Introduction
Ischemic heart disease remains the leading cause of
death and is a critical threat to public health worldwide (1). Considerable advancements have been
made in the acute care of patients with ischemic heart disease,
particularly timely reperfusion interventions, through either
percutaneous coronary intervention (PCI) or coronary artery bypass
graft surgery (CABG) (2). However,
the clinical outcome of these therapeutic developments remains
unsatisfactory (3), which is
largely due to myocardial damage, including ischemia/reperfusion
(I/R) injury brought about by timely reperfusion treatment
(4).
Myocardial I/R is known to result in myocardial
damage, including necrosis and/or tissue degradation, heart
failure, myocardial stunning, no-reflow and reperfusion arrhythmia
(5). These factors have a
significant impact on the clinical outcomes of patients with
ischemic heart disease, including myocardial infarct size and
dysfunction (6). Although the
underlying mechanisms of I/R-induced myocardial injury are not
completely understood (7), various
studies have reported that a series of pathophysiological
alterations are involved in this process (8). Among these, cellular
hypoxia/reoxygenation may simulate a state of I/R (9), where cellular oxidative stress serves
an important role in the activation of diverse pathways resulting
in myocardial injury (10).
Oxidative stress has been reported to impair mitochondrial function
(11) and promote the excessive
production of reactive oxygen species (ROS) (12). This results in an imbalance between
ROS production and radical scavenging, and thus a disturbed redox
status (13). It is evident that
this altered redox status may activate a series of cellular
processes, including apoptosis, which is reported to be critically
involved in I/R-associated myocardial injury (14).
In this context, in order to improve the clinical
outcome of ischemic heart disease, measures that counteract the
cellular mechanisms of I/R-induced myocardial damage are of great
importance. In previous decades, effective measures for protecting
the myocardium from I/R injury have been investigated (5). Pre- and post-conditioning with
physical or pharmacological strategies have been extensively
researched (15), and although the
results of experimental studies appear promising, those of clinical
trials have been disappointing (16). Therefore, the investigation of
effective treatments to protect against myocardial I/R injury are
of considerable scientific and clinical significance.
Dimethyl fumarate (DMF) is derived from fumarate
acid esters and has historically been used to treat patients with
relapsing-remitting multiple sclerosis (17). In addition, DMF has been reported
to exert protective effects against I/R injury in the brain
(18). DMF is recognized as a
potent antioxidant (19) that
exerts profound effects on the modulation of apoptosis (20). It has also been shown to activate
the nuclear factor erythroid 2-related factor 2 (Nrf2) signaling
pathway. Nrf2 is located in the cytoplasm and is bound to the
cytosolic protein Kelch-like ECH-associated protein 1 (Keap1),
which subsequently regulates its activity. The Nrf2/Keap1 complex
is rapidly recycled via a series of ubiquitination and proteasomal
degradation events, and reactive sulfhydryls that act on Keap1 are
readily oxidized by ROS and electrophiles, thereby releasing Nrf2,
and allowing it to translocate into the nucleus. Here, Nrf2
activates target genes that possess antioxidant response elements
(AREs) in their promoter regions (21), and that regulate downstream
mitochondria-mediated apoptosis via caspase-3 (22). Therefore, DMF may exert protective
effects against myocardial I/R injury. However, to the best of our
knowledge, this has not previously been reported. The present study
aimed to investigate the potential protective effects of DMF on
I/R-induced oxidative damage and signaling in a cardiomyocyte
culture setting.
Materials and methods
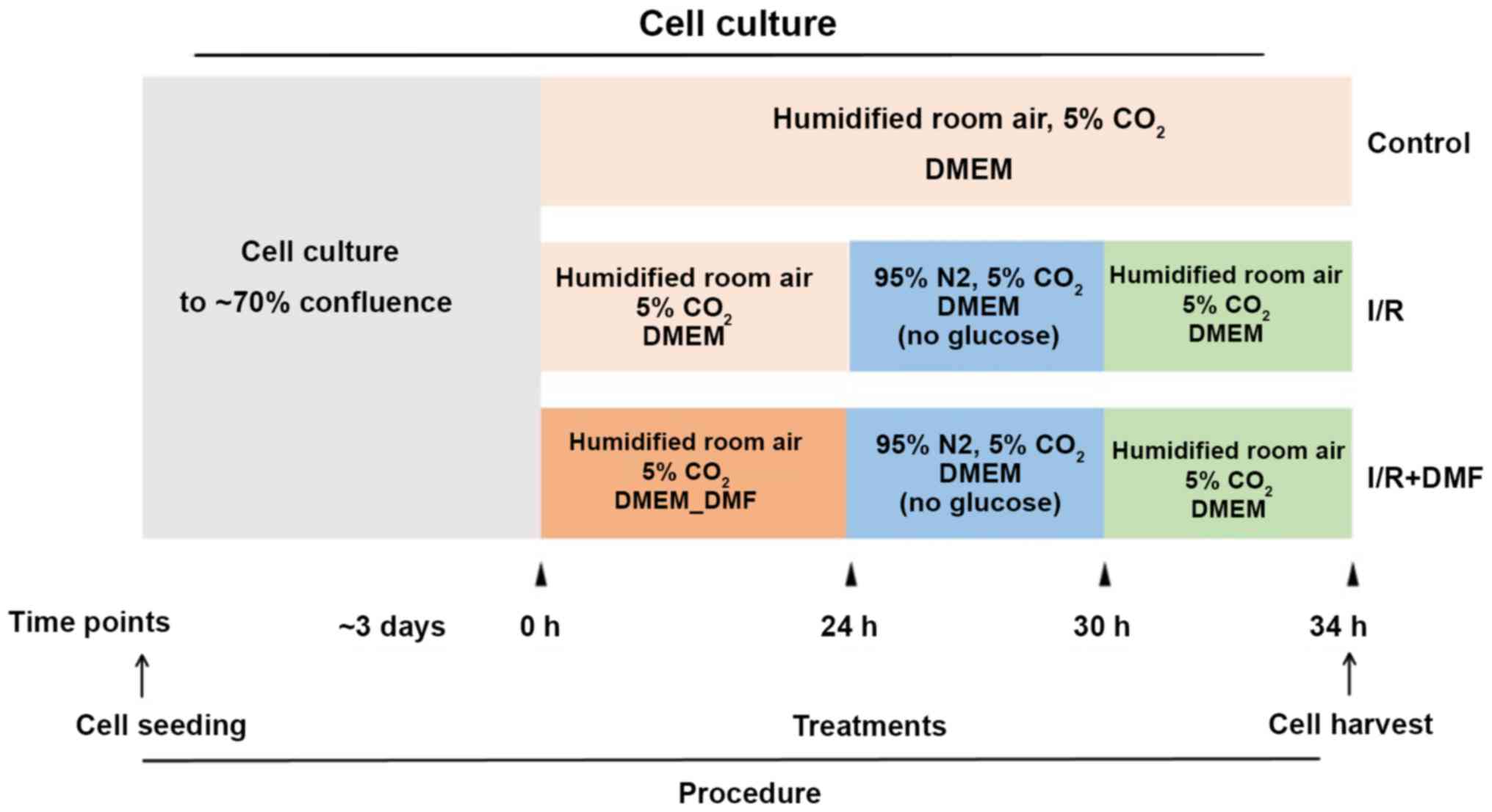
Experimental protocol
The experimental protocol is depicted in Fig. 1. H9c2 cells were cultured to 70%
confluence and grouped according to the associated treatment
conditions. At the indicated time points, the media and culture
conditions were changed accordingly.
