Introduction
Multiple acyl-CoA dehydrogenase deficiency (MADD), a
rare autosomal recessive disease, is a disorder of fatty acid
oxidation caused by defects in electron transfer flavoprotein (ETF)
or ETF dehydrogenase (ETFDH) (1).
MADD can be divided into two subtypes, namely the neonatal and
late-onset types, according to their clinical manifestations. The
symptoms of late-onset MADD vary significantly, with muscle
weakness being the most common symptom, while its most frequent
pathological feature is lipid deposition in the muscle tissue.
Late-onset MADD is the leading cause of lipid storage myopathy, and
treatment with riboflavin has been considered to be effective
(2–4). Several muscle diseases, such as
inflammatory myopathies, metabolic myopathies and progressive
muscular dystrophies, may also present with muscle weakness,
therefore, MADD may be easily misdiagnosed as another type of
muscle disease (5).
MADD was first described by Przyrembel et al
(6) in 1976. The prevalence of
MADD in the general population is estimated to be ~9/1,000,000
worldwide (7). Traditionally,
muscle biopsy combined with biochemical tests to detect
acylcarnitines and urine organic acids have been applied to
diagnose MADD. However, gene sequencing can more precisely detect
causative gene mutations. To date, published literature has
reported >160 mutation sites in the ETFDH gene, but only a few
repeat mutations (8). The present
study reported the case of a patient who presented with muscle
weakness and was diagnosed with MADD following genetic testing.
Furthermore, the genetic analysis of the patient's parents
identified a novel c.1514T>C mutation located in the 4Fe4S
domain of the ETFDH gene. Following timely and reasonable
treatment, the patient's condition was significantly improved.
Materials and methods
Subjects and ethical approval
A 35-year-old female patient and her asymptomatic
parents were enrolled at the First Hospital of Lanzhou University
in February 2019. The present study was conducted with the approval
of the Ethics Committee of the First Hospital of Lanzhou University
(Lanzhou, China; approval no. LDYYLL2020-248). Written informed
consent was obtained from the patient and her parents for
participation in the study and publication of case details. The
present study was performed in accordance with The Declaration of
Helsinki.
Laboratory tests and muscle
biopsy
Blood and urine samples were collected from the
proband to determine the levels of acylcarnitines and organic
acids, respectively. Urinary organic acids and blood acylcarnitines
were analyzed by gas chromatography-mass spectrometry (GCMS-QP2010
analyzer; Shimadzu Corporation) and tandem mass spectrometry (ABI
2000; Applied Biosystems; Thermo Fisher Scientific, Inc.),
respectively. For each examination, three scanning modes were used.
For amino acids, the neutral loss scan was used (m/z 102) with the
scanning range m/z 140–280. For acylcarnitines, the precursor scan
of the product ion m/z 85 was used with the scanning range m/z
210–502. For glycine, ornithine, arginine and citrulline, multiple
reaction monitoring (MRM) was used. GC-MS analysis was performed on
an GCMS-QP2010 analyzer system. High-purity helium (99.9996%) was
used as the carrier gas at a constant flow rate of 1.2 ml/min. The
sample injection volume was 1 µl and the split ratio was 10:1. The
injection temperature was 300°C and the transfer line temperature
was maintained at 280°C. The column temperature was initially set
at 70°C for 3 min and then increased to 300°C at 5°C/min and held
for 5 min. The mass scan was set from 33 to 600 with a scan speed
of 2 scans/sec. The diagnostic biopsy was performed on the left
musculus biceps brachii. Briefly, following local infiltration
anesthesia with 5 ml of 2% lidocaine hydrochloride, an incision
~2.5 cm in length was made at the maximum contraction of the muscle
and a piece of muscle tissue 0.5×0.8×0.5 cm in size was harvested.
