Introduction
Cardiac fibrosis is a worldwide health issue
associated with nearly all forms of heart disease, such as
ventricular dilation and heart failure (1), which accounts for nearly 31% of all
deaths each year (2). Fibrosis
pathogenesis mainly involves inappropriate fibroblast accumulation
and excess collagen deposition in the myocardium (3), which is accompanied by excessive
oxidative stress and apoptosis or necrosis of cardiomyocytes
(4). Acute myocardial infarction,
ageing, pressure overload, volume overload, hypertrophic
cardiomyopathy and post-viral dilated cardiomyopathy are common
pathophysiological conditions that induce cardiac fibrosis
(5). Moreover, metabolic disorders
such as diabetes and obesity are involved in the formation of
fibrosis in the myocardium of patients (6,7). A
previous study reported that cardiac fibrosis is closely associated
with the progression of diabetic cardiomyopathy, which is a major
cause of mortality in patients with diabetes (8). Thus, it is crucial to develop novel
targets to suppress apoptosis of cardiomyocytes, and to prevent the
formation of diabetic cardiac fibrosis and cardiomyopathy.
MicroRNAs (miRNAs) are small non-coding RNAs that
are involved in the regulation of gene expression at the
post-transcriptional level (9). It
has been demonstrated that miRNAs serve a significant role in
cardiovascular diseases (10); for
example, miR-155 dysregulation is essential for cardiac
pathophysiology, including hypertrophy, remodeling and fibrosis.
Loss of miR-155 can inhibit pathological cardiac hypertrophy
(11), whereas cardiac fibrosis is
usually associated with elevated miR-155 expression levels
(12). However, the downstream
signaling pathways of miR-155 that regulate myocardial fibrosis
have not been fully elucidated.
Nuclear factor erythroid-2-related factor 2 (Nrf2)
is a transcription factor with a basic-leucine zipper domain that
can regulate various antioxidant proteins (13). The Nrf2 signaling pathway is
implicated in the amelioration of cardiac fibrosis by regulating
oxidative stress and cell apoptosis (14,15).
Heme oxygenase-1 (HO-1) is an antioxidative gene, and its
expression is initiated and promoted by Nrf2 (16). The HO-1 protein degrades heme into
biliverdin, ferrous iron and carbon monoxide, and thus is able to
ameliorate cellular injury by exerting antioxidant effects
(17). Nrf2/HO-1 signaling was
demonstrated to be increased in an overpressure-induced cardiac
fibrosis model (15) and in a
doxorubicin-induced oxidative injury model (18), and this enhanced anti-oxidative
effect is essential for inhibiting fibrosis and injury. Although
previous studies have found that miR-155 modulates oxidative stress
by targeting the Nrf2-mediated signaling pathway in various
diseases (19–21), the specific association between
miR-155 and the Nrf2/HO-1 signaling pathway in the progression of
cardiac fibrosis remains largely unknown.
The present study established a high glucose
(HG)-induced cardiac fibrosis cell model by culturing H9C2 cells
with 30 mM glucose, which mimics the diabetes-induced
cardiomyopathy condition (22). It
has been reported that HG treatment can increase the activity of
the transcriptional co-regulator p300 and also enhance transforming
growth factor-β (TGF-β) signaling through SMAD2 acetylation
(23), which then promotes the
formation of cardiac fibrotic tissue (11). Results from the present study
suggested that miR-155 impaired the Nrf2/HO-1 signaling pathway to
induce oxidative stress, promote mitochondrial injury and
cardiomyocyte apoptosis, and increase extracellular matrix
accumulation in the cardiac fibrosis model. To the best of our
knowledge, the present study is the first to indicate that miR-155
targeting the Nrf2/HO-1 signaling pathway may regulate cardiac
fibrosis. Moreover, this mechanism may be a potential therapeutic
target for cardiac fibrosis treatment.
Materials and methods
Cell culture
H9C2 rat cardiomyocytes (American Type Culture
Collection) were cultured in DMEM (Gibco; Thermo Fisher Scientific,
Inc.) supplemented with 10% FBS (Gibco; Thermo Fisher Scientific,
Inc.) and 1% penicillin and streptomycin (Gibco; Thermo Fisher
Scientific, Inc.), and were maintained at 37°C in a humidified
incubator with 5% CO2. To induce HG-mediated fibrosis,
the H9C2 cells were cultured with 30 mM glucose (Sigma-Aldrich;
Merck KGaA) at 37°C for 24 or 48 h.
Cell transfection
H9C2 cells (2×105) were seeded in 6-well
plates and cultured at 37°C for 24 h. Before transfection, the DMEM
was replaced with serum-free DMEM medium (Gibco; Thermo Fisher
Scientific, Inc.) for a further 24 h of culturing at 37°. Then,
miR-155 inhibitor (5′-ACCCCUAUCACGAUUAGCAUUAA-3′) or miR-155 mimics
(sense 5′-UUAAUGCUAAUCGUGAUAGGGGUU-3′, antisense
5′-CCCCUAUCACGAUUAGCAUUAAUU-3′) miRNAs (10 nM, Invitrogen; Thermo
Fisher Scientific, Inc.) were transfected for 6 h into H9C2 cells
at 37°C using Lipofectamine® RNAi MAX transfection
reagent (Invitrogen; Thermo Fisher Scientific, Inc.), followed by
exposure to normal glucose (NG; 5 mM) or HG (30 mM) treatment for
24 or 48 h at 37°C. The cells were subsequently collected for
western blot analysis, reverse transcription-quantitative PCR
(RT-qPCR), mitochondrial membrane potential measurement, and
oxidative stress and cell apoptosis assays.
Plasmid construction for short hairpin
RNA (shRNA)-mediated knockdown
H9C2 cells (2×105) were seeded in 6-well
plates and cultured at 37°C for 24 h. A pGPH1 plasmid expressing
shRNA targeting Nrf2 (shNrf2,
5′-CCGGAGTTTGGGAGGAGCTATTATCCTCGAGGATAATAGCTCCTCCCAAACTTTTTTG-3′)
and a scrambled control shRNA-expressing pGPH1 plasmid (shNC,
5′-CCGGCCTAAGGTTAAGTCGCCCTCGCTCGAGCGAGGGCGACTTAACCTTAGGTTTTTG-3′)
were purchased from Shanghai GenePharma Co., Ltd. Lipofectamine
2000 (3 µl/well; Thermo Fisher Scientific, Inc.) and 1 µg/well
pGPH1 plasmids were mixed and added to cells according to the
manufacturer's instructions. Then the cells were cultured for
another 48 h at 37°C before subsequent experiments.
Apoptosis analysis with Annexin V/PI
staining
Cell apoptosis was measured using an Annexin
V-FITC/PI dual staining kit (Thermo Fisher Scientific, Inc.)
according to the manufacturer's instructions. H9C2 cells were
trypsinized, collected and washed once with cold PBS. After
centrifugation at 4°C, 200 × g for 5 min, the cells were collected
and concentrated to 1×105 cells per ml. A 0.1 ml sample
solution was mixed with 5 µl of Annexin V-FITC and 5 µl of
propidium iodide (PI) solution, and incubated for 15 min at room
temperature. After mixing, the cell samples were subjected to flow
cytometry (FACSCalibur; BD Biosciences) analysis. A total of 10,000
cells were analyzed by flow cytometry. The apoptotic cell rate was
analyzed via FlowJo software (version 10; FlowJo LLC) and
calculated by adding the proportions of early apoptotic cells
(Annexin V-FITC+PI−) and late apoptotic cells
(Annexin V-FITC+PI+).
Reactive oxygen species (ROS)
analysis
The level of intracellular ROS was determined using
a dichloro-dihydro-fluorescein diacetate (DCFH-DA) assay (Nanjing
Jiancheng Bioengineering Institute) and a ROS assay kit (Nanjing
Jiancheng Bioengineering Institute). First, H9C2 cells were
cultured with 10 µM DCFH-DA at 37°C for 30 min, and then harvested
by trypsinization and washed once with PBS. Next, the cells were
resuspended in PBS and the cell density was adjusted to
1×106 cells/ml. The fluorescence intensity of the DCF
was measured with a Synergy MX Multi-Mode microplate reader (BioTek
Instruments, Inc.) with the excitation wavelength at 480 nm and the
emission wavelength at 525 nm, which were used to determine the
intracellular ROS levels.
Superoxide dismutase (SOD) and
malonaldehyde (MDA) measurements
The activities of MDA and SOD in H9C2 cells were
also assessed using Malondialdehyde (MDA) and Superoxide Dismutase
(SOD) assay kit (Nanjing Jiancheng Bioengineering Institute). H9C2
cells were collected using a rubber scraper and homogenized in cold
buffer (10 mM Tris-HCl; 0.25 M sucrose; and 25 mM
phenylmethylsulfonyl fluoride, pH 7.4). A total of 0.1 ml
homogenate was mixed with 1 ml MDA working solution and incubated
at 95°C for 40 min. The samples were cooled and centrifuged at 200
× g, 22°C for 10 min, and then 250 µl of supernatant was loaded
into 96-well plate. The optical density (OD) of each well was
measured at 530 nm wavelength (OD530) with a microplate reader. For
the SOD assay, 0.1 ml of the homogenate was sequentially mixed with
20 µl enzyme working liquid and 200 µl substrate reaction solution,
according to the manufacturer's instructions, and then incubated at
37°C for 40 min. SOD activity was then assessed by measuring the
OD550 with a spectrophotometer. The values of MDA and SOD were used
as indicators of lipid superoxide and oxygen free radical in the
cardiomyocytes, respectively.
Mitochondrial membrane potential
measurement by JC-1 assay
The mitochondrial membrane potential was measured
using a JC-1 Mitochondrial Membrane Potential assay kit (Cayman
Chemical Company). H9C2 cells were seeded (1×105
cells/well) in a 24-well plate in DMEM with NG or HG for 24 or 48
h. Next, JC-1 staining solution (100 µl/ml of medium/well) was
loaded into each well of the plate and mixed for 10 sec at 25°C,
and the cells were cultured in a CO2 incubator at 37°C
for 15 min. Subsequently, the plate was centrifuged at 400 × g at
25°C for 5 min and the supernatant was discarded. The plate was
washed twice with 500 µl of assay buffer (supplied as part of the
JC-1 kit), and then the cells were covered with 250 µl of assay
buffer. The cells were then analyzed with a TS100 fluorescence
microscope (magnification, ×200; Nikon Corporation). Normal cells
(cells that did not receive treatment) with aggregated JC-1 were
captured with the fluorescence microscopy filter set for red
fluorescent dye (excitation/emission=540/570 nm), and apoptotic
cells with monomeric JC-1 were detected with the fluorescence
microscopy filter set for green fluorescent dye
(excitation/emission =485/535 nm).