Cell culture and treatments
Rat H9c2 cardiomyoblast cells were purchased from
the American Type Culture Collection and cultured in Dulbecco's
modified Eagle's medium (DMEM) supplemented with 10% fetal bovine
serum and penicillin/streptomycin (100 U/ml/100 mg/ml; all NCM
Biotech) at 37°C (5% CO2) in a humidified incubator.
Firstly, the rat H9C2 cardiomyocytes were randomly divided into 5
groups: i) Control group; ii) 5 µM DMF; iii) 10 µM DMF; iv) 20 µM
DMF; v) 40 µM DMF. Cell Counting Kit-8 (CCK-8; BioTool Service
GmbH) was used determine the ideal concentration of DMF that was
not toxic to cells, it was found that the safest concentration of
DMF was 20 µM. Secondly, at 70% confluence, the cells were
categorized into different groups (n=3 wells/group) according to
treatment. The control group cells were cultured for a further 34
h; the I/R group cells were cultured for a further 24 h and were
then transferred to glucose-free DMEM and cultured in an anaerobic
chamber (95% N2, 5% CO2) for 6 h, followed by 4 h
culture under the same conditions as the control group; the DMF
group was treated in the same way as the I/R group with the
exception of DMF (20 µM) pretreatment (Sigma-Aldrich; Merck KGaA)
for 24 h. At the end of the culture period, the cells and media
were harvested for experimentation.
Cell viability
Cell viability was determined using the CCK-8
(BioTool Service GmbH). The cultured cells were washed three times
with PBS and 10 µl CCK-8 solution was added to each well prior to
incubation for 1 h at 37°C. Absorbance was measured at 450 nm using
a microplate reader (Tecan Group, Ltd.). Absorbance values were
recorded relative to the control group, which was set at 100%.
Lactate dehydrogenase (LDH)
release
The harvested cell culture media were collected and
LDH levels were quantified using a LDH colorimetric assay kit
(Beyotime Institute of Biotechnology), according to the
manufacturer's protocol. Briefly, 120 µl cell culture medium was
mixed with 60 µl LDH working solution and incubated at 25°C for 30
min. The absorbance at 490 nm was determined using a microplate
reader (as aforementioned).
ROS production
ROS production was determined using a ROS Assay kit
(Beyotime Institute of Biotechnology), according to the
manufacturer's protocol. DCFH-DA was diluted in serum-free medium
(1:1,000) to a working concentration of 10 µM. The cells
(1×106) were incubated at 37°C for 20 min, and then
washed three times with serum-free cell culture medium to remove
excess DCFH-DA. The fluorescence intensity of the stained cells was
observed using an inverted fluorescence microscope (magnification,
×200).
Flow cytometric analysis
Cell apoptotic rates were detected by flow cytometry
using an Annexin V/propidium iodide (PI) apoptosis detection kit
(Hanteng Biology). Annexin V is a sensitive indicator of early
apoptosis (23), and PI is a
nucleic acid-binding dye that detects late apoptotic and dead cells
(23). Samples (1×104)
were double-stained with Annexin V (2 µl) and PI (5 µl) for 15 min
at 25°C in the dark, and then evaluated using a flow cytometer (BD
FACSCanto II; Becton, Dickinson and Company) and FlowJo v10.4
software (FlowJo, LLC). Through flow cytometry, cellular vital
conditions could be assessed, and cells were differentiated into
dead, late apoptotic, early apoptotic and living cells (24).
TdT-mediated dUTP-biotin nick end
labeling (TUNEL) analysis
Cellular apoptosis was also determined at the
nuclear level using the TUNEL method. TUNEL staining was performed
using an In Situ Cell Death Detection kit (Roche
Diagnostics), according to the manufacturer's protocol.
TUNEL-positive cells were detected under an inverted fluorescence
microscope and analyzed using ImageJ Software 1.8.0 (National
Institutes of Health), and the optical staining values were
calculated.
Western blot analysis
The harvested cells were homogenized in ice-cold
lysis buffer (SDS-PAGE Sample Loading buffer; Beyotime Institute of
Biotechnology) for protein extraction. Sample protein
concentrations were determined using a BCA Protein Assay kit
(Beyotime Institute of Biotechnology). Proteins (20 µg) were loaded
and separated by SDS-PAGE (10 and 12% gels), and were then
transferred onto PVDF membranes, which were blocked with 4% BSA
(Sigma-Aldrich; Merck KGaA) at 25°C and 1 h. Membranes were then
incubated at 4°C for 12 h with primary antibodies against the
following proteins: GAPDH (1:1,000; cat. no. GB12002; Wuhan
Servicebio Technology Co., Ltd.), cleaved caspase-3 (1:500; cat.
no. 9661T; Cell Signaling Technology, Inc.), B-cell lymphoma 2
(Bcl-2; 1:500; cat. no. GTX100064; Genetex, Inc.), Bcl-2-associated
X protein (Bax; 1:500; cat. no. 2772T; Cell Signaling Technology,
Inc.), AKT (1:1,000; cat. no. 4691T; Cell Signaling Technology,
Inc.), phosphorylated (p)-AKT (1:1,000; cat. no. 4060T; Cell
Signaling Technology, Inc.), Nrf2 (1:500; cat. no. GTX103322;
Genetex, Inc.), NAD(P)H quinone dehydrogenase 1 (NQO1; 1:500; cat.
no. GTX100235; Genetex, Inc.) and heme oxygenase 1 (HO-1; 1:500;
cat. no. GTX101147; Genetex, Inc.). Blots were then incubated at
25°C for 1 h with a horseradish peroxidase-conjugated secondary
antibody (1:1,000; cat. no. GB23302; Wuhan Servicebio Technology
Co., Ltd.) and underwent chemiluminescence detection with the
Molecular Imager ChemiDoc XRS system (Bio-Rad Laboratories, Inc.).
Densitometric semi-quantification was performed using Image Lab 3.0
(Bio-Rad Laboratories, Inc.).
Statistical analysis
Data are presented as the mean ± standard deviation.
Results were analyzed by one-way ANOVA followed by Tukey's post hoc
test using SPSS version 20 (IBM Corp.). P<0.05 was considered to
indicate a statistically significant difference.
