Introduction
Non-small cell lung carcinoma (NSCLC) accounts for
~85% of all lung cancer cases (1);
lung adenocarcinoma (LUAD) is the main pathological type of NSCLC
(2). The prognosis of lung cancer
is related to the rate of recurrence, metastasis and chemotherapy
resistance (3). The 5-year
survival rate is 51.4% for patients with adenocarcinoma (4). The poor prognosis of lung cancer
highlights the requirement for the development of novel biomarkers
for the early diagnosis of the disease (5,6).
Thus, there is an urgent need to improve the diagnosis and
management of NSCLC.
Protein arginine methyltransferases (PRMTs) can be
divided into three types: Asymmetrically, symmetrically and
monomethylate protein arginines (type I/II/III, respectively)
(7–9). Histone protein arginine methylation,
catalyzed by PRMTs serves a crucial role in gene regulation
(10). In addition, several
non-histone substrates have also been discovered to be involved in
gene transcription and protein translation (11). PRMTs were revealed to be widely
expressed and activated in gastric and prostate cancer, as well as
myeloid leukemia, where they were involved in cell growth,
differentiation and apoptosis (12–15).
In fact, the disruption of the modification catalyzed by PRMTs
suppressed tumor development, indicating that PRMTs may be used as
a potential therapeutic target for cancer (16). However, to the best of our
knowledge, only a few studies have reported the dysregulation of
PRMTs in lung cancer. For example, PRMT1 and PRMT4 were identified
to be involved in the regulation of proliferation in lung cancer
(17); and PRMT1 and PRMT5 were
discovered to regulate apoptosis induced by doxorubicin or
pemetrexed by affecting cellular FADD-like IL-1β-converting
enzyme-inhibitory protein in NSCLC cells (18). In addition, enolase 1 methylation
by PRMT5 was discovered to be critical for lung cancer cell
invasion (19). Interestingly, to
the best of our knowledge, no other PRMT members and their
dysregulation were reported to be associated with lung cancer.
PRMT6, a type I arginine methyltransferase, has high
affinity for the arginine-2 of Histone H3, specifically catalyzing
Histone H3 asymmetric demethylation at arginine 2 (H3R2me2a)
(10). PRMT6 was first identified
to modify the glycine-and arginine-rich motifs (13), and subsequently reported to target
histones and non-histones (20).
However, the role of PRMT6 in human cancer remains controversial.
The downregulation of PRMT6 expression levels has been reported in
melanoma (21), while the
upregulation of PRMT6 expression levels was reported in the bladder
(13), liver (22) and prostate (14). Interestingly, in one study, PRMT6
upregulation contributed to global DNA hypomethylation in
colorectal and lung adenocarcinoma (23).
At present, studies on PRMT6 are mainly focused on
its function in the nucleus, while the biological function and
important target proteins of PRMT6 in human cancer remain unclear.
The present study aimed to determine the association between PRMT6
expression levels and clinicopathological characteristics of LUAD
via analyzing the putative oncogenic role and the potential
underlying mechanism of PRMT6 in LUAD. The present study
demonstrated that PRMT6 expression levels were markedly upregulated
LUAD.
Materials and methods
Tissue microarray
The LUAD tissue microarray (cat. no. HlugA180Su05),
including 85 pairs of LUAD tissues and matched normal adjacent
tissues (NAT) with clinicopathological data, was provided by
Shanghai Outdo Biotech Co. Ltd. (Outdo Biotech). All patients were
classified according to the tumor-node-metastasis (TNM)
classification by the American Joint Commission of Cancer (24). Lymph node metastasis and the depth
of invasion were classified using the 7th edition of the
International Union Against Cancer TNM staging system (25). The survival time was set as the
time from the day of pathological diagnosis to the day of last
contact or the date of death.
Patient studies
Fresh LUAD tissues and matched NAT from 7 LUAD
patients [3 male and 4 female; age, 47.4±5.2 years (mean ± SD)]
were obtained from The Second Affiliated Hospital of Nanjing
University of Chinese Medicine (Nanjing, China) from March 2018 to
February 2019. Patients included in the study had neither received
chemotherapy nor undergone surgery. Combined with lung disease
already known, other tumor and autoimmune diseases as exclusion
criteria. The study was approved by The Ethics Committee of The
Second Affiliated Hospital of Nanjing University of Chinese
Medicine (Nanjing, China). Written informed consent was obtained
from the patients for the use of fresh lung tissues.
Hematoxylin & Eosin (H&E)
staining and immunohistochemistry
Lung tissues were fixed in 4% paraformaldehyde for
24 h at room temperature before paraffin embedding. Sections (3.5
µm) cut from paraffin-embedded specimens were deparaffinised in
xylene and rehydrated through graded alcohol series. The sections
were first processed for hematoxylin and eosin (H&E) staining
according to standard method (26). Then, antigen retrieval was
conducted using the 1X Diva Decloaker antigen retrieval solution
(Biocare Medical, LLC). at 95°C for 15 min, and then blocking
non-specific sites with 10% goat serum (cat. no. C0265; Beyotime
Institute of Biotechnology) in PBS for 1 h at room temperature,
Subsequently, the sections were incubated with an anti-PRMT6
primary antibody (1:250; cat. no. 14641; Cell Signaling Technology,
Inc.), anti-p18 (1:250; cat. no. A8751; Abclonal Biotech Co., Ltd.)
and anti-Ki67 (1:500; cat. no. A2094; Abclonal Biotech Co., Ltd.)
at 4°C for overnight. Following the primary antibody incubation,
the slides were then incubated with a horseradish peroxidase
(HRP)-conjugated anti-rabbit secondary antibody (1:1,000; cat. no.
AS038; Abclonal Biotech Co., Ltd.) at 25°C for 1 h. The slides were
subsequently stained with a DAB substrate kit (Dako; Agilent
Technologies, Inc.) and counterstained with hematoxylin at 25°C for
20 sec. The immunostaining was detected using an Aperio Digital
Pathology Slide scanner (Leica Biosystems, Wetzlar, Germany).
The nuclear staining of PRMT6/p18/Ki67 was analyzed
using the H-score system. Nuclear staining results were analyzed
using Hscore using Zeiss microscope at a ×100 magnification.
Positive cells were analyzed according to the staining intensity on
a scale of 0–3 (0 = negative, 1 = weak, 2 = moderate, 3 = strong).
H-scores were calculated as the sum of the intensity score (i)
multiplied by the percentage of cells at each intensity (Pi).
H-score =Σ [Pi(i)] ×100. Score values range between 0 and 300.
Cell culture
A549 cells were purchased from the Cell Bank of Type
Culture Collection of the Chinese Academy of Sciences and cultured
in high glucose DMEM (Gibco; Thermo Fisher Scientific, Inc.),
supplemented with 10% FBS (Gibco; Thermo Fisher Scientific, Inc.)
and 1% penicillin streptomycin combination (Gibco; Thermo Fisher
Scientific, Inc.). Cells were maintained at 37°C in an atmosphere
containing 5% CO2.
Reverse transcription-quantitative PCR
(RT-qPCR)
Total RNA was extracted from the adherent cells and
tissues using TRIzol® reagent (Invitrogen; Thermo Fisher
Scientific, Inc.) and cDNA was synthesized using HiScript III 1st
Strand cDNA Synthesis Kit for qPCR (cat. no. R312; Vazyme Biotech
Co., Ltd.). The thermocycling conditions of the RT were as follows:
Remove genomic DNA at 42°C for 2 min; first strand cDNA synthesised
at 25°C for 5 min, 37°C for 45 sec and 85°C for 5 sec. qPCR was
subsequently performed using the ChamQ Universal SYBR qPCR Master
Mix (cat. no. Q711 Vazyme Biotech Co., Ltd.) and the primers
provided in Table I on an ABI 7500
Real-Time PCR machine (Applied Biosystems Inc.). The thermocycling
conditions of the qPCR were as follows: Denaturation at 95°C for 5
min; 40 cycles at 95°C for 10 sec and 60°C for 30 sec; and a final
dissociation stage (95°C for 15 sec, 60°C for 60 sec and 95°C for
15 sec) was added at the end of the amplification procedure. The
data were analyzed using the ABI 7500 SDS software (Version 2.0.6,
Applied Biosystems Inc.). The relative mRNA expression levels were
calculated using the 2−ΔΔCq method (27) and normalized to the GAPDH reference
gene.
 | Table I.Sequences of the primers used for
reverse transcription-quantitative PCR. |
Table I.