Muscle specimens were frozen in isopentane that was precooled in
liquid nitrogen and stored at −80°C. Furthermore, for histological
examination, serial frozen sections (8-µm thick) were stained with
hematoxylin and eosin, Oil Red O (ORO), modified Gomori trichrome
and periodic acid Schiff. Briefly, cells were fixed with 10%
formalin for 10 min at 25°C and treated with Oil Red O working
solution for 5 min at 25°C. Hematoxylin was added to the slide and
incubated for 1 min at 25°C, and then the slide was rinsed with tap
water. Gomori staining solution was added for 20 min at 25°C,
followed by rinsing with tap water. Schiff's solution was stained
for 10 min at 25°C and rinsed with water for 5 min. Morphological
alterations to muscle fibers were observed under a light microscope
(magnification, ×40).
Gene sequencing
Peripheral blood samples were collected from the
proband and her parents in EDTA-treated tubes. The genomic DNA was
extracted using the Blood Genome Column Medium Extraction kit
(CWBio), according to the manufacturer's instructions. The
extracted DNA was then subjected to the quality control process
using a Qubit 2.0 fluorimeter and 0.8% agarose gel electrophoresis.
Subsequently, protein-coding exome enrichment was performed using
the xGen Exome Research Panel v1.0 (Integrated DNA Technologies,
Inc.). This panel consists of 429,826 individually synthesized and
quality-controlled probes that target 39 Mb protein-coding regions
(19,396 genes) of the human genome and cover 51 Mb of end-to-end
tiled probe space. Furthermore, high-throughput sequencing was
carried out on a NovaSeq 6000 series sequencer (PE150; Illumina,
Inc.), and >99% of the target sequences were sequenced. The
sequencing process was performed by Chigene Translational Medicine
Research Center Co., Ltd. A search was conducted in the HGMDpro
database for the novel mutation (https://digitalinsights.qiagen.com/products-overview/clinical-insights-portfolio/human-gene-mutation-database/).
Bioinformatics analysis
Raw data were processed using fastp (version 0.18.1)
(9) to remove adapter sequences
and filter low quality reads. The Burrows-Wheeler Aligner (version
0.7.8) was used to align the paired-end reads to the Ensembl
GRCh37/hg19 reference genome. Furthermore, the base quality score
recalibration together with single nucleotide polymorphism (SNP)
and short small insertions and deletion (indel) calling was
performed using GATK (version 3.8) (10). SNPs and indels were screened to
obtain high quality and reliable variants, depending on the
sequencing depth and variant quality. In addition, an online
system, independently developed by Chigene (www.chigene.org), was used to annotate database-based
minor allele frequencies (MAFs), and the ACMG practice
guideline-based pathogenicity of every yielded gene variant. The
system also provided several software packages for the conservative
analysis and protein product structure prediction. Several
databases for MAF annotation were used, including 1000 genomes
(www.1000genomes.org), dbSNP (http://www.ncbi.nlm.nih.gov/SNP), ESP (http://evs.gs.washington.edu/EVS/), ExAC
(http://exac.broadinstitute.org/about), and the Chigene
in-house MAF database. Additionally, for the prediction of the
protein product structure variation, the following software
packages were used: PROVEAN (version 1.1.3), SIFT (http://sift.jcvi.org/www/SIFT_seq_submit2.html),
Polypen2_hdiv, Polypen2_hvar (http://genetics.bwh.harvard.edu/pph2/index.shtml),
Mutationtaster2, M-Cap (version 1.4) and REVEL. To assess the
pathogenicity of the genetic variation according to the ACMG
guidelines, OMIM (http://www.ncbi.nlm.nih.gov/omim/limits), HGMD
(http://www.hgmd.cf.ac.uk/ac/index.php) and ClinVar
databases (https://www.ncbi.nlm.nih.gov/clinvar/) were used.
Finally, to predict the functional changes of splice site variants,
the MaxEntScan (http://genes.mit.edu/burgelab/maxent/Xmaxentscan_scoreseq.html),
dbscSNV (11) and GTAG (www.chigene.org) software packages were applied,
instead of the product structure prediction software. Computational
mutation prediction of conserved domain analysis of the ETFDH gene
was performed using NCBI/Structure/Cdd (https://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi).
