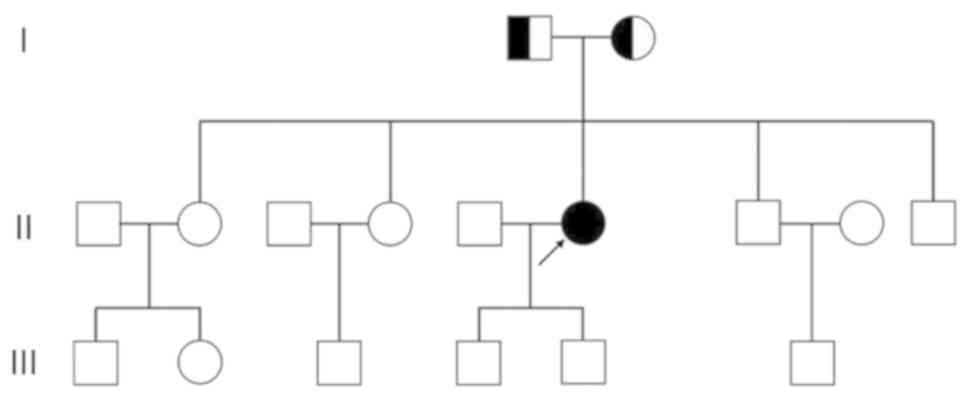
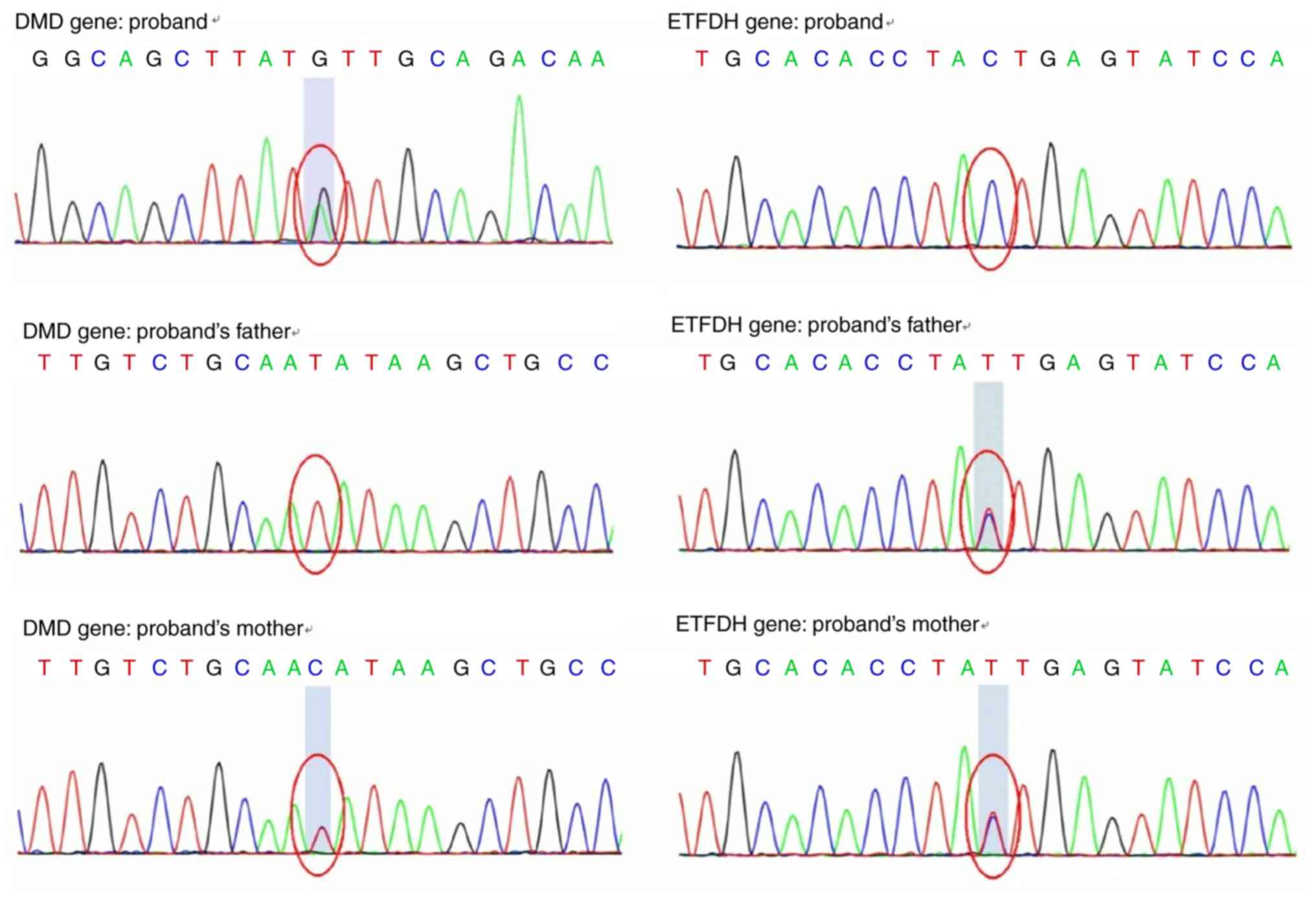
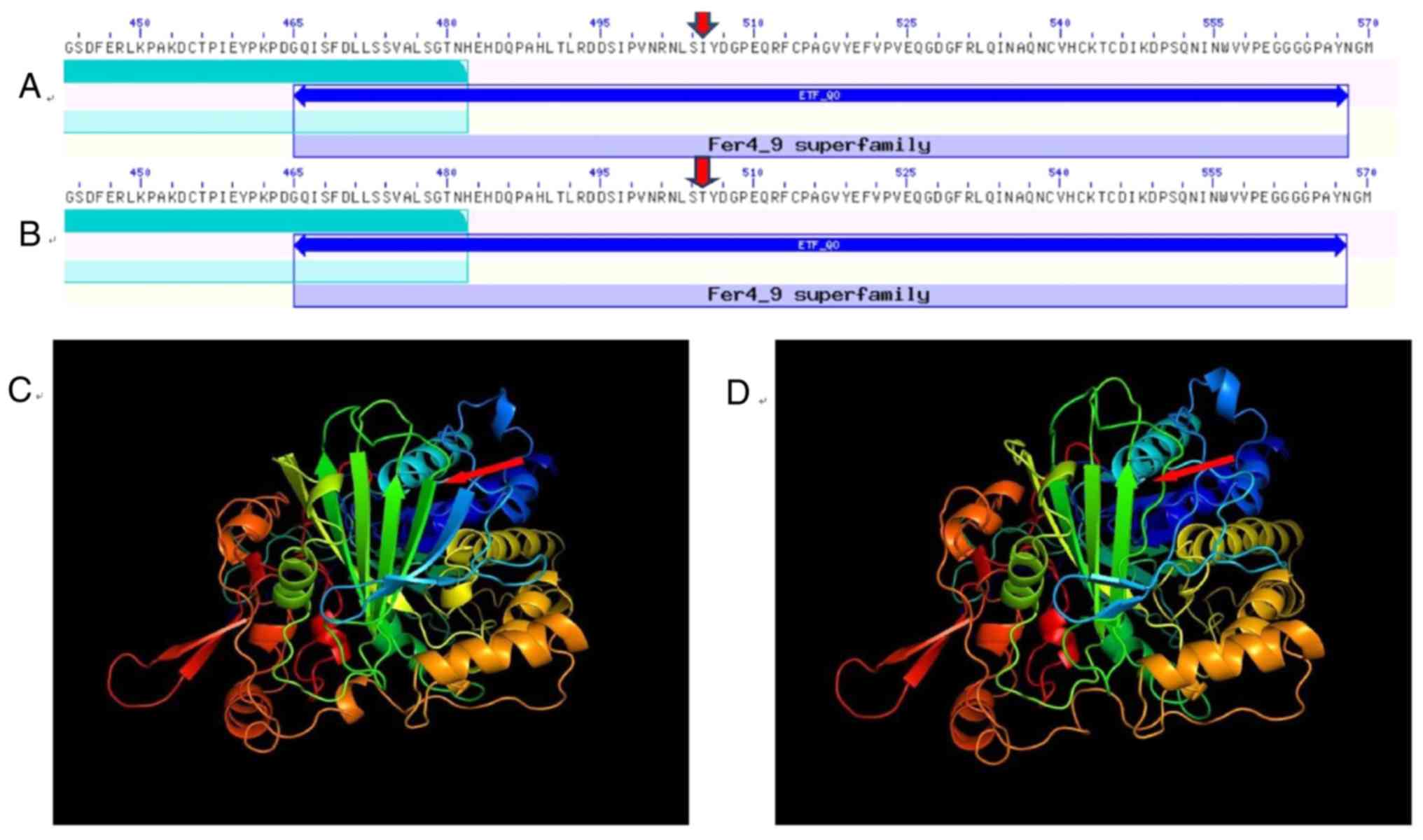
|
1
|
Frerman FE and Goodman SI: Deficiency of
electron transfer flavoprotein or electron transfer flavoprotein:
Ubiquinone oxidoreductase in glutaric acidemia type II fibroblasts.
Proc Natl Acad Sci USA. 82:4517–4520. 1985. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Vergani L, Barile M, Angelini C, Burlina
AB, Nijtmans L, Freda MP, Brizio C, Zerbetto E and Dabbeni-Sala F:
Riboflavin therapy. Biochemical heterogeneity in two adult lipid
storage myopathies. Brain. 122:2401–2411. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Wen B, Dai T, Li W, Zhao Y, Liu S, Zhang
C, Li H, Wu J, Li D and Yan C: Riboflavin-responsive lipid-storage
myopathy caused by ETFDH gene mutations. J Neurol Neurosurg