RT-qPCR
TRIzol reagent (Invitrogen; Thermo Fisher
Scientific, Inc.) was used to extract the total RNA from H9C2
cells. After quantification using a NanoDrop ND-1000
spectrophotometer (NanoDrop Technologies; Thermo Fisher
Scientific), 2 µg total RNA was applied to generate the
first-strand cDNA at 25°C for 5 min, 37°C for 30 min and 85°C for 5
sec with First Strand cDNA Synthesis kit (Sigma-Aldrich; Merck
KGaA) according to the manufacturer's instructions. qPCR
experiments were performed on an ABI 7300 thermocycler (Applied
Biosystems; Thermo Fisher Scientific, Inc.) with SYBR-Green dye
(Invitrogen; Thermo Fisher Scientific, Inc.). The thermocycling
conditions were as follows: Initial denaturation, 95°C for 5 sec;
followed by 35 cycles of denaturation at 94°C for 15 sec, annealing
at 55°C for 25 sec and extension at 70°C for 30 sec. GAPDH was used
as the internal reference for mRNA, and U6 small nuclear RNA (U6
snRNA) was used as the miRNA control. The relative expression of a
target gene was calculated with the 2−ΔΔCq method
(24). The specific primers used
for qPCR are listed in Table
I.
 | Table I.Primers sequences used for reverse
transcription-quantitative PCR. |
Table I.
Primers sequences used for reverse
transcription-quantitative PCR.
| Gene | Primer sequence
(5′→3′) |
|---|
| GAPDH | F:
CGCTAACATCAAATGGGGTG |
|
| R:
TTGCTGACAATCTTGAGGGAG |
| U6 | F:
CTCGCTTCGGCAGCACA |
|
| R:
AACGCTTCACGAATTTGCGT |
| MicroRNA-155 | F:
TGCCTCCAACTGACTCCTAC |
|
| R:
GCCAGCAGAATAATACGAC |
| α-smooth muscle
actin | F:
AGCATCCGACCTTGCTAACG |
|
| R:
TGAGTCACGCCATCTCCAGAG |
| Collagen I | F:
TTTAATGGATAGGGACTTGTGTGAA |
|
| R:
GAGAGAGAGAGAAGCTGAGGGTAGG |
Endonuclear, cytoplasmic and
mitochondrial protein extraction
Endonuclear proteins and cytoplasmic proteins were
obtained using Cell Nuclear and Cytoplasmic Protein Extraction kit
(Beyotime Institute of Biotechnology), according to the
manufacturer's protocol. After being washed with PBS,
1×107 H9C2 cells were detached from the dish using a
rubber scraper, and then collected and centrifuged at 4°C, 200 × g
for 5 min. After the supernatant was aspirated, the cell pellet was
resuspended with cytoplasmic protein extraction reagent A
[supplemented with 1 mM PMSF (Sigma-Aldrich; Merck KGaA)], vortexed
for 5 sec and incubated on ice for 15 min. Then, the cell samples
were mixed with cytoplasmic protein extraction reagent B, vortexed
for 5 sec and incubated on ice for 1 min. Cells were vortexed again
for 5 sec, centrifuged at 4°C at 16,000 × g for 5 min and the
supernatant, which contained the cytoplasmic proteins, was
collected and transferred to a new tube. The remaining pellet in
the tube was then re-suspended with nuclear protein extraction
reagent (supplemented with 1 mM PMSF) and further homogenized by
cycles of vigorous vortex mixing for 15–30 sec followed by
incubation on ice for 1–2 min for 30 min. After centrifugation at
16,000 × g at 4°C for 10 min, the supernatant containing the
nuclear proteins was transferred to a cold tube.
Mitochondrial and cytoplasmic proteins were obtained
with a Cell Mitochondria Isolation kit (cat. no. C3601; Beyotime
Institute of Biotechnology), following the manufacturer's protocol.
H9C2 cells were trypsinized, washed once with cold PBS and
resuspended in mitochondria isolation buffer (supplied as part of
the Cell Mitochondria Isolation kit) supplemented with 1 mM PMSF
and incubated on ice for 15 min. Next, the samples were homogenized
and centrifuged at 4°C at 600 × g for 15 min. The supernatant was
collected and transferred to a clean tube, which was then
centrifuged at 4°C at 11,000 × g for 15 min. The supernatant, which
contained the cytoplasmic proteins, was collected in a new 1.5 ml
eppendorf tube and the pellet, which contained the isolated
mitochondria, was subjected to mitochondria lysis buffer
supplemented with 1 mM PMSF. Following the lysis of mitochondria,
all the mitochondrial proteins were released in the lysis buffer.
The endonuclear, cytoplasmic and mitochondrial proteins were then
analyzed by western blotting, as described in the following
method.
Western blot analysis
A total of 1×107 cells were harvested and
lysed in RIPA cell lysis buffer (150 mM NaCl; 50 mM Tris-HCl; 0.5%
sodium deoxycholate; 1% NP-4; 0.1% SDS, pH 7.4; and 1X protease
inhibitor cocktail; Selleck Chemicals] and rotated for 1 h at 4°C.
Next, the cell lysates were centrifuged at 4°C at 1,200 × g for 10
min, and the supernatant was collected. Protein concentrations were
determined with a commercial bicinchoninic acid protein assay kit
(Tiangen Biotech Co., Ltd.). 30 µg protein samples were subjected
to 10% SDS-PAGE and transferred onto PVDF membranes (EMD
Millipore). After blocking with 5% skim milk (EMD Millipore) for 1
h at 25°C, the PVDF membranes were probed with primary antibodies
against α-SMA (1:1,000; cat. no. ab5694; Abcam), collagen I
(1:2,000; cat. no. ab34710; Abcam), Nrf2 (1:1,000; cat. no.
ab62352; Abcam), HO-1 (1:1,000; cat. no. ab 13248; Abcam),
cytochrome-c (Cyt-c; 1:1,000; cat. no.
ab13575; Abcam), cytochrome-c oxidase subunit IV (COX IV;
1:1,000; cat. no. ab33985; Abcam), Lamin B1 (1:5,000; cat. no.
ab16048; Abcam), cleaved Caspase-3 (1:2,000; cat. no. 9661; Cell
Signaling Technology, Inc.), cleaved Caspase-9 (1:1,000; cat. no.
7237; Cell Signaling Technology, Inc.), poly (ADP-ribose)
polymerase (PARP; 1:2,000; cat. no. 9532; Cell Signaling
Technology, Inc.), Bax (1:2,000; cat. no. 5023; Cell Signaling
Technology, Inc.), Bcl-2 (1:2,000; cat. no. 12789-1-AP; ProteinTech
Group, Inc.) and GAPDH (1:5,000; cat. no. 60004-1-Ig ProteinTech
Group, Inc.) at 4°C overnight. After washing three times with TBST
(20 mM Tris, 137 mM NaCl, 0.1% Tween-20), the membranes were then
probed with horseradish peroxidase-conjugated goat anti-mouse or
anti-rabbit (both 1:5,000; cat. nos. 7076 and 7074, respectively;
both Cell Signaling Technology, Inc.) antibody for 1 h at room
temperature. Protein bands were visualized using an ECL reagent
(EMD Millipore), captured using a LAS-3000 imager (Fuji Film
Corporation) and semi-quantified using ImageJ software (version
1.52, National Institutes of Health). The protein expression levels
were normalized to the controls.
Statistical analysis
Each experiment was performed at least times, and
representative data from one replicate are shown in the figures.
Data are presented as the mean ± SD. Statistical analysis was
performed using a one-way ANOVA followed by Tukey's post hoc test,
using SPSS software version 13.0 (SPSS, Inc.). P<0.05 was
considered to indicate a statistically significant difference.
Results
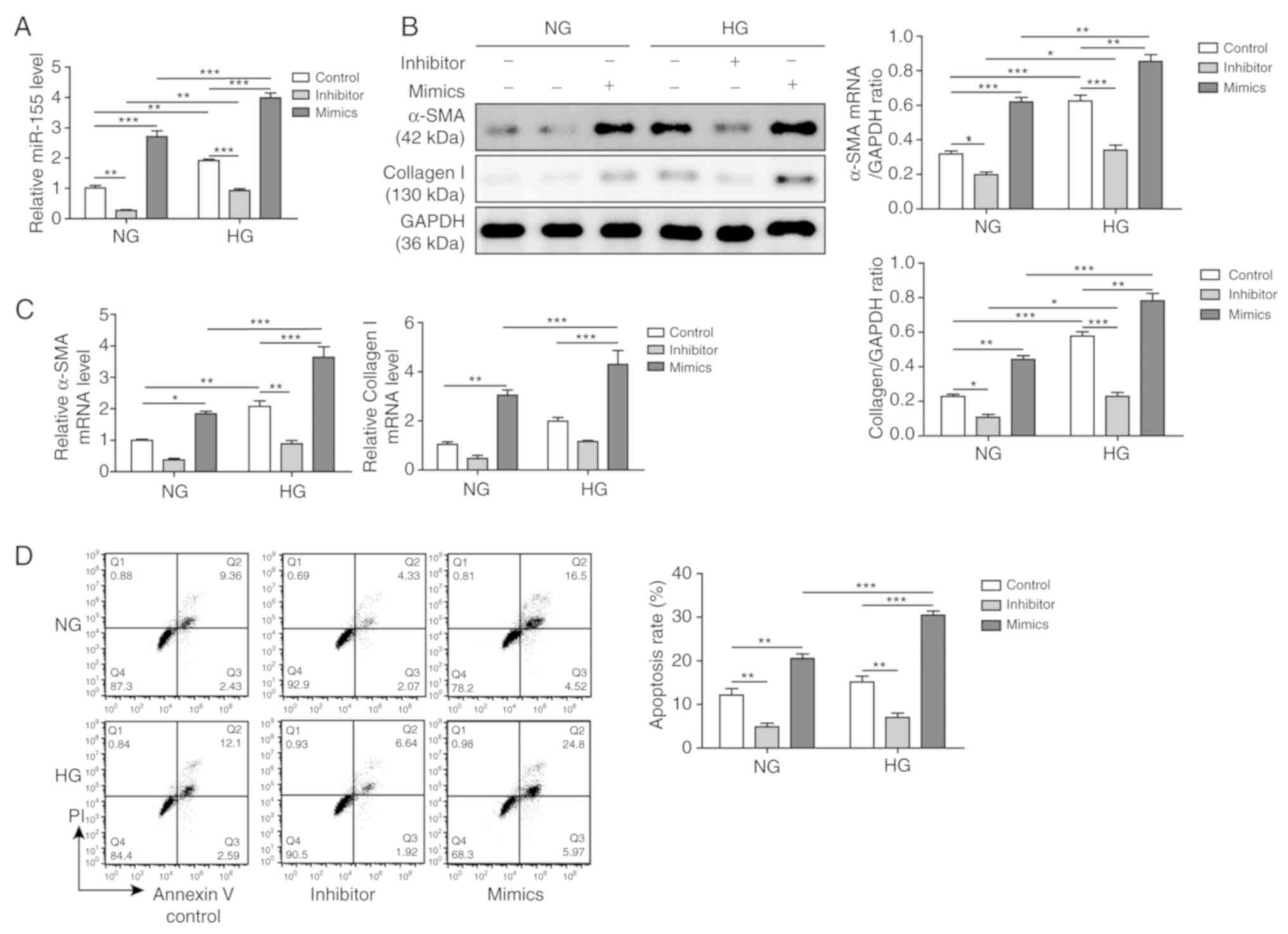
Effects of miR-155 in the HG-induced
cardiac fibrosis model
H9C2 cells were treated with a high concentration of
glucose (30 mM) for 24 h to establish a cardiac fibrosis model at
the cellular level. The expression level of miR-155 was assessed
using RT-qPCR, which demonstrated that HG treatment resulted in
increased miR-155 expression compared with NG-treated cells
(Fig. 1A). In addition, the
effects of miR-155 inhibitors and miR-155 mimics in both NG- and
HG-treated H9C2 cells were examined. It was shown that the miR-155
inhibitor suppressed miR-155 expression levels, whereas the miR-155
mimics significantly increased miR-155 expression levels,
regardless of NG or HG treatment (Fig.