Sequences of the primers used for
reverse transcription-quantitative PCR.
| Gene | Primer sequence
(5′→3′) |
|---|
| Protein arginine
methyltransferase 6 | F:
ACGAGTGCTACTCGGACGTT |
|
| R:
AGTTCCGAAGGATACCCAGG |
| p21 | F:
TACCCTTGTGCCTCGCTCAG |
|
| R:
CGGCGTTTGGAGTGGTAGA |
| p27 | F:
GGAGCAATGCGCAGGAATAA |
|
| R:
TGGGGAACCGTCTGAAACAT |
| p18 | F:
ACTGGTTTCGCTGTCATTCA |
|
| R:
GCAGGTTCCCTTCATTATCC |
| CDK inhibitor
3 | F:
TCCGGGGCAATACAGACCAT |
|
| R:
CAGCTAATTTGTCCCGAAACTC |
| CDK4 | F:
ATGGCTACCTCTCGATATGAGC |
|
| R:
CATTGGGGACTCTCACACTCT |
| CDK6 | F:
GCTGACCAGCAGTACGAATG |
|
| R:
GCACACATCAAACAACCTGACC |
| Cyclin D1 | F:
GCTGCGAAGTGGAAACCATC |
|
| R:
CCTCCTTCTGCACACATTTGAA |
| Cyclin E1 | F:
ACTCAACGTGCAAGCCTCG |
|
| R:
GCTCAAGAAAGTGCTGATCCC |
| S-phase
kinase-associated protein 2 | F:
ATGCCCCAATCTTGTCCATCT |
|
| R:
CACCGACTGAGTGATAGGTGT |
| GAPDH | F:
GAGCCACATCGCTCAGACAC |
|
| R:
CATGTAGTTGAGGTCAATGAAGG |
Western blotting
Total protein was extracted from tissues and LUAD
cells using RIPA lysis buffer (Beyotime Institute of
Biotechnology), supplemented with protease and phosphatase
inhibitor cocktails (CST Biological Reagents Co., Ltd.). Nuclear
protein extracts were obtained using the Nuclear Extract kit (cat.
no. P0027; Beyotime Institute of Biotechnology). Core histones were
extracted from the nuclear extracts of the LUAD cells using an
acid-extraction method as previously described (28). Total protein was quantified using a
bicinchoninic acid assay kit (cat. no. 23227; Pierce; Thermo Fisher
Scientific, Inc.) and 25 µg total protein extracts/lane and 10 µg
nuclear extracts/lane were separated via 12% SDS-PAGE.
Subsequently, the separated proteins were transferred onto PVDF
membranes (Roche Diagnostics) and probed with specific primary
antibodies in 5% skimmed milk in PBST (PBS with 0.1% Tween-20)
overnight at 4°C. The membranes were then incubated with rabbit-or
mouse-specific HRP-conjugated secondary antibodies for 2 h at room
temperature. The following primary antibodies were used: Anti-GAPDH
(1:10,000; cat. no. M171-3; MBL Co., Ltd.), anti-PRMT6 (1:1,000;
cat. no. A7814; Abclonal Biotech Co., Ltd.), anti-p18 (1:1,000;
cat. no. A8751; Abclonal Biotech Co., Ltd.), anti-Lamin B1
(1:1,000; cat. no. A11495 Abclonal Biotech Co., Ltd.), anti-Histone
H3 (1:1,000; cat. no. A2348; Abclonal Biotech Co., Ltd.) and
anti-Histone H3R2me2a (1:1,000; cat. no. A3155; Abclonal Biotech
Co., Ltd.). Goat HRP-conjugated anti-rabbit immunoglobulin G (IgG;
1:1,000; cat. no. AS014; Abclonal Biotech Co., Ltd.) and
HRP-conjugated goat anti-mouse IgG (1:1,000; cat. no. AS003;
Abclonal Biotech Co., Ltd.) secondary antibodies were used. Lamin
B1 was used as a loading control for nuclear proteins and GAPDH was
used as a loading control for total proteins. Antibody binding was
detected using an ECL detection system (cat. no. 32106; Thermo
Fisher Scientific, Inc.).
Chromatin immunoprecipitation
(ChIP)
The ChIP assay was performed as previously described
(29). Briefly, cells were
crosslinked by 1% formaldehyde (Sigma-Aldrich) in PBS for 10 min at
25°C. Formaldehyde was quenched by the addition of glycine (Beijing
Solarbio Science & Technology Co., Ltd.) to a final
concentration of 125 µM. Then, 1×106 cells were
collected by centrifugation at 300 × g for 3 min at 25°C and washed
with pre-cooled PBS twice. The immunoprecipitation of crosslinked
100 µg DNA (using a spectrophotometer at 260 nm)/protein was
performed using 2 µg anti-H3R2me2a (1 µg/µl, H3R2me2a; cat. no.
A3155; Abclonal Biotech Co., Ltd.), anti-histone H3 lysine 4
trimethylation (1 µg/µl, H3K4me3; cat. no. A2357; Abclonal Biotech
Co., Ltd.) or anti-mouse/rabbit IgG (1 µg/µl, cat. no. A7028 and
A7016; Beyotime Institute of Biotechnology) antibodies for a 2-h
incubation at 4°C. The immunoprecipitated DNA was purified using a
ChIP DNA purification kit (cat. no. D0033; Beyotime Institute of
Biotechnology) and amplified by qPCR as described above. The chip
primers for the detection of H3R2me2a/H4K4me3 enrichment on p18
promotor as follows: Forward, 5′-GTCTTAAATAACAAACCCCTGTC-3′ and
reverse, 5′-CTCCTCCCGTCAAGTCTCTCGCG-3′.
Vectors, transfections and
infections
pLKO lentiviral vectors for gene knockdown and the
pLKO-scrambled (Scr) short hairpin (sh)RNA vector (control) were
obtained from Sigma-Aldrich; Merck KGaA. The shRNA sequences for
the pLKO lentiviral vector constructions are listed in Table II. A total of 6×106
293T cells were seeded into 100-mm cell culture dishes and
incubated at 37°C and 5% CO2 for 16 h. When the cultured
cells reached 85% confluence, cells were co-transfected with 3 µg
of lentiviral expression constructs pLKO.1-shRNA, pMD2.G and psPAX2
(Sigma- Aldrich; Merck KGaA) using Lipofectamine® 2000
(Invitrogen; Thermo Fisher Scientific, Inc.), according to the
manufacturer's protocol. Viral supernatants were collected by
centrifugation at 800 × g for 5 min at 25°C and filtered through a
0.22-µm membrane filter 48 h post-transfection and stored at −80°C.
A549 cells (5×106 cells per 100-mm culture dish) were
seeded and incubated overnight at 37°C prior to infection. Medium
was then replaced with 1:1 diluted viral supernatant supplemented
with 8 µg/ml polybrene and incubated for 24 h at 37°C, followed by
replacement with normal growth medium. Stable cell lines with shRNA
were selected by puromycin (Clontech Laboratories, Inc.) at a final
concentration of 2 µg/ml in A549 cells.
| Table II.shRNA sequence used for pLKO
lentiviral vectors construction. |
Table II.
shRNA sequence used for pLKO
lentiviral vectors construction.
| shRNA | Sequence
(5′→3′) |
|---|
| PRMT6 sh1 |
CCGGCACCGGCATTCTGAGCATCTTCTCGAGAAGATGCTCAGAATGCCGGTGTTTTTG |
| PRMT6 sh2 |
CCGGCACGGACGTTTCAGGAGAGATCTCGAGATCTCTCCTGAAACGTCCGTGTTTTTG |
| p18 sh1 |
CCGGTGGATTTGGAAGGACTGCGCTCTCGAGAGCGCAGTCCTTCCAAATCCATTTTTG |
| p18 sh2 |
CCGGACTGGTTTCGCTGTCATTCATCTCGAGATGAATGACAGCGAAACCAGTTTTTTG |
| Scr |
CCTAAGGTTAAGTCGCCCTCGCTCGAGCGAGGGCGACTTAACCTTAGG |
Cell proliferation assay
For the cell proliferation assay, 2×103
A549 cells/well were seeded into 96-well plates in triplicate. Cell
proliferation was analyzed using a Cell Counting Kit-8 (CCK-8; cat.
no. A311; Vazyme Biotech Co., Ltd.) assay, according to
manufacturer's protocol. Briefly, cells were plated and incubated
for 24 h in 96-well plates prior to test, 10 µl CCK8 solution was
added to each well and incubated at 37°C for 2 h. The optical
density (OD) was read at an absorbance of 450 nm using a
multifunction microplate reader (Safire, TECAN) for 4 continuous
days.
For the colony formation assay, 500 viable A549
cells per well were seeded into 6-well plates in triplicate.
Following incubation at 37°C for 10 days, the colonies were fixed
with methanol at room temperature for 30 min, stained with 0.05%
crystal violet (Beyotime Institute of Biotechnology) for 60 min at
25°C, washed with running water to remove the excessive dye and
imaged with an Epson Perfection V550 Photo scanner (Seiko Epson
Corporation). Number of colonies (>50 cells) was calculated
using ImageJ software version 1.45 (National Institutes of Health,
Bethesda, MD, USA).
Cell cycle assay
A total of 1×104 cells were collected by
centrifugation at 300 × g for 3 min at 25°C and permeabilized with
ice-cold 70% ethanol overnight at 4°C. Then, the cells were
collected by centrifugation at 300 × g for 3 min at 4°C and stained
with 50 ug/ml propidium iodide in ice-cold PBS supplemented with
0.25 mg/ml RNase A at 4°C for 30 min (cat. no. KGA214-10; Nanjing
KeyGen Biotech Co., Ltd.). The cells were analyzed using a
FACSCalibur (BD Biosciences) flow cytometer and FlowJo X V10.0.7
software (FlowJo LLC) was used to analyze the data.