Results
Effects of DMF on I/R-induced cellular
damage
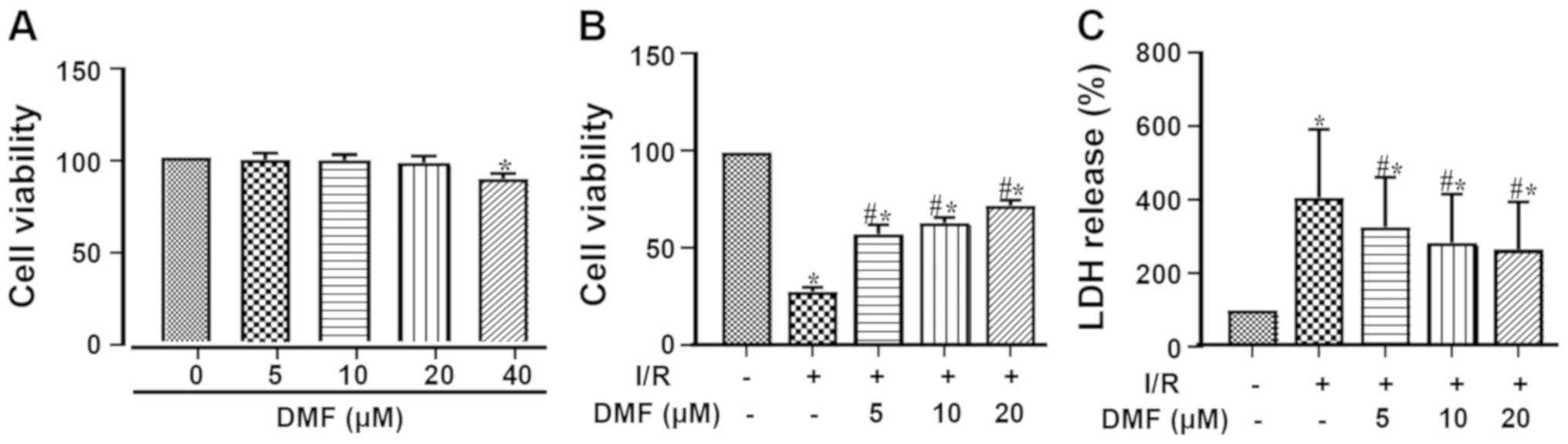
The cellular toxicity of different concentrations of
DMF was assessed using the CCK-8 assay (Fig. 2A). Treatment with 40 µM DMF
resulted in a significant decrease in cell viability; therefore, 20
µM DMF was used for subsequent experimentation. Compared with the
control group, I/R injury markedly decreased cell viability
(Fig. 2B). Treatment with DMF
significantly attenuated this effect in a dose-dependent manner,
such that the viability of the DMF-treated cells was significantly
higher than that of the I/R-treated cells. Furthermore, LDH release
into the culture media was significantly elevated by I/R induction
(Fig. 2C) and was dose-dependently
attenuated by DMF.
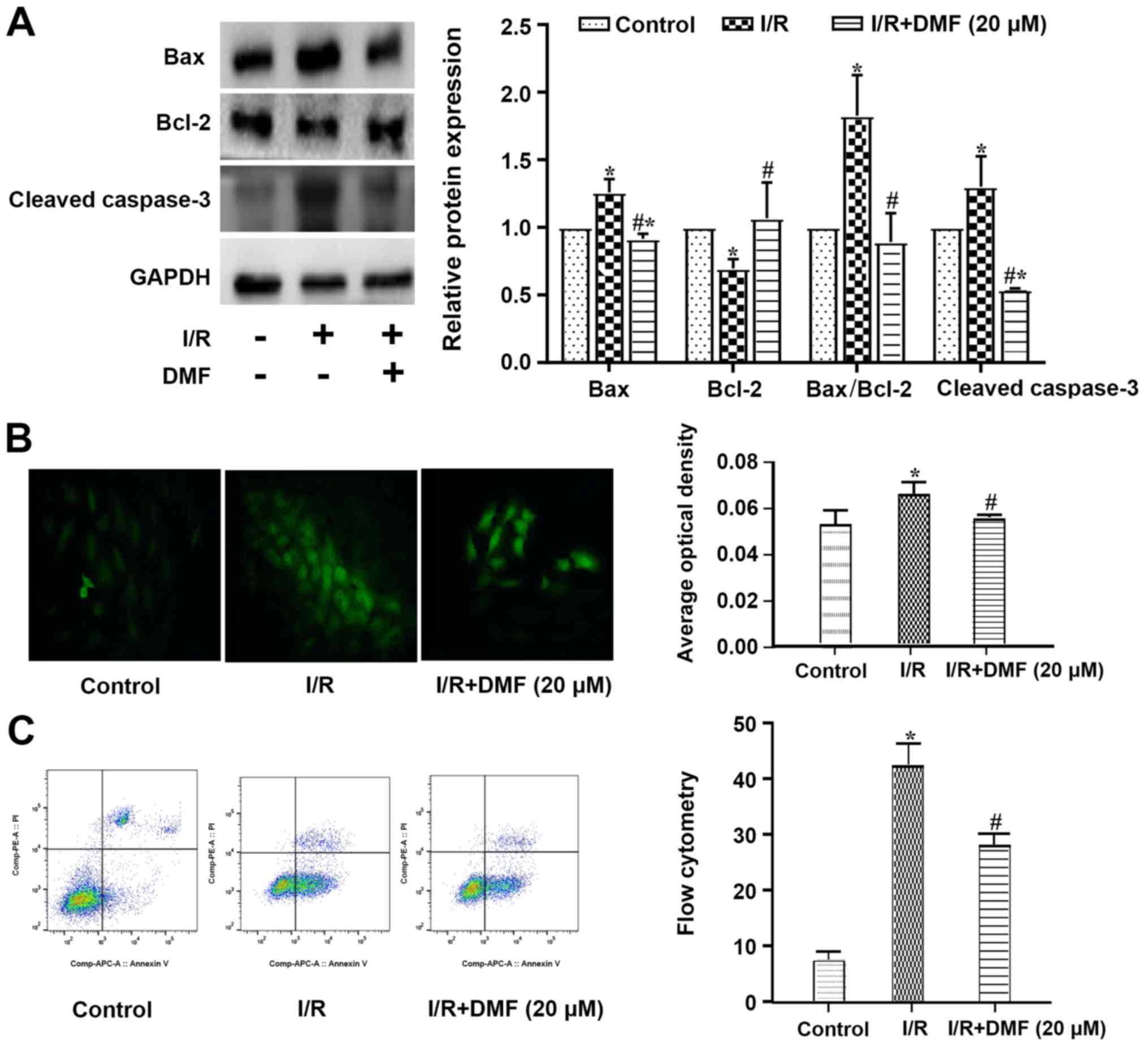
Effects of DMF on apoptosis
Compared with the control cells, a significant
increase in Bax and a decrease in Bcl-2 were observed in
I/R-treated cells, such that the apoptotic Bax/Bcl-2 index was
significantly elevated (Fig. 3A).
The expression levels of cleaved caspase-3 were also increased in
the I/R-treated cells compared with those in the control group. The
addition of DMF to the I/R-treated cells significantly decreased
Bax, cleaved caspase-3 and the apoptotic index, and increased the
expression levels of Bcl-2.