Results
Clinical characteristics and
laboratory analysis
A 35-year-old woman was admitted to The First
Hospital of Lanzhou University with progressively worsening limb
weakness for >2 years. Since January 2016, the patient had been
prone to fatigue without any obvious inducement. The female patient
felt obvious weakness when she was going upstairs, which could be
relieved after resting. The weakness in both lower limbs was
aggravated in January 2017, and gradually she was not able to walk
continuously, even for 20 meters. Gradually she exhibited weakness
in both upper limbs and the nuchal region, and difficulty in
raising her head, therefore, she was not capable for daily work.
Subsequently, these symptoms were progressively aggravated. In July
2018, she was not able to speak and relied on a wheelchair for
movement. During this period, she visited local hospitals several
times, while the laboratory tests revealed elevated myozyme serum
levels (Table I). However, she was
misdiagnosed with polymyositis and progressive muscular dystrophy,
and she underwent treatment with poor efficacy. She did not exhibit
muscle twitching or muscle pain, choking cough when drinking water,
or dyspnea. Her parents and siblings had no similar clinical
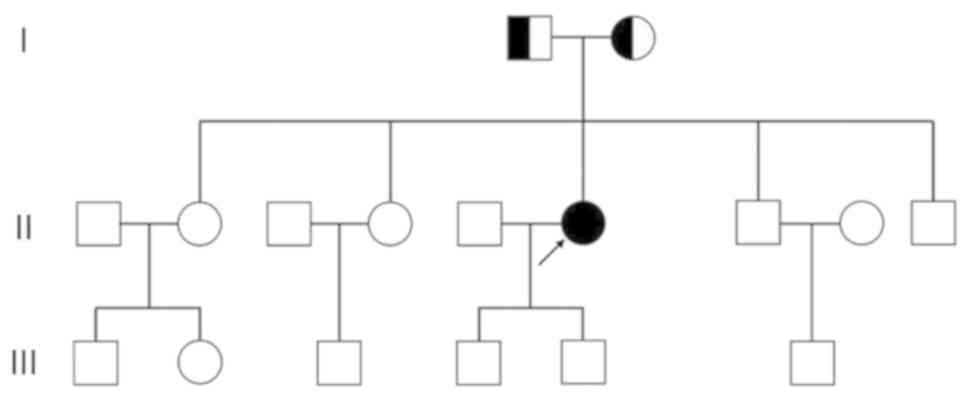
symptoms. The pedigree of the proband is shown in Fig. 1. Neurological examination revealed
that the muscle strength of the proximal limbs was grade IV, the
distal muscle strength was grade IV, the neck muscle force was
grade IV (Medical Research Council scale) (12), whereas the muscle tension was
normal. The physiological reflexes were normal, while pathological
ones were not elicited.
 | Table I.Results of multiple muscle enzymes in
this patient serum. |
Table I.
Results of multiple muscle enzymes in
this patient serum.
| Date | AST (normal range
1–49 U/l) | ALT (normal range
1–49 U/l) | LDH (normal range
125–240 U/l) | α-HBDH (normal range
72–182 U/l) | CK (normal range
38–240 U/l) | CK-MB (normal range
0–25 U/l) |
|---|
| 28/07/2017 | 29 | / | 385 | 427 | 281 | 18 |
| 07/08/2017 | 88 | 81 | 917 | 1,132 | 1,499 | 66 |
| 11/08/2017 | 32 | / | 591 | 658 | 442 | 23 |
| 30/04/2018 | 53 | / | 614 | 707 | 904 | 52 |
| 19/06/2018 | 47 | / | 556 | 635 | 551 | 36 |
| 28/07/2018 | 78 | 74 | 1,230 | 1,371 | 1,091 | / |
| 15/08/2018 | 35 | 41 | 178 | 96 | 107 | 20 |
As shown in Table
I, multiple muscle enzymes were elevated in the serum. Blood
tandem mass spectrometry analysis revealed elevated C4, C8 and
C14:1 acylcarnitine levels, whereas, urine gas chromatography-mass
spectrometry showed increased urinary excretion of
2-hydroxyglutaric acid, 3-hydroxyglutaric acid, 2-hydroxyisovaleric
acid and ethylmalonic acid (Table
II). Furthermore, blood glucose levels, electrolytes,
infectious indicators, including C-reactive protein, procalcitonin
and erythrocyte sedimentation rate, immunoglobulins, rheumatoid
factors, autoantibody profiles, serum protein electrophoresis and
endocrine hormone levels showed no significant abnormalities.