In vivo tumor models
The animal studies were performed according to the
guidelines of the National Institutes of Health for the Care and
Use of Laboratory Animals (30).
The protocols were approved by The Institute of Animal Care and Use
Committee of Nanjing University of Chinese Medicine (Nanjing,
China).
In order to establish a subcutaneous tumor model, 18
female BALB/c nu/nu mice (6-week-old; weight, 18–20 g) were
obtained from the Model Animal Research Center of Nanjing
University of Chinese Medicine, and maintained under specific
pathogen-free conditions at The Animal Experiment Center of The
Second Affiliated Hospital of Nanjing University of Chinese
Medicine (Nanjing, China). Mice were housed 6 per cage at 25±2°C
with 50±10% humidity and on a 12-hour light–dark cycle with free
access to pellet food and water. They were given a minimum
acclimation period of 1 week before subcutaneous tumor
implantation. A549 cells were first transduced with either Scr,
PRMT6 sh1 or combined PRMT6 sh1 and p18 sh1 lentiviruses to
establish stable cell lines for in vivo studies.
Subsequently, 2×106 cells in 200 µl DMEM supplemented
with 50% Matrigel (2 mg/ml; BD Biosciences) were inoculated
subcutaneously into the right flank of 8-week-old BALB/c nude mice
(6 mice/group). Tumor growth rate was monitored by measuring tumor
diameters every 4 days. Both length and width (W) of the tumor were
measured using a slide caliper, and the tumor volume was calculated
as (Lengthxwidthxwidth)/2.
The sizes of the tumors were measured every 3 days
from injection and tumor volumes were calculated using the formula:
Volume (cm3)=0.52×(lengthxwidth2). All
animals were euthanized under general anesthesia with carbon
dioxide when max tumor volumes reached humane endpoints (~1,000
mm3). The flow rate displaced 10–30% of the chamber
volume/minute. The animals that lost consciousness and muscle
activity were identified as deceased.
Statistical analysis
Data are presented as the mean ± SD of three
independent experiments. Statistical significances were determined
using a two-tailed Student's paired t-test, or a one-way ANOVA,
followed by a Bonferroni's multiple comparisons post hoc test.
Receiver operating characteristic (ROC) curve analysis was
performed to determine the diagnostic value of PRMT6 expression
levels in LUAD. Kaplan-Meier estimates for the primary end point
were calculated and compared using a log-rank (Mantel-Cox) test of
equality. The correlation between PRMT6 and p18 expression levels
was analyzed using Pearson's correlation analysis. All the
statistical analyses were conducted using GraphPad Prism 7 software
(GraphPad Software, Inc.). P<0.05 was considered to indicate a
statistically significant difference.
Results
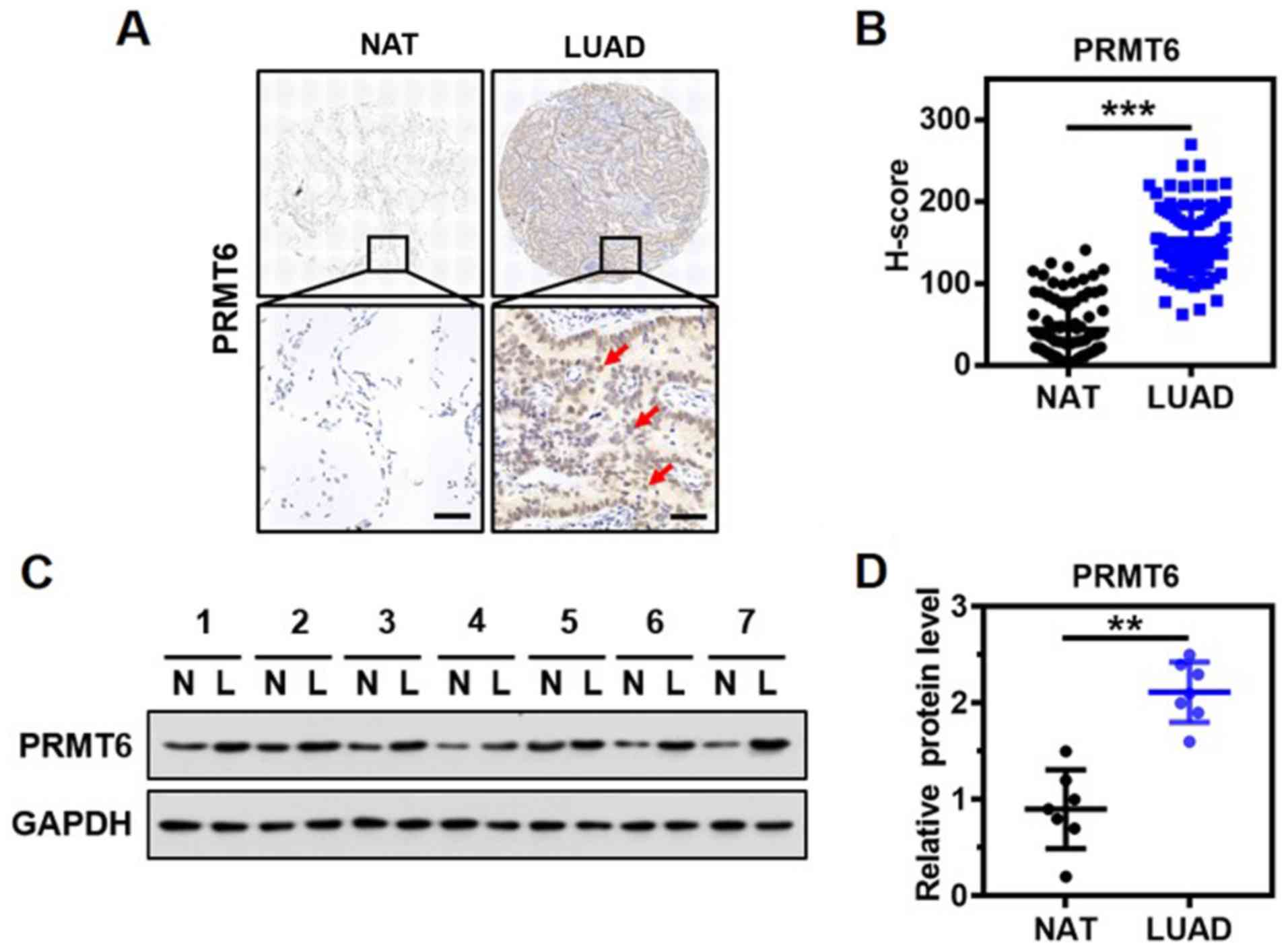
PRMT6 is overexpressed in LUAD
To determine the clinical significance of PRMT6
expression in LUAD, the expression levels of PRMT6 in the lung
tissue from a cohort of 85 patients with LUAD were investigated
using immunohistochemistry and a specific anti-PRMT6 antibody. The
protein was discovered to be predominantly localized in the nucleus
of the glandular epithelium of malignant tissues (Fig. 1A). Notably, significantly
upregulated expression levels of PRMT6 were observed in the tumor
tissues of patients with LUAD compared with the matched NAT
(Fig. 1B). Western blotting
results confirmed that the expression levels of PRMT6 in tumor
tissues from 7 patients with LUAD were significantly upregulated
compared with in NAT (Fig. 1C and
D). Thus, these findings suggested that PRMT6 expression levels
may be significantly upregulated in LUAD.
