Introduction
Lipid accumulation in the arterial walls is one of
the hallmarks of atherosclerosis (AS) (1), which is an important pathological
manifestation of cardiovascular disease and a notable cause of
mortality in numerous countries, such as the UK, Australia and USA
(2). It has been demonstrated that
monolayer endothelial cell (EC) dysfunction on the inner wall of
arterial vessels is the initial step in the process of
atherosclerotic plaque formation (3,4). EC
dysfunction can be caused by oxidized low-density lipoprotein
(ox-LDL), which is often induced by oxidative stress that can be
caused by reactive oxygen species, smoking, diabetes and obesity
(5–8). Under normal conditions, ECs can
export hydrolysed LDL-derived cholesterol into extracellular
high-density lipoprotein (HDL), but cannot efficiently efflux
ox-LDL-derived cholesterol, resulting in lipid accumulation in ECs
(9). Furthermore, the accumulation
of ox-LDL in the endothelium has been significantly associated with
the dysfunction and apoptosis of ECs (10,11).
Autophagy is a process of evolutionarily conserved
catabolism, which degrades a number of cellular components,
including long-lived proteins and various organelle components
(12,13). A high LC3-II to LC3-I ratio
reflects increasing autophagosome formation, whereas low p62
protein levels are associated with high autophagy flux (14,15).
LAMP1 is an essential component of the lysosomal membrane, serving
as a lysosomal biomarker with a key role in autophagy and lysosomal
fusion (16). It has been
demonstrated that autophagy can protect ECs from oxidative
stress-induced cell damage (17).
Since ox-LDL has been reported to be involved in the development of
AS and the rupture of atherosclerotic plaques by inhibiting
autophagy (18), it was
hypothesized that endothelial autophagy serves a key role in
limiting the accumulation of ox-LDL-associated lipids within vessel
walls (19). An association
between ox-LDL-associated autophagy and endothelial lipid
accumulation has been identified; however, the mechanism remains
unclear.
The specific degradation of lipids by autophagy
(also known as lipophagy) was first described in the liver, where
genetic defects (mutations in RNAi-ATG5 and ATG7) of
macro-autophagy led to the accumulation of lipid droplets (LDs)
(20). Lipophagy serves a critical
role in maintaining overall lipid homeostasis (21). It has been demonstrated that mice
bearing autophagy-related 5-deficient macrophages exhibit increased
plaque formation due to an impairment of lipophagy (22,23).
Therefore, the present study hypothesized that the decrease in
endothelial autophagy induced by ox-LDL may be due to impaired
lipophagy, resulting in intracellular lipid accumulation and EC
damage.
The current study investigated the association
between autophagy and lipid accumulation in human umbilical vein
endothelial cells (HUVECs) treated with ox-LDL. The results provide
evidence that lipophagy may be attenuated in HUVECs with prolonged
exposure to ox-LDL and may provide novel insights into the
prevention and treatment of atherosclerotic cardiovascular
disease.
Materials and methods
Cell culture and treatments
HUVECs were purchased from The Cell Bank of Type
Culture Collection of the Chinese Academy of Sciences and were
maintained at 37°C with 5% CO2 in high glucose DMEM
supplemented with 10% FBS (both Gibco; Thermo Fisher Scientific,
Inc.), 100 U/ml penicillin and 100 µg/ml streptomycin. HUVECs were
treated with PBS (basal), LDL (50 µg/ml; control) or ox-LDL (100
µg/ml) for 6, 12, 24 and 48 h.
Cell viability assay
An MTT assay was used to determine the viability of
HUVECs. Cells were seeded into 96-well microplates at a density of
4×104 cells/well and cultured 37°C for 24 h.
Subsequently, HUVECs were treated with different concentrations of
ox-LDL (25, 50 or 100 µg/ml) for 6, 12, 24 and 48 h, and 20 µl MTT
solution (5 mg/ml; Beyotime Institute of Biotechnology) was added
to each well. The plates were incubated at 37°C for 4 h and then
DMSO was added to dissolve the purple formazan products. The
optical density value at 490 nm was measured using a microplate
reader spectrophotometer (Molecular Devices, LLC).
Transmission electron microscopy
(TEM)
After removing the culture medium, the HUVECs were
washed twice with ice-cold PBS and fixed with 2.5% glutaraldehyde
overnight at 4°C. The cells were post-fixed in 2% osmium tetroxide
at 4°C for 1 h. Dehydration was subsequently performed in 50–90%
ethanol, and the samples were embedded in epoxy resin at 45°C for
12 h. Ultrathin sections (70-nm) were cut using an ultramicrotome
(Leica Microsystems GmbH) and counterstained with uranyl acetate
for 10 min and lead citrate for 30 min at RT. The sections were
examined using a Jeol JEM SX 100 electron microscope
(magnification, ×1,200) (JEOL, Ltd.).
Oil Red O staining
The HUVECs were seeded into 6-well plates
(2.5×105 cells/well) and were treated as aforementioned.
The cells were washed twice with PBS and fixed with 4%
paraformaldehyde for 20 min at room temperature. Subsequently, the
HUVECs were stained with a 0.3% Oil Red O staining solution for 8
min at room temperature and washed with 60% isopropanol for 5 sec.
The cells were stained with haematoxylin (Sigma-Aldrich; Merck
KGaA) for 10 sec at RT prior to obtaining images using a light
microscope (magnification, ×40) (Nikon Corporation).
Western blot analysis
Total protein was extracted from HUVECs using a
lysis buffer (20 mM Tris-HCl pH 7.5, 150 mM NaCl, 1% Triton X-100,
1 mM phenylmethylsulphonyl fluoride, 1 mM
Na3VO4, leupeptin and EDTA) and was
quantified using a bicinchoninic acid kit (Beijing ComWin Biotech
Co., Ltd.). Equal amounts of proteins (30 µg/lane) were separated
using 8–12% SDS-PAGE and transferred onto polyvinylidene difluoride
membranes (EMD Millipore). Following blocking with 5% BSA
(Sigma-Aldrich; Merck KGaA) for 1.5 h at room temperature, the
membranes were incubated with the following primary antibodies (all
1:1,000) at 4°C overnight: Anti-LC3B (cat. no. 3868S; Cell
Signaling Technology, Inc.), anti-p62 (cat. no. ab91526; Abcam),
anti-lysosomal-associated membrane protein 1 (LAMP1; cat. no.
ab24170; Abcam) and anti-β-actin (cat. no. 4970S; Cell Signaling
Technology, Inc.). Subsequently, the membranes were incubated with
an anti-rabbit IgG HRP-linked secondary antibody (1:5,000; cat. no.