1A). The expression levels of collagen I and α-SMA in NG- and
HG-treated H9C2 cells, both of which were positively associated
with fibrosis, were also examined. Western blot analysis results
indicated that both collagen I and α-SMA protein expression levels
were significantly increased after HG treatment (Fig. 1B). In addition, inhibiting miR-155
expression led to significantly reduced collagen I and α-SMA
expression levels, whereas overexpression of miR-155 increased the
expression levels. Moreover, the mRNA expression levels of α-SMA
and collagen I were also downregulated when miR-155 expression
levels were impaired, although there were no significant
differences in collagen I expression levels in the NG and HG
groups, and α-SMA expression levels in the NG group. However,
miR-155 overexpression upregulated α-SMA and collagen I mRNA
expression levels in both the NG and HG groups (Fig. 1C). Therefore, these results
suggested that the cardiac fibrosis model was successfully
established.
In addition, apoptotic rates were examined by flow
cytometry. It was demonstrated that the miR-155 mimics further
increased the number of apoptotic cells, whereas the miR-155
inhibitor suppressed apoptosis compared with the Control (Fig. 1D). Collectively, the results
suggested that miR-155 was upregulated in the HG-induced cardiac
fibrosis cell model, which indicated that miR-155 may be involved
in cardiac fibrosis, extracellular matrix deposition and
cardiomyocyte apoptosis. In addition, it was shown that the miR-155
mimics contributed to the fibrotic phenotypes, whereas the miR-155
inhibitor prevented the acquisition of the fibrotic phenotypes.
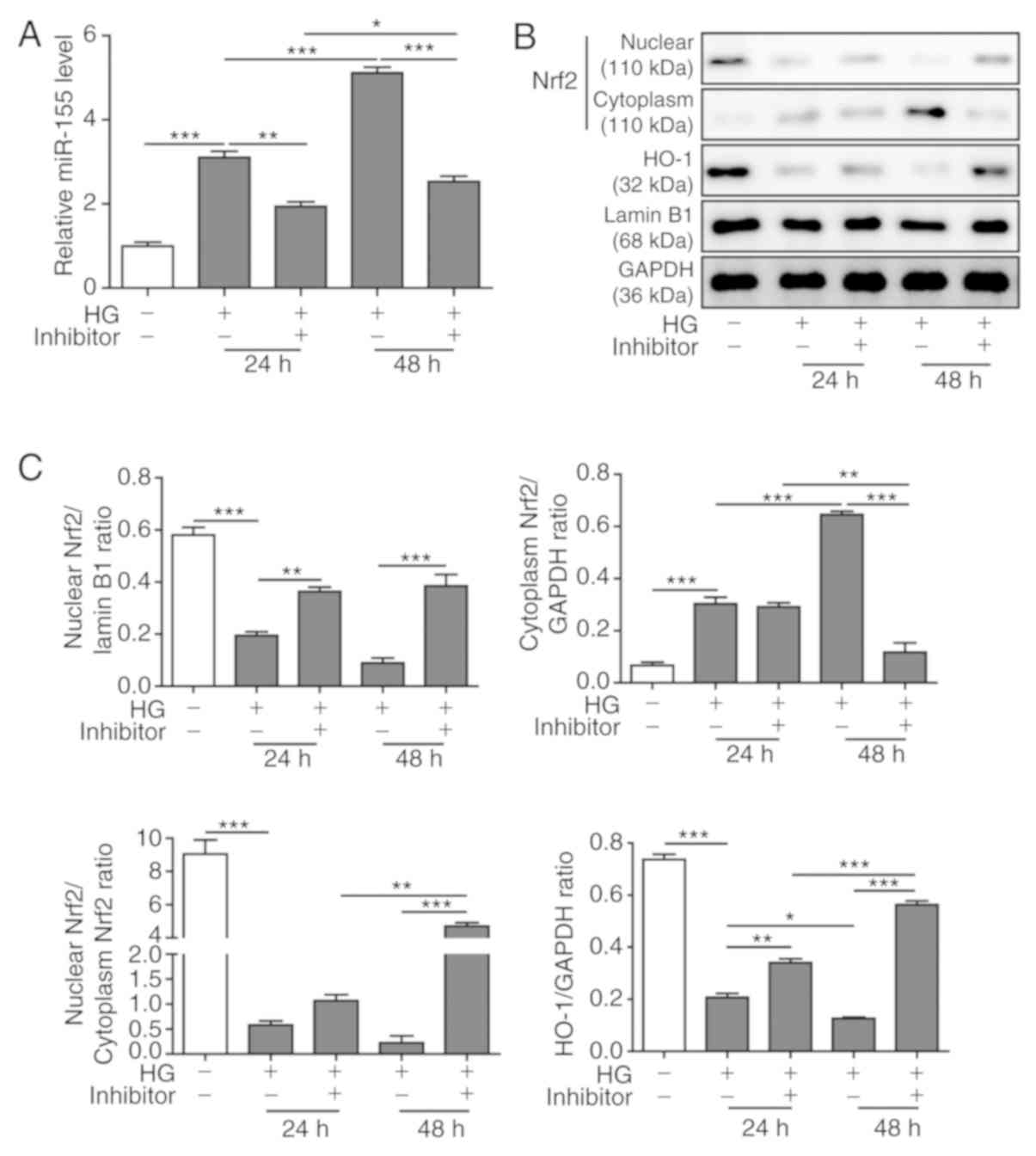
miR-155 inhibition partially restores
the Nrf2/HO-1 signaling pathway induced by HG
The role of the miR-155 inhibitor on the Nrf2/HO-1
signaling pathway in the HG-induced cardiac fibrosis model was
investigated. It was demonstrated that HG treatment for 48 h
increased the expression levels of miR-155 in H9C2 cells compared
with the levels produced after 24 h of treatment. miR-155 inhibitor
transfection significantly reduced miR-155 expression levels that
had been induced by HG at both 24 and 48 h (Fig. 2A). The effects of the miR-155
inhibitor on the expression levels of Nrf2 and HO-1 in HG-treated
H9C2 cells were also examined. Western blot analysis results
indicated that HG treatment downregulated endonuclear Nrf2
expression level and upregulated cytoplasmic Nrf2 expression level
compared with the control group (Fig.
2B and C). As a downstream antioxidative protein induced after
endonuclear Nrf2-mediated transcriptional activation (16), HO-1 was identified to have similar
expression levels as endonuclear Nrf2. However, the miR-155
inhibitor reversed these changes, most notably in HG-cells treated
for 48 h (Fig. 2B and C). It was
demonstrated that miR-155 inhibition enhanced nuclear translocation
of Nrf2 and increased the expression level of HO-1 in HG-treated
H9C2 cells (Fig. 2C).
Collectively, the present results indicated that miR-155 may
modulate HG-induced cardiac fibrosis by targeting the Nrf2/HO-1
signaling pathway.
Inhibiting miR-155 reduces oxidative
stress and mitochondrial damage induced by HG
Since oxidative stress and mitochondrial injury are
the main manifestations of cardiac fibrosis (25), and as miR-155 regulates oxidative
stress by targeting the Nrf2-mediated signaling pathway in some
diseases (16–18), the present study examined miR-155
regulation of oxidative stress and mitochondrial injury in
HG-treated H9C2 cells. It was demonstrated that HG treatment
significantly increased ROS and MDA expression levels, and
decreased SOD expression levels in H9C2 cells compared with control
cells at both 24 and 48 h (Fig.
3A-C, respectively), which indicated that substantial oxidative
stress occurred. However, miR-155 inhibition reversed the increased
ROS and MDA levels and the decreased SOD levels, suggesting that an
antioxidative response may be triggered and enhanced by miR-155
attenuation (Fig. 3A-C).
Cyt-c is an important component of the electron transport
chain, and is localized in the spaces between the intermembrane and
the cristae of mitochondria (26).
Generally, ROS production in mitochondria results in the release of
Cyt-c from the mitochondria into the cytosol and initiates
cell apoptosis (27). Therefore,
the present study assessed the content of mitochondrial and
cytoplasmic Cyt-c in HG-treated H9C2 cells to investigate
the effect of miR-155 on mitochondrial damage. The western blot
analysis results indicated that HG treatment induced the release of
Cyt-c from the mitochondria into the cytosol, particularly
in the H9C2 cells that were treated for 48 h (Fig. 3D and E). However, miR-155
inhibition partially prevented the release of Cyt-c into the
cytosol, as determined by increased protein expression levels of
mitochondrial Cyt-c and decreased levels of cytoplasmic
Cyt-c (Fig. 3D and E).
