Introduction
Pulmonary stenosis (PS) is a congenital heart
disease (CHD) caused by abnormal development of the fetal heart
during the first eight weeks of pregnancy, accounting for ~6.2% of
all CHD cases worldwide (1). PS
can be classified into valvular, subvalvular and supravalvular
subtypes (2). The symptoms of PS
can be mild, moderate or severe (3), and patients with severe PS require
therapy due to possibly life-threatening complications. The
etiology and pathogenesis of PS remain poorly understood (2). Current diagnostic methods for PS
include electrocardiography, echocardiography, magnetic resonance
imaging, multislice computed tomography, cardiac catheterization
and angiography (2). However,
biomarkers for early prediction and diagnosis remain unavailable.
Several risk factors are associated with the onset of CHD,
including genetic, epigenetic and environmental factors (4,5), and
this may also be the case for PS. However, to the best of the
authors knowledge, no genetic assessment for fetal PS during
pregnancy has been conducted.
Extracellular vesicles (EVs) are secreted as
exosomes after fusion of endosomes with the plasma membrane or shed
from the plasma membrane as microvesicles (6). EVs are involved in cell-to-cell
communication between tumor cells and surrounding cells in the
primary tumor microenvironment (7). Moreover, previous studies indicated
that EVs play an important role in the maternal system as part of
the physiological changes taking place during pregnancy. Cell-free
DNA fragments released from the fetus are found in maternal
circulation, and EVs can also be released from the fetus, placenta
and maternal body into fetal-maternal circulation (8). EVs are detectable in maternal
circulation as early as the sixth week of pregnancy (8). The concentration of EVs in
circulation is higher in pregnant females compared with
non-pregnant females and increases with severity of
pregnancy-related disease, such as gestational diabetes mellitus,
fetal growth restriction and preterm birth, and oxidative stress
(9).
Various functional proteins, RNAs, lipids and
metabolites are detectable in different classes of EVs (6). Since microenvironmental parameters
such as acidosis, hypoxia and elevated interstitial fluid pressure
can influence the release of EVs, optimized in vitro models
that mimic in vivo cellular environments are required for
the study of EVs (10,11). EVs can contain several types of
RNA, including mRNA, circular RNA (circRNA) and long non-coding RNA
(lncRNA) (12). Functional
extracellular vesicle long RNAs (EVLRs) found in peripheral blood
could represent biomarkers for the diagnosis of a variety of
diseases, including hepatic tumors, gastric cancer, kidney cancer
and breast cancer (13,14). However, the characteristics of
EVLRs in pregnant females with PS infants remain largely unknown.
Therefore, understanding the roles of plasma EVLRs in pregnant
females may provide new predictive strategies for the diagnosis of
fetal PS in pregnant females.
In the present study, EVLRs from pregnant females
who had healthy infants or infants with PS were analyzed by RNA
sequencing and evaluated for their potential use as diagnostic
markers for PS during pregnancy. The present study provides a
comprehensive analysis of hub genes associated with EVLRs. The
identification of these hub genes may provide insight into improved
diagnostic approaches for PS during pregnancy.
Materials and methods
Sample collection
Peripheral plasma samples from pregnant females were
provided by Guangdong Provincial People's Hospital for
high-throughput sequencing. All subjects gave their verbal informed
consent before enrollment. The study was conducted in accordance
with the Declaration of Helsinki, and the protocol was approved by
the Ethics Committee of Guangdong Provincial People's Hospital.
Blood samples were obtained between September 2017 and November
2018 from pregnant females with children affected by PS, as well as
pregnant females carrying unaffected fetuses (n=6 in each group).
All blood samples were drawn 1–2 days before delivery. PS was
diagnosed using a postnatal cardiac color ultrasound. Both groups
consisted of three male infants and three female infants. The mean
age of pregnant females in the PS and control groups was 33.3 and
29 years, respectively.
Isolation of EVs
Plasma EVs were extracted using an exoRNeasy
Serum/Plasma Midi kit (Qiagen GmbH) and ~1 ml plasma was
centrifuged at 16,000 × g for 10 min at 4°C. Cellular materials and
coagulated proteins were removed, and the supernatant was
transferred into a new tube by careful aspiration. The re-suspended
EV liquid was subsequently used for characterization of EVs.
Nanoparticle tracking analysis
The size distribution of the isolated EVs was
analyzed using NanoSight NS300 (Malvern Panalytical). Particles
were automatically tracked and sized based on Brownian motion and
the diffusion coefficient. After isolation, EVs were diluted in 1
ml of exosome-free PBS, and the mixture was slowly injected into a
clean particle-free sample pool to avoid formation of bubbles. The
sample pool was covered and placed into the instrument.
Manipulations were performed according to the manufacturers
instructions. Three recordings were carried out for each sample and
results are presented as an average of these recordings.
Transmission electron microscopy
(TEM)
For TEM, 3 µl of EV pellet were placed on 200-mesh
EM copper grids for 5 min, incubated for 5 min at room temperature,
and then subjected to standard uranyl acetate staining at room
temperature for 1–2 min. The grids were then washed three times
with PBS and allowed to semi-dry at room temperature. Subsequently,
the grids were visualized with a transmission electron microscope
(H7650; Hitachi, Ltd.) at magnification, ×300, using digital
micrograph software (v3.8; Gatan, Inc.).
Western blotting
EV lysate supernatants were prepared, total protein
was extracted using Exosome cracking solution (Shanghai Umibio
Biotechnology), and protein concentrations were determined using a
bicinchoninic acid protein assay kit (Thermo Fisher Scientific,
Inc.). Proteins (60 µl) were separated via 10% SDS-PAGE, and
subsequently transferred onto PVDF membranes (EMD Millipore). Then,
5% non-fat milk in TBS with 0.05% Tween-20 (TBST) buffer was used
to block the membranes for 1 h at room temperature. The membranes
were incubated with rabbit anti-CD63 (1:500; cat. no. sc-5275;
Santa Cruz Biotechnology, Inc.), anti-CD9 (1:1,000; cat. no.
ab263019; Abcam) and anti-calnexin antibodies (1:1,000; cat. no.
ab22595; Abcam) at 4°C overnight, washed with TBST, then incubated
with horseradish peroxidase-conjugated goat anti-rabbit secondary
antibody (1:5,000; cat. no. 7074S; Cell Signaling Technology,
Inc.). Protein bands were visualized with an ECL reagent (Merck
KGaA) using an automatic imager (GE Healthcare).