7074; Cell Signalling Technology) for 45 min at room temperature.
The bands were visualized using an enhanced chemiluminescence
reagent (EMD Millipore). The densitometry of each band was analysed
using the Sigma Scan Pro5 software (version 5.0; Systat Software,
Inc.) and normalized to β-actin values.
Immunofluorescence staining
The HUVECs were fixed with 4% paraformaldehyde (pH
7.4) for 20 min at RT, washed three times with PBS, blocked with 5%
BSA at RT for 2 h, and then incubated with the following rabbit
primary antibodies (all 1:200) overnight at 4°C: Anti-LAMP 1 (cat.
no. ab24170; Abcam) and anti-LC3B (cat. no. 3868S; Cell Signaling
Technology, Inc.). Subsequently, the cells were incubated with
anti-rabbit IgG (H+L), F(ab′)2 Fragment (Alexa Fluor®
647 Conjugate) (red) antibodies (1:500; cat. no. 4414; Cell
Signaling Technology) for 1 h at 37°C. LDs were stained with 1
µg/ml Bodipy 493/503 (Invitrogen; Thermo Fisher Scientific, Inc.)
for 30 min at RT in the dark, and with 10 mg/ml DAPI (cat. no.
H-1200; Vector Laboratories, Inc.) at RT for 5 min. The cells were
examined under a confocal laser scanning microscope (magnification,
×400) (LSCM 510 Meta; Zeiss AG). The relative colocalization
coefficient between LC3, LAMP1 and LD was analysed via ImageJ
(version no. 1.52P) software (24).
Statistical analysis
All values were presented as the mean ± SD (n≥3).
The differences between groups were analysed via one-way ANOVA and
post hoc Dunnett's multiple comparisons test by GraphPad Prism
(version 6; GraphPad Software, Inc.). P<0.05 was considered to
indicate a statistically significant difference.
Results
Lipid accumulation in HUVECs induced
by ox-LDL
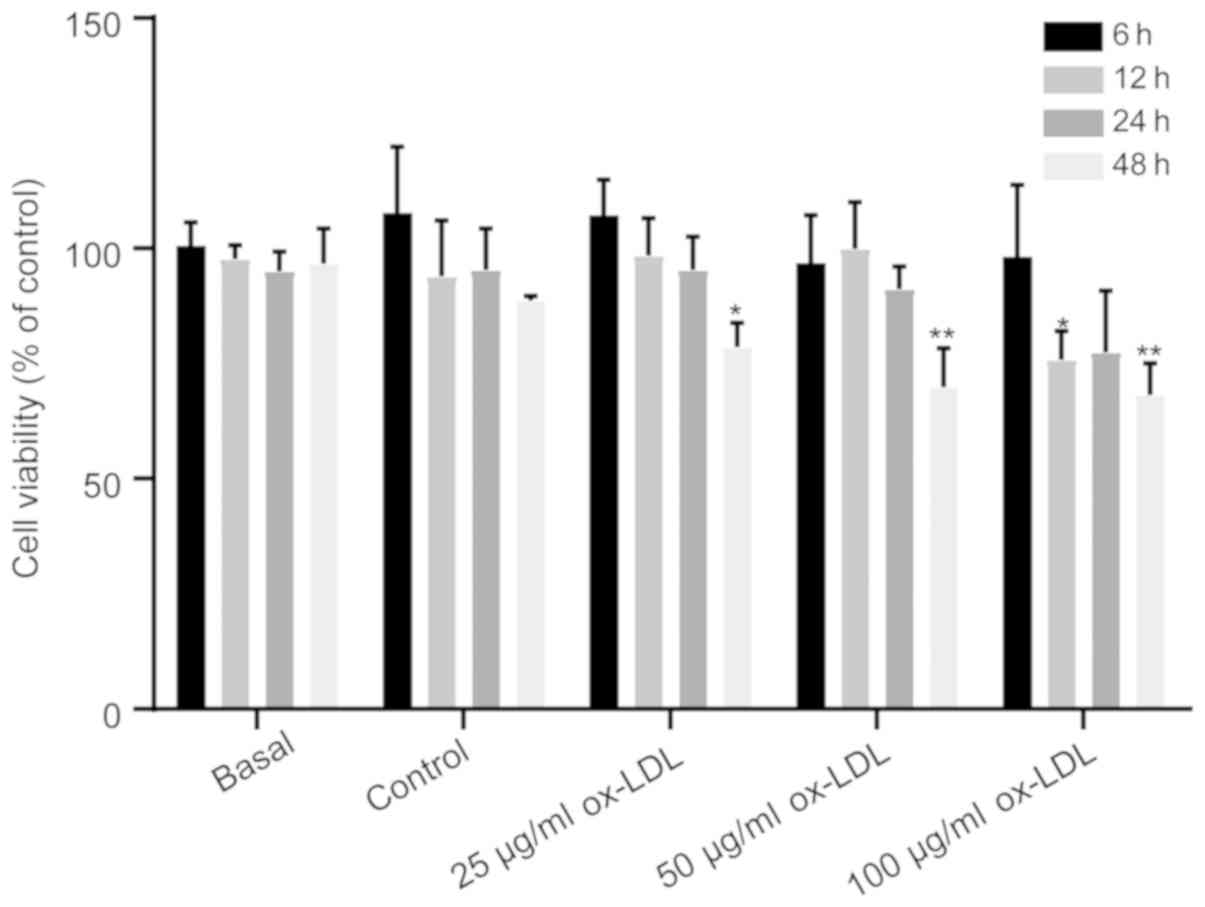
To determine the appropriate concentration of
ox-LDL, as an oxidative stress factor on ECs, the HUVECs were
treated with different concentrations of ox-LDL (25, 50 or 100
µg/ml) for 6, 12, 24 and 48 h, and the cell viability was detected
using the MTT assay. The results demonstrated that there was no
difference in cell viability in HUVECs treated with 25 µg/ml ox-LDL
for 6, 12, and 24 h, whereas compared with corresponding control
group, 50 µg/ml ox-LDL for 48 h inhibited cell growth (Fig. 1). Furthermore, when treated with
100 µg/ml ox-LDL, the cell viability significantly decreased at 12
and 48 h compared with the control group (Fig. 1), which is consistent with the
finding that 100 µg/ml ox-LDL decreased HUVEC viability when
investigating the effect of ox-LDL on autophagy (25). Therefore, 100 µg/ml ox-LDL was
chosen as the oxidative stress factor for all subsequent
experiments.