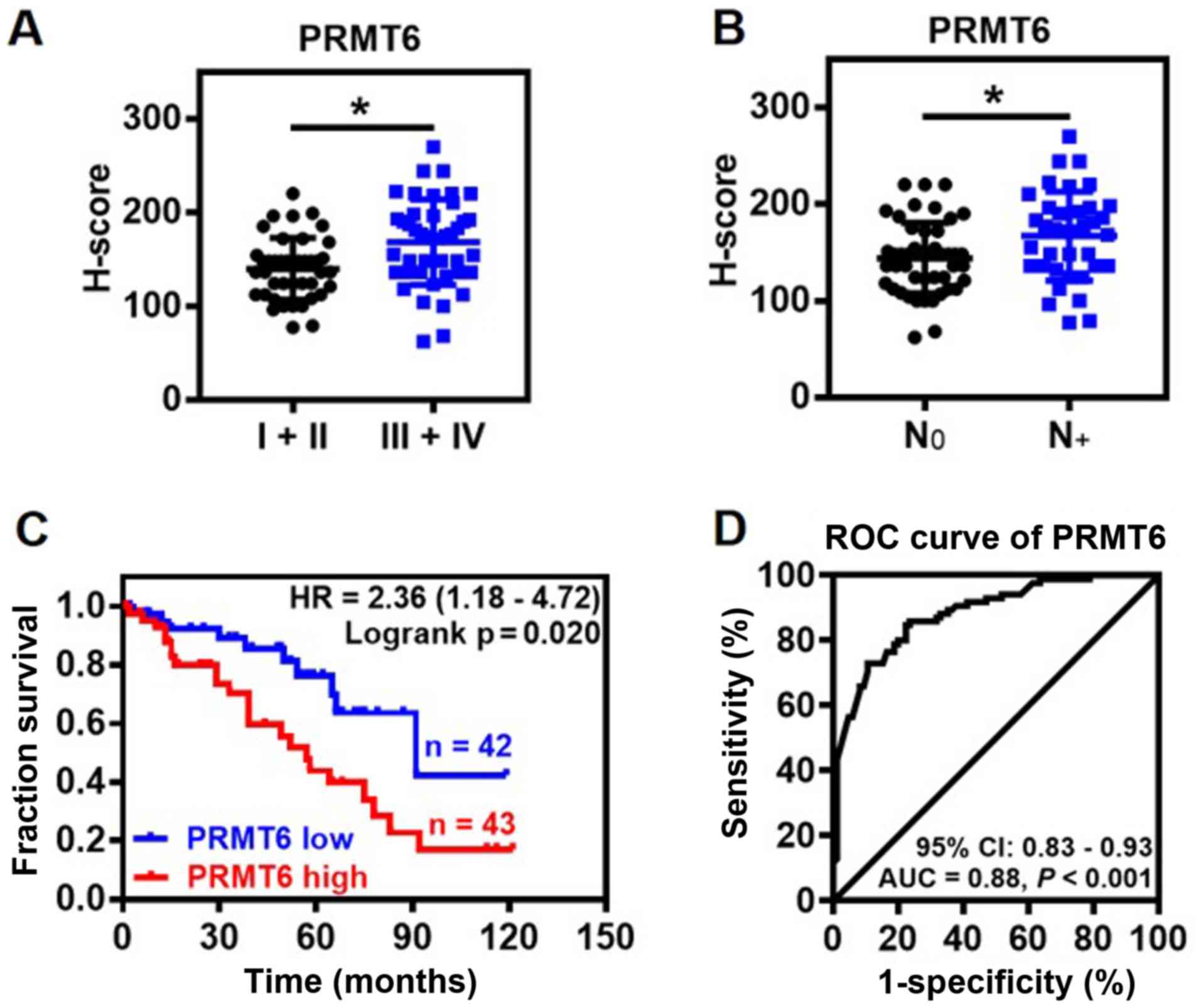
Association between PRMT6 expression
levels and clinicopathological features
The association between PRMT6 expression levels and
clinicopathological features of LUAD was further investigated. The
expression levels of PRMT6 protein were significantly upregulated
in patients with advanced clinical stages (III and IV) and lymph
node metastasis compared with the patients with non-advanced
clinical stages (I and II) and no lymph nodes metastasis,
respectively (Fig. 2A and B).
Notably, the expression levels of PRMT6 were not related to the
age, sex, tumor size, differentiation or local invasion of patients
with LUAD (data not shown). Kaplan-Meier survival analysis
indicated that high expression levels of PRMT6 protein were linked
to a significantly poorer prognosis in patients with LUAD compared
with patients with low expression levels of PRMT6 (Fig. 2C). Furthermore, the predictive
value of PRMT6 was evaluated using ROC curve analysis. The results
indicated that the area under the curve (AUC) was 0.88 [95%
confidence interval (CI), 0.83-0.93] between LUAD tissues and NAT
(Fig. 2D). In the ROC curve
analysis, H-score 97 was set as the cut-off for the expression
levels, based on which, the tumor tissues were discriminated from
NAT with high sensitivity (84.71%) and specificity (76.47%;
Fig. 2D). These findings suggested
that PRMT6 may be used as a novel diagnostic biomarker for
LUAD.
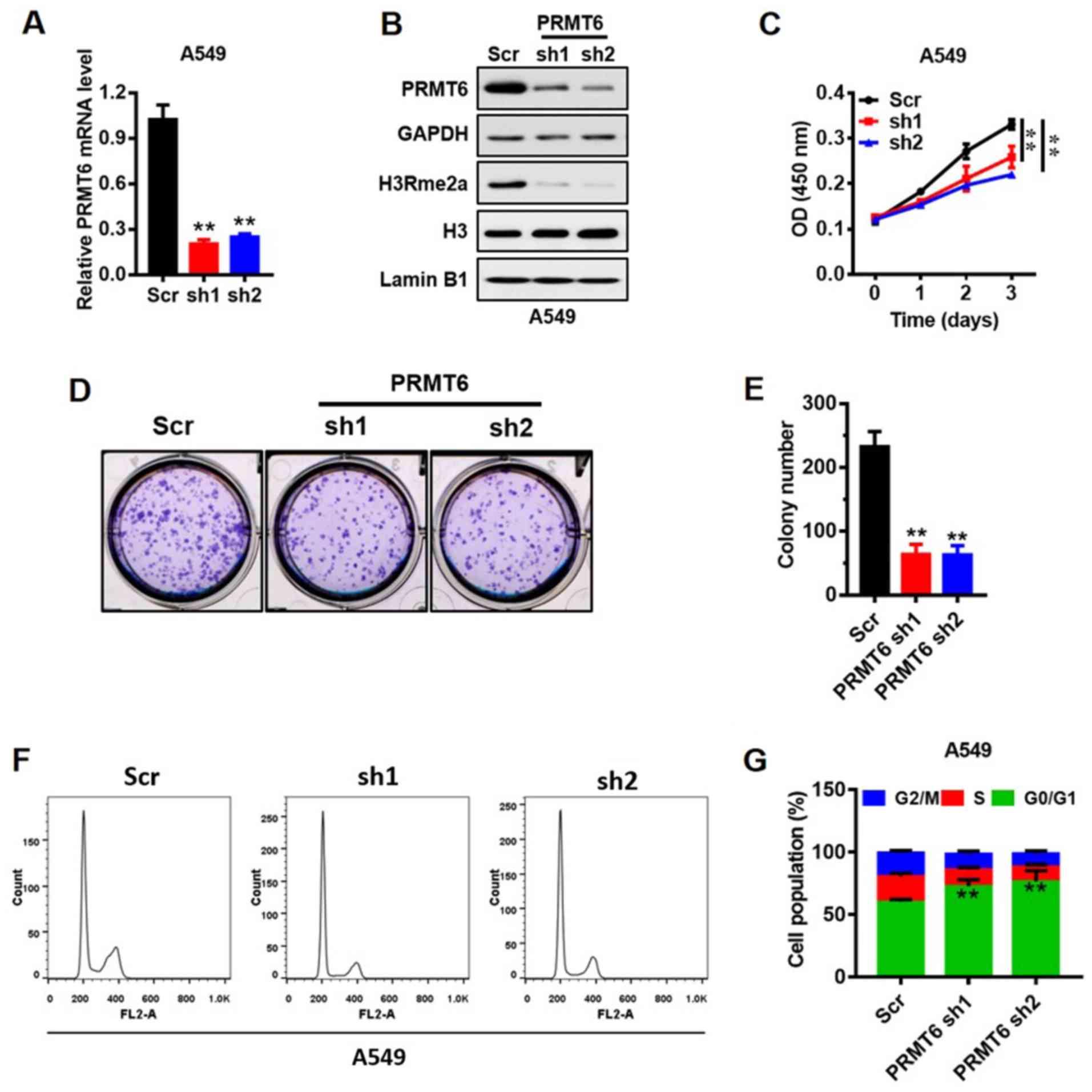
Knockdown of PRMT6 suppresses LUAD
cell growth in vitro through G1/S arrest
A549 cells are the most frequently used cells to
study LUAD (31), thus, the
present study used the A549 cell line to represent LUAD. The
expression of PRMT6 in mRNA (Fig.
3A) and protein (Fig. 3B)
levels was reduced in A549 cell lines using shRNAs (sh1 and sh2)
mediated by lentivirus. PRMT6 was previously demonstrated to
mediate the H3R2me2a modification (9). Herein, the expression levels of
global H3R2me2a were markedly downregulated in PRMT6
sh1/2-transfected cells compared with Scr-transfected cells
(Fig. 3B). Subsequently, the
effects of PRMT6 on the proliferation of A549 cells in vitro
were analyzed using a CCK-8 assay. Notably, the stable knockdown of
PRMT6 significantly suppressed the proliferation of A549 cells
compared with the Scr cells (Fig.
3C). In addition, the number of cell colonies formed were
significantly decreased in the knockdown cells compared with in the
Scr-transfected cells (Fig. 3D and
E).