A greater number of TUNEL-positive cells was
observed in the I/R-treated group than in the control group
(Fig. 3B), and this was perturbed
by treatment with 20 µM DMF. The original flow cytometric data are
depicted in Fig. 3C. The
proportion of cells in quadrant 3 was significantly decreased in
I/R + DMF-treated cells compared with in the I/R group.
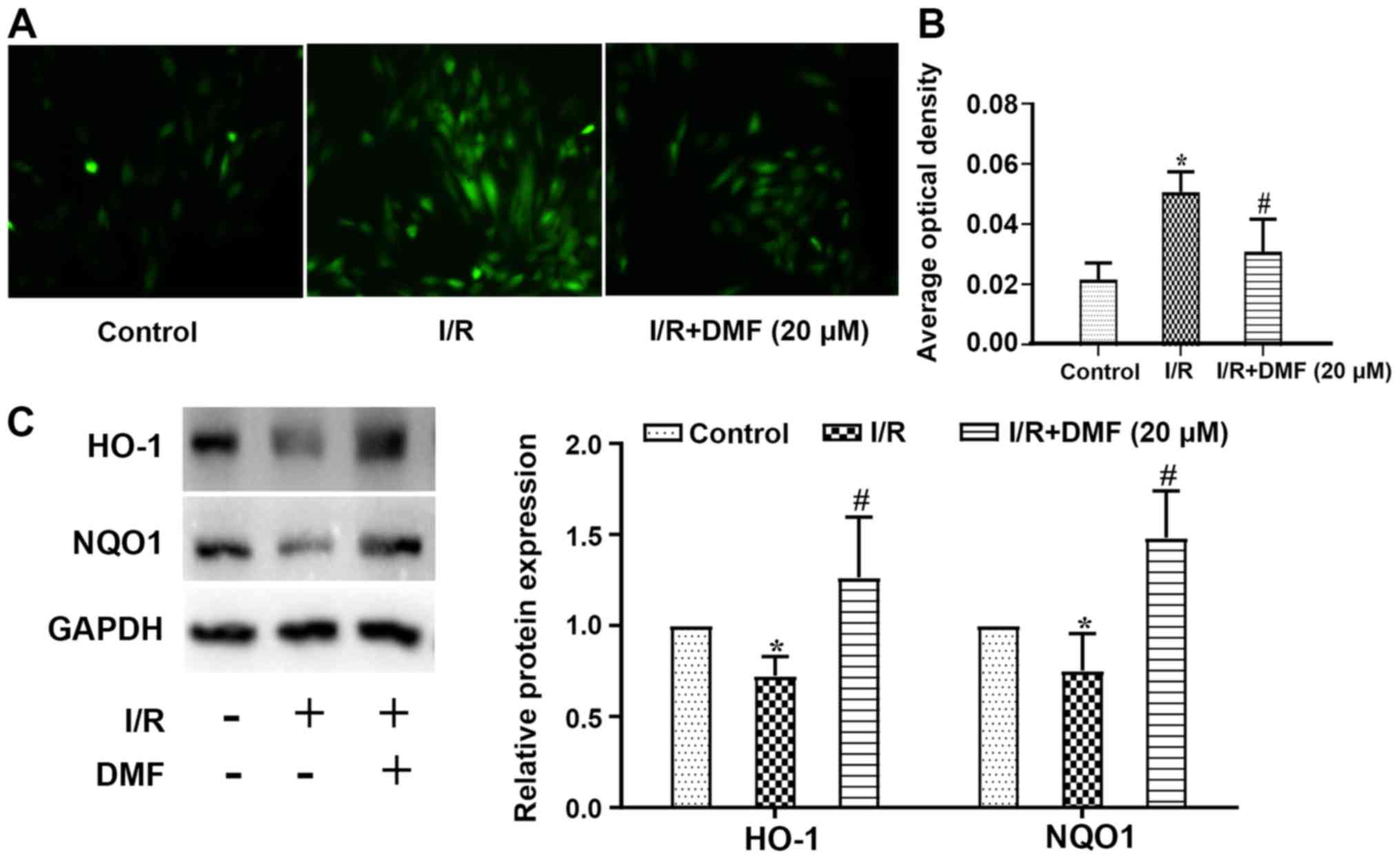
Effects of DMF on ROS production
Compared with in the control cells, ROS levels in
I/R-treated cells were increased, whereas DMF treatment attenuated
this effect (Fig. 4A).
Semi-quantitative analysis revealed that in comparison with the
control group, the signal intensity of DCFH-DA staining was
significantly amplified in the I/R group (P<0.05; Fig. 4B). The addition of DMF to the
I/R-treated cells significantly reduced the signal intensity, which
was lower than that in I/R-treated cells (P<0.05). Furthermore,
the expression levels of HO-1 were lower in I/R-treated cells than
in control cells (P<0.05), whereas DMF administration
significantly increased HO-1 expression (Fig. 4C). Similar results were obtained
regarding NQO1 protein expression.