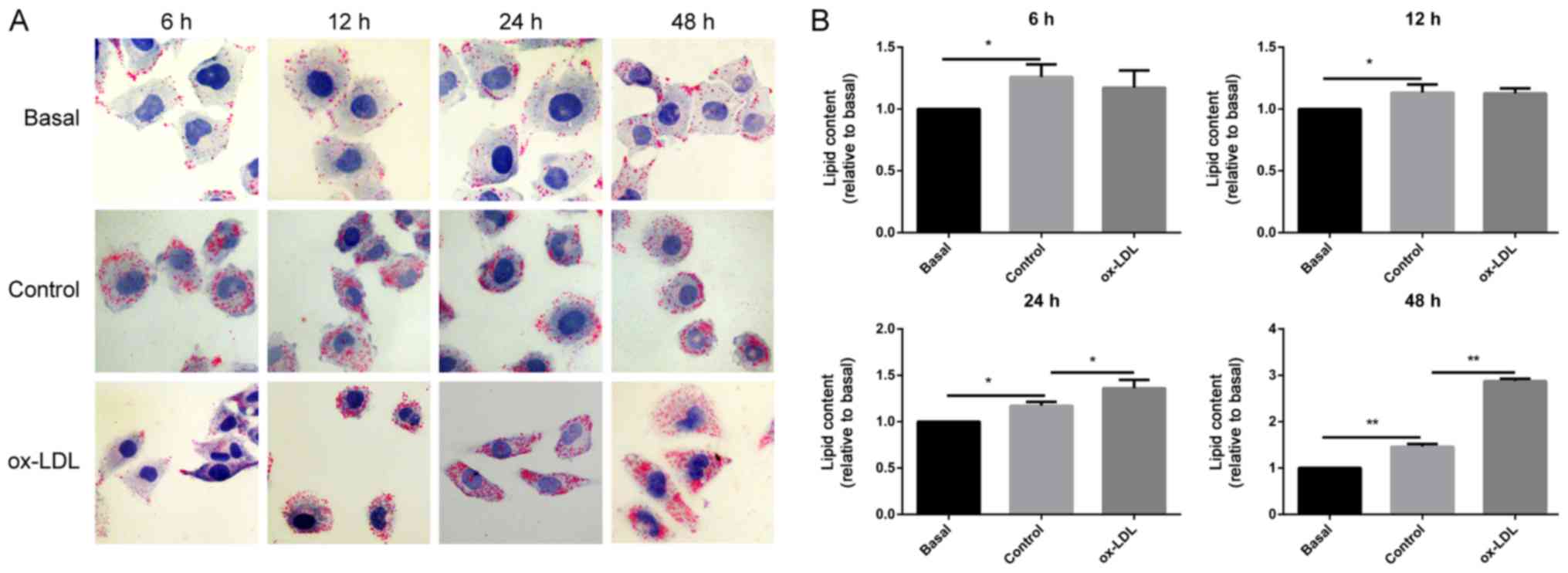
To investigate the deposited lipid effect on the ECs
under oxidative stress, the LDs were stained with Oil Red O in
HUVECs that had been exposed to 100 µg/ml ox-LDL. As shown in
Fig. 2, the results demonstrated
that compared with the basal group, lipid droplets in HUVECs
treated with LDL for 6, 12, 24 and 48 h were significantly
increased, which indicates that HUVECs were lipid-loaded. Lipid
staining in HUVECs showed a decreased tendency without statistical
difference following treatment with ox-LDL for 6 h and 12 h
compared with that in cells treated with LDL (control). However,
when the HUVECs were treated with ox-LDL for 24 and 48 h, lipid
deposition in the cells was significantly increased compared with
that in the control group. The present data indicated that
prolonged exposure to ox-LDL led to elevated lipid accumulation in
the ECs.
Impaired autophagy in HUVECs due to
ox-LDL
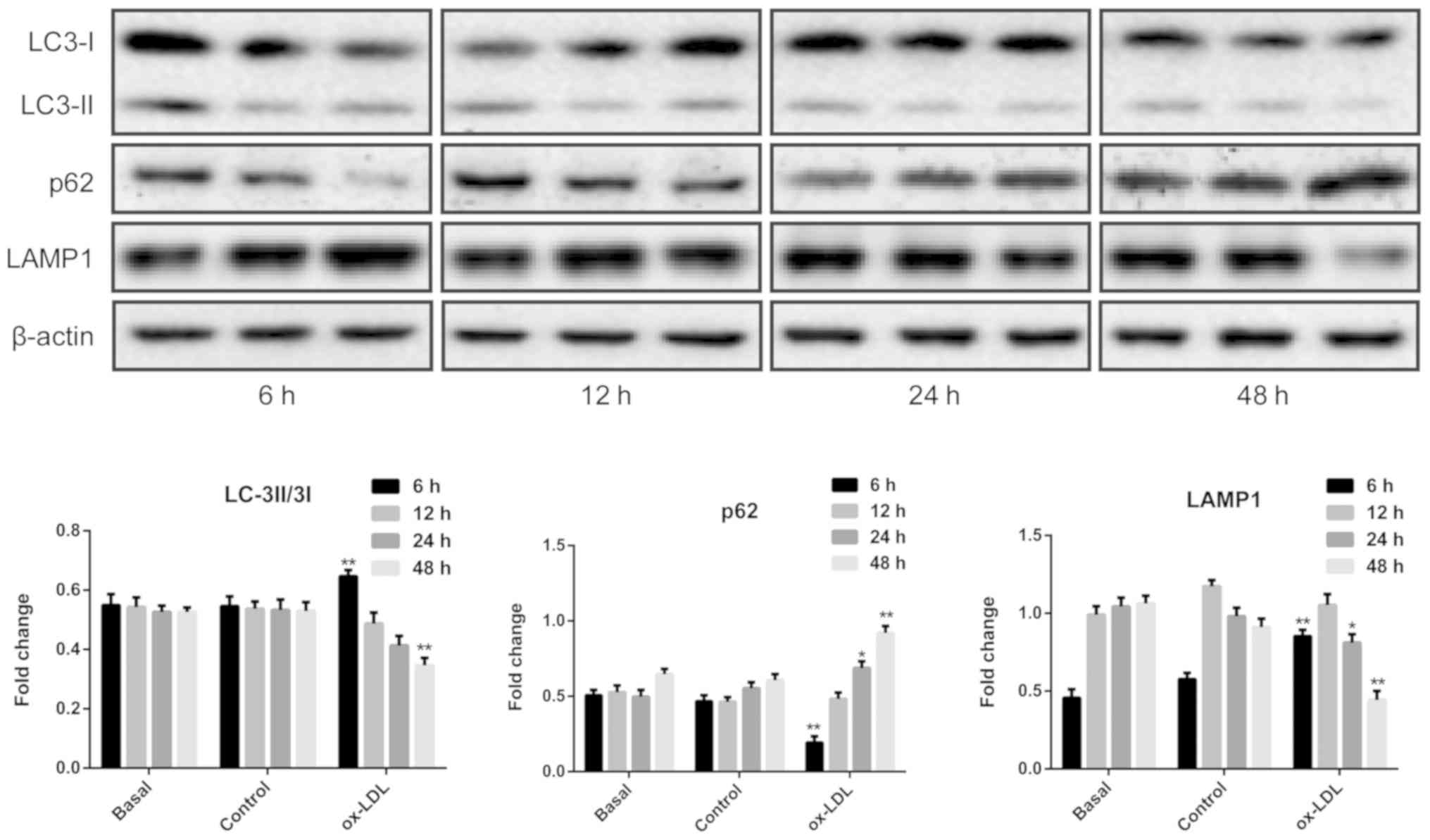
To investigate the effect of ox-LDL on autophagy in
HUVECs, the ratio of LC3-II to LC3-I and the p62 protein expression
levels, which serve as well-known biomarkers of autophagy (26), were detected using western blot