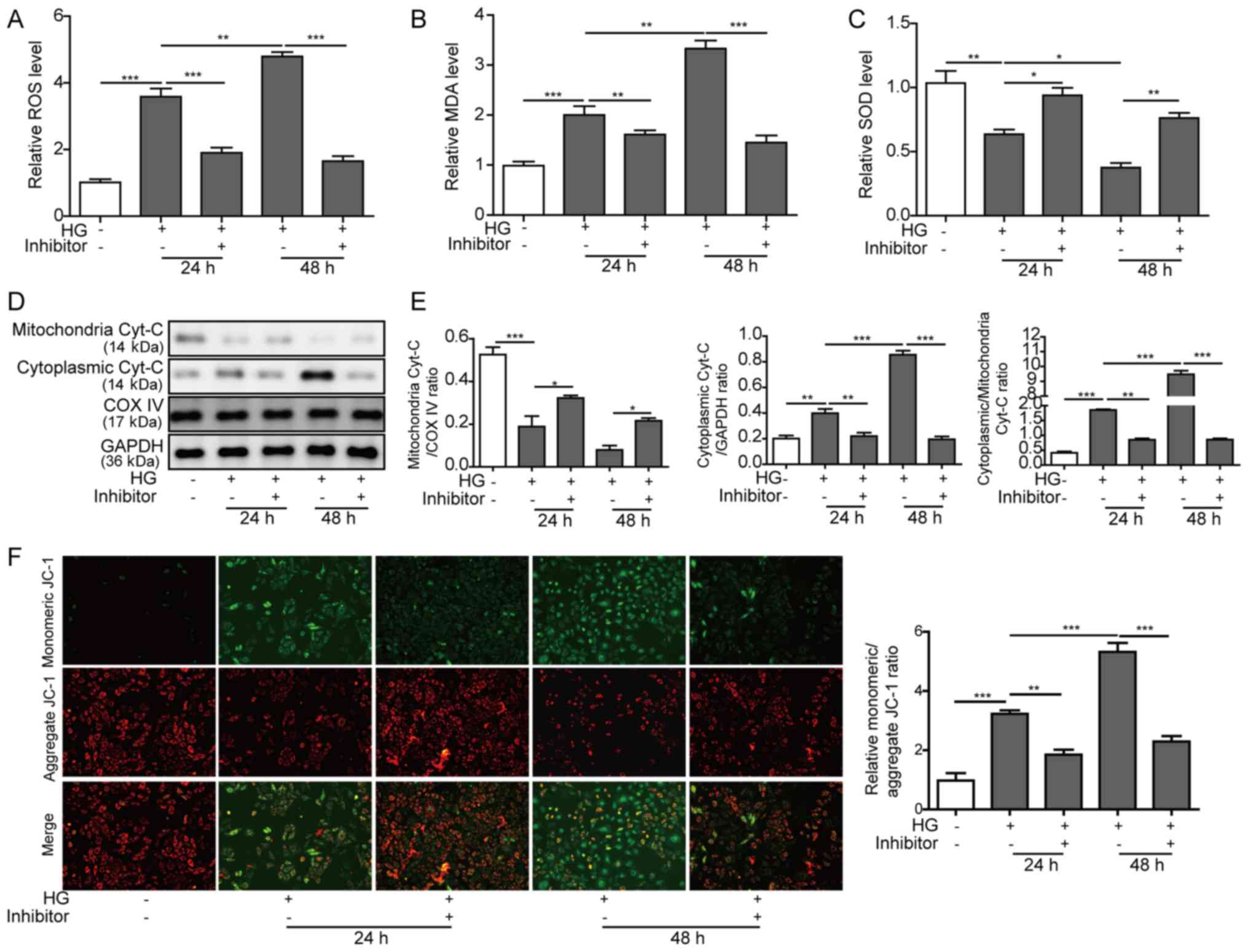 | Figure 3.Effects of the miR-155 inhibitor on
the oxidative stress and mitochondrial damage induced by HG. H9C2
cells were treated with HG and co-transfected with or without
miR-155 inhibitor for 24 and 48 h. (A) Intracellular ROS levels
were measured with the DCFH-DA assay. (B) MDA levels were measured
with an MDA assay kit. (C) SOD levels were measured with a SOD
assay kit. (D) Protein expression levels of mitochondrial and
cytoplasmic Cyt-c were assessed by western blot analysis;
COX IV and GAPDH were used as the loading controls for
mitochondrial and cytoplasmic fractions, respectively. (E) Relative
expression levels of mitochondrial and cytoplasmic Cyt-c,
and the ratio of the normalized cytoplasmic/mitochondrial
Cyt-c levels presented. (F) Mitochondrial membrane potential
of was measured by JC-1 assay. The ratios of monomeric
JC-1/aggregated JC-1 are presented on the right. Data are presented
as the mean ± SD from three independent experiments. *P<0.05,
**P<0.01 and ***P<0.001. COX IV, cytochrome-c oxidase
subunit IV; Cyt-c, cytochrome-c; HG, high glucose;
MDA, malonaldehyde; miR-155, microRNA 155; ROS, reactive oxygen
species; SOD, Superoxide dismutase. |
In addition, mitochondria membrane potential was
examined using a JC-1 assay. It was found that HG treatment
impaired the integrity of mitochondria in H9C2 cells, which led to
a reduced membrane potential as indicated by elevated JC-1 monomer
and decreased JC-1 aggregation (Fig.
3F). In addition, miR-155 inhibitor transfection significantly
repressed the downregulation of the membrane potential of the
mitochondria in H9C2 cells after HG treatment, which suggested that
miR-155 inhibition may aid in relieving mitochondrial damage under
HG conditions. Overall, these results indicated that inhibition of
miR-155 suppressed oxidative stress and mitochondrial damage in the
HG-induced cardiac fibrosis model.
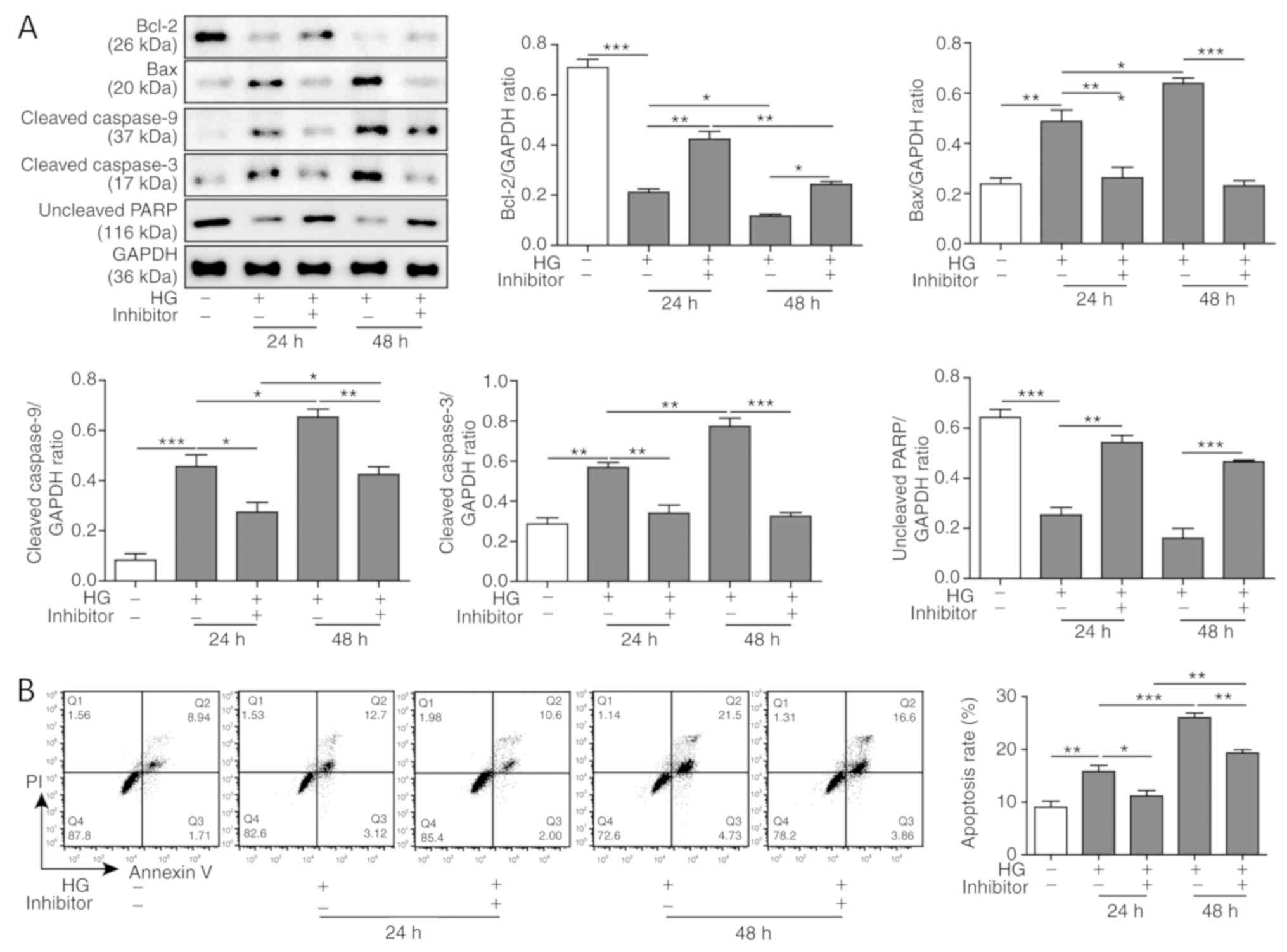
miR-155 inhibition suppresses the
apoptosis of cardiomyocytes induced by HG
Excessive oxidative stress and mitochondrial damage
lead to cell apoptosis, which is one of the causes of cardiac
fibrosis (28). To investigate the
function of miR-155 on the apoptosis of cardiomyocytes, western
blotting was performed to assess the protein expression levels of
apoptosis-associated proteins in HG-treated H9C2 cells. The present
results suggested that the protein expression levels of Bcl-2, an
anti-apoptotic protein (29), were
downregulated, and Bax, a pro-apoptotic protein (30), were upregulated after HG treatment
(Fig. 4A). Moreover, the protein
expression levels of the activated caspases, cleaved caspase-3 and
caspase-9, were increased, and the uncleaved PARP expression levels
were decreased (Fig. 4A). This
phenotype indicated that HG-induced cardiomyocyte apoptosis may be
mediated by the caspase-9-dependent mitochondrial damage pathway.
In addition, the decreased expression levels of anti-apoptotic
proteins and the enhanced expression levels of pro-apoptotic
proteins had a time-dependent effect (Fig. 4A). However, miR-155 inhibition
reversed the expression levels of the apoptosis-associated proteins
in H9C2 cells (Fig. 4A). In
addition, it was demonstrated that HG treatment increased the
number of apoptotic cells compared with the control treated cells,
and this result had a time-dependent trend (Fig. 4B). Additionally, miR-155 inhibition
reduced the total apoptotic rate. Taken together, these results
suggested that miR-155 inhibition suppressed HG-induced apoptosis
in cardiomyocytes.