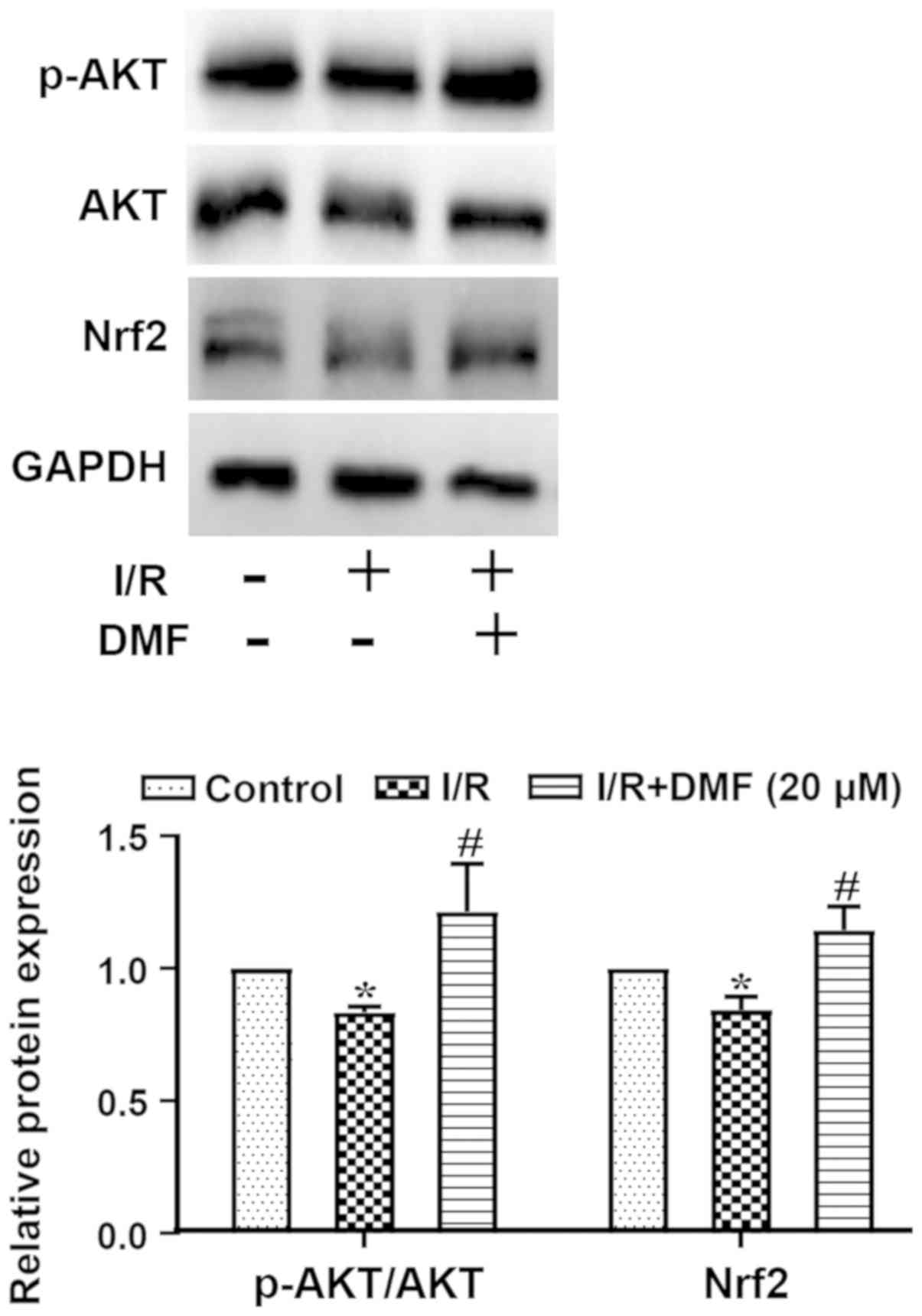
Effects of DMF on AKT/Nrf2
signaling
The ratio of p-AKT/AKT and the protein expression
levels of Nrf2 are shown in Fig.
5. Compared with in the control group, I/R-treated cells
exhibited a significantly decreased p-AKT/AKT ratio, which was
markedly elevated above I/R levels by the addition of DMF. The
expression levels of Nrf2 were decreased in I/R-treated cells
compared with in the control group (P<0.05), but were
significantly elevated by DMF administration (P<0.05).
Discussion
Coronary artery disease is characterized by
myocardial ischemia and is currently the leading cause of death in
the United States (25). Prolonged
severe ischemia results in irreversible myocardial injury (26), which should be prevented where
possible. Therefore, the potential for immediate/early
interventions for myocardial reperfusion are important for the
clinical outcome of ischemic heart disease (27). In previous decades, advancements in
timely reperfusion have been achieved (5,26),
which have significantly improved the clinical outcome of patients
with ischemic heart disease (26).
However, a number of studies have revealed that timely reperfusion
of the myocardium may also bring about serious cardiac damage,
termed myocardial I/R injury (4),
which can at least partly undermine the beneficial effects of early
reperfusion, and thus worsen clinical outcomes (28). Notably, timely reperfusion therapy
itself has been reported as a cause of I/R, through occlusion of
the coronary branch by balloon inflation and deflation during PCI,
or aorta clamping and unclamping during CABG. Therefore, the ideal
therapeutic strategy to manage ischemic heart disease would include
both timely reperfusion and the prevention of I/R damage to the
myocardium (29).
In the last decade, great improvements in timely
perfusion have been achieved through PCI or CABG (2); however, treatments to prevent or
reduce myocardial I/R injury are less than optimal (30). The results of several experimental
studies appear promising (31,32);
however, such results are rarely translated into clinical studies;
thus to date, there are a lack of established treatments to improve
the clinical outcome of ischemic heart disease via the prevention
or reduction of myocardial I/R injury (4,26,30).
In this context, the exploration of potential treatments to improve
myocardial I/R injury is of great significance. The present study
aimed to investigate the effects and underlying mechanisms of DMF
on I/R damage in vitro.
Although the mechanisms responsible for I/R injury
are not completely understood, a great deal of evidence has
indicated that ROS production and apoptosis may have important
roles in this event (33). DMF has
historically been used to treat relapsing-remitting multiple
sclerosis (34), and is currently
regarded as an antioxidant that can modulate ROS production
(35). Increasing evidence has
suggested that DMF may have a profound impact on ROS production and
apoptosis (36), and could exert
protective effects against reperfusion injury to the liver
(17) and brain (18). However, to the best of our
knowledge, there are currently no studies reporting the effects of
DMF on myocardial I/R injury.
The present study revealed that I/R treatment
significantly decreased cell viability and simultaneously increased
LDH release into the culture media, a clear indication of cellular
damage. These results are in accordance with previously reported
findings (37,38), which demonstrated the successful
establishment of an I/R injury model. Notably, pretreatment with
DMF significantly improved viability and reduced LDH release in
I/R-treated cells in a dose-dependent manner, although
I/R-associated damage could not be completely prevented. These
findings suggested that DMF exerted distinct cellular protection
against I/R injury.
The mechanisms underlying I/R-induced cellular
injury have been extensively investigated (39). It is evident that apoptosis may
serve a determining role in cellular damage, and augmented
apoptotic activity is a well-known cause of I/R-associated
myocardial injury (40,41). In the present study, I/R clearly
upregulated Bax and downregulated Bcl-2 expression, with a
reflected increase in the apoptotic index (Bax/Bcl-2 ratio) and
cleaved caspase-3 expression. Cleaved caspase-3 plays an important
role in apoptosis as it is the aggregation point of a number of
apoptotic stimulating signaling pathways, such as the mitochondrial
pathway, death receptor pathway and endoplasmic reticulum stress
pathway (42). Cleaved caspase-3
has been reported to break down various functional proteins to
induce apoptosis; therefore, its activation is regarded as a sign
of the irreversible stage of apoptosis after activation (43). The present study revealed that
compared with in the control group, the expression levels of
cleaved caspase-3 were increased in the I/R group and were
decreased by 24-h pretreatment with DMF; these results are
consistent with those derived from TUNEL staining. The levels of
these apoptotic indicators were comparable between the control and
I/R + DMF groups, suggesting that DMF may have powerful
anti-apoptotic effects. This was further supported by flow
cytometric analysis, which indicated that the number of early
apoptotic cells was significantly increased in the I/R-treated
group compared with in the control group, whereas the proportion of
living cells was notably decreased. DMF treatment also
significantly decreased the number of early apoptotic cells and
increased the proportion of living cells. Collectively, these
results suggested that along with protection against I/R-induced
cellular damage, DMF may significantly inhibit apoptotic activity,
and that the protective effects of DMF against I/R-induced cellular
damage could be associated with inhibition of apoptosis.