analysis. As shown in Fig. 3, the
results demonstrated that compared with LDL exposure, the ratio of
LC3II/LC3I was significantly upregulated in HUVECs exposed to
ox-LDL for 6 h, along with significantly increased protein
expression levels of LAMP1 and decreased protein levels of p62,
suggesting that autophagy events may be triggered in the early
period of oxidative stress exposure in ECs. However, compared with
the control group, decreased protein levels of LAMP1 were observed
in HUVECs exposed to ox-LDL for 24 h, which was accompanied by
increased p62 expression. Furthermore, there was a significant
decrease in both the ratio of LC3II/LC3I and the protein expression
levels of LAMP1, along with a significant increase in p62
expression, in HUVECs treated with ox-LDL for 48 h, indicating that
autophagy may be inhibited with prolonged exposure of ECs to
ox-LDL.
Suppression of lipophagy in HUVECs
treated with ox-LDL
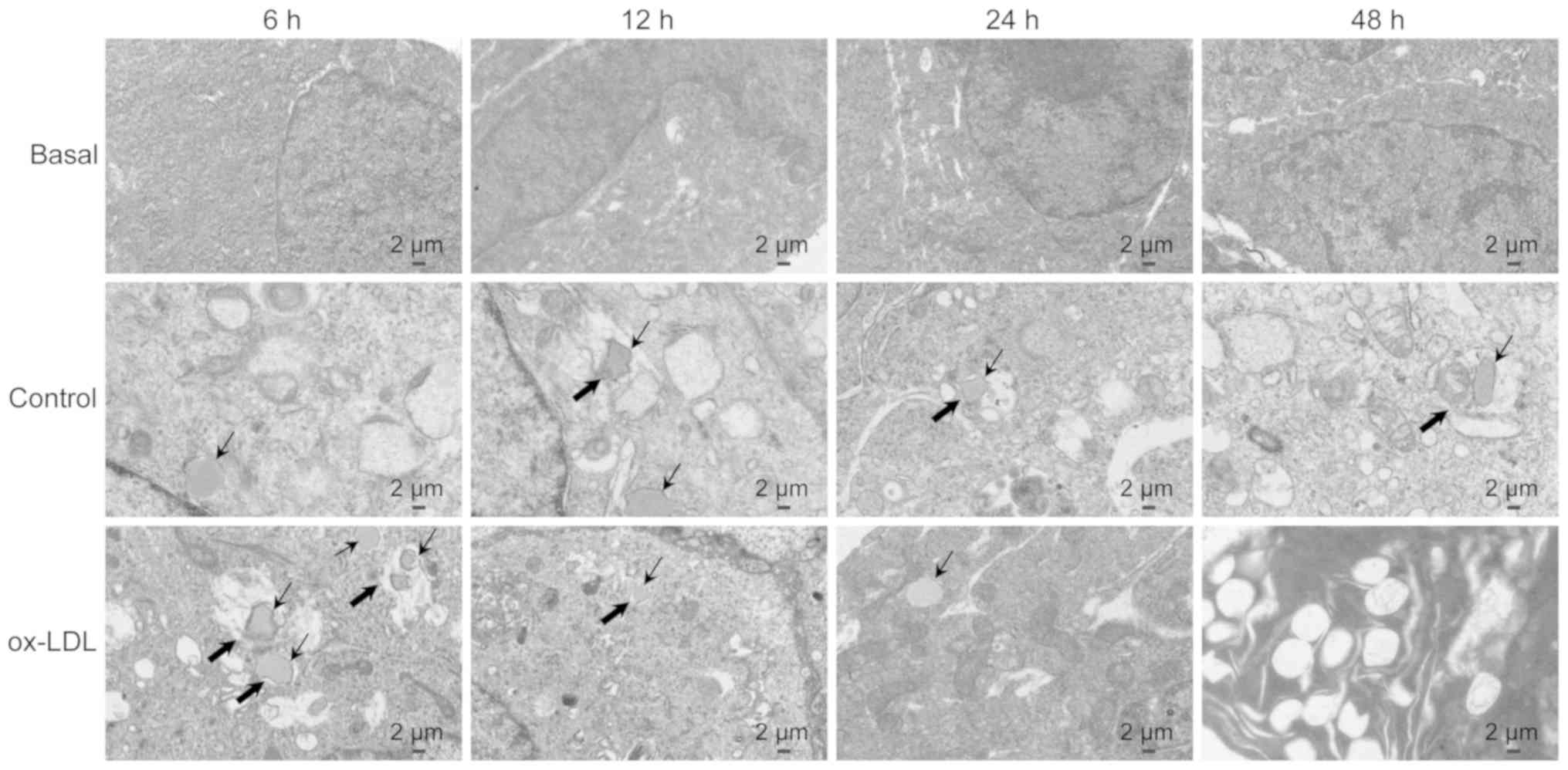
To clarify the association between ox-LDL-induced
autophagy deficiency and lipid accumulation in ECs, the changes in
the number of autolipophagosomes (ALPs) were detected using TEM.
