong AS,
Gastineau DA, Hiddinga HJ, Jahangir A, Eberhardt NL and Kudva YC:
Single dose streptozotocin-induced diabetes: Considerations for
study design in islet transplantation models. Lab Anim. 45:131–140.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Tonne JM, Sakuma T, Deeds MC, Munoz-Gomez
M, Barry MA, Kudva YC and Ikeda Y: Global gene expression profiling
of pancreatic islets in mice during streptozotocin-induced β-cell
damage and pancreatic Glp-1 gene therapy. Dis Model Mech.
6:1236–1245. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Junod A, Lambert AE, Stauffacher W and
Renold AE: Diabetogenic action of streptozotocin: Relationship of
dose to metabolic response. J Clin Invest. 48:21291969. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Rösen P, Nawroth P, King G, Möller W,
Tritschler HJ and Packer L: The role of oxidative stress in the
onset and progression of diabetes and its complications: A summary
of a Congress Series sponsored byUNESCO-MCBN, the American Diabetes
Association and the German Diabetes Society. Diabetes Metab Res
Rev. 17:189–212. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Nahdi AMTA, John A and Raza H: Elucidation
of molecular mechanisms of streptozotocin-induced oxidative stress,
apoptosis, and mitochondrial dysfunction in Rin-5F pancreatic
β-cells. Oxid Med Cell Longev. 2017:70542722017. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Gehrmann W, Elsner M and Lenzen S: Role of
metabolically generated reactive oxygen species for lipotoxicity in
pancreatic β-cells. Diabetes Obes Metab. 12:149–158. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Mandrup-Poulsen T: Apoptotic signal
transduction pathways in diabetes. Biochem Pharmacol. 66:1433–1440.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Ates O, Cayli SR, Yucel N, Altinoz E,
Kocak A, Durak MA, Turkoz Y and Yologlu S: Central nervous system
protection by resveratrol in streptozotocin-induced diabetic rats.
J Clin Neurosci. 14:256–260. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Rhee SG: Overview on peroxiredoxin. Mol
Cells. 39:1–5. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Pacifici F, Arriga R, Sorice GP, Capuani
B, Scioli MG, Pastore D, Donadel G, Bellia A, Caratelli S, Coppola
A, et al: Peroxiredoxin 6, a novel player in the pathogenesis of
diabetes. Diabetes. 63:3210–3220. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Wolf G, Aumann N, Michalska M, Bast A,
Sonnemann J, Beck JF, Lendeckel U, Newsholme P and Walther R:
Peroxiredoxin III protects pancreatic ss cells from apoptosis. J
Endocrinol. 207:163–175. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Zhao F and Wang Q: The protective effect
of peroxiredoxin II on oxidative stress induced apoptosis in
pancreatic β-cells. Cell Biosci. 2:222012. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Maris M, Waelkens E, Cnop M, D'Hertog W,
Cunha DA, Korf H, Koike T, Overbergh L and Mathieu C:
Oleate-induced beta cell dysfunction and apoptosis: A proteomic
approach to glucolipotoxicity by an unsaturated fatty acid. J
Proteome Res. 10:3372–3385. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Maris M, Robert S, Waelkens E, Derua R,
Hernangomez MH, D'Hertog W, Cnop M, Mathieu C and Overbergh L: Role
of the saturated nonesterified fatty acid palmitate in beta cell
dysfunction. J Proteome Res. 12:347–362. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Elghazi L, Balcazar N and Bernal-Mizrachi
E: Emerging role of protein kinase B/Akt signaling in pancreatic
beta-cell mass and function. Int J Biochem Cell Biol. 38:157–163.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Franke TF: PI3K/Akt: Getting it right
matters. Oncogene. 27:6473–6488. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Sarbassov DD, Guertin DA, Ali SM and
Sabatini DM: Phosphorylation and regulation of Akt/PKB by the
rictor-mTOR complex. Science. 307:1098–1101. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Cho H, Mu J, Kim JK, Chu Q, Crenshaw EB
III, Kaestner KH, Bartolomei MS, Shulman GI and Birnbaum MJ:
Insulin resistance and a diabetes mellitus-like syndrome in mice
lacking the protein kinase Akt2 (PKB beta). Science. 292:1728–1731.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Dan HC, Sun M, Kaneko S, Feldman RI,
Nicosia SV, Wang HG, Tsang BK and Cheng JQ: Akt phosphorylation and
stabilization of X-linked inhibitor of apoptosis protein (XIAP). J
Biol Chem. 279:5405–5412. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Huo X, Liu S, Shao T, Hua H, Kong Q, Wang
J, Luo T and Jiang Y: GSK3 protein positively regulates type I
insulin-like growth factor receptor through forkhead transcription
factors FOXO1/3/4. J Biol Chem. 289:24759–24770. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Li Y, Hansotia T, Yusta B, Ris F, Halban
PA and Drucker DJ: Glucagon-like peptide-1 receptor signaling
modulates β cell apoptosis. J Biol Chem. 278:471–478. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Mishra R: Glycogen synthase kinase 3 beta:
Can it be a target for oral cancer. Mol Cancer. 9:1442010.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Parida S, Pal I, Parekh A, Thakur B,
Bharti R, Das S and Mandal M: GW627368X inhibits proliferation and
induces apoptosis in cervical cancer by interfering with EP4/EGFR
interactive signaling. Cell Death Dis. 7:e21542016. View Article : Google Scholar : PubMed/NCBI
|