Psychiatry. 81:231–236. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Nilipour Y, Fatehi F, Sanatinia S,
Bradshaw A, Duff J, Lochmüller H, Horvath R and Nafissi S: Multiple
acyl-coenzyme A dehydrogenase deficiency shows a possible founder
effect and is the most frequent cause of lipid storage myopathy in
Iran. J Neurol Sci. 411:1167072020. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Fu HX, Liu XY, Wang ZQ, Jin M, Wang DN, He
JJ, Lin MT and Wang N: Significant clinical heterogeneity with
similar ETFDH genotype in three Chinese patients with late-onset
multiple acyl-CoA dehydrogenase deficiency. Neurol Sci.
37:1099–1105. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Przyrembel H, Wendel U, Becker K, Bremer
HJ, Bruinvis L, Ketting D and Wadman SK: Glutaric aciduria type II:
Report on a previously undescribed metabolic disorder. Clin Chim
Acta. 66:227–239. 1976. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Chautard R, Laroche-Raynaud C, Lia AS,
Chazelas P, Derouault P, Sturtz F, Baaj Y, Veauville-Merllié A,
Acquaviva C, Favreau F and Faye PA: A case report of a mild form of
multiple acyl-CoA dehydrogenase deficiency due to compound
heterozygous mutations in the ETFA gene. BMC Med Genom. 13:122020.
View Article : Google Scholar
|
|
8
|
Lucas TG, Henriques BJ and Gomes CM:
Conformational analysis of the riboflavin-responsive
ETF:QO-p.Pro456Leu variant associated with mild multiple acyl-CoA
dehydrogenase deficiency. Biochim Biophys Acta Proteins Proteom.
1868:1403932020. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Chen S, Zhou Y, Chen Y and Gu J: fastp: An
ultra-fast all-in-one FASTQ preprocessor. Bioinformatics.