Although the factors that initiate apoptosis are not
yet fully understood, a number of studies have shown that excessive
ROS production may serve a critical role in the activation of
apoptosis (44). ROS are
hypothesized to mediate myocardial injury by inducing mitochondrial
permeability transition pore expression (45) and dysfunction of the sarcoplasmic
reticulum (46). In the present
study, ROS production was significantly elevated by I/R-treatment,
which, to a great extent, was counteracted by DMF, indicating that
this treatment markedly suppressed ROS production. Furthermore, I/R
significantly decreased the expression levels of the free radical
scavengers HO-1 and NQO1, whereas their expression was completely
restored by DMF. Therefore, on one hand, DMF may suppress ROS
production, and on the other hand, it may augment the expression
levels of free radical scavengers, which would beneficially
modulate redox status (47) and
further inhibit apoptosis (48).
The results of the present study suggested that the protective
effects of DMF on I/R-induced cellular damage may be associated
with redox status modulation, which could lead to attenuation of
apoptosis in response to I/R insult.
It is well known that a series of pathways are
involved in the regulation of redox status; among others, AKT and
Nrf2 have been extensively investigated (49). In Nrf2-knockout mice, DMF failed to
deliver protection against I/R injury, which strongly indicated
that DMF may act downstream of Nrf2 (18). Furthermore, previous studies have
shown that Nrf2 can be activated via phosphorylation of AKT pathway
components (50), which also
upregulated HO-1 and NQO1 (51).
In the present study, the p-AKT/AKT ratio was significantly
decreased in I/R-treated cells, which was accompanied by a decrease
in Nrf2 expression. Treatment with DMF markedly elevated the levels
of p-AKT/AKT and Nrf2, indicating that DMF activity could be
attributed to activation of the AKT/Nrf2 pathway.
Nrf2 is a putative transcription factor, and the
suppression of Nrf2 activity is associated with an increase in
Keap1, which sequesters Nrf2 in the cytoplasm and regulates its
ubiquitin-dependent degradation (52). Upon activation by small molecules,
the interaction between Keap1 and Nrf2 is disrupted. Nrf2 protein
turnover is thereby attenuated, and the transcription factor
translocates to the nucleus where it modulates transcription
through AREs. Thus, a limitation of the present study is that it
did not involve isolating nuclear proteins and comparing the
expression of Nrf2 in the nuclear fraction of cells; this will be
addressed in the future to support the data of the present
study.
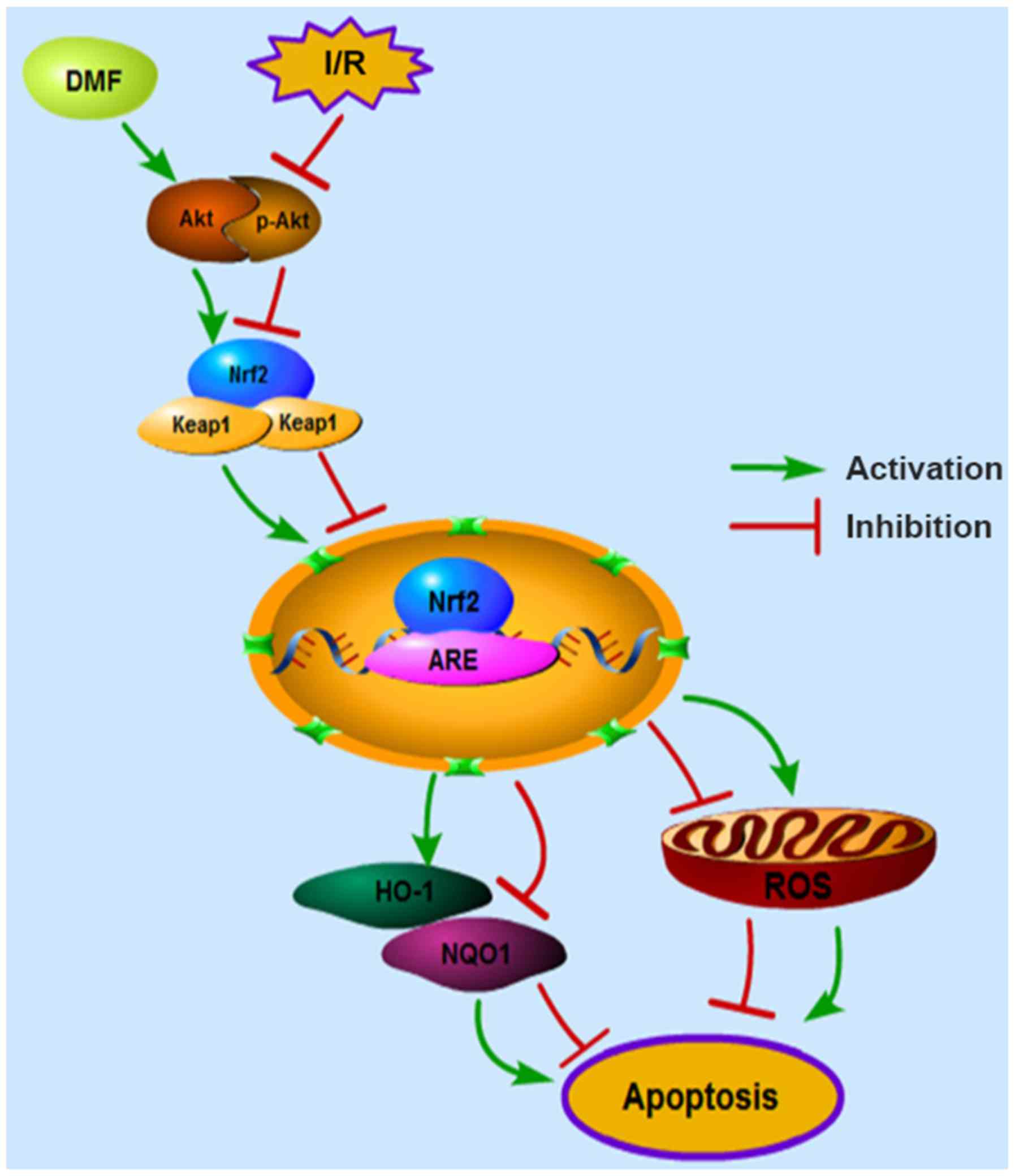
In conclusion, the myocardial protective functions
of DMF were confirmed in a myocardial I/R model, and Nrf2
modulation was validated as a primary mechanism for inhibiting
oxidative stress and apoptosis (Fig.
6). Currently, DMF has been confirmed to suppress
cardiovascular diseases, such as pulmonary hypertension and
diabetic cardiomyopathy. Based on the present findings, modulation
of the AKT/Nrf2 pathway by DMF may be a promising treatment option
for patients with acute ischemic heart disease. With continuous
in-depth research on DMF, it is expected to become a future
treatment for myocardial I/R injury and pave the way for improved
clinical applications.
Acknowledgements
Not applicable.
Funding
The present study was supported by grants from the
National Natural Science Foundation of China (grant nos. 81370250
and 81974026) and the Hunan Provincial Natural Science Foundation
of China (grant no. 2017SK50108).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
QM conceived the study and designed the experiments.