The results in Fig. 4 revealed
that ALP formation in the ox-LDL treatment group was higher
compared with that in the LDL treatment group following 6 h of
treatment. However, this ultra-structural conformation was
decreased with exposure to ox-LDL for 24 and 48 h compared with
that in LDL-treated HUVECs. In addition, following exposure to
ox-LDL for 48 h, the signs of damage was observed in ECs by the
presence of vacuole-like structures. The present results suggested
that lipophagy may be impaired after prolonged exposure to
oxidative stress.
Impaired lipophagy may be associated
with lipid accumulation in ECs
To further elucidate the localization of LDs in
HUVECs treated with ox-LDL, the co-localization of LDs with LC3 or
LAMP1 was measured using confocal microscopy, as previously
described (27). Compared with the
basal group, the co-localization of LDs with LC3 was significantly
increased in HUVECs treated with LDL for 24 h (Fig. 5C). In addition, this
co-localization was increased with exposure to ox-LDL for 6 and 12
h compared with that in the control group, (Fig. 5A and B); however, decreased
co-localization of LDs with LC3 was observed with exposure to
ox-LDL for 24 and 48 h (Fig. 5C and
D), indicating that oxidative stress may reduce lipophagy in
HUVECs when treated with ox-LDL for a longer time.
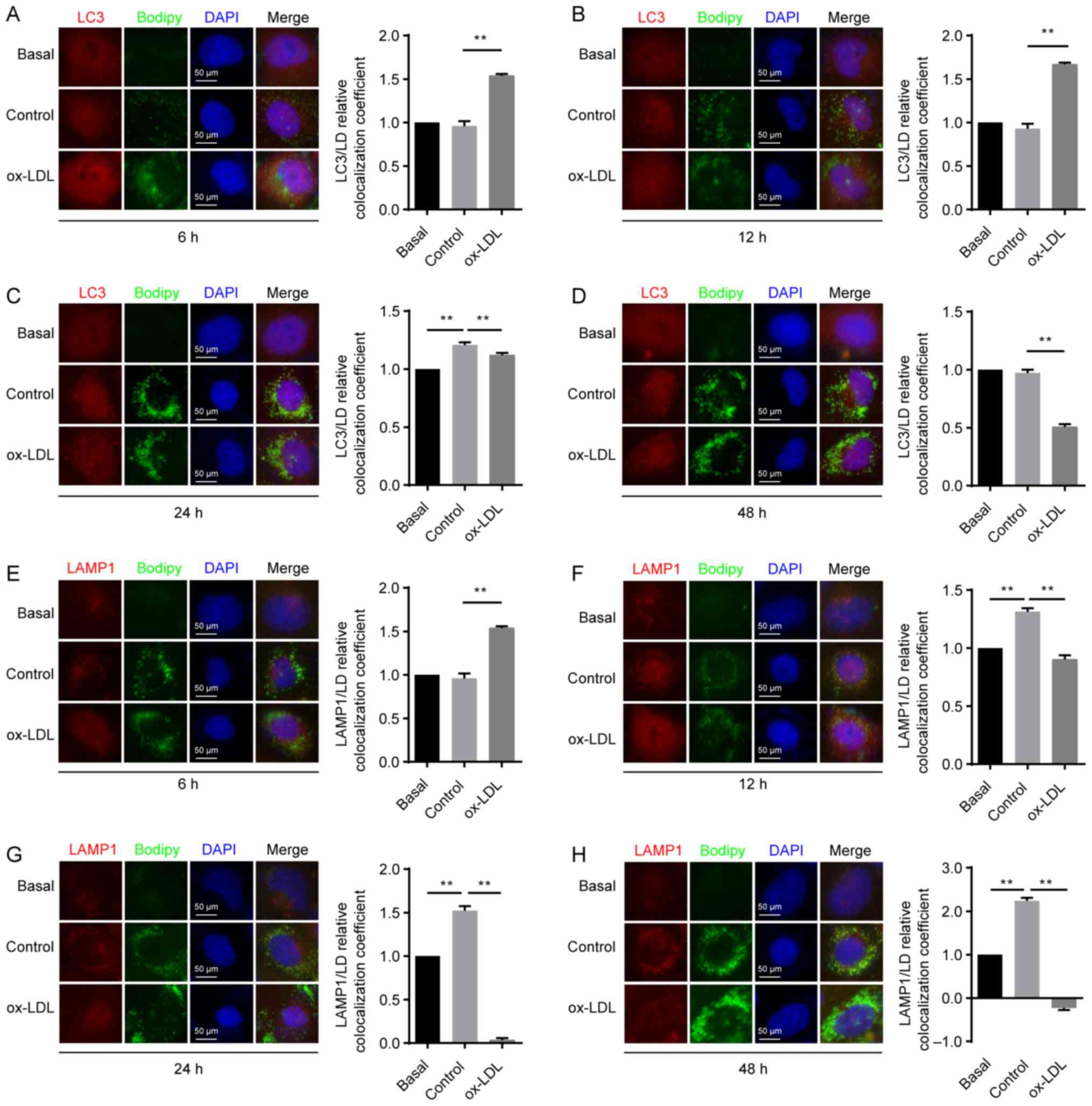 | Figure 5.LD delivery to lysosomes for
degradation is impaired in HUVECs exposed to ox-LDL. HUVECs were
untreated (basal) or treated with either 50 µg/ml LDL (control
group) or 100 µg/ml ox-LDL for 6, 12, 24 or 48 h. Co-localization
of LC3 (red) and LDs (green) in HUVECs following exposure to ox-LDL
for (A) 6, (B) 12, (C) 24 and (D) 48 h. Co-localization of LAMP1
(red) and LDs (green) in HUVECs following exposure to ox-LDL for
(E) 6, (F) 12, (G) 24 and (H) 48 h. Magnification, ×400.
**P<0.01 (n=3). HUVEC, endothelial cell; ox-LDL, oxidized
low-density lipoprotein; LD, lipid droplet; LAMP1,
lysosomal-associated membrane protein 1. |
As the exposure time of HUVECs to LDL was increased,
the co-localization of LDs with LAMP1 was significantly increased
at both 12 and 24 h compared with that in the basal group (Fig. 5E-H), which demonstrated that
treatment with natural LDL may accelerate the process of lipid
degradation in the lysosomes. Compared with the control group, the
co-localization of LDs with LAMP1 was significantly increased in
ECs treated with ox-LDL for 6 h (Fig.