Long RNA sequencing and data
processing
Long RNAs from EVs were amplified by Epi™ longRNA
Ampli kit (Epibiotek). RNA sequencing libraries were constructed
using SMARTer Stranded Total RNA-Seq kit (cat. no. 634413; Clontech
Laboratories, Inc.). Adapters and low-quality bases were removed
using Trimmomatic software (v0.36) (15). Clean RNA sequencing data were
aligned to the GRCh38 human genome (hg38) downloaded from Ensembl
(www.ensembl.org) using HISAT2 software (v2.1.0)
(16). Genes were annotated using
GENCODE annotation (v25; www.gencodegenes.org), and the read count for each
gene was obtained using featureCounts (v1.6.3;
subread.sourceforge.net). Clean read counts were further normalized
using the fragments per kilobase per million mapped reads method.
Moreover, underexpressed genes in any sample were filtered out.
Cluster analysis was performed using the flashClust R (v1.01-2-2)
package (17).
Construction of co-expression modules
of CHD
In weighted gene co-expression network analysis
(WGCNA), the connectivity between two genes is the β power of their
correlation coefficient. The β value determined both the scale-free
topology fitting index and the mean connectivity of genes. The β
value for scale-free topology fitting index was ≥0.8 by plotting
the index against soft thresholds, which indicated that the
topology of the network was scale-free, or independent, and
simultaneously, the mean connectivity of genes was as high as
possible, and it may result in several modules in the subsequent
analysis for an insignificant mean connectivity. To understand
relationship between them, the softConnectivity function from WGCNA
package (v1.63) (18) was used,
and the number of randomly selected genes was set to 5,000, while
other parameters were set as default. The power was calculated by
the pickSoftThreshold function in the WGCNA package. The expression
values were compiled using the collapseRows function in the WGCNA
package. Cluster analysis was subsequently performed using the
flashClust function.
Detection of hub genes and functional
enrichment analysis
Hub genes are defined as genes with high correlation
in candidate modules. In the present study, genes in the same
co-expression modules with module membership ≥0.8 were considered
as hub genes. Gene Ontology (GO; www.geneontology.org) enrichment analysis was carried
out for each module and the corresponding gene data were mapped to
the Database for Annotation, Visualization, and Integrated
Discovery (DAVID; david.ncifcrf.gov/summary.jsp). A corrected
P-value <0.05 was used as threshold. The most favorable module
in the current study was visualized by Cytoscape v3.6.6 software
(www.cytoscape.org), and the maximum intramodular
connectivity of genes was considered as intramodular hub genes.
Results
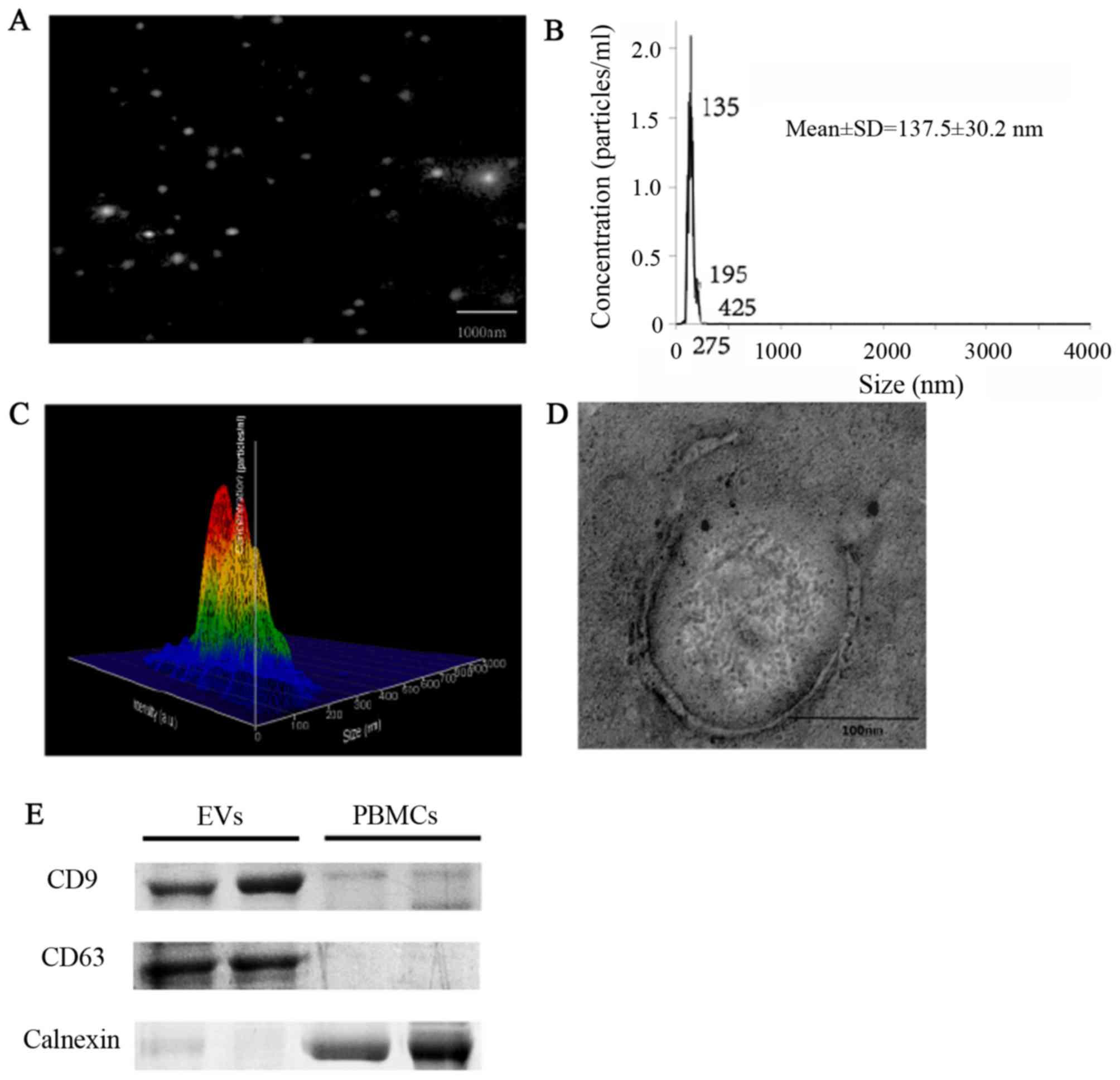
Isolation and characterization of
EVs
EVs were isolated and characterized morphologically
and phenotypically. Measurements demonstrated that the
concentration of the isolated particles was
9.55×109±3.27×107 particles/ml (data not
shown), and the mean diameter of the particles was 137.8±30.2 nm
(Fig. 1A-C). EVs were round-shaped
and membrane-enclosed (Fig. 1D).