YK, YZ and ZX performed the experiments. LX and PW analyzed the
data and drafted the manuscript. All authors read and approved the
final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Yoon J, Seo H, Oh IH and Yoon SJ: The
non-communicable disease burden in Korea: Findings from the 2012
Korean burden of disease study. J Korean Med Sci. 31 (Suppl
2):2783–S167. 2016. View Article : Google Scholar
|
|
2
|
Yetgin T, Manintveld OC, Boersma E,
Kappetein AP, van Geuns RJ, Zijlstra F, Duncker DJ and van der
Giessen WJ: Remote ischemic conditioning in percutaneous coronary
intervention and coronary artery bypass grafting. Circ J.
76:2392–2404. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bompotis GC, Deftereos S, Angelidis C,
Choidis E, Panagopoulou V, Kaoukis A, Vassilikos VP, Cleman MW and
Giannopoulos G: Altered calcium handling in reperfusion injury. Med
Chem. 12:114–130. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ibáñez B, Heusch G, Ovize M and Van de
Werf F: Evolving therapies for myocardial ischemia/reperfusion
injury. J Am Coll Cardiol. 65:1454–1471. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Binder A, Ali A, Chawla R, Aziz HA, Abbate
A and Jovin IS: Myocardial protection from ischemia-reperfusion
injury post coronary revascularization. Expert Rev Cardiovasc Ther.
13:1045–1057. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kern KB, Hanna JM, Young HN, Ellingson CJ,
White JJ, Heller B, Illindala U, Hsu CH and Zuercher M: Importance
of both early reperfusion and therapeutic hypothermia in limiting
myocardial infarct size post-cardiac arrest in a porcine model.
JACC Cardiovasc Interv. 9:2403–2412. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wang S, Zhang F, Zhao G, Cheng Y, Wu T, Wu
B and Zhang YE: Mitochondrial PKC-ε deficiency promotes
I/R-mediated myocardial injury via GSK3β-dependent mitochondrial
permeability transition pore opening. J Cell Mol Med. 21:2009–2021.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Neri M, Riezzo I, Pascale N, Pomara C and
Turillazzi E: Ischemia/reperfusion injury following acute
myocardial infarction: A critical issue for clinicians and forensic
pathologists. Mediators Inflamm. 2017:70183932017. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Granger DN and Kvietys PR: Reperfusion
injury and reactive oxygen species: The evolution of a concept.
Redox Biol. 6:524–551. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Sanderson TH, Reynolds CA, Kumar R,
Przyklenk K and Hüttemann M: Molecular mechanisms of
ischemia-reperfusion injury in brain: Pivotal role of the
mitochondrial membrane potential in reactive oxygen species
generation. Mol Neurobiol. 47:9–23. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Sifuentes-Franco S, Pacheco-Moisés FP,
Rodríguez-Carrizalez AD and Miranda-Díaz AG: The role of oxidative
stress, mitochondrial function, and autophagy in diabetic
polyneuropathy. J Diabetes Res. 2017:16730812017. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Bhat AH, Dar KB, Anees S, Zargar MA,
Masood A, Sofi MA and Ganie SA: Oxidative stress, mitochondrial
dysfunction and neurodegenerative diseases; a mechanistic insight.
Biomed Pharmacother. 74:101–110. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Sinha K, Das J, Pal PB and Sil PC:
Oxidative stress: The mitochondria-dependent and
mitochondria-independent pathways of apoptosis. Arch Toxicol.
87:1157–1180. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Boshra V and Atwa A: Effect of
cerebrolysin on oxidative stress-induced apoptosis in an
experimental rat model of myocardial ischemia. Physiol Int.
103:310–320. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Herr DJ, Aune SE and Menick DR: Induction
and assessment of ischemia-reperfusion injury in
Langendorff-perfused rat hearts. J Vis Exp. 101:e529082015.
|
|
16
|
Heusch G and Gersh BJ: The pathophysiology
of acute myocardial infarction and strategies of protection beyond
reperfusion: A continual challenge. Eur Heart J. 38:774–784.
2017.PubMed/NCBI
|
|
17
|
Takasu C, Vaziri ND, Li S, Robles L, Vo K,
Takasu M, Pham C, Farzaneh SH, Shimada M, Stamos MJ, et al:
Treatment with dimethyl fumarate ameliorates liver
ischemia/reperfusion injury. World J Gastroenterol. 23:4508–4516.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Yao Y, Miao W, Liu Z, Han W, Shi K, Shen
Y, Li H, Liu Q, Fu Y, Huang D, et al: Dimethyl fumarate and
monomethyl fumarate promote post-ischemic recovery in mice. Transl
Stroke Res. 7:535–547. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Li J, Ma J, Lacagnina MJ, Lorca S, Odem
MA, Walters ET, Kavelaars A and Grace PM: Oral dimethyl fumarate
reduces peripheral neuropathic pain in rodents via NFE2L2
antioxidant signaling. Anesthesiology,. 132:343–356. 2020.
View Article : Google Scholar
|
|
20
|
Ghadiri M, Rezk A, Li R, Evans A, Luessi
F, Zipp F, Giacomini PS, Antel J and Bar-Or A: Dimethyl
fumarate-induced lymphopenia in MS due to differential T-cell
subset apoptosis. Neurol Neuroimmunol Neuroinflamm. 4:e3402017.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Belcher JD, Chen C, Nguyen J, Zhang P,
Abdulla F, Nguyen P, Killeen T, Xu P, O'Sullivan G, Nath KA, et al:
Control of Oxidative Stress and Inflammation in Sickle Cell Disease
with the Nrf2 Activator Dimethyl Fumarate. Antioxid Redox Signal.
26:748–762. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Sghaier R, Nury T, Leoni V, Caccia C, Pais
De Barros JP, Cherif A, Vejux A, Moreau T, Limem K, et al: Dimethyl
fumarate and monomethyl fumarate attenuate oxidative stress and
mitochondrial alterations leading to oxiapoptophagy in 158N murine
oligodendrocytes treated with 7β-hydroxycholesterol. J Steroid
Biochem Mol Biol. 194:1054322019. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Crowley LC, Marfell BJ, Scott AP and
Waterhouse NJ: Quantitation of apoptosis and necrosis by Annexin V
binding, propidium iodide uptake, and flow cytometry. Cold Spring
Harb Protoc. 2016:953–957. 2016. View Article : Google Scholar
|
|
24
|
Koç E, Çelik-Uzuner S, Uzuner U and Çakmak
R: The detailed comparison of cell death detected by Annexin V-PI
counterstain using fluorescence microscope, flow cytometry and
automated cell counter in mammalian and microalgae cells. J
Fluoresc. 28:1393–1404. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Dalen JE, Alpert JS, Goldberg RJ and
Weinstein RS: The epidemic of the 20(th) century: Coronary heart
disease. Am J Med. 127:807–812. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Chi HJ, Chen ML, Yang XC, Lin XM, Sun H,
Zhao WS, Qi D, Dong JL and Cai J: Progress in therapies for
myocardial ischemia reperfusion injury. Curr Drug Targets.