Besides, a lower limb muscle magnetic resonance imaging scan
indicated possible muscle inflammation. Additionally,
electromyography showed myogenic damage, whereas abdominal
ultrasound, echocardiography and craniocerebral computed tomography
did not reveal any abnormalities (data not shown).
| Table II.Blood acylcarnitine and urine organic
acid test results of the proband. |
Table II.
Blood acylcarnitine and urine organic
acid test results of the proband.
| Item | Before
treatment | After
treatment | Reference
range |
|---|
| Blood acylcarnitine
(µmol/l) |
|
|
|
| C4 | 2.10 | 0.39 | 0.17–0.91 |
| C8 | 1.76 | 0.08 | 0.04–0.28 |
|
C14:1 | 0.43 | 0.03 | 0-0.15 |
| Urinary organic
acids (mmol/mol Cr) |
|
|
|
|
2-hydroxyglutaric acid | 14.70 | 6.70 | <9.10 |
|
3-hydroxyglutaric acid | 2.18 | 0.10 | <0.40 |
|
2-hydroxyisovaleric acid | 3.90 | 1.25 | <2.26 |
|
Ethylmalonic acid | 7.62 | 0.48 | <4.14 |
Muscle biopsy specimens exhibited a significant
increase of the lipid droplets within the muscle fibers, which was
consistent with the pathological characteristics of lipid storage
myopathy. In addition, a few scattered atrophic myofibers were
observed, 12–30 µm in diameter. Small vacuoles in uniform size were
observed in several muscle fibers, while some vacuolar muscle
fibers were slightly basophilic. Furthermore, ORO staining revealed
a massive accumulation of lipid droplets within the vacuolar muscle
fibers, with some of them fused into sheets. The aforementioned
pathological changes may appear in MADD, therefore, its further
diagnosis and classification needs to be determined by biochemical