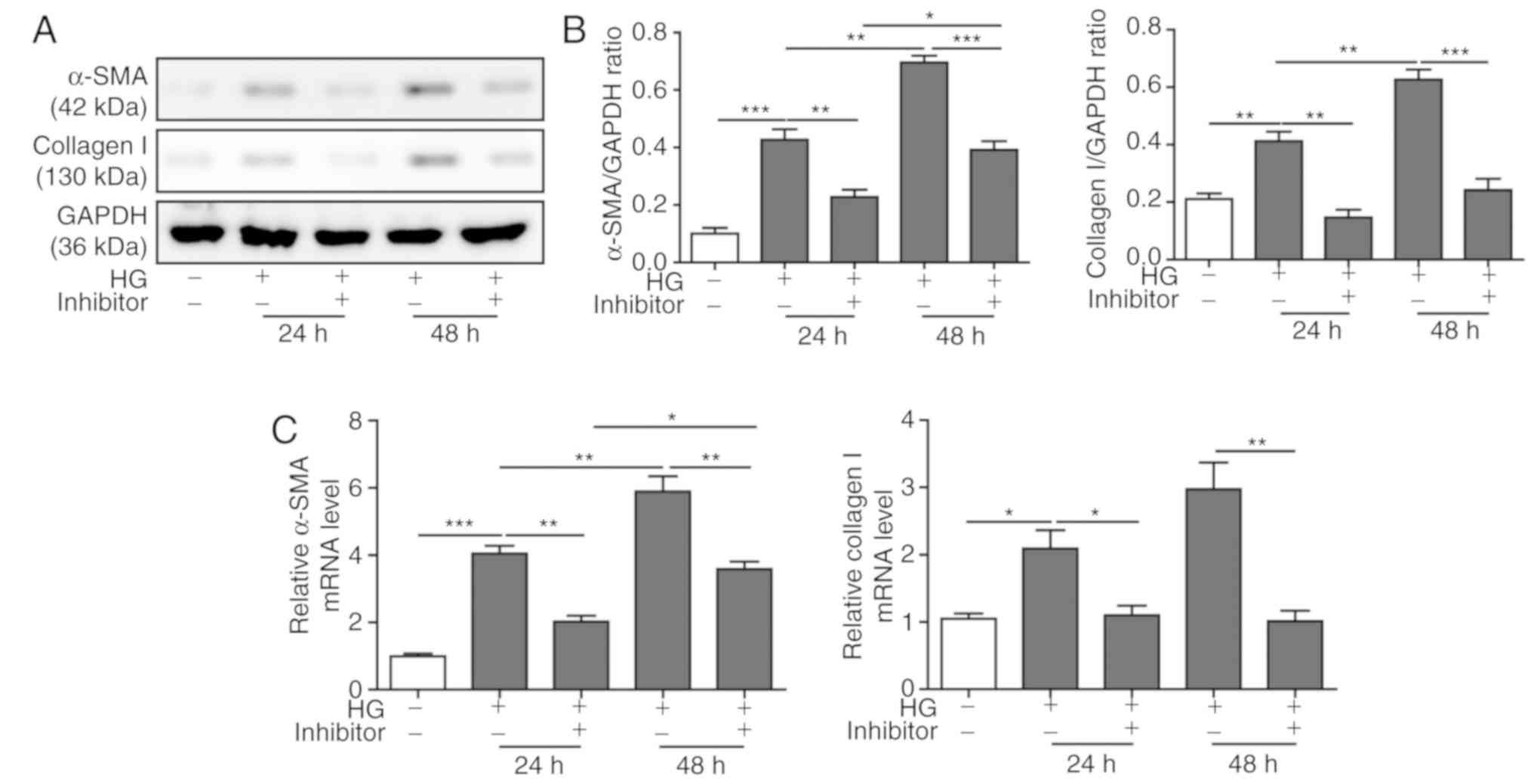
miR-155 inhibition mitigates cardiac
fibrosis induced by HG
The progression of cardiac fibrosis is characterized
by the abnormal expression and secretion of collagen I and α-SMA in
cardiomyocytes (31). To
investigate the effect of miR-155 inhibition on cardiac fibrosis,
western blot analyses was performed to assess the protein
expression levels of collagen I and α-SMA in HG-treated H9C2 cells
with or without miR-155 inhibitor transfection. It was demonstrated
that HG treatment increased the protein expression levels of
collagen I and α-SMA in a time-dependent manner, and miR-155
inhibition decreased these expression levels (Fig. 5A and B). The mRNA expression levels
of collagen I and α-SMA after HG treatment and miR-155 inhibitor
transfection were consistent with the protein expression levels
described above (Fig. 5C).
Collectively, the present results indicated that the miR-155
inhibitor may ameliorate cardiac fibrosis induced by HG.
Nrf2/HO-1 signaling is required to
maintain the homeostasis of mitochondria in cardiomyocytes after HG
treatment
To further examine the role of the Nrf2/HO-1
signaling pathway in the homeostasis of mitochondrial function,
Nrf2 expression was knocked down using shRNA in HG-treated H9C2
cells transfected with the miR-155 inhibitor. To assess the
transfection efficiency of shNrf2, western blotting was used to
examine the protein expression level of Nrf2. It was shown that the
protein expression level of Nrf2 was significantly decreased after
shNrf2 transfection for 48 h (Fig.
6A). The protein expression levels of the Nrf2 and HO-1 were
then examined in H9C2 cells at 48 h after transfection of miR-155
inhibitor and shNrf2. It was found that both Nrf2 and HO-1 protein
expression levels were reduced in H9C2 cells after HG treatment,
whereas miR-155 inhibition partially rescued their expression
(Fig. 6B). However, Nrf2 knockdown
significantly suppressed the protein expression levels of Nrf2 and
HO-1 compared with the HG+miR-155 inhibitor+shNC group (Fig. 6B).
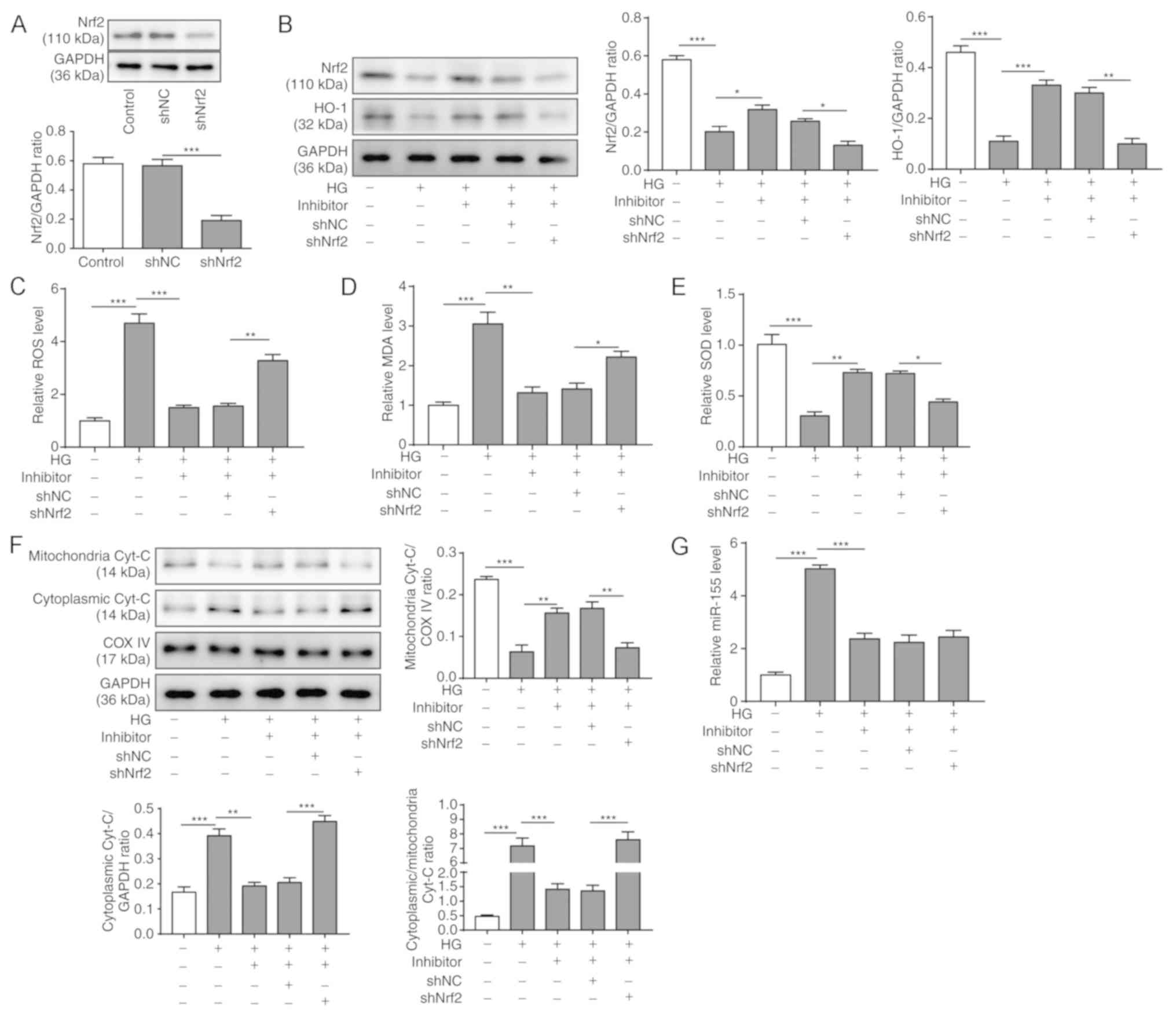 | Figure 6.Nrf2/HO-1 signaling is required to
maintain the homeostasis of mitochondria in cardiomyocytes after HG
treatment. HG-treated H9C2 cells were co-transfected with or
without miR-155 inhibitor and with or without sh-Nrf2 for 48 h. (A)
Transfection efficiency of shNC and shNrf2 in H9C2 cells was
assessed by western blot analyses. (B) Protein expression levels of
Nrf2 and HO-1 in H9C2 cells were measured by western blot analyses;
GAPDH was used as the loading control. Intracellular (C) ROS, (D)
MDA and (E) SOD expression levels were measured with a DCFH-DA
assay kit, an MDA assay kit and a SOD assay kit, respectively. (F)
Protein expression levels of mitochondrial and cytoplasmic
Cyt-c were assessed by western blot analyses; COX IV and
GAPDH were used as the loading controls for the mitochondrial and
cytoplasmic fractions, respectively. (G) Relative mRNA expression
levels of miR-155 were measured by reverse
transcription-quantitative PCR. Data are presented as the mean ± SD
from three independent experiments. *P<0.05, **P<0.01 and
***P<0.001. COX IV, cytochrome c oxidase subunit IV;
Cyt-c, cytochrome-c; DCFH-DA,
dichloro-dihydro-fluorescein diacetate; HG, high glucose; HO-1,
heme oxygenase-1; MDA, malonaldehyde; miR-155, microRNA 155; NC,
negative control; Nrf2, nuclear factor erythroid-2-related factor
2; sh, short hairpin RNA; ROS, reactive oxygen species; SOD,
superoxide dismutase. |
To assess the homeostasis of mitochondria, ROS, MDA
and SOD levels were measured in HG-treated H9C2 cells at 48 h. It
was shown that HG treatment significantly increases ROS and MDA
expression levels and decreased SOD levels in H9C2 cells, whereas
miR-155 inhibition reversed these phenotypes (Figs. 3A-C and 6C-E). Nrf2 knockdown reversed the effects
of the miR-155 inhibitor, leading to elevated ROS and MDA
expression levels, and reduced SOD levels (Fig. 6C-E), which suggested that Nrf2 may
be required for mitochondrial homeostasis.