5E); however, this co-localization was significantly decreased
when increasing the exposure time from 12 to 48 h (Fig. 5F, G and H). The present results
demonstrated that ox-LDL induced by oxidative stress may hinder
lipid degradation in the ECs lysosomes.
Overall, the increased lipophagy resulting from
exposure to ox-LDL for 6 to 12 h may be associated with enhanced
degradation of ox-LDL via autophagy-lysosomal pathways, and the
decreased lipophagy observed with exposure to ox-LDL for 24 and 48
h may be associated with attenuated degradation of ox-LDL via
autophagy-lysosomal pathways. This phenomenon can be explained by
the fact that the lysosomal pathway is associated with decreased
ox-LDL degradation, while lipids are degraded in
autophagy-lysosomal pathways (28).
Discussion
AS is a major cause of cardiovascular diseases and
typically leads to stroke, myocardial infarction and coronary heart
disease (2). Damaged ECs lose the
barrier function of the arterial endothelium, accelerating the
development of AS (29).
Furthermore, ECs are highly sensitive to ox-LDL-induced oxidative
damage (30), which is associated
with the risk of coronary heart disease and myocardial infarction
(31). In addition to being an
oxidative stress factor, ox-LDL particles are recognized and
captured by scavenger receptors on ECs, rather than by LDL
receptors, via a reverse cholesterol transport mechanism (32). It has been previously demonstrated
that ox-LDL promotes apoptosis, which disrupts the integrity of the
arterial endothelium (33).
The present study demonstrated that exposure to a
high concentration of ox-LDL (100 µg/ml) decreased the viability of
HUVECs and increased lipid deposition, whereas natural LDL had no
effect on the activity of HUVECs. The present results suggested
that oxidative stress may damage ECs, further leading to lipid
accumulation. It has been established that 7-keto cholesterol
(7KC), one of the components of ox-LDL which has a similar effect
on ECs to total ox-LDL (34), is
also found in human atherosclerotic plaques and serves an active
role in plaque development via inducing vascular EC apoptosis
(35). Therefore, it is important
to develop strategies to reduce the lipid accumulation induced by
ox-LDL.
Both autophagy and apoptosis are associated with
maintaining normal cell functions (36). Autophagy inhibits apoptosis to
ensure cell survival and it is also an alternative mechanism for
cell death in the absence of cell apoptosis (37). In addition, autophagy can protect
various vascular cells, such as macrophages and vascular smooth
muscle cells, against oxidative stress and inflammation, slowing
down atherosclerotic processes (38). It has been demonstrated that
autophagy can be induced by 7KC to inhibit the death of smooth
muscle cells and to avoid thinning of the fibrous cap (39). Furthermore, it has been revealed
that ox-LDL inhibits the autophagy of ECs, serving an essential
role in the development of atherosclerotic lesions (12). However, the precise mechanism by
which ox-LDL inhibits autophagy in ECs has not yet been elucidated.
In the present study, the potential role of autophagy in regulating
ox-LDL-induced ECs was investigated. It was revealed that when
treating HUVECs with ox-LDL for 24 and 48 h, the ratio of LC3-II/I
protein expression levels were significantly decreased, but the
protein expression levels of p62 were significantly increased,
indicating that autophagy was significantly impaired. After
autophagy is induced, autophagosomes are fused with lysosomes to
form autolysosomes, in which the cargo of the autophagosomes is
ultimately degraded (40). LAMP1
is an essential factor that mediates the fusion between
autophagosomes and lysosomes (16). Treatment with ox-LDL for 24 and 48
h revealed that LAMP1 expression was reduced in HUVECs, which is
consistent with previous research (25). Conversely, previous studies
demonstrate that autophagy induced by rapamycin can decrease ox-LDL
accumulation in ECs (41–43). Overall, the present data indicate
that autophagy may be inhibited by ox-LDL in ECs.
Previous studies have revealed that autophagy and
lipid accumulation serve important roles in the development of
atherosclerotic vascular diseases (44,45).
Of note, lipid accumulation has been revealed to accelerate AS
development (45); however, the
exact mechanism of this process remains unclear. In the present
study, the underlying role of autophagy in regulating
ox-LDL-induced EC lipid accumulation was explored, and the
intermolecular interactions were investigated. Increased lipid
staining was observed around the nucleus of HUVECs treated with
ox-LDL for 24 and 48 h, suggesting that there may be an association
between autophagy and lipid accumulation in ECs exposed to
ox-LDL.
Lipophagy is a special form of autophagy (46), which may be involved in
transporting LDs from cells to extracellular HDL. To investigate
whether lipophagy was impaired under ox-LDL prolonged exposure,
numerous ultrastructural changes in ECs were observed using TEM.
Compared with control cells, fewer ALPs were observed in cells
following treatment with ox-LDL for 24 and 48 h, after which they
also exhibited features of cell damage, such as an excess in the
number of vacuoles. The present results suggested that lipophagy
may be attenuated by ox-LDL, which subsequently promoted lipid
accumulation in ECs.
To confirm the impaired lipophagy, the
co-localization of LC3, LDs and LAMP1 were assessed using
immunofluorescence staining in HUVECs. Compared with the basal
group, the co-localization of LDs with LC3 was not significantly
different in HUVECs treated with LDL for 6, 12, and 48 h, which
suggested that lipophagy occurs under normal LDL exposure. Whereas
following treatment with ox-LDL for 24 and 48 h, the
co-localization of LC3 and LDs was significantly decreased, as well
as the co-localization of LAMP1 and LDs. The present result
suggested that endothelial lipophagy may partially maintain
vascular lipid homeostasis on autophagosomal-mediated delivery of
lipids to the lysosome for degradation (19). Furthermore, lipophagy accelerates
lipid hydrolysis, which indirectly promotes the release of free
cholesterol to extracellular Apolipoprotein (Apo) AI receptors
(47,48).
In conclusion, compared with the control group,
lipophagy was impaired in HUVECs treated with ox-LDL, resulting in
lipid accumulation. It was hypothesized that lipophagy may be
attenuated by ox-LDL, resulting in excess cholesterol deposition in
ECs, which may be attributed to inhibiting cholesterol efflux to
Apo AI receptors. Overall, the regulation of lipophagy may be an
attractive therapeutic strategy to limit atherosclerotic disease in
the future.
Acknowledgements
The authors would like to thank Dr Zhi-xin Huang
(Guangdong Second Provincial General Hospital, Guangzhou, China)
for assisting with statistical analysis; Dr Hong An (University of
Missouri, Columbia, MO, USA) for helping to modify the manuscript;
Dr Tan Tan and Dr Xiao-bo Hu (University of South China, Hengyang,
China) for technical support.