The exosomal markers CD63 and CD9 were detected in the isolated
vesicles, but not in peripheral blood mononuclear cells, whereas
the expression of calnexin, the marker of peripheral blood
mononuclear cells, showed the opposite results (Fig. 1E).
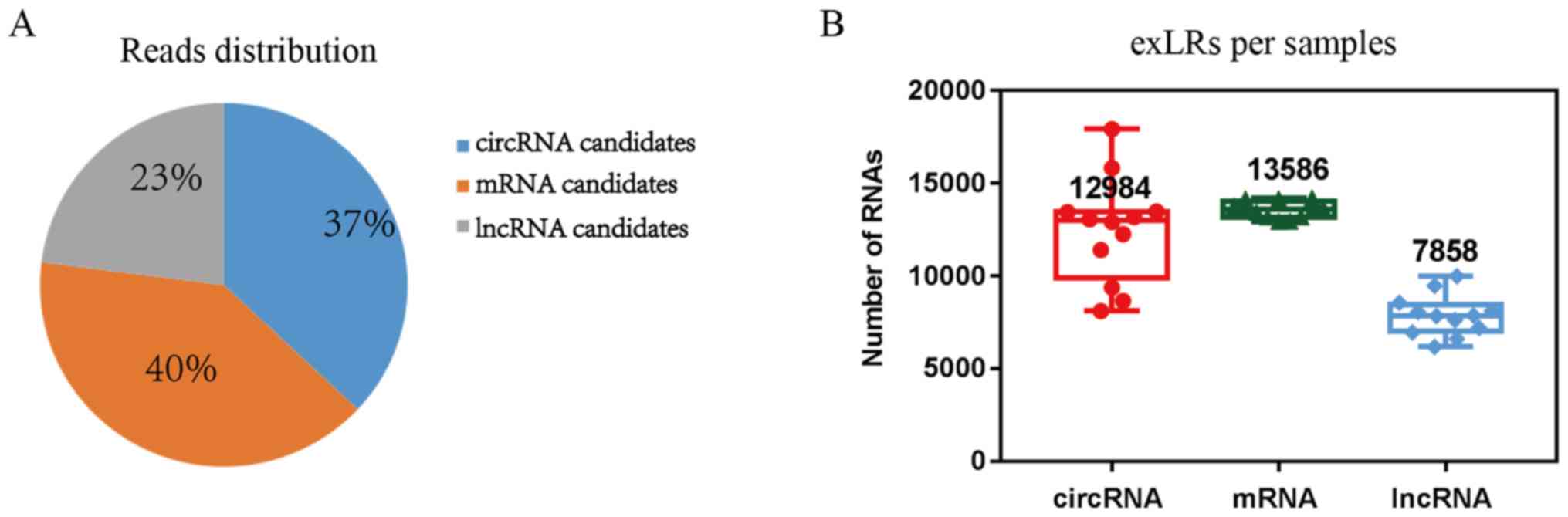
EVLR-sequencing
EVLR-sequencing yielded a median read count of 27.98
million mapped reads/sample. Overall, mRNA constituted 40% of total
mapped reads. Other types of RNA included 37% circRNAs and 23%
lncRNAs (Fig. 2A). On average,
13,586 mRNAs, 12,984 circRNAs, and 7,858 lncRNAs were detected from
the 12 samples (Fig. 2B). In
total, 19,391 genes were detected by EVLR-sequencing in the 12
samples. Moreover, 2,997 genes were filtered out as their
expression was too low in any sample. Thus, expression levels of
16,394 genes in 12 samples were used to construct the co-expression
network by the WGCNA package, of these 40% were associated with
mRNA, 37% with circRNA and 23% with lncRNA.