To further investigate the molecular mechanism
underlying the action of PRMT6 in the proliferation of LUAD cells,
the cell cycling patterns of Scr- and PRMT6 sh1/2-transfected cells
were determined using flow cytometry (Fig. 3F and G). The number of PRMT6
knockdown cells in the G0/1 phase was significantly increased
compared with the Scr-transfected cells. By contrast, the number of
PRMT6 knockdown cells in the S and G2/M phases was significantly
decreased compared with the Scr-transfected cells (Fig. 3F and G). These results suggested
that the downregulation of PRMT6 may induce G1/S phase arrest in
A549 cells, which may subsequently inhibit PRMT6-mediated
proliferation.
p18 is a direct target of PRMT6 and
interferes with G1/S phase arrest in LUAD
To investigate the mechanism underlying cell cycle
arrest induced by the knockdown of PRMT6 in LUAD cells, the mRNA
expression levels of important regulatory genes involved in the
G1/S transition or switch, including p21, p27, p18, CDK inhibitor 3
(CDKN3), CDK4, CDK6, cyclin D1 (CCND1), cyclin E1 (CCNE1) and
S-phase kinase-associated protein 2 (SKP2), were analyzed. RT-qPCR
results demonstrated that the expression levels of p18 were
significantly upregulated in PRMT6 knockdown cells compared with in
Scr-transfected cells, whereas no significant differences were
observed in the remaining genes between the groups (Fig. 4A). The effect of PRMT6 on p18
protein expression levels was also confirmed by western blotting,
where a similar trend was observed (Fig. 4B). Altogether, these findings
indicated that PRMT6 may negatively regulate p18 gene expression
levels.
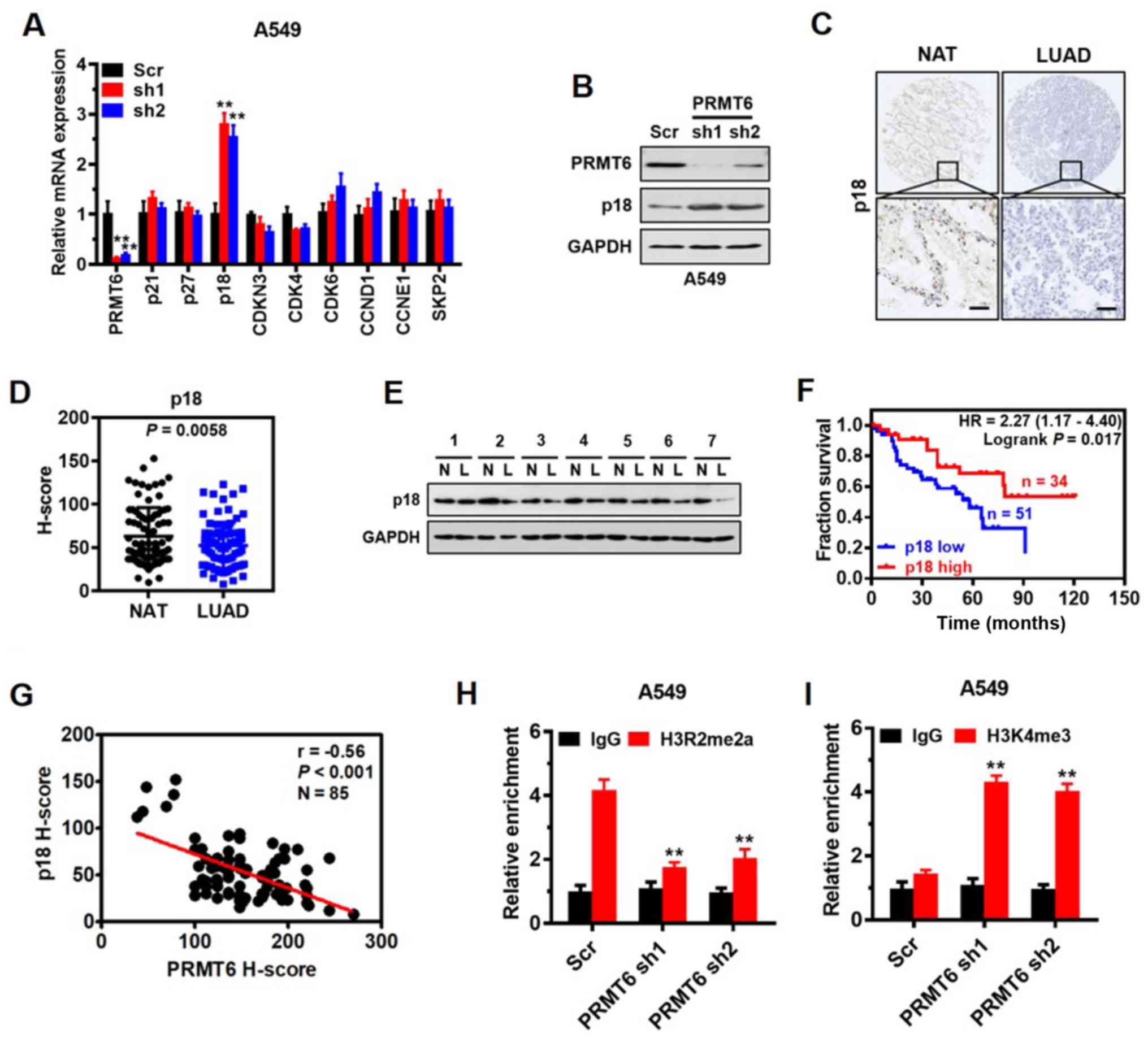 | Figure 4.p18 is a direct target of PRMT6 and
interferes with G1/S phase arrest in LUAD. (A) Reverse
transcription-quantitative PCR analysis was used to determine the
expression levels of important regulatory genes involved in the
G1/S phase in A549 cells infected with Scr, or PRMT6 sh1or sh2. **P
<0.01 vs. Scr group. (B) Western blotting was used to analyze
p18 and PRMT6 expression levels in A549 cells following PRMT6
knockdown. (C) Representative immunohistochemistry images of p18
expression levels in LUAD tissues and matched NAT from 85 patients
with LUAD. Scale bar, 50 µm. (D) Quantitative analysis of
immunohistochemistry scores of p18 protein expression levels in
LUAD tissues and matched NAT from part (C). (E) Western blotting
was used to determine the expression levels of p18 in LUAD tissues
and matched NAT (n=7). GAPDH served as the normalization control.
(F) Kaplan-Meier survival curves were used to determine the overall
survival according to low (n=51) and high (n=34) expression levels
of PRMT6 using immunohistochemistry scores from 85 patients with
LUAD. (G) Correlation between PRMT6 and p18 expression levels was
evaluated using Pearson's correlation analysis. Chromatin
immunoprecipitation assays were used to determine the effects of
PRMT6 knockdown on (H) H3R2me2a and (I) H3K4me3 enrichment in the
p18 promoter of A549 cells. Normalized inputs of A549 chromatin DNA
were pulled down by antibodies against H3R2me2a, H3K4me3 or
negative IgG. **P <0.01 vs. IgG group. PRMT6, protein arginine
methyltransferase 6; LUAD/L, lung adenocarcinoma; NAT/N, matched
normal adjacent tissues; sh; short hairpin RNA; Scr, scramble; IgG,
immunoglobulin G; CCND1, cyclin D1; CCNE1, cyclin E1; SKP2, S-phase
kinase-associated protein 2; CDKN3, CDK inhibitor 3; H3R2me2a,
Histone H3 asymmetric demethylation at arginine 2; H3K4me3, histone
H3 lysine 4 trimethylation. |
p18 INK4c (commonly referred to as p18) is a member
of the INK4 family of CDK inhibitors, which interacts with CDK4/6
and suppresses its activation, functions as a cell growth regulator
of G1/S cell cycle progression and serves as a tumor suppressor
(32,33). However, to the best of our
knowledge, the expression pattern of p18 in LUAD and its
association with patient prognosis remains to be determined. Thus,
the present study investigated the expression levels of p18 in LUAD
tissue arrays using immunohistochemistry. The results demonstrated
that p18 was mainly localized in the nucleus of the glandular
epithelium of LUAD tissues (Fig.
4C) and the protein expression levels of p18 were significantly
downregulated in the LUAD tissues compared with the NAT (Fig. 4D). Western blotting also confirmed
that the expression levels of p18 in tumor tissues from seven
patients with LUAD were downregulated compared with the NAT
(Fig. 4E). Kaplan-Meier survival
analysis demonstrated that lower expression levels of p18 protein
were associated with a significantly poorer prognosis in patients
with LUAD (Fig. 4F). In addition,
a significantly negative correlation was established between PRMT6
and p18 expression levels in vivo by Pearson's correlation
analysis (r=−0.56; Fig. 4G).
PRMT6 functions as a transcriptional repressor by
generating H3R2me2a (34). Thus,
to determine whether PRMT6 directly regulated p18, the enrichment
of H3R2me2a on the p18 promoter was analyzed using a ChIP assay. A
prominent enrichment of H3R2me2a was noted in the gene promoter of
p18 in Scr-transfected cells, which was significantly decreased
when PRMT6 was knocked down in A549 cells (Fig. 4H). The results of ChIP assay are
consistent with the inhibitory effect of PRMT6 on p18 gene
expression. Also, a significant increase was observed in the
enrichment of H3K4me3 in the promoter of p18 when PRMT6 was knocked
down in A549 cells (Fig. 4I),
indicating a potential crosstalk between H3R2me2a and H3K4me3 to
enhance p18 gene expression. These findings indicated that p18 may
be a downstream target of PRMT6 and interfere with G1/S phase
arrest in LUAD cells.