Funding
The present study was supported by the National
Nature Science Fund of China (grant nos. 81600291 and 31871169),
the Nature Science Fund of Hunan province (grant nos. 2018JJ2346
and 2018JJ2348) and the Hunan Provincial Innovation Foundation for
Postgraduate (grant no. CX2018B62).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
SYL, MZ and CPZ conceptualized and designed the
experiments. XXD, TT and CPZ performed the experiments. BJL, CYW
and SSJ analyzed and interpreted the data. JQS and YLY aquired the
data. CPZ and XXD wrote the manuscript. YT made substantial
contributions to conception of the study and critically revised the
manuscript for important intellectual content. All authors read and
approved the final version of the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
ALPs
|
autolipophagosomes
|
|
AS
|
atherosclerosis
|
|
ECs
|
endothelial cells
|
|
HUVECs
|
human umbilical vein endothelial
cells
|
|
7KC
|
7-keto cholesterol
|
|
LAMP1
|
lysosomal-associated membrane protein
1
|
|
LDs
|
lipid droplets
|
|
ox-LDL
|
oxidized low-density lipoprotein
|
|
TEM
|
transmission electron microscopy
|
References
|
1
|
Libby P, Ridker PM and Hansson GK:
Progress and challenges in translating the biology of
atherosclerosis. Nature. 473:2665–325. 2011. View Article : Google Scholar
|
|
2
|
Reamy BV, Williams PM and Kuckel DP:
Prevention of cardiovascular disease. Prim Care. 45:25–44. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Yamagishi SI and Matsui T:
Anti-atherothrombogenic properties of PEDF. Curr Mol Med.
10:284–291. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Tsaousi A, Mill C and George SJ: The Wnt
pathways in vascular disease: Lessons from vascular development.
Curr Opin Lipidol. 22:350–357. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ding L, Biswas S, Morton RE, Smith JD, Hay
N, Byzova TV, Febbraio M and Podrez EA: Akt3 deficiency in
macrophages promotes foam cell formation and atherosclerosis in
mice. Cell Metab. 15:861–872. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Williams HJ, Fisher EA and Greaves DR:
Macrophage differentiation and function in atherosclerosis:
Opportunities for therapeutic intervention? J Innate Immun.
4:498–508. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Hellings WE, Peeters W, Moll FL, Piers SR,
van Setten J, Van der Spek PJ, de Vries JP, Seldenrijk KA, De Bruin
PC, Vink A, et al: Composition of carotid atherosclerotic plaque is
associated with cardiovascular outcome: A prognostic study.
Circulation. 121:1941–1950. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Mimura J and Itoh K: Role of Nrf2 in the
pathogenesis of atherosclerosis. Free Radic Biol Med. 88:221–232.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Voloshyna I, Hai O, Littlefield MJ,
Carsons S and Reiss AB: Resveratrol mediates anti-atherogenic
effects on cholesterol flux in human macrophages and endothelium
via PPARg and adenosine. Eur J Pharmacol. 698:299–309. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wu CY, Zhou ZF, Wang B, Ke ZP, Ge ZC and
Zhang XJ: MicroRNA-328 ameliorates oxidized low-density
lipoprotein-induced endothelial cells injury through targeting
HMGB1 in atherosclerosis. J Cell Biochem. Oct 15–2018.(Epub ahead
of print).
|
|
11
|
Demers A, Samami S, Lauzier B, Des Rosiers
C, Ngo Sock ET, Ong H and Mayer G: PCSK9 induces CD36 degradation
and affects long-chain fatty acid uptake and triglyceride
metabolism in adipocytes and in mouse liver. Arterioscler Thromb
Vasc Biol. 35:2517–2525. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Che J, Liang B, Zhang Y, Wang Y, Tang J
and Shi G: Kaempferol alleviates ox-LDL-induced apoptosis by
up-regulation of autophagy via inhibiting PI3K/Akt/mTOR pathway in
human endothelial cells. Cardiovasc Pathol. 31:57–62. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Boya P, Reggiori F and Codogno P: Emerging
regulation and functions of autophagy. Nat Cell Biol. 15:713–720.
2013. View
Article : Google Scholar : PubMed/NCBI
|
|
14
|
Li X, Li Y, Zhao J, Li L, Wang Y, Zhang Y,
Li Y, Chen Y, Liu W and Gao L: Administration of ketamine causes
autophagy and apoptosis in the rat fetal hippocampus and in PC12
cells. Front Cell Neurosci. 12:212018. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wang L, Kim D, Wise JTF, Shi X, Zhang Z
and DiPaola RS: p62 as a therapeutic target for inhibition of
autophagy in prostate cancer. Prostate. 78:390–400. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Fortunato F, Burgers H, Bergmann F, Rieger
P, Buchler MW, Kroemer G and Werner J: Impaired autolysosome
formation correlates with Lamp-2 depletion: Role of apoptosis,
autophagy, and necrosis in pancreatitis. Gastroenterology.
137:350–360.e1-5. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Nowicki M, Zabirnyk O, Duerrschmidt N,
Borlak J and Spanel-Borowski K: No upregulation of lectin-like
oxidized low-density lipoprotein receptor-1 in serum-deprived
EA.hy926 endothelial cells under oxLDL exposure, but increase in
autophagy. Eur J Cell Biol. 86:605–616. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Mollace V, Gliozzi M, Musolino V, Carresi
C, Muscoli S, Mollace R, Tavernese A, Gratteri S, Palma E, Morabito
C, et al: Oxidized LDL attenuates protective autophagy and induces
apoptotic cell death of endothelial cells: Role of oxidative stress
and LOX-1 receptor expression. Int J Cardiol. 184:152–158. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Torisu K, Singh KK, Torisu T, Lovren F,
Liu J, Pan Y, Quan A, Ramadan A, Al-Omran M, Pankova N, et al:
Intact endothelial autophagy is required to maintain vascular lipid
homeostasis. Aging Cell. 15:187–191. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Singh R: Autophagy and regulation of lipid
metabolism. Results Probl Cell Differ. 52:35–46. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Jarc E and Petan T: Lipid droplets and the
management of cellular stress. Yale J Biol Med. 92:435–452.
2019.PubMed/NCBI
|
|
22
|
Liao X, Sluimer JC, Wang Y, Subramanian M,
Brown K, Pattison JS, Robbins J, Martinez J and Tabas I: Macrophage
autophagy plays a protective role in advanced atherosclerosis. Cell
Metab. 15:545–553. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Sergin I and Razani B: Self-eating in the
plaque: What macrophage autophagy reveals about atherosclerosis.