Moreover, western blotting was used to investigate
the mitochondrial and cytoplasmic expression levels of Cyt-c
in H9C2 cells after the treatment (Fig. 6F). The results revealed that
mitochondrial Cyt-c was reduced by HG treatment, increased
by miR-155 inhibition, and subsequently reduced by Nrf2 knockdown,
whereas the cytoplasmic Cyt-c expression levels were
opposite (Fig. 6F). The changes in
mitochondrial and cytoplasmic Cyt-c expression following
Nrf2 knockdown further demonstrated that Nrf2 may be essential for
maintaining the normal function of mitochondria under HG
conditions. Furthermore, RT-qPCR results demonstrated that the
miR-155 inhibitor suppressed the expression of miR-155 in the H9C2
cells after HG treatment, and subsequent Nrf2 knockdown had no
significant effect on miR-155 expression level (Fig. 6G). Taken together, these results
demonstrated that the Nrf2/HO-1 signaling pathway may be required
for the homeostasis of the mitochondria in cardiomyocytes after HG
treatment.
Nrf2/HO-1 signaling is essential for
the effects of the miR-155 inhibitor on the apoptosis and cardiac
fibrosis induced by HG
To investigate whether the Nrf2/HO-1 signaling
pathway is required for miR-155 inhibitor-induced suppression of
apoptosis and/or fibrosis of cardiomyocytes, Nrf2 was knocked down
by shRNA in H9C2 cells transfected with miR-155 inhibitor and
treated with HG for 48 h. The western blotting results demonstrated
that the expression level of the anti-apoptotic protein Bcl-2 was
reduced by HG treatment, increased by miR-155 inhibition and
decreased after Nrf2 knockdown (Fig.
7A). By contrast, the expression levels of the pro-apoptotic
protein Bax were increased by HG treatment, reduced by miR-155
inhibition and increased following Nrf2 knockdown. Cleaved
caspase-3 and cleaved caspase-9 protein expression levels were
increased in H9C2 cells after Nrf2 knockdown, whereas uncleaved
PARP expression levels were decreased (Fig. 7A). miR-155 inhibition significantly
decreased the apoptotic rate of H9C2 cells after HG treatment,
whereas Nrf2 knockdown enhanced the apoptosis of H9C2 cells
(Fig. 7B).
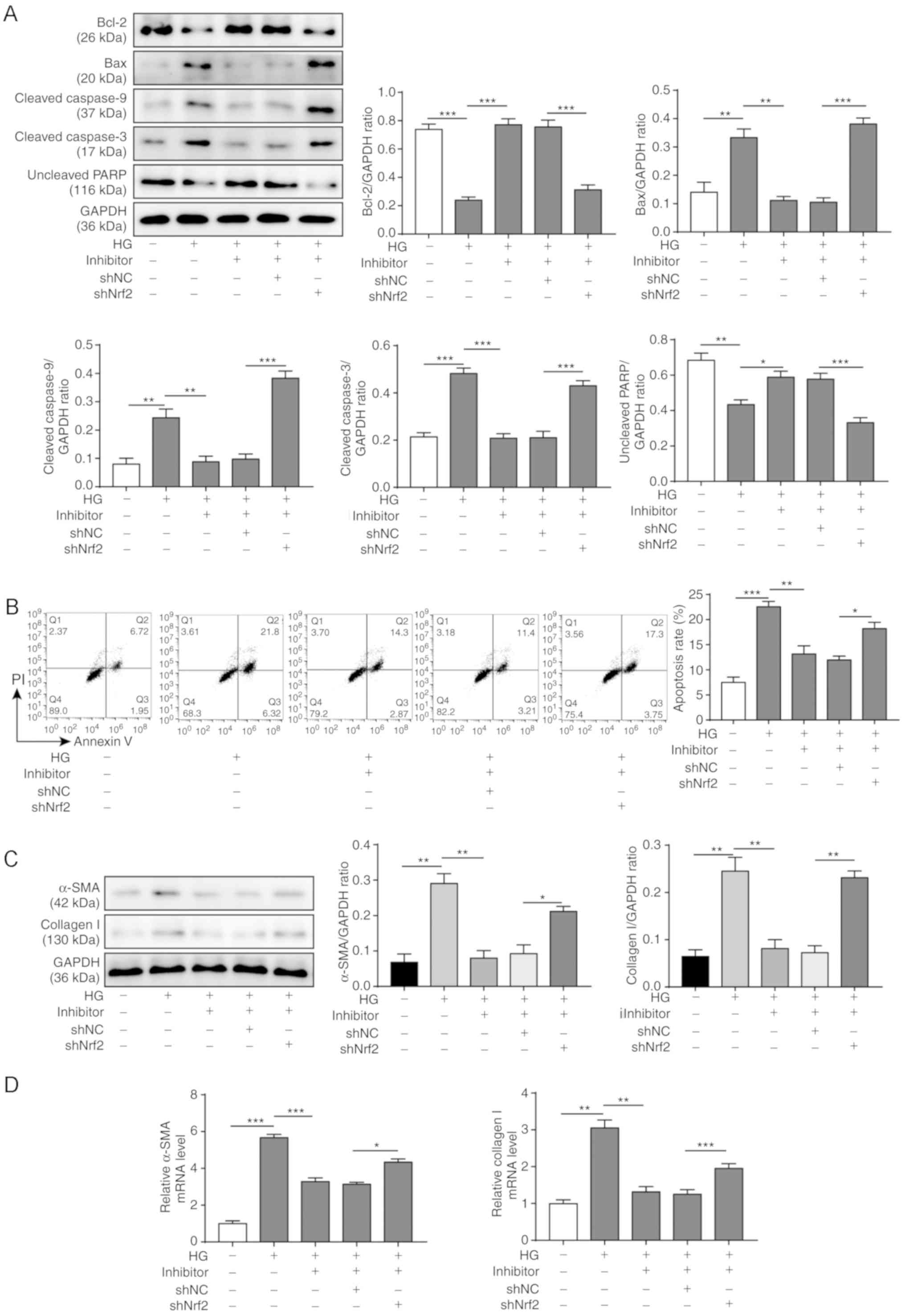 | Figure 7.Effects of miR-155 inhibitor on
Nrf2/HO-1 signaling and HG-induced apoptosis and cardiac fibrosis.
HG-treated H9C2 cells were co-transfected with or without miR-155
inhibitor and with or without sh-Nrf2 for 48 h. (A) Expression
levels of apoptosis-related proteins were assessed by western blot
analyses; GAPDH was used as the loading control. (B) Apoptotic
rates of H9C2 cells were examined by Annexin V-FITC/PI staining and
flow cytometry. (C) Protein expression levels of α-SMA and collagen
I were analyzed by western blotting; GAPDH was used as the loading
control. (D) Relative mRNA expression levels of a-SMA and collagen
I RNA were measured by reverse transcription-quantitative PCR. Data
are presented as the mean ± SD from three independent experiments.
*P<0.05, **P<0.01 and ***P<0.001. α-SMA, α-smooth muscle
actin; HG, high glucose; HO-1, heme oxygenase-1; miR-155, microRNA
155; NC, negative control; Nrf2, nuclear factor erythroid-2-related
factor 2; sh, short hairpin RNA; PARP, poly (ADP-ribose)
polymerase; PI, propidium iodide. |
To evaluate the effect of Nrf2 knockdown on the
extent of miR-155-induced fibrosis of the cardiomyocytes, the
expression levels of a-SMA and collagen I were measured by western
blotting and RT-qPCR (Fig. 7C and
D, respectively). The results showed that both a-SMA and
collagen I were upregulated in HG-treated H9C2 cells and were
downregulated after miR-155 inhibition; however, Nrf2 knockdown led
to increased expression of a-SMA and collagen I at both the protein
and mRNA level. Collectively, these results indicated that the
Nrf2/HO-1 signaling pathway may be the underlying mechanism for
miR-155 inhibitor-induced inhibition of apoptosis and fibrosis of
the cardiomyocytes in response to HG treatment.
Discussion
Progressive cardiac fibrosis is associated with
myocardial infarction, hypertrophy, hypertension, heart failure and
other cardiovascular diseases (1).
However, current therapies for cardiac fibrosis are severely
limited and inefficient (32). To
develop strategies against cardiac fibrosis, the pathogenesis and
the complex regulatory network of these pathologies must be
investigated in greater depth. To the best of our knowledge, the
present study is the first to report the role of miR-155 in
regulating oxidative stress, mitochondrial injury and myocardial
apoptosis via the Nrf2/HO-1 signaling pathway in cardiac fibrosis,
using HG-induced H9C2 rat cardiomyocytes as an in vitro
model.
A previous clinical study has shown that myocardial
damage associated with diabetic cardiomyopathy is related to
glucose level (33). In addition,
dysregulation of miRNAs is associated with cardiac fibrosis
(34). To investigate whether
miR-155 affected oxidative stress, mitochondrial damage, apoptosis,
as well as extracellular matrix accumulation, the present study
aimed to determine the underlying mechanisms of these processes in
cardiomyocytes under HG conditions. It was demonstrated that
expression levels of collagen I and α-SMA were significantly
upregulated after HG treatment. In addition, miR-155 expression
level was increased in cardiomyocytes by induced glucotoxicity; a
finding that was consistent with previous studies on miR-155
expression in human renal glomerular endothelial cells and
endothelial progenitor cells in HG microenvironments (35,36).