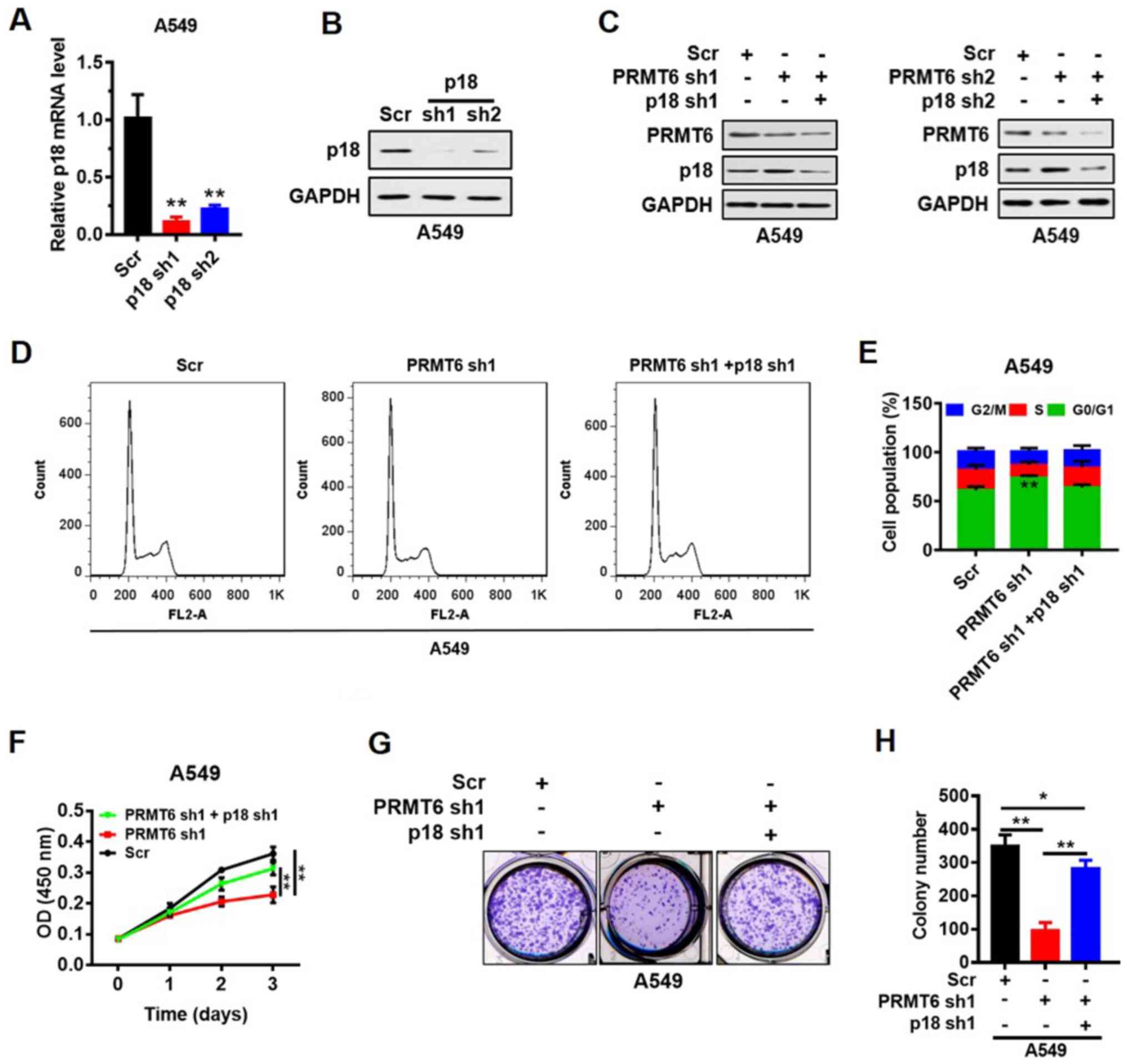
PRMT6 knockdown suppresses LUAD cell
growth by activating p18 expression in vitro
p18 was knocked down by shRNAs (sh1 and sh2) in the
A549 cell line and the transfection efficiency was analyzed using
RT-qPCR and western blotting. The results indicated that the
expression levels of p18 mRNA (Fig.
5A) and protein (Fig. 5B) were
downregulated in p18 shRNA-transfected cells compared with the Scr
shRNA-transfected cells. To verify the suppression of LUAD cell
proliferation mediated by p18 re-activation, stable cells with p18
and PRMT6 double knockdown were constructed. Western blot analyses
indicated that p18 protein expression levels were not recovered by
the knockdown of p18 and PRMT6 simultaneously (Fig. 5C). Furthermore, the G0/1 phase
arrest induced by PRMT6 knockdown was abrogated by further
knockdown of p18 (Fig. 5D and E).
In addition, the PRMT6 knockdown-induced suppression of cell
proliferation (Fig. 5F) and colony
formation (Fig. 5G and H) was
significantly reversed by p18 knockdown. These results indicated
that PRMT6 knockdown may suppress LUAD cell growth by activating
p18 expression in vitro.
PRMT6 knockdown suppresses LUAD
development by activating p18 expression in vivo
So far, the present study demonstrated that PRMT6
served an important role in the regulation of LUAD cell growth
in vitro by regulating the expression levels of p18.
However, whether this regulatory mechanism was reciprocated in
vivo remained to be clarified. Stable cell lines transfected
with Scr, PRMT6 sh1 or PRMT6 sh1 and p18 sh1 were used to create
subcutaneous xenografts in BALB/c nude mice. The present study
observed that the knockdown of PRMT6 expression levels
significantly inhibited tumor growth compared with the Scr group,
whereas the combined downregulation with p18 significantly reversed
the inhibition of tumor growth mediated by PRMT6 knockdown in
vivo (Fig. 6A-C). However, the
body weight of the nude mice was not altered between the groups
(Fig. 6D). In addition, the
present study further analyzed the changes in the PRMT6 and p18
protein expression levels in the xenograft tumor tissues. Western
blotting data revealed that the protein expression levels of p18
were markedly upregulated in the PRMT6 knockdown group compared
with the Scr group; however, this regulation was abrogated by
further knockdown of p18 (Fig.
6E). Similar results were obtained through using
immunohistochemistry analysis to determine the levels of Ki67,
PRMT6 and p18 in xenograft tumor tissues (Fig. 6F). These results suggested that the
knockdown of PRMT6 may suppress LUAD development by activating p18
expression in vivo.
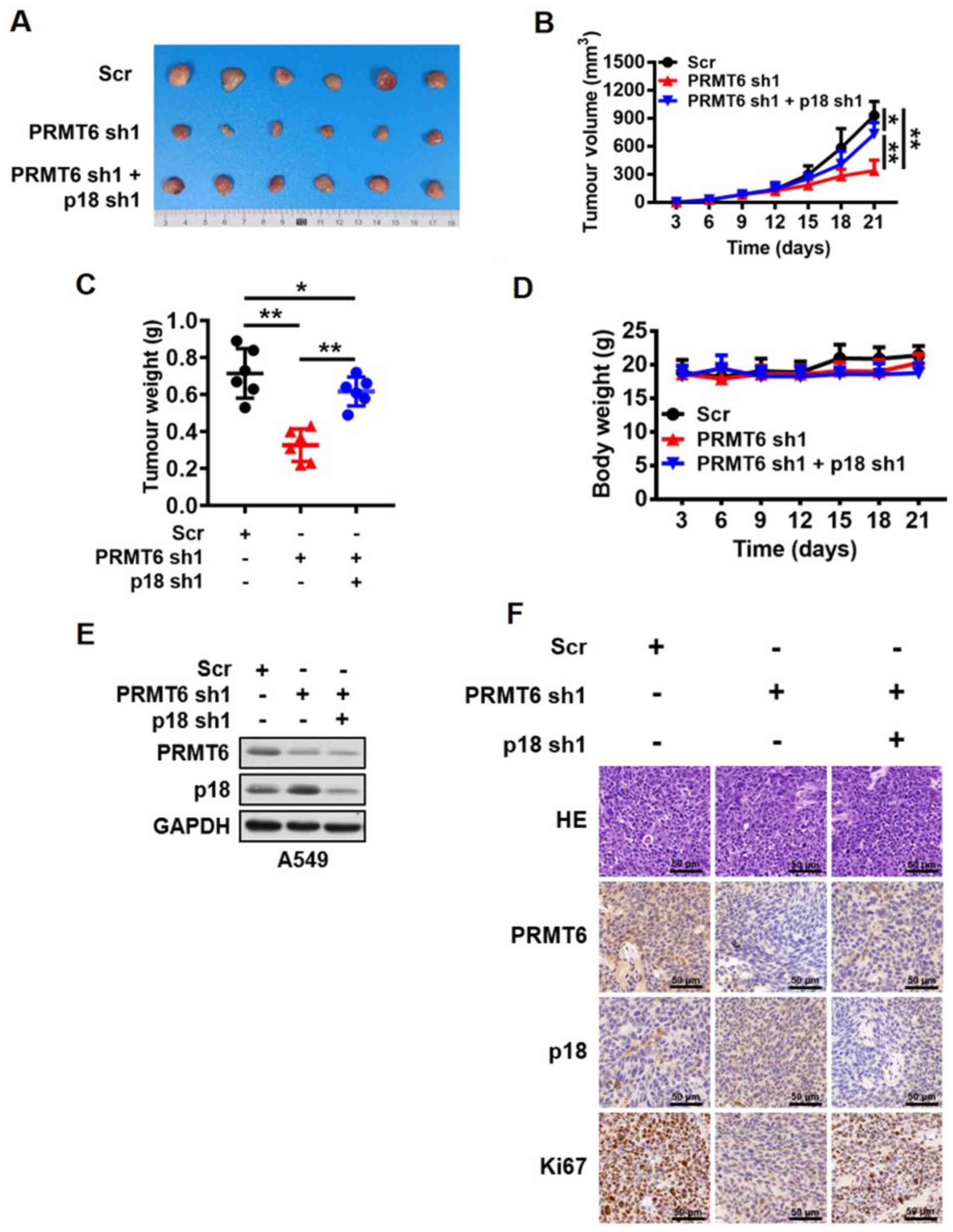 | Figure 6.Knockdown of PRMT6 suppresses lung
adenocarcinoma development by activating p18 expression in vivo.