34:i884–i890. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
McKenna A, Hanna M, Banks E, Sivachenko A,
Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly
M and DePristo MA: The Genome Analysis Toolkit: A MapReduce
framework for analyzing next-generation DNA sequencing data. Genome
Res. 20:1297–1303. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Jian X, Boerwinkle E and Liu X: In silico
prediction of splice-altering single nucleotide variants in the
human genome. Nucleic Acids Res. 42:13534–13544. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Paternostro-Sluga T, Grim-Stieger M, Posch
M, Schuhfried O, Vacariu G, Mittermaier C, Bittner C and
Fialka-Moser V: Reliability and validity of the Medical Research
Council (MRC) scale and a modified scale for testing muscle
strength in patients with radial palsy. J Rehabil Med. 40:665–671.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Koenig M, Beggs AH, Moyer M, Scherpf S,
Heindrich K, Bettecken T, Meng G, Müller CR, Lindlöf M, Kaariainen
H, et al: The molecular basis for Duchenne versus Becker Muscular
Dystrophy: Correlation of severity with type of deletion. Am J Hum
Genet. 45:498–506. 1989.PubMed/NCBI
|
|
14
|
Moser H: Duchenne muscular dystrophy:
Pathogenetic aspects and genetic prevention. Human Genetics.
66:17–40. 1984. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Goodman SI and Frerman FE: Glutaric
acidaemia type II (multiple Acyl-Coa dehydrogenation deficiency). J
Inherit Metab Dis. 7:33–37. 1984. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Gordon N: Glutaric aciduria types I and
II. Brain Dev. 28:136–140. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
al-Essa MA, Rashed MS, Bakheet SM, Patay
ZJ and Ozand PT: Glutaric aciduria type II: Observations in seven
patients with neonatal- and late-onset disease. J Perinatol.
20:120–128. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Schiff M, Froissart R, Olsen RK, Acquaviva
C and Vianey-Saban C: Electron transfer flavoprotein deficiency:
Functional and molecular aspects. Mol Genet Metab. 88:153–158.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhang J, Frerman FE and Kim JJ: Structure
of electron transfer flavoprotein-ubiquinone oxidoreductase and
electron transfer to the mitochondrial ubiquinone pool. Proc Natl
Acad Sci USA. 103:16212–16217. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Angelini C, Tavian D and Missaglia S:
Heterogeneous phenotypes in lipid storage myopathy Due to ETFDH
gene mutations. JIMD Rep. 38:33–40. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Champion M: An approach to the diagnosis
of inherited metabolic disease. Arch Dis Child Educ Pract Ed.
95:40–46. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Li Q, Wei S, Wu D, Wen C and Zhou J:
Urinary metabolomics study of patients with gout using gas
chromatography-mass spectrometry. Biomed Res Int. 2018:34615722018.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wang ZQ, Chen XJ, Murong SX, Wang N and Wu
ZY: Molecular analysis of 51 unrelated pedigrees with late-onset
multiple acyl-CoA dehydrogenation deficiency (MADD) in southern
China confirmed the most common ETFDH mutation and high carrier
frequency of c.250G>A. J Mol Med (Berl). 89:569–576. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Usselman RJ, Fielding AJ, Frerman FE,
Watmough NJ, Eaton GR and Eaton SS: Impact of mutations on the
midpoint potential of the [4Fe-4S]+1,+2 cluster and on catalytic
activity in electron transfer flavoprotein-ubiquinone
oxidoreductase (ETF-QO). Biochemistry. 47:92–100. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Olsen RKJ, Olpin SE, Andresen BS,
Miedzybrodzka ZH, Pourfarzam M, Merinero B, Frerman FE, Beresford
MW, Dean JC, Cornelius N, et al: ETFDH mutations as a major cause
of riboflavin-responsive multiple acyl-CoA dehydrogenation
deficiency. Brain. 130:2045–2054. 2007. View Article : Google Scholar : PubMed/NCBI
|