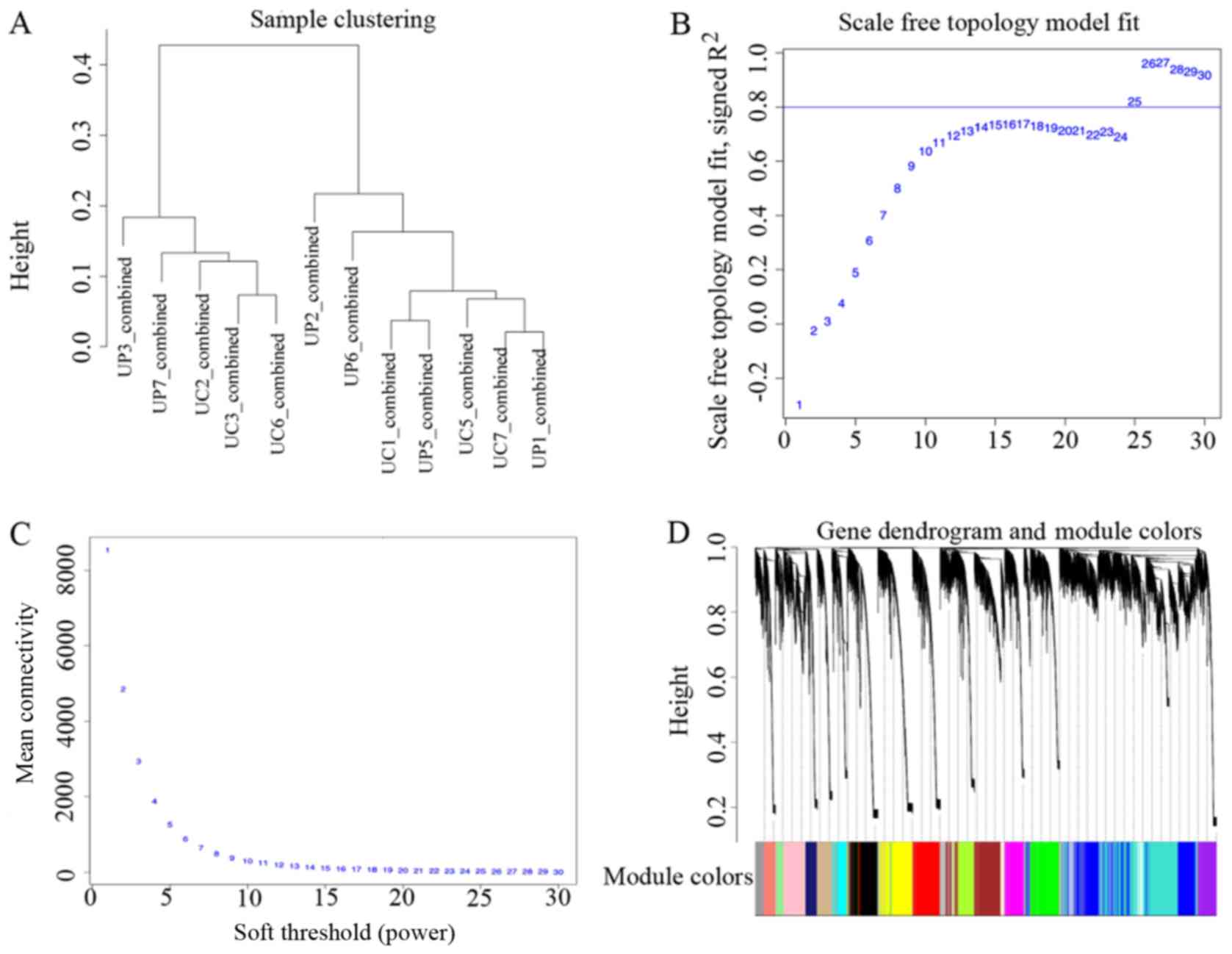
Construction of co-expression
modules
Cluster analysis was conducted on the mRNAs detected
in all 12 samples, using the flashClust package (Fig. 3A). Hub genes are displayed in gene
dendrograms, in which they positively correlated together in
different samples. Different soft-thresholding power values were
analyzed. When the power value reached 25, the scale-free topology
fitting index was ≥0.08 (Fig. 3B),
and as the power value increased, the connectivity declined
(Fig. 3C). Therefore, the power
value used to construct co-expression module was 25. A total of 26
distinct gene co-expression modules were constructed altogether
(Fig. 3D). The number of genes in
each module is listed in Table
I.
 | Table I.Number of genes in the 26
modules. |
Table I.
Number of genes in the 26
modules.
| Module colors | Frequency |
|---|
| Black | 808 |
| Blue | 1,909 |
| Brown | 1,187 |
| Cyan | 407 |
| Dark green | 68 |
| Dark grey | 56 |
| Dark brown | 124 |
| Dark turquoise | 61 |
| Green | 997 |
| Green-yellow | 606 |
| Grey | 1,153 |
| Grey 60 | 284 |
| Light cyan | 404 |
| Light green | 246 |
| Light yellow | 208 |
| Magenta | 648 |
| Midnight blue | 404 |
| Orange | 54 |
| Pink | 781 |
| Purple | 640 |
| Red | 943 |
| Royal blue | 176 |
| Salmon | 411 |
| Tan | 515 |