Trends Endocrinol Metab. 25:225–234. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Schindelin J, Arganda-Carreras I, Frise E,
Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S,
Schmid B, et al: Fiji: An open-source platform for biological-image
analysis. Nat Methods. 9:676–682. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zhang YL, Cao YJ, Zhang X, Liu HH, Tong T,
Xiao GD, Yang YP and Liu CF: The autophagy-lysosome pathway: A
novel mechanism involved in the processing of oxidized LDL in human
vascular endothelial cells. Biochem Biophys Res Commun.
394:377–382. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Gómez-Sánchez R, Yakhine-Diop SM,
Rodríguez-Arribas M, Bravo-San Pedro JM, Martínez-Chacón G,
Uribe-Carretero E, Pinheiro de Castro DC, Pizarro-Estrella E,
Fuentes JM and González-Polo RA: mRNA and protein dataset of
autophagy markers (LC3 and p62) in several cell lines. Data Brief.
7:641–647. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Singh R and Cuervo AM: Autophagy in the
cellular energetic balance. Cell Metab. 13:495–504. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Horie S, Hiraishi S, Hirata Y, Kazama M
and Matsuda J: Oxidized low-density lipoprotein impairs the
anti-coagulant function of tissue-factor-pathway inhibitor through
oxidative modification by its high association and accelerated
degradation in cultured human endothelial cells. Biochem J.
352:277–285. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Maracle CX, Agca R, Helder B, Meeuwsen
JAL, Niessen HWM, Biessen EAL, de Winther MPJ, de Jager SCA,
Nurmohamed MT and Tas SW: Noncanonical NF-κB signaling in
microvessels of atherosclerotic lesions is associated with
inflammation, atheromatous plaque morphology and myocardial
infarction. Atherosclerosis. 270:33–41. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Chen B, Meng L, Shen T, Gong H, Qi R, Zhao
Y, Sun J, Bao L and Zhao G: Thioredoxin attenuates oxidized
low-density lipoprotein induced oxidative stress in human umbilical
vein endothelial cells by reducing NADPH oxidase activity. Biochem
Biophys Res Commun. 490:1326–1333. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Mitra S, Goyal T and Mehta JL: Oxidized
LDL, LOX-1 and atherosclerosis. Cardiovasc Drugs Ther. 25:419–429.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Matsuura E, Hughes GR and Khamashta MA:
Oxidation of LDL and its clinical implication. Autoimmun Rev.
7:558–566. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Suc I, Escargueil-Blanc I, Troly M,
Salvayre R and Nègre-Salvayre A: HDL and ApoA prevent cell death of
endothelial cells induced by oxidized LDL. Arterioscler Thromb Vasc
Biol. 17:2158–2166. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Brown AJ and Jessup W: Oxysterols and
atherosclerosis. Atherosclerosis. 142:1–28. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Sasaki H, Watanabe F, Murano T, Miyashita
Y and Shirai K: Vascular smooth muscle cell apoptosis induced by
7-ketocholesterol was mediated via Ca2+ and inhibited by
the calcium channel blocker nifedipine. Metabolism. 56:357–362.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ali ES, Rychkov GY and Barritt GJ:
Metabolic disorders and cancer: Hepatocyte store-operated
Ca2+ channels in nonalcoholic fatty liver disease. Adv
Exp Med Biol. 993:595–621. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Wei DH, Jia XY, Liu YH, Guo FX, Tang ZH,
Li XH, Wang Z, Liu LS, Wang GX, Jian ZS and Ruan CG: Cathepsin L
stimulates autophagy and inhibits apoptosis of ox-LDL-induced
endothelial cells: Potential role in atherosclerosis. Int J Mol
Med. 31:400–406. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Miyazaki T and Miyazaki A: Defective
protein catabolism in atherosclerotic vascular inflammation. Front
Cardiovasc Med. 4:792017. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
He C, Zhu H, Zhang W, Okon I, Wang Q, Li
H, Le YZ and Xie Z: 7-Ketocholesterol induces autophagy in vascular
smooth muscle cells through Nox4 and Atg4B. Am J Pathol.
183:626–637. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Gatica D, Lahiri V and Klionsky DJ: Cargo
recognition and degradation by selective autophagy. Nat Cell Biol.
20:233–242. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Zhang Y, Han Q, You S, Cao Y, Zhang X, Liu
H, Hu L and Liu CF: Rapamycin promotes the autophagic degradation
of oxidized low-density lipoprotein in human umbilical vein
endothelial cells. J Vasc Res. 52:210–219. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Zhou YD, Cao XQ, Liu ZH, Cao YJ, Liu CF,
Zhang YL and Xie Y: Rapamycin inhibits oxidized low density
lipoprotein uptake in human umbilical vein endothelial cells via
mTOR/NF-κB/LOX-1 pathway. PLoS One. 11:e01467772016. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Sun JJ, Yin XW, Liu HH, Du WX, Shi LY,
Huang YB, Wang F, Liu CF, Cao YJ and Zhang YL: Rapamycin inhibits
ox-LDL-induced inflammation in human endothelial cells in vitro by
inhibiting the mTORC2/PKC/c-Fos pathway. Acta Pharmacol Sin.
39:336–344. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Evans TD, Jeong SJ, Zhang X, Sergin I and
Razani B: TFEB and trehalose drive the macrophage
autophagy-lysosome system to protect against atherosclerosis.
Autophagy. 14:724–726. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Michiels CF, Kurdi A, Timmermans JP, De
Meyer GRY and Martinet W: Spermidine reduces lipid accumulation and
necrotic core formation in atherosclerotic plaques via induction of
autophagy. Atherosclerosis. 251:319–327. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Liu K and Czaja MJ: Regulation of lipid
stores and metabolism by lipophagy. Cell Death Differ. 20:3–11.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Zhu X, Xiong T, Liu P, Guo X, Xiao L, Zhou
F, Tang Y and Yao P: Quercetin ameliorates HFD-induced NAFLD by
promoting hepatic VLDL assembly and lipophagy via the IRE1a/XBP1s
pathway. Food Chem Toxicol. 114:52–60. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Leng S, Iwanowycz S, Saaoud F, Wang J,
Wang Y, Sergin I, Razani B and Fan D: Ursolic acid enhances
macrophage autophagy and attenuates atherogenesis. J Lipid Res.
57:1006–1016. 2016. View Article : Google Scholar : PubMed/NCBI
|