(A) Images of tumors excised from BALB/c nude mice in the Scr,
PRMT6 knockdown, and PRMT6 and p18 knockdown groups. (B) Tumor
volume of xenograft tumors from mice in the Scr, PRMT6 knockdown,
and PRMT6 and p18 knockdown groups were measured every 3 days.
*P<0.05, **P<0.01. (C) Weight of xenograft tumors excised
from mice in the Scr, PRMT6 knockdown, and PRMT6 and p18 knockdown
groups were measured at the end of the experiment. *P<0.05,
**P<0.01. (D) Body weight of A549 tumor-bearing mice were
measured every 3 days. (E) Western blotting analysis of PRMT6 and
p18 protein expression levels in xenograft tumors excised from mice
in the Scr, PRMT6 knockdown and, PRMT6 and p18 knockdown groups.
(F) Representative pathological images of PRMT6, p18 and Ki67
expression levels in xenograft tumors excised from mice in the Scr,
PRMT6 knockdown and, PRMT6 and p18 knockdown groups. Scale bar, 50
µm. PRMT6, protein arginine methyltransferase 6; sh; short hairpin
RNA; Scr, scramble; HE, hematoxylin and eosin; NS,
non-significant. |
Discussion
The current study aimed to investigate the
biological effects of PRMT6 and its potential mechanism of action
in LUAD. The results of the present study demonstrated that the
proliferation of LUAD cells was significantly suppressed by
silencing PRMT6 expression both in vitro and in vivo.
In addition, PRMT6 knockdown decreased the enrichment of H3R2me2a
in the promoter region of the p18 gene, thereby activating the
expression of the gene. G1/S phase arrest was also induced,
resulting in the inhibition of cell proliferation. These results
strongly indicated that PRMT6 may serve a prooncogenic role in the
progression of LUAD through the epigenetic suppression of p18
expression. Thus, these findings study may provide a novel
potential target for the treatment of LUAD.
PRMTs, which specialize in methylating both histone
and non-histone proteins, have been discovered to be involved in
numerous biological processes, such as cell growth, metabolism and
signal transduction, among others (35,36).
However, to the best of our knowledge, the role of PRMT6 in human
LUAD remains unknown. Considering that the detection of protein
expression levels in tissue microarrays containing clinical samples
are more accurate and reliable than the gene expression levels
found in RNA-seq databases, and the observation time of the
patients in tissue microarrays was longer compared with the
patients with LUAD listed in the RNA-seq database, RNA-seq database
analysis was not performed in the present study. In fact, to the
best of our knowledge, the investigations of the current study were
the first to analyze the expression levels of the proteins in fresh
clinical tissues samples. In the present research, the expression
levels of PRMT6 were discovered to be negatively associated with
the clinical staging, lymph node metastasis and clinical prognosis
of patients with LUAD, indicating that PRMT6 may serve an oncogenic
role in LUAD development. Furthermore, silencing PRMT6 suppressed
the cell proliferation of LUAD cells in vivo and in
vitro, which was ascribed to G1/S cell cycle arrest. These data
suggested that PRMT6 may serve a pivotal role in the G1/S phase
transition of LUAD cells. Interestingly, differences were noted in
the shRNA transfection efficiency in A549 cells between Figs. 3B and 4B; in Fig.
3B, sh2 appeared more effective, whereas in Fig. 4B, sh1 exhibited nearly 100%
efficiency. The differences in the shRNA transfection efficiency
may be related to the semi-quantification of protein samples and/or
the transfer efficiency of western blotting. Nevertheless, PRMT6
was effectively knocked down in the present study and the
differences in the shRNA transfection efficiencies did not affect
the experimental conclusion.
It is well known that cancers are considered to be a
disease of cell cycle disorder, which is accompanied by the
abnormal regulation of cell cycle regulatory proteins (37). The arginine methylation of a
protein is a post-translational modification, which has been
contributed to the disorder of the cell cycle in melanoma (38). PRMTs have been reported to
methylate several regulatory proteins of the cell cycle, such as
p21, p53, cyclin D1 and phosphorylated Rb (39). Previous studies have also
identified that p21 and p27 were direct target genes of PRMT6,
which received H3R2me2a modifications and promoted cell cycle
progression through CDK1/2 in U2OS and breast cancer cells
(34,40,41).
In the present study, it was hypothesized that the downregulation
of PRMT6 expression levels may inhibit the proliferation of LUAD
cells through p18 activation. Thus, to investigate whether p18 was
a direct target gene of PRMT6. ChIP analysis was performed in LUAD
cells, which confirmed that H3R2me2a was significantly enriched in
the promoter of the p18 gene. These findings indicated that PRMT6
may serve a prooncogenic function in the development of LUAD via
the epigenetic suppression of p18 expression.
PRMT6 has been reported to be responsible for
H3R2me2a; it has been associated with the inactive promoters of
mammalian (42). For example, the
H3R2me2a modification has been identified to prevent MLL/SET lysine
methyltransferase complexes from binding to H3 (43) and PRMT6 action was discovered to
impede the deposition of H3K4me3 (44). In the present study, H3R2me2a was
enriched at the p18 gene promoter in the control cells.
Correspondingly, the study further confirmed that the enrichment of
H3K4me3 on the p18 promoter was significantly increased following
the knockdown of PRMT6, indicating crosstalk between H3R2me2a and
H3K4me3 and the enhancement of p18 gene repression.
In conclusion, the findings of the current study
suggested that PRMT6 may serve as a novel potential diagnostic
biomarker for LUAD through acting as an oncogene in the disease,
epigenetically suppressing p18 expression. Taken together, these
findings may offer novel opportunities for the treatment, diagnosis
and management of this major subtype of NSCLC.
Acknowledgements
Not applicable.
Funding
The present study was supported by the Project of
The Second Affiliated Hospital of Nanjing University of Chinese
Medicine (grant no. SEZJJP2018039), The General Project of Jiangsu
Natural Science Foundation (grant no. BK20161605) and The Science
and Technology Bureau of Nanjing City (grant no. 201803060).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
JT performed the experiments; QM and RS analyzed the
data; and YX designed the experiments, interpreted the data and
wrote the manuscript. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
This study was approved by The Ethics Committee of
the Second Affiliated Hospital of Nanjing University of Chinese
Medicine (approval no. 2019-010-054). Written informed consent was
obtained from the patients for the use of lung tissue. The animal
experiments were approved by The Institute of Animal Care and Use
Committee of Nanjing University of Chinese Medicine.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Molina JR, Yang P, Cassivi SD, Schild SE
and Adjei AA: Non-small cell lung cancer: Epidemiology, risk
factors, treatment, and survivorship. Mayo Clin Proc. 83:3161–594.
2008. View Article : Google Scholar
|
|
2
|
Chen Z, Fillmore CM, Hammerman PS, Kim CF
and Wong KK: Non-small-cell lung cancers: A heterogeneous set of
diseases. Nat Rev Cancer. 14:535–546. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Wood SL, Pernemalm M, Crosbie PA and
Whetton AD: The role of the tumor-microenvironment in lung
cancer-metastasis and its relationship to potential therapeutic
targets. Cancer Treat Rev. 40:558–566. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
de Perrot M, Fadel E, Mussot S, de Palma
A, Chapelier A and Dartevelle P: Resection of locally advanced (T4)
non-small cell lung cancer with cardiopulmonary bypass. Ann Thorac
Surg. 79:1691–1697. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wu H, Zhou J, Mei S, Wu D, Mu Z, Chen B,
Xie Y, Ye Y and Liu J: Circulating exosomal microRNA-96 promotes
cell proliferation, migration and drug resistance by targeting
LMO7. J Cell Mol Med. 21:1228–1236. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Joshi P, Jeon YJ, Laganà A, Middleton J,
Secchiero P, Garofalo M and Croce CM: MicroRNA-148a reduces
tumorigenesis and increases TRAIL-induced apoptosis in NSCLC. Proc
Natl Acad Sci USA. 112:8650–8655. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Fedoriw A, Rajapurkar SR, O'Brien S,
Gerhart SV, Mitchell LH, Adams ND, Rioux N, Lingaraj T, Ribich SA,
Pappalardi MB, et al: Anti-tumor activity of the type I PRMT
inhibitor, GSK3368715, synergizes with PRMT5 inhibition through
MTAP loss. Cancer Cell. 36:100–114.e25. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Bedford MT and Clarke SG: Protein arginine
methylation in mammals: Who, what, and why. Mol Cell. 33:1–13.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Pahlich S, Zakaryan RP and Gehring H:
Protein arginine methylation: Cellular functions and methods of
analysis. Biochim Biophys Acta. 1764:1890–1903. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Blanc RS and Richard S: Arginine
methylation: The coming of age. Mol Cell. 65:8–24. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Bowitch A, Michaels KL, Yu MC and Ferkey
DM: The protein arginine methyltransferase PRMT-5 regulates SER-2
tyramine receptor-mediated behaviors in caenorhabditis elegans. G3
(Bethesda). 8:2389–2398. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Altan B, Yokobori T, Ide M, Mochiki E,
Toyomasu Y, Kogure N, Kimura A, Hara K, Bai T, Bao P, et al:
Nuclear PRMT1 expression is associated with poor prognosis and
chemosensitivity in gastric cancer patients. Gastric Cancer.