| Turquoise | 2,242 |
| Yellow | 1,062 |
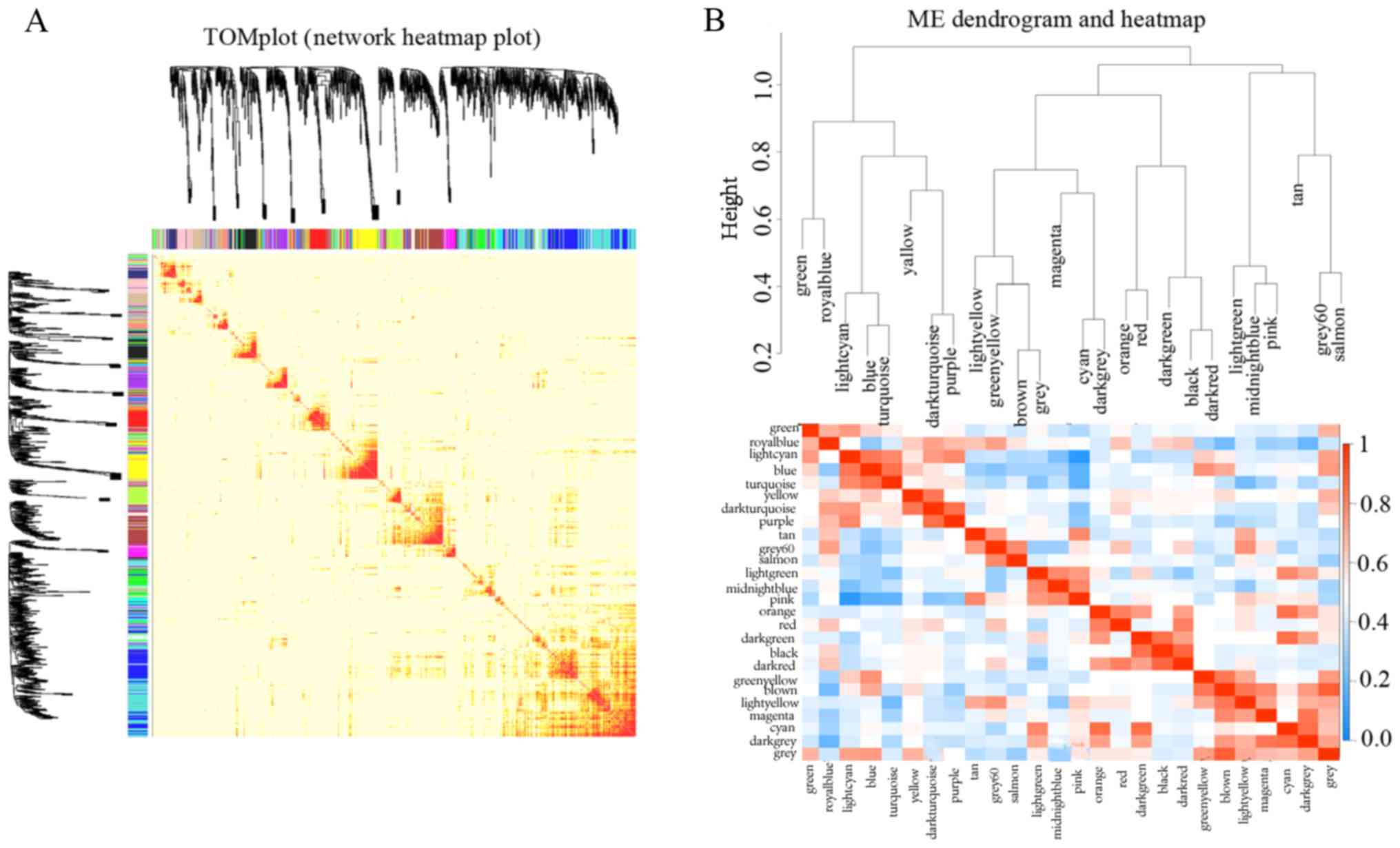
Analysis of co-expression modules
As shown on the network heatmap plot (Fig. 4A), each module showed an
independent expression to each other. Hence, we calculated
eigengenes of each module, so as to quantify their co-expression
similarity. Modules were clustered on the basis of their
correlation with each other, which was consistent with the heatmap
plot of the adjacencies (Fig.
4B).
Functional enrichment analysis and
module visualization
Results of GO enrichment analysis are summarized in
Table II. Biological process
involved in fetal cardiovascular malformations, such as regulation
of glucose transport and apoptotic signaling pathway were included
in the blue module. Furthermore, the expression of each module in
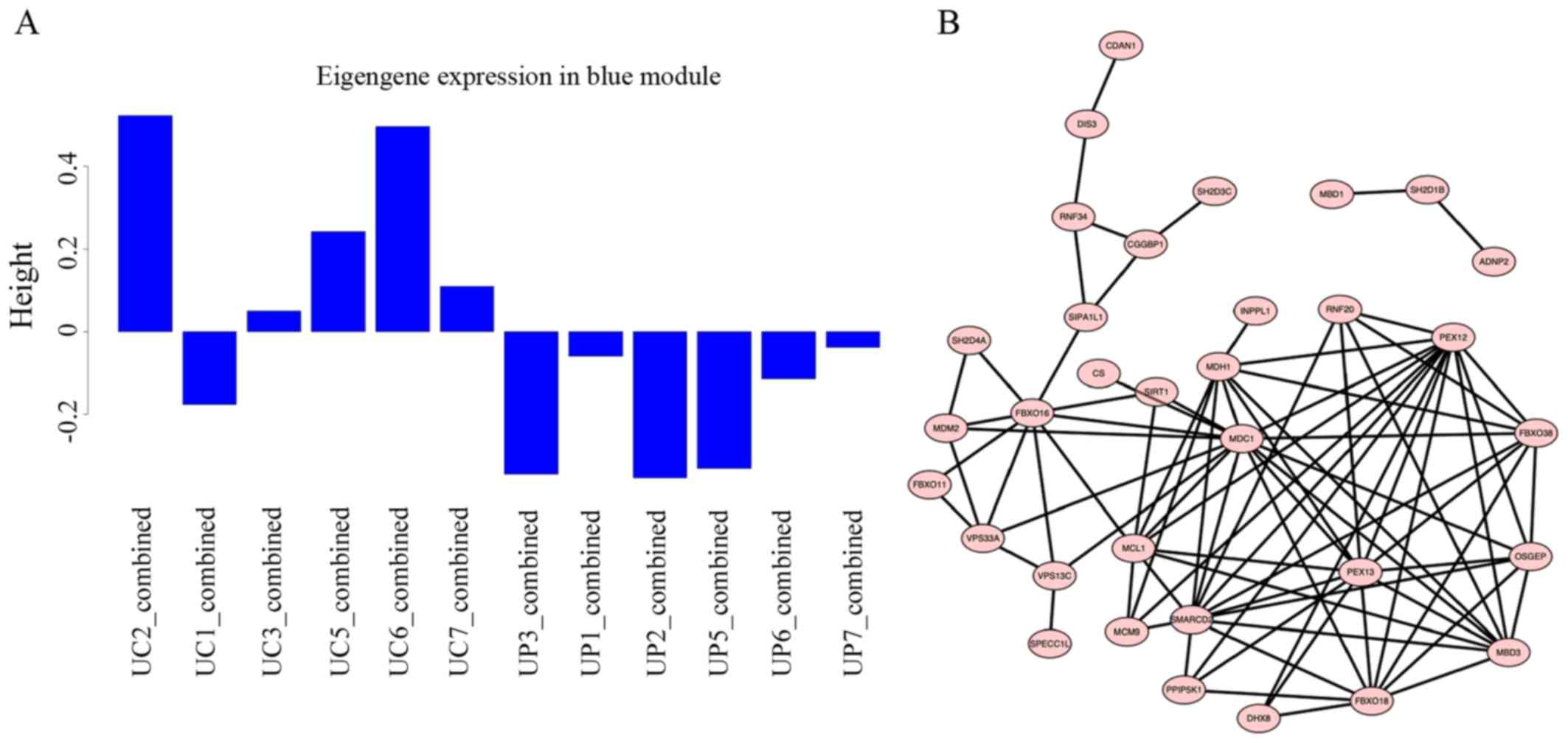
all samples was analyzed. Among all the 26 modules, the blue module
markedly differed between the two groups (Fig. 5A). Additionally, a total of 735
genes were involved in the blue module. These genes were enriched
in ‘regulation of glucose transport’, ‘glucose transport’,
‘glucocorticoid receptor signaling pathway’, ‘ATP-dependent
chromatin remodeling’, ‘histone deacetylation’, ‘histone H3-K4
methylation’, ‘DNA methylation’ and ‘apoptotic signaling pathways’.