18:1712–1721. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kleinbongard P, Skyschally A and Heusch G:
Erratum to: Cardioprotection by remote ischemic conditioning and
its signal transduction. Pflugers Arch. 469:8432017. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Heusch G: Molecular basis of
cardioprotection: Signal transduction in ischemic pre-, post-, and
remote conditioning. Circ Res. 116:674–699. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Bernink FJ, Timmers L, Beek AM, Diamant M,
Roos ST, Van Rossum AC and Appelman Y: Progression in attenuating
myocardial reperfusion injury: An overview. Int J Cardiol.
170:261–269. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Hausenloy DJ and Yellon DM: Myocardial
ischemia-reperfusion injury: A neglected therapeutic target. J Clin
Invest. 123:92–100. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
31
|
Ndrepepa G, Colleran R and Kastrati A:
Reperfusion injury in ST-segment elevation myocardial infarction:
The final frontier. Coron Artery Dis. 28:253–262. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Frank A, Bonney M, Bonney S, Weitzel L,
Koeppen M and Eckle T: Myocardial ischemia reperfusion injury: From
basic science to clinical bedside. Semin Cardiothorac Vasc Anesth.
16:123–132. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Guo W, Liu X, Li J, Shen Y, Zhou Z, Wang
M, Xie Y, Feng X, Wang L and Wu X: Prdx1 alleviates cardiomyocyte
apoptosis through ROS-activated MAPK pathway during myocardial
ischemia/reperfusion injury. Int J Biol Macromol. 112:608–615.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Huisman E, Papadimitropoulou K, Jarrett J,
Bending M, Firth Z, Allen F and Adlard N: Systematic literature
review and network meta-analysis in highly active
relapsing-remitting multiple sclerosis and rapidly evolving severe
multiple sclerosis. BMJ Open. 7:e0134302017. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Akino N, Wada-Hiraike O, Terao H, Honjoh
H, Isono W, Fu H, Hirano M, Miyamoto Y, Tanikawa M, Harada M, et
al: Activation of Nrf2 might reduce oxidative stress in human
granulosa cells. Mol Cell Endocrinol. 470:96–104. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ohl K, Tenbrock K and Kipp M: Oxidative
stress in multiple sclerosis: Central and peripheral mode of
action. Exp Neurol. 277:58–67. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Chen Y, Wang H, Zhang Y, Wang Z, Liu S and
Cui L: Pretreatment of ghrelin protects H9c2 cells against
hypoxia/reoxygenation-induced cell death via PI3K/AKT and AMPK
pathways. Artif Cells Nanomed Biotechnol. 47:2179–2187. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Fan L, Zhou W, Zhang L, Jiang D, Zhao Q
and Liu L: Sitagliptin protects against hypoxia/reoxygenation
(H/R)-induced cardiac microvascular endothelial cell injury. Am J
Transl Res. 11:2099–2107. 2019.PubMed/NCBI
|
|
39
|
Moe GW and Marín-García J: Role of cell
death in the progression of heart failure. Heart Fail Rev.
21:157–167. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Qian W, Wang Z, Xu T and Li D:
Anti-apoptotic effects and mechanisms of salvianolic acid A on
cardiomyocytes in ischemia-reperfusion injury. Histol Histopathol.
34:223–231. 2019.PubMed/NCBI
|
|
41
|
Lejay A, Fang F, John R, Van JA, Barr M,
Thaveau F, Chakfe N, Geny B and Scholey JW: Ischemia reperfusion
injury, ischemic conditioning and diabetes mellitus. J Mol Cell
Cardiol. 91:11–22. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Chen Y, Feng X, Hu X, Sha J, Li B, Zhang H
and Fan H: Dexmedetomidine ameliorates acute stress-induced kidney
injury by attenuating oxidative stress and apoptosis through
inhibition of the ROS/JNK signaling pathway. Oxid Med Cell Longev.
2018:40353102018. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Crowley LC and Waterhouse NJ: Detecting
cleaved caspase-3 in apoptotic cells by flow cytometry. Cold Spring
Harb Protoc. Nov 1–2016.(Epub ahead of print). View Article : Google Scholar
|
|
44
|
Liu YF, Chu YY, Zhang XZ, Zhang M, Xie FG,
Zhou M, Wen HH and Shu AH: TGFβ1 protects myocardium from apoptosis
and oxidative damage after ischemia reperfusion. Eur Rev Med
Pharmacol Sci. 21:1551–1558. 2017.PubMed/NCBI
|
|
45
|
Cadenas S: ROS and redox signaling in
myocardial ischemia-reperfusion injury and cardioprotection. Free
Radic Biol Med. 117:76–89. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Hall AR, Burke N, Dongworth RK, Kalkhoran
SB, Dyson A, Vicencio JM, Dorn GW II, Yellon DM and Hausenloy DJ:
Hearts deficient in both Mfn1 and Mfn2 are protected against acute
myocardial infarction. Cell Death Dis. 7:e22382016. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Carlström KE, Ewing E, Granqvist M,
Gyllenberg A, Aeinehband S, Enoksson SL, Checa A, Badam TVS, Huang
J, Gomez-Cabrero D, et al: Therapeutic efficacy of dimethyl
fumarate in relapsing-remitting multiple sclerosis associates with
ROS pathway in monocytes. Nat Commun. 10:30812019. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Han G and Zhou Q: Dimethylfumarate induces
cell cycle arrest and apoptosis via regulating intracellular redox
systems in HeLa cells. In Vitro Cell Dev Biol Anim. 52:1034–1041.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Hu YR, Ma H, Zou ZY, He K, Xiao YB, Wang
Y, Feng M, Ye XL and Li XG: Activation of Akt and JNK/Nrf2/NQO1
pathway contributes to the protective effect of coptisine against
AAPH-induced oxidative stress. Biomed Pharmacother. 85:313–322.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Ci X, Zhou J, Lv H, Yu Q, Peng L and Hua
S: Betulin exhibits anti-inflammatory activity in LPS-stimulated
macrophages and endotoxin-shocked mice through an
AMPK/AKT/Nrf2-dependent mechanism. Cell Death Dis. 18(8):
e27982017. View Article : Google Scholar
|
|
51
|
Wu PS, Ding HY, Yen JH, Chen SF, Lee KH
and Wu MJ: Anti-inflammatory activity of 8-hydroxydaidzein in
LPS-stimulated BV2 microglial cells via activation of
Nrf2-antioxidant and attenuation of Akt/NF-κB-inflammatory
signaling pathways, as well as inhibition of COX-2 activity. J
Agric Food Chem. 66:5790–5801. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Bellezza I, Giambanco I, Minelli A and
Donato R: Nrf2-Keap1 signaling in oxidative and reductive stress.
Biochim Biophys Acta Mol Cell Res. 1865:721–733. 2018. View Article : Google Scholar : PubMed/NCBI
|