19:789–797. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Yoshimatsu M, Toyokawa G, Hayami S, Unoki
M, Tsunoda T, Field HI, Kelly JD, Neal DE, Maehara Y, Ponder BA, et
al: Dysregulation of PRMT1 and PRMT6, Type I arginine
methyltransferases, is involved in various types of human cancers.
Int J Cancer. 128:562–573. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Almeida-Rios D, Graça I, Vieira FQ,
Ramalho-Carvalho J, Pereira-Silva E, Martins AT, Oliveira J,
Gonçalves CS, Costa BM, Henrique R and Jerónimo C: Histone
methyltransferase PRMT6 plays an oncogenic role of in prostate
cancer. Oncotarget. 7:53018–53028. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Greenblatt SM, Man N, Hamard PJ, Asai T,
Karl D, Martinez C, Bilbao D, Stathias V, Jermakowicz AM, Duffort
S, et al: CARM1 is essential for myeloid leukemogenesis but
dispensable for normal hematopoiesis. Cancer Cell. 35:1562019.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Cheung N, Chan LC, Thompson A, Cleary ML
and So CW: Protein arginine-methyltransferase-dependent
oncogenesis. Nat Cell Biol. 9:1208–1215. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Elakoum R, Gauchotte G, Oussalah A,
Wissler MP, Clément-Duchêne C, Vignaud JM, Guéant JL and Namour F:
CARM1 and PRMT1 are dysregulated in lung cancer without
hierarchical features. Biochimie. 97:210–218. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Li M, An W, Xu L, Lin Y, Su L and Liu X:
The arginine methyltransferase PRMT5 and PRMT1 distinctly regulate
the degradation of anti-apoptotic protein CFLARL in
human lung cancer cells. J Exp Clin Cancer Res. 38:642019.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zakrzewicz D, Didiasova M, Krüger M,
Giaimo BD, Borggrefe T, Mieth M, Hocke AC, Zakrzewicz A, Schaefer
L, Preissner KT and Wygrecka M: Protein arginine methyltransferase
5 mediates enolase-1 cell surface trafficking in human lung
adenocarcinoma cells. Biochim Biophys Acta Mol Basis Dis.
1864:1816–1827. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Frankel A, Yadav N, Lee J, Branscombe TL,
Clarke S and Bedford MT: The novel human protein arginine
N-methyltransferase PRMT6 is a nuclear enzyme displaying unique
substrate specificity. J Biol Chem. 277:3537–3543. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Limm K, Ott C, Wallner S, Mueller DW,
Oefner P, Hellerbrand C and Bosserhoff AK: Deregulation of protein
methylation in melanoma. Eur J Cancer. 49:1305–1313. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Chan LH, Zhou L, Ng KY, Wong TL, Lee TK,
Sharma R, Loong JH, Ching YP, Yuan YF, Xie D, et al: PRMT6
regulates RAS/RAF binding and MEK/ERK-mediated cancer stemness
activities in hepatocellular carcinoma through CRAF methylation.
Cell Rep. 25:690–701 e698. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Veland N, Hardikar S, Zhong Y, Gayatri S,
Dan J, Strahl BD, Rothbart SB, Bedford MT and Chen T: The arginine
methyltransferase PRMT6 regulates DNA methylation and contributes
to global DNA hypomethylation in cancer. Cell Rep. 21:3390–3397.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Sellers AH: The clinical classification of
malignant tumours: The TNM system. Can Med Assoc J. 105:836passim.
1971.PubMed/NCBI
|
|
25
|
Wohlschläger J, Wittekind C and Theegarten
D: New TNM classification of malignant lung tumours. Pathologe.
31:355–360. 2010.(In German). View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Feldman AT and Wolfe D: Tissue processing
and hematoxylin and eosin staining. Methods Mol Biol. 1180:31–43.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Shechter D, Dormann HL, Allis CD and Hake
SB: Extraction, purification and analysis of histones. Nat Protoc.
2:1445–1457. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zhao Q, Rank G, Tan YT, Li H, Moritz RL,
Simpson RJ, Cerruti L, Curtis DJ, Patel DJ, Allis CD, et al:
PRMT5-mediated methylation of histone H4R3 recruits DNMT3A,
coupling histone and DNA methylation in gene silencing. Nat Struct
Mol Biol. 16:304–311. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Clark JD, Gebhart GF, Gonder JC, Keeling
ME and Kohn DF: Special report: The 1996 Guide for the Care and Use
of Laboratory Animals. ILAR J. 38:41–48. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Gong S, Qu X, Yang S, Zhou S, Li P and
Zhang Q: RFC3 induces epithelialmesenchymal transition in lung
adenocarcinoma cells through the Wnt/β-catenin pathway and
possesses prognostic value in lung adenocarcinoma. Int J Mol Med.
44:2276–2288. 2019.PubMed/NCBI
|
|
32
|
Sherr CJ and Roberts JM: CDK inhibitors:
Positive and negative regulators of G1-phase progression. Genes
Dev. 13:1501–1512. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Guan KL, Jenkins CW, Li Y, Nichols MA, Wu
X, O'Keefe CL, Matera AG and Xiong Y: Growth suppression by p18, a
p16INK4/MTS1- and p14INK4B/MTS2-related CDK6 inhibitor, correlates
with wild-type pRb function. Genes Dev. 8:2939–2952. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Stein C, Riedl S, Ruthnick D, Nötzold RR
and Bauer UM: The arginine methyltransferase PRMT6 regulates cell
proliferation and senescence through transcriptional repression of
tumor suppressor genes. Nucleic Acids Res. 40:9522–9533. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Auclair Y and Richard S: The role of
arginine methylation in the DNA damage response. DNA Repair (Amst).
12:459–465. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Biggar KK and Li SS: Non-histone protein
methylation as a regulator of cellular signalling and function. Nat
Rev Mol Cell Biol. 16:5–17. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Malumbres M and Carnero A: Cell cycle
deregulation: A common motif in cancer. Prog Cell Cycle Res.
5:5–18. 2003.PubMed/NCBI
|
|
38
|
AbuHammad S, Cullinane C, Martin C,
Bacolas Z, Ward T, Chen H, Slater A, Ardley K, Kirby L, Chan KT, et
al: Regulation of PRMT5-MDM4 axis is critical in the response to
CDK4/6 inhibitors in melanoma. Proc Natl Acad Sci USA.
116:17990–18000. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Raposo AE and Piller SC: Protein arginine
methylation: An emerging regulator of the cell cycle. Cell Div.
13:32018. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Kleinschmidt MA, de Graaf P, van Teeffelen
HA and Timmers HT: Cell cycle regulation by the PRMT6 arginine
methyltransferase through repression of cyclin-dependent kinase
inhibitors. PLoS One. 7:e414462012. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Phalke S, Mzoughi S, Bezzi M, Jennifer N,
Mok WC, Low DH, Thike AA, Kuznetsov VA, Tan PH, Voorhoeve PM and
Guccione E: p53-independent regulation of p21Waf1/Cip1 expression
and senescence by PRMT6. Nucleic Acids Res. 40:9534–9542. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Guccione E, Bassi C, Casadio F, Martinato
F, Cesaroni M, Schuchlautz H, Lüscher B and Amati B: Methylation of
histone H3R2 by PRMT6 and H3K4 by an MLL complex are mutually
exclusive. Nature. 449:933–937. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Hyllus D, Stein C, Schnabel K, Schiltz E,
Imhof A, Dou Y, Hsieh J and Bauer UM: PRMT6-mediated methylation of
R2 in histone H3 antagonizes H3 K4 trimethylation. Genes Dev.
21:3369–3380. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Kirmizis A, Santos-Rosa H, Penkett CJ,
Singer MA, Vermeulen M, Mann M, Bähler J, Green RD and Kouzarides
T: Arginine methylation at histone H3R2 controls deposition of H3K4
trimethylation. Nature. 449:928–932. 2007. View Article : Google Scholar : PubMed/NCBI
|