These biological processes were not enriched in other modules. The
top 33 hub genes in the blue module, which have a high degree of
correlation, are represented in an interaction network using
Cytoscape software (Fig. 5B).
| Table II.GO enrichment analysis in blue, light
cyan and grey 60 co-expression modules. |
Table II.
GO enrichment analysis in blue, light
cyan and grey 60 co-expression modules.
| Module | GO ID | GO Term | Difgene | GeneInGO | P-value |
|---|
| Blue | GO:0010827 | regulation of
glucose transport | 10 | 31 |
1.76×10−7 |
|
| GO:0043044 | ATP-dependent
chromatin remodeling | 8 | 23 |
1.61×10−6 |
|
| GO:0016575 | histone
deacetylation | 8 | 34 |
4.06×10−5 |
|
| GO:0042921 | glucocorticoid
receptor signaling pathway | 4 | 8 |
1.47×10−4 |
|
| GO:0051568 | histone H3-K4
methylation | 6 | 23 |
2.07×10−4 |
|
| GO:0015758 | glucose
transport | 10 | 65 |
2.12×10−4 |
|
| GO:0006306 | DNA
methylation | 6 | 26 |
4.28×10−4 |
|
| GO:0097190 | apoptotic signaling
pathway | 13 | 130 |
1.86×10−3 |
| Light cyan | GO:0006413 | translational
initiation | 53 | 157 |
2.18×10−67 |
|
| GO:0006415 | translational
termination | 42 | 89 |
9.93×10−61 |
| Grey 60 | GO:0030097 | Hemopoiesis | 7 | 85 |
1.30×10−6 |
|
| GO:0048821 | erythrocyte
development | 7 | 19 |
6.02×10−6 |
Discussion
WGCNA is a method used to investigate the
relationship between gene expression and phenotype (18). In particular, WGCNA transforms gene
expression data into co-expression modules, providing insight into
signaling networks that may be responsible for phenotypic traits of
interest (19). Not only can it
illustrate how modules differ between control and experimental
groups, it can also be used to examine the functions of genes
within a particular module. WGCNA has been applied in the context
of several diseases, such as osteosarcoma, glioma and renal cell
carcinoma (20–23). In the present study, 26
co-expression modules were extracted from 16,394 genes using this
method. Among these, one particular module, referred to as the blue
module, displayed marked dissimilarity between PS group and normal
group. The blue module was enriched in a variety of biological
processes, which may influence fetal heart development, including
‘glucose transport’, ‘ATP-dependent chromatin remodeling’, ‘histone
deacetylation’, ‘histone H3-K4 methylation’, ‘DNA methylation’,
‘apoptotic signaling pathways’ and ‘glucocorticoid receptor
signaling pathway’.
Maternal glycemia is a risk factor for the
development of CHD in the fetus (24–27).
A previous study indicated that maternal blood glucose levels were
strongly associated with odds of tetralogy of Fallot (28). This highlights the need for further
epidemiological and mechanistic investigation into the risk
conferred by insulin signaling and glucose metabolism during early
pregnancy. In the present research, genes of glucose transport in
PS group were markedly downregulated compared with those in normal
group.
A previous study suggested that maternal epigenetic
changes were related to fetal cardiovascular malformations
(29). Another study used
genome-wide DNA methylation assays on placental samples and
identified a total of 80 highly accurate potential CpG sites for
detection of ventricular septal defects (30). These differentially methylated
genes were previously known to be associated with cardiovascular
development (31). Moreover, DNA
methylation of the glucocorticoid receptor gene promoter is an
epigenetic mechanism that can lead to abnormal development of the
fetal heart (32). MicroRNA
(miR)-29c-3p overexpression in the serum of pregnant females
inhibited embryonic P19 cell proliferation, promoted cell apoptosis
and differentiation and is associated with fetal CHD (33). Altogether, these studies suggested
that maternal epigenetic changes may play a pivotal role in heart
development and pathogenesis of fetal PS.
Glucocorticoid signaling plays an important role in
cardiac physiology (34). The
effects of glucocorticoids are mediated classically by the
glucocorticoid receptor (35).
Previous studies indicated that the glucocorticoid receptor in
cardiomyocytes is critical for normal development and function of
the heart (36–38). Maternal hypoxia caused a
significant increase of global methylation in the fetal heart,
which was sustained in 4-week-old human infants (30). Maternal hypoxia induced miR-210
production via hypoxia inducible factor-1α, which reduced the
expression of glucocorticoid receptors in the fetal heart, and led
to cardiomyocyte death (39). In
the present study, the expression of the glucocorticoid receptor
signaling pathway was reduced in the EVs of pregnant women carrying
fetuses with PS offspring.