and genetic analyses (data not shown).
Genetic analysis
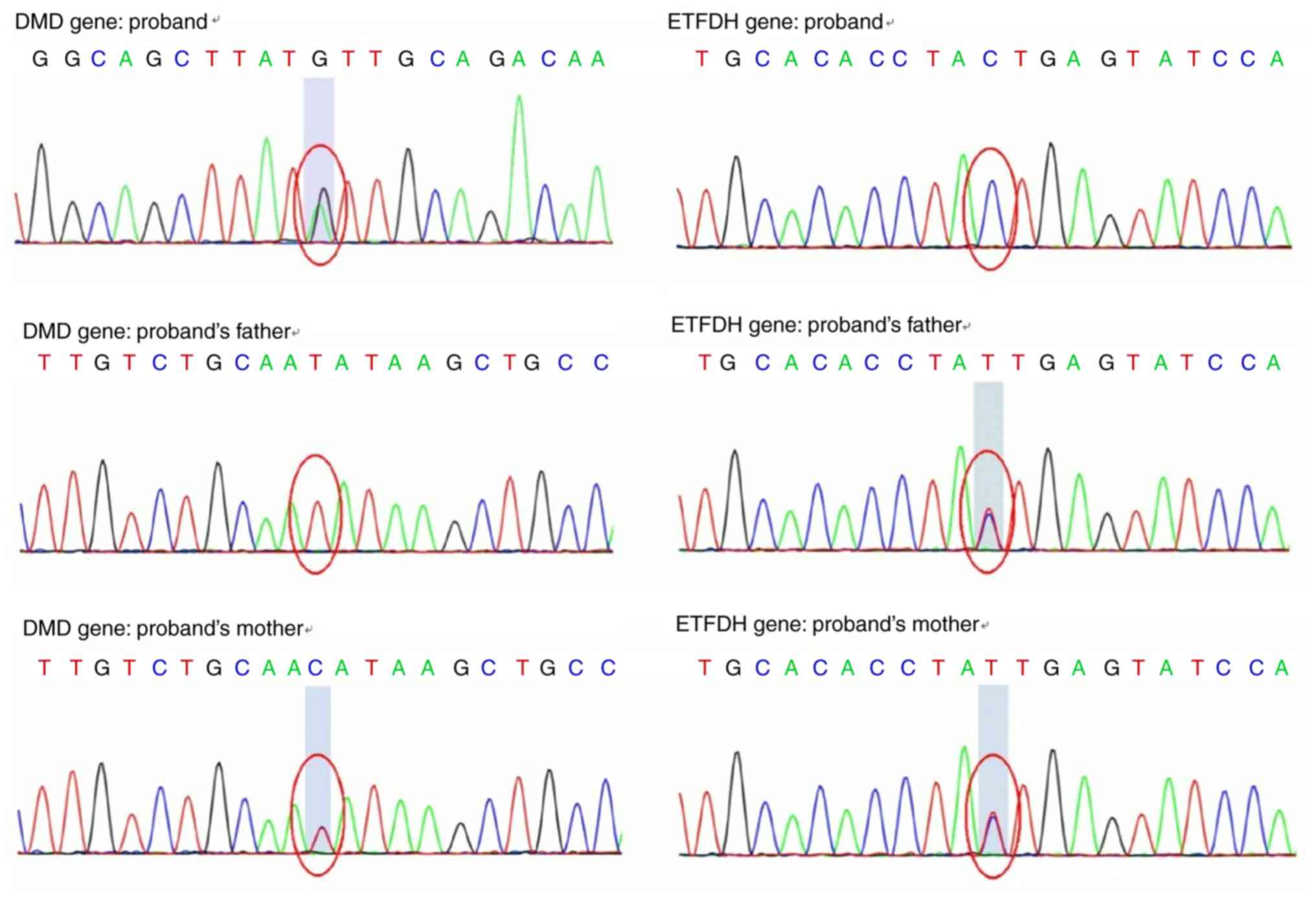
Whole exon sequencing revealed that the proband
carried a c.4189A>G (p.I1397V) heterozygous mutation and a
c.1514T>C (p.I505T) homozygous mutation, located in the
dystrophin (DMD) and ETFDH genes, respectively (Fig. 2). No pathogenic mutations were
detected in the ETFα and ETFβ genes.
The patient harbored a heterozygous mutation,
c.4189A>G, in the 30th exon region of the DMD gene, resulting in
the amino acid substitution p.I1397V (isoleucine > valine), as
shown in Fig. 2. For the female
patient, the carrier status of the single heterozygous mutation
should be classified as not pathogenic. Furthermore, DMD/Becker
muscular dystrophy (BMD) gene mutations did not affect fatty acid
metabolism and led to elevated serum levels of acylcarnitines and
urine organic acids (13,14), therefore, they were excluded.
The ETFDH gene exhibits autosomal recessive
inheritance (1), therefore,
pathogenic mutations on both alleles are likely to be pathogenic
(homozygous or compound heterozygous mutations). A homozygous
mutation, c.1514T>C, was identified in the 12th exon region of
the ETFDH gene of the proband, leading to the amino acid
substitution p.I505T (isoleucine > threonine). The homozygous
mutation originated from her parents (Fig. 2) and was consistent with an
autosomal recessive mode of inheritance. Mutations in the ETFDH
gene are often associated with impaired fatty acid oxidation
(3), which is consistent with the
laboratory findings of this patient.