Moreover, the present results indicated that inhibiting miR-155
suppressed glucotoxicity-induced apoptosis of cardiomyocytes,
whereas overexpression of miR-155 increased the number of apoptotic
cells. Similar phenotypes have also been reported in HG-treated
endothelial progenitor cells and acute myeloid leukemia cells
(36,37). The present study demonstrated that
miR-155 may be implicated in HG-induced cardiac fibrosis and is
positively associated with apoptosis.
The present results showed that the Nrf2/HO-1
signaling pathway was impaired under HG conditions in a
time-dependent manner, as indicated by decreased endonuclear Nrf2
and HO-1 expression levels but increased cytoplasmic Nrf2
expression; this has also been reported in previous studies
(38–41). However, another previous study
found that Nrf2 expression and nuclear translocation are enhanced
after HG exposure (42). These
contradicting conclusions indicated that the details of Nrf2
functions in cardiomyocytes are complex. Excessive Nrf2 protein can
be ubiquitinated and degraded through the ubiquitin-proteasome
system, which is regulated by the miR-29/Kelch Like ECH Associated
Protein 1 axis (43). We
hypothesized that, over a short time period, such as <24 h after
the initial HG challenge, cardiomyocytes show increased Nrf2
expression levels to counteract the induced oxidative stress and
protect themselves from oxidant-induced damage; however, over a
longer period of oxidative stress, such as 48 h, Nrf2 is
ubiquitinated and degraded. Therefore, a precise tuning mechanism
likely maintains the homeostasis of Nrf2 and protects
cardiomyocytes from oxidative stress-induced apoptosis. HG-induced
impairment of the Nrf2/HO-1 pathway is implicated in the disruption
of homeostasis by oxidative stress in cardiomyocytes. However, the
complex regulatory network behind cardiac fibrosis also comprises
other pathways, such as the TGFb1/SMAD2 signaling pathway, as
reported in a diabetes-induced cardiac fibrosis in mice (11). The present results suggested that
the miR-155 inhibitor partially rescued the impaired Nrf2/HO-1
signaling pathway induced by HG, which was reflected by the
significant increase in nuclear translocation of Nrf2 and the
upregulated expression level of HO-1. Therefore, miR-155 may play a
pivotal role in the regulation of the Nrf2/HO-1 signaling pathway.
Previous studies have demonstrated that miR-155 directly targets
Nrf2 in septic liver injury and diabetic peripheral neuropathy
(44,45). However, the precise mechanism of
miR-155 in the regulation of the Nrf2/HO-1 pathway in cardiac
fibrosis requires further investigation.
Previous studies have shown that the Nrf2/HO-1
pathway is extensively involved in the regulation of oxidative
stress (46), mitochondrial injury
(47) and apoptosis (48), and these phenotypes contribute to
the pathogenesis of cardiac fibrosis (46,49).
Thus, the present study focused on oxidative stress, mitochondrial
injury and apoptosis in subsequent experiments. In the present
study, H9C2 cells treated with HG showed enhanced accumulation of
ROS and MDA, and lower expression levels of SOD, which indicated
that after HG treatment cardiomyocytes were under substantial
oxidative stress. Previous studies have also revealed that HG
induces oxidative stress and apoptosis in cardiac microvascular
endothelial cells (50) and in
human umbilical vein endothelial cells (51). In addition, the increased ratio of
cytoplasmic Cyt-c to mitochondrial Cyt-c indicated
the presence of mitochondrial injury upon HG induction, which
coincides with a previous study showing that HG and high-fat
conditions lead to mitochondrial dysfunction and apoptosis in
cardiomyocytes (52). Directly
measuring the mitochondrial ROS level could reveal the oxidative
stress accumulation in mitochondria upon HG treatment; however,
this measurement was not performed in the present study due to the
facility limitations. Moreover, the dissipation of mitochondrial
membrane potential in the H9C2 cells further supported the
hypothesis that mitochondrial function was impaired upon HG
treatment, and this phenomenon has also been reported in previous
studies with respect to 3T3-L1 adipocytes and human peritoneal
mesothelial cells (53,54). Elevated levels of oxidative stress
and mitochondrial injury resulted in increased numbers of apoptotic
cells, as indicated in the present study by increased expression
levels of pro-apoptotic proteins and reduced expression levels of
anti-apoptotic proteins. The results also demonstrated that
apoptosis induced by HG is caspase-9-dependent, which is consistent
with previous studies investigating human endothelial cells and
periodontal ligament fibroblasts under HG conditions (55,56).
Moreover, miR-155 inhibition significantly suppressed oxidative
stress, mitochondrial injury and apoptosis. Therefore, miR-155 may
exert its pro-fibrosis function by impairing the Nrf2/HO-1
signaling pathway to cause oxidative stress, mitochondrial injury
and myocardial apoptosis. Moreover, the importance of Nrf2/HO-1
signaling in the homeostasis of mitochondria upon HG treatment was
indicated in subsequent experiments, in which Nrf2 was knocked
down. However, further investigation is required to obtain more
details on this mechanism.
The present study also investigated the function of
miR-155 attenuation on cardiac fibrosis. High expression levels of
α-SMA and collagen I were significantly decreased after the miR-155
inhibition. Wei et al (9)
found that miR-155 deficiency ameliorated the progression of
angiotensin II-induced cardiac fibrosis in mice. During the wound
healing process, acute reduction in miR-155 expression resulted in
a milder fibrosis phenotype, which was acquired by a suppressed
inflammatory response, and also accelerated the healing rate
(57). Therefore, targeting
miR-155 may ameliorate cardiac fibrosis caused by HG. In addition,
the present study found that miR-155 may exert its effect partially
via Nrf2/HO-1 signaling, as indicated by the results from Nrf2
knockdown experiments, which showed that Nrf2 knockdown compromised
the effects of miR-155 inhibition on oxidative stress and cell
apoptosis, as well as a-SMA and collagen I accumulation in
HG-treated H9C2 cells.
In conclusion, miR-155 may regulate the pathogenesis
of cardiac fibrosis via the Nrf2/HO-1 signaling pathway. The
present study results not only elucidated a novel pathway for the
pathogenesis of cardiac fibrosis induced by HG, but also identified
the potential of miR-155 as a therapeutic for cardiac fibrosis.
However, this conclusion is based mainly on in vitro
cellular assays, thus in vivo experiments are required to
further investigate these effects. Therefore, generating specific
transgenic mice and evaluating the clinical relevance of miR-155
using human samples are required in follow-up studies.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
YL conceptualized and designed the study, drafted
and revised the manuscript, acquired funding and supervised the
research group. CQW analyzed the data. JZD, YL and QH performed the
experiments and analyzed the data. YL and CQW edited the
manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Worke LJ, Barthold JE, Seelbinder B, Novak
T, Main RP, Harbin SL and Neu CP: Densification of type I collagen
matrices as a model for cardiac fibrosis. Adv Healthc Mater.
6:17001142017. View Article : Google Scholar
|
|
2
|
Murtha LA, Schuliga MJ, Mabotuwana NS,
Hardy SA, Waters DW, Burgess JK, Knight DA and Boyle AJ: The
processes and mechanisms of cardiac and pulmonary fibrosis. Front
Physiol. 8:7772017. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Louridas GE and Lourida KG: Systems
biology and biomechanical model of heart failure. Curr Cardiol Rev.
8:220–230. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Zhang WW, Bai F, Wang J, Zheng RH, Yang
LW, James EA and Zhao ZQ: Edaravone inhibits pressure
overload-induced cardiac fibrosis and dysfunction by reducing
expression of angiotensin II AT1 receptor. Drug Des Devel Ther.
11:3019–3033. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Michael LH, Taffet GE, Entman ML, Reddy
AK, Hartley CJ and Frangogiannis NG: Chapter 2.6-The Cardiovascular
System. The Laboratory Mouse. Second Edition. Hedrich HJ: Academic
Press; Boston: pp. 241–270. 2012, View Article : Google Scholar
|
|
6
|
Asbun J and Villarreal FJ: The
pathogenesis of myocardial fibrosis in the setting of diabetic
cardiomyopathy. J Am Coll Cardiol. 47:693–700. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Bharati S and Lev M: Cardiac conduction
system involvement in sudden death of obese young people. Am Heart
J. 129:273–281. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Dei Cas A, Khan SS, Butler J, Mentz RJ,
Bonow RO, Avogaro A, Tschoepe D, Doehner W, Greene SJ, Senni M, et
al: Impact of diabetes on epidemiology, treatment, and outcomes of
patients with heart failure. JACC Heart Fail. 3:136–145. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wei Y, Yan X, Yan L, Hu F, Ma W, Wang Y,
Lu S, Zeng Q and Wang Z: Inhibition of microRNA-155 ameliorates
cardiac fibrosis in the process of angiotensin II-induced cardiac
remodeling. Mol Med Rep. 16:7287–7296. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Quiat D and Olson EN: MicroRNAs in
cardiovascular disease: From pathogenesis to prevention and
treatment. J Clin Invest. 123:11–18. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhang D, Cui Y, Li B, Luo X, Li B and Tang
Y: miR-155 regulates high glucose-induced cardiac fibrosis via the
TGF-β signaling pathway. Mol Biosyst. 13:215–224. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Seok HY, Chen J, Kataoka M, Huang ZP, Ding
J, Yan J, Hu X and Wang DZ: Loss of MicroRNA-155 protects the heart
from pathological cardiac hypertrophy. Circ Res. 114:1585–1595.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Tran PL, Tran PT, Tran HNK, Lee S, Kim O,
Min BS and Lee JH: A prenylated flavonoid, 10-oxomornigrol F,
exhibits anti-inflammatory effects by activating the Nrf2/heme
oxygenase-1 pathway in macrophage cells. Int Immunopharmacol.
55:165–173. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Chen RR, Fan XH, Chen G, Zeng GW, Xue YG,
Liu XT and Wang C: Irisin attenuates angiotensin II-induced cardiac
fibrosis via Nrf2 mediated inhibition of ROS/ TGFβ1/Smad2/3
signaling axis. Chem Biol Interact. 302:11–21. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Meng Z, Li HY, Si CY, Liu YZ and Teng S:
Asiatic acid inhibits cardiac fibrosis throughNrf2/HO-1 and
TGF-β1/Smads signaling pathways in spontaneous hypertension rats.