The results of the present study suggested that
EVLRs may be used for the diagnosis of fetal PS. Hub genes involved
in ‘regulation of glucose transport’, ‘glucose transport’,
‘ATP-dependent chromatin remodeling’, ‘histone deacetylation’,
‘histone H3-K4 methylation’, ‘DNA methylation’, ‘apoptotic
signaling pathway’, and ‘glucocorticoid receptor signaling pathway’
were identified, which could represent potential biomarkers for PS.
The use of EVLR as a diagnostic would be non-invasive to the fetus.
Moreover, hub genes could be easily identified by reverse
transcription-quantitative PCR or targeted analysis sequencing from
the perinatal blood of pregnant females. Similar to cell-free RNA
tests, the latter is a non-invasive blood test for fetal
development (40). However, WGCNA
can only provide a cluster of hub genes rather than specific genes
related to disease. Thus, the hub genes identified in the present
study require further individual validation. Additionally, the
cohort enrolled in the present study was relatively small, and
further extensive validation is still needed to confirm the present
results.
In summary, a profile of EVLR was established for
healthy pregnant females and pregnant females with PS, and a gene
co-expression network was constructed to predict a cluster of
candidate genes involved in the diagnosis of fetal PS. The present
findings may provide a theoretical basis for novel, non-invasive
detection methods of aberrant fetal heart development.
Acknowledgements
The authors would like to thank Mr Xuemin Yang
(Guangzhou Epibiotek Co., Ltd.) for assisting in sequencing and
bioinformatics analysis.
Funding
The present study was supported by The Natural
Science Foundation of Guangdong (grant no. 2018A0303130266),
National Key R&D Program of China (grant no. 2018YFC1002600),
the Scientific and Technological Projects of Guangdong (grant nos.
2017A070701013 and 2017B090904034) and the Science and
Technological Program of Guangzhou (grant no. 201704020126).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors contributions
ZH, XiaohL, JZ and JimC conceived the study. ZH, MT,
FH and XiaohL designed the method. FH provided samples. ZH, XiaohL,
MQ, JinC, YO, XiaoqL, CZ and HY prepared the software and performed
data analysis. ZH wrote the original draft. XiaohL reviewed and
edited the draft. JZ and JimC supervised and administrated the
project. XiaohL, JZ and JimC provided funding. All authors read and
approved the final manuscript.
Ethics approval and consent to
participate
The present study was approved by The Ethics
Committee of Guangdong Provincial People's Hospital and conducted
in accordance with the Declaration of Helsinki. All participants
gave their informed oral consent for participation in the
study.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Liu Y, Chen S, Zühlke L, Black GC, Choy
MK, Li N and Keavney BD: Global birth prevalence of congenital
heart defects 1970–2017: Updated systematic review and
meta-analysis of 260 studies. Int J Epidemiol. 48:455–463. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Cuypers JA, Witsenburg M, van der Linde D
and Roos-Hesselink JW: Pulmonary stenosis: Update on diagnosis and
therapeutic options. Heart. 99:339–347. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Patel AB, Ratnayaka K and Bergersen L: A
Review: Percutaneous pulmonary artery stenosis therapy:
state-of-the-art and look to the future. Cardiol Young. 29:93–99.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Webber DM, MacLeod SL, Bamshad MJ, Shaw
GM, Finnell RH, Shete SS, Witte JS, Erickson SW, Murphy LD and
Hobbs C: Developments in our understanding of the genetic basis of
birth defects. Birth Defects Res A Clin Mol Teratol. 103:680–691.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Bahado-Singh RO, Zaffra R, Albayarak S,
Chelliah A, Bolinjkar R, Turkoglu O and Radhakrishna U: Epigenetic
markers for newborn congenital heart defect (CHD). J Matern Fetal
Neonatal Med. 29:1881–1887. 2016.PubMed/NCBI
|
|
6
|
van Niel G, DAngelo G and Raposo G:
Shedding light on the cell biology of extracellular vesicles. Nat
Rev Mol Cell Biol. 19:213–228. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Shah R, Patel T and Freedman JE:
Circulating extracellular vesicles in human disease. N Engl J Med.
379:958–966. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Tong M, Kleffmann T, Pradhan S, Johansson
CL, DeSousa J, Stone PR, James JL, Chen Q and Chamley LW: Proteomic
characterization of macro-, micro- and nano-extracellular vesicles
derived from the same first trimester placenta: Relevance for
feto-maternal communication. Hum Reprod. 31:687–699. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Adam S, Elfeky O, Kinhal V, Dutta S, Lai
A, Jayabalan N, Nuzhat Z, Palma C, Rice GE and Salomon C: Review:
Fetal-maternal communication via extracellular vesicles -
Implications for complications of pregnancies. Placenta. 54:83–88.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Colombo M, Raposo G and Théry C:
Biogenesis, secretion, and intercellular interactions of exosomes
and other extracellular vesicles. Annu Rev Cell Dev Biol.
30:255–289. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Jin J and Menon R: Placental exosomes: A
proxy to understand pregnancy complications. Am J Reprod Immunol.
79:e127882018. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zhou R, Chen KK, Zhang J, Xiao B, Huang Z,
Ju C, Sun J, Zhang F, Lv XB and Huang G: The decade of exosomal
long RNA species: An emerging cancer antagonist. Mol Cancer.
17:752018. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Del Re M, Biasco E, Crucitta S, Derosa L,
Rofi E, Orlandini C, Miccoli M, Galli L, Falcone A, Jenster GW, et
al: The detection of androgen receptor splice variant 7 in
plasma-derived exosomal RNA strongly predicts resistance to
hormonal therapy in metastatic prostate cancer patients. Eur Urol.
71:680–687. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Li Y, Zhao J, Yu S, Wang Z, He X, Su Y,
Guo T, Sheng H, Chen J, Zheng Q, et al: Extracellular vesicles long
RNA sequencing reveals abundant mRNA, circRNA, and lncRNA in human
blood as potential biomarkers for cancer diagnosis. Clin Chem.
65:798–808. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Bolger AM, Lohse M and Usadel B:
Trimmomatic: A flexible trimmer for Illumina sequence data.
Bioinformatics. 30:2114–2120. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kim D, Langmead B and Salzberg SL: HISAT:
A fast spliced aligner with low memory requirements. Nat Methods.
12:357–360. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Langfelder P, Horvath S and Fast R: Fast R
functions for robust correlations and hierarchical clustering. J
Stat Softw. 46:i112012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Langfelder P and Horvath S: WGCNA: An R
package for weighted correlation network analysis. BMC
Bioinformatics. 9:5592008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Shi Z, Derow CK and Zhang B: Co-expression
module analysis reveals biological processes, genomic gain, and
regulatory mechanisms associated with breast cancer progression.
BMC Syst Biol. 4:742010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Liu X, Hu AX, Zhao JL and Chen FL:
Identification of key gene modules in human osteosarcoma by
co-expression analysis weighted gene co-expression network analysis
(WGCNA). J Cell Biochem. 118:3953–3959. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Deng J, Kong W, Mou X, Wang S and Zeng W:
Identifying novel candidate biomarkers of RCC based on WGCNA
analysis. Per Med. 15:381–394. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Maertens A, Tran V, Kleensang A and
Hartung T: Weighted Gene correlation network analysis (WGCNA)
Reveals novel transcription factors associated with Bisphenol a
dose-response. Front Genet. 9:5082018. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Xi X, Chu Y, Liu N, Wang Q, Yin Z, Lu Y
and Chen Y: Joint bioinformatics analysis of underlying potential
functions of hsa-let-7b-5p and core genes in human glioma. J Transl
Med. 17:1292019. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Cypryk K, Bartyzel L, Zurawska-Klis M,
Mlynarski W, Szadkowska A, Wilczynski J, Nowakowska D, Wozniak LA
and Fendler W: Continuous glucose monitoring in type 1 diabetes
pregnancy shows that fetal heart rate correlates with maternal
glycemia. Diabetes Technol Ther. 17:619–624. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Øyen N, Diaz LJ, Leirgul E, Boyd HA,
Priest J, Mathiesen ER, Quertermous T, Wohlfahrt J and Melbye M:
Prepregnancy diabetes and offspring risk of congenital heart
disease: A nationwide cohort study. Circulation. 133:2243–2253.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Hoang TT, Marengo LK, Mitchell LE,
Canfield MA and Agopian AJ: Original findings and updated
meta-analysis for the association between maternal diabetes and
risk for congenital heart disease phenotypes. Am J Epidemiol.
186:118–128. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Helle EIT, Biegley P, Knowles JW, Leader
JB, Pendergrass S, Yang W, Reaven GR, Shaw GM, Ritchie M and Priest
JR: First trimester plasma glucose values in women without diabetes
are associated with risk for congenital heart disease in offspring.
J Pediatr. 195:275–278. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Priest JR, Yang W, Reaven G, Knowles JW
and Shaw GM: Maternal Midpregnancy glucose levels and risk of
congenital heart disease in offspring. JAMA Pediatr. 169:1112–1116.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Moore-Morris T, van Vliet PP, Andelfinger
G and Puceat M: Role of epigenetics in cardiac development and
congenital diseases. Physiol Rev. 98:2453–2475. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Radhakrishna U, Albayrak S, Zafra R, Baraa
A, Vishweswaraiah S, Veerappa AM, Mahishi D, Saiyed N, Mishra NK,
Guda C, et al: Placental epigenetics for evaluation of fetal
congenital heart defects: Ventricular Septal Defect (VSD). PLoS
One. 14:e02002292019. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Muntean I, Togănel R and Benedek T:
Genetics of congenital heart disease: Past and Present. Biochem
Genet. 55:105–123. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Song R, Hu XQ and Zhang L: Glucocorticoids
and programming of the microenvironment in heart. J Endocrinol.
242:T121–T133. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Chen T, Li SJ, Chen B, Huang Q, Kong XY,
Shen C, Gu HT and Wang XW: Akt3 is a target of miR-29c-3p and
serves an important function in the pathogenesis of congenital
heart disease. Int J Mol Med. 43:980–992. 2019.PubMed/NCBI
|
|
34
|
Fowden AL and Forhead AJ: Glucocorticoids
as regulatory signals during intrauterine development. Exp Physiol.
100:1477–1487. 2015. View
Article : Google Scholar : PubMed/NCBI
|
|
35
|
Weikum ER, Knuesel MT, Ortlund EA and
Yamamoto KR: Glucocorticoid receptor control of transcription:
Precision and plasticity via allostery. Nat Rev Mol Cell Biol.
18:159–174. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Rog-Zielinska EA, Thomson A, Kenyon CJ,
Brownstein DG, Moran CM, Szumska D, Michailidou Z, Richardson J,
Owen E, Watt A, et al: Glucocorticoid receptor is required for
foetal heart maturation. Hum Mol Genet. 22:3269–3282. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Rog-Zielinska EA, Craig MA, Manning JR,
Richardson RV, Gowans GJ, Dunbar DR, Gharbi K, Kenyon CJ, Holmes
MC, Hardie DG, et al: Glucocorticoids promote structural and
functional maturation of foetal cardiomyocytes: A role for PGC-1α.
Cell Death Differ. 22:1106–1116. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Oakley RH and Cidlowski JA: Glucocorticoid
signaling in the heart: A cardiomyocyte perspective. J Steroid
Biochem Mol Biol. 153:27–34. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Martinez SR, Ma Q, Dasgupta C, Meng X and
Zhang L: MicroRNA-210 suppresses glucocorticoid receptor expression
in response to hypoxia in fetal rat cardiomyocytes. Oncotarget.
8:80249–80264. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Ngo TTM, Moufarrej MN, Rasmussen MH,
Camunas-Soler J, Pan W, Okamoto J, Neff NF, Liu K, Wong RJ, Downes
K, et al: Noninvasive blood tests for fetal development predict
gestational age and preterm delivery. Science. 360:1133–1136. 2018.
View Article : Google Scholar : PubMed/NCBI
|