Int Immunopharmacol. 74:1057122019. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Jeong JY, Cha HJ, Choi EO, Kim CH, Kim GY,
Yoo YH, Hwang HJ, Park HT, Yoon HM and Choi YH: Activation of the
Nrf2/HO-1 signaling pathway contributes to the protective effects
of baicalein against oxidative stress-induced DNA damage and
apoptosis in HEI193 Schwann cells. Int J Med Sci. 16:145–155. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Abraham NG and Kappas A: Heme oxygenase
and the cardiovascular-renal system. Free Radic Biol Med. 39:1–25.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Chen M, Samuel VP, Wu Y, Dang M, Lin Y,
Sriramaneni R, Sah SK, Chinnaboina GK and Zhang G: Nrf2/HO-1
mediated protective activity of genistein against
doxorubicin-induced cardiac toxicity. J Environ Pathol Toxicol
Oncol. 38:143–152. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Chen C, Jiang X, Gu S and Zhang Z:
MicroRNA-155 regulates arsenite-induced malignant transformation by
targeting Nrf2-mediated oxidative damage in human bronchial
epithelial cells. Toxicol Lett. 278:38–47. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wan C, Han R, Liu L, Zhang F, Li F, Xiang
M and Ding W: Role of miR-155 in fluorooctane sulfonate-induced
oxidative hepatic damage via the Nrf2-dependent pathway. Toxicol
Appl Pharmacol. 295:85–93. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Gu S, Lai Y, Chen H, Liu Y and Zhang Z:
miR-155 mediates arsenic trioxide resistance by activating Nrf2 and
suppressing apoptosis in lung cancer cells. Sci Rep. 7:121552017.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Hu M, Ye P, Liao H, Chen M and Yang F:
Metformin protects H9C2 cardiomyocytes from high-glucose and
hypoxia/reoxygenation injury via inhibition of reactive oxygen
species generation and inflammatory responses: Role of AMPK and
JNK. J Diabetes Res. 2016:29619542016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Bugyei-Twum A, Advani A, Advani SL, Zhang
Y, Thai K, Kelly DJ and Connelly KA: High glucose induces Smad
activation via the transcriptional coregulator p300 and contributes
to cardiac fibrosis and hypertrophy. Cardiovasc Diabetol.
13:892014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ahmad A and Abdel Moneim AE: Indigofera
oblongifolia prevents lead acetate-induced hepatotoxicity,
oxidative stress, fibrosis and apoptosis in rats. PLoS One.
11:e01589652016. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Hüttemann M, Pecina P, Rainbolt M,
Sanderson TH, Kagan VE, Samavati L, Doan JW and Lee I: The multiple
functions of cytochrome c and their regulation in life and death
decisions of the mammalian cell: From respiration to apoptosis.
Mitochondrion. 11:369–381. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Garrido C, Galluzzi L, Brunet M, Puig PE,
Didelot C and Kroemer G: Mechanisms of cytochrome c release from
mitochondria. Cell Death Differ. 13:1423–1433. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Tsutsui H, Kinugawa S and Matsushima S:
Mitochondrial oxidative stress and dysfunction in myocardial
remodelling. Cardiovasc Res. 81:449–456. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Opferman JT and Kothari A: Anti-apoptotic
BCL-2 family members in development. Cell Death Differ. 25:37–45.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Czabotar PE, Lessene G, Strasser A and
Adams JM: Control of apoptosis by the BCL-2 protein family:
Implications for physiology and therapy. Nat Rev Mol Cell Biol.
15:49–63. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Ma ZG, Yuan YP, Wu HM, Zhang X and Tang
QZ: Cardiac fibrosis: New insights into the pathogenesis. Int J
Biol Sci. 14:1645–1657. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Fang L, Murphy AJ and Dart AM: A Clinical
perspective of anti-fibrotic therapies for cardiovascular disease.
Front Pharmacol. 8:186. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Brahma MK, Pepin ME and Wende AR: My
sweetheart is broken: Role of glucose in diabetic cardiomyopathy.
Diabetes Metab J. 41:1–9. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Costantino S, Paneni F, Lüscher TF and
Cosentino F: MicroRNA profiling unveils hyperglycaemic memory in
the diabetic heart. Eur Heart J. 37:572–576. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Huang Y, Liu Y, Li L, Su B, Yang L, Fan W,
Yin Q, Chen L, Cui T, Zhang J, et al: Involvement of
inflammation-related miR-155 and miR-146a in diabetic nephropathy:
implications for glomerular endothelial injury. BMC Nephrol.
15:1422014. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Gao J, Zhao G, Li W, Zhang J, Che Y, Song
M, Gao S, Zeng B and Wang Y: miR-155 targets PTCH1 to mediate
endothelial progenitor cell dysfunction caused by high glucose. Exp
Cell Res. 366:55–62. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Palma CA, Al Sheikha D, Lim TK, Bryant A,
Vu TT, Jayaswal V and Ma DD: MicroRNA-155 as an inducer of
apoptosis and cell differentiation in Acute Myeloid Leukaemia. Mol
Cancer. 13:792014. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Song Y, Wen L, Sun J, Bai W, Jiao R, Hu Y,
Peng X, He Y and Ou S: Cytoprotective mechanism of ferulic acid
against high glucose-induced oxidative stress in cardiomyocytes and
hepatocytes. Food Nutr Res. 60:303232016. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
You S, Qian J, Sun C, Zhang H, Ye S, Chen
T, Xu Z, Wang J, Huang W and Liang G: An Aza resveratrol-chalcone
derivative 6b protects mice against diabetic cardiomyopathy by
alleviating inflammation and oxidative stress. J Cell Mol Med.
22:1931–1943. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Ying Y, Jin J, Ye L, Sun P, Wang H and
Wang X: Phloretin prevents diabetic cardiomyopathy by dissociating
Keap1/Nrf2 complex and inhibiting oxidative stress. Front
Endocrinol (Lausanne). 9:7742018. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Li L, Luo W, Qian Y, Zhu W, Qian J, Li J,
Jin Y, Xu X and Liang G: Luteolin protects against diabetic
cardiomyopathy by inhibiting NF-κB-mediated inflammation and
activating the Nrf2-mediated antioxidant responses. Phytomedicine.
59:1527742019. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Tsai CY, Wen SY, Cheng SY, Wang CH, Yang
YC, Viswanadha VP, Huang CY and Kuo WW: Nrf2 activation as a
protective feedback to limit cell death in high glucose-exposed
cardiomyocytes. J Cell Biochem. 118:1659–1669. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Zhou L, Xu DY, Sha WG, Shen L, Lu GY, Yin
X and Wang MJ: High glucose induces renal tubular epithelial injury
via Sirt1/NF-kappaB/microR-29/Keap1 signal pathway. J Transl Med.
13:3522015. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Yang ZB, Chen WW, Chen HP, Cai SX, Lin JD
and Qiu LZ: MiR-155 aggravated septic liver injury by oxidative
stress-mediated ER stress and mitochondrial dysfunction via
targeting Nrf-2. Exp Mol Pathol. 105:387–394. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Chen J, Li C, Liu W, Yan B, Hu X and Yang
F: miRNA-155 silencing reduces sciatic nerve injury in diabetic
peripheral neuropathy. J Mol Endocrinol. 63:227–238. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Kosuru R, Kandula V, Rai U, Prakash S, Xia
Z and Singh S: Pterostilbene decreases cardiac oxidative stress and
inflammation via activation of AMPK/Nrf2/HO-1 pathway in
fructose-fed diabetic rats. Cardiovasc Drugs Ther. 32:147–163.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Holmström KM, Kostov RV and
Dinkova-Kostova AT: The multifaceted role of Nrf2 in mitochondrial
function. Curr Opin Toxicol. 1:80–91. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Liang J, Li L, Sun Y, He W, Wang X and Su
Q: The protective effect of activating Nrf2/HO-1 signaling pathway
on cardiomyocyte apoptosis after coronary microembolization in
rats. BMC Cardiovasc Disord. 17:2722017. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Zhang YF, Meng NN, Li HZ, Wen YJ, Liu JT,
Zhang CL, Yuan XH and Jin XD: Effect of naringin on oxidative
stress and endoplasmic reticulum stress in diabetic cardiomyopathy.
Zhongguo Zhong Yao Za Zhi. 43:596–602. 2018.(In Chinese).
PubMed/NCBI
|
|
50
|
Peng C, Ma J, Gao X, Tian P, Li W and
Zhang L: High glucose induced oxidative stress and apoptosis in
cardiac microvascular endothelial cells are regulated by FoxO3a.
PLoS One. 8:e797392013. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Wang R, Lu L, Guo Y, Lin F, Chen H, Chen W
and Chen M: Effect of glucagon-like peptide-1 on
high-glucose-induced oxidative stress and cell apoptosis in human
endothelial cells and its underlying mechanism. J Cardiovasc
Pharmacol. 66:135–140. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Zhang M, Niu X, Hu J, Yuan Y, Sun S, Wang
J, Yu W, Wang C, Sun D and Wang H: Lin28a Protects against
hypoxia/reoxygenation induced cardiomyocytes apoptosis by
alleviating mitochondrial dysfunction under high glucose/high fat
conditions. PLoS One. 9:e1105802014. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Gao CL, Zhu C, Zhao YP, Chen XH, Ji CB,
Zhang CM, Zhu JG, Xia ZK, Tong ML and Guo XR: Mitochondrial
dysfunction is induced by high levels of glucose and free fatty
acids in 3T3-L1 adipocytes. Mol Cell Endocrinol. 320:25–33. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Hung KY, Liu SY, Yang TC, Liao TL and Kao
SH: High-dialysate-glucose-induced oxidative stress and
mitochondrial-mediated apoptosis in human peritoneal mesothelial
cells. Oxid Med Cell Longev. 2014:642793. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Ho FM, Liu SH, Liau CS, Huang PJ and
Lin-Shiau SY: High glucose-induced apoptosis in human endothelial
cells is mediated by sequential activations of c-Jun NH(2)-terminal
kinase and caspase-3. Circulation. 101:2618–2624. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Liu J, Wu Y, Wang B, Yuan X and Fang B:
High levels of glucose induced the caspase-3/PARP signaling
pathway, leading to apoptosis in human periodontal ligament
Fibroblasts. Cell Biochem Biophys. 66:229–237. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Yang LL, Liu JQ, Bai XZ, Fan L, Han F, Jia
WB, Su LL, Shi JH, Tang CW and Hu DH: Acute downregulation of
miR-155 at wound sites leads to a reduced fibrosis through
attenuating inflammatory response. Biochem Biophys Res Commun.
453:153–159. 2014. View Article : Google Scholar : PubMed/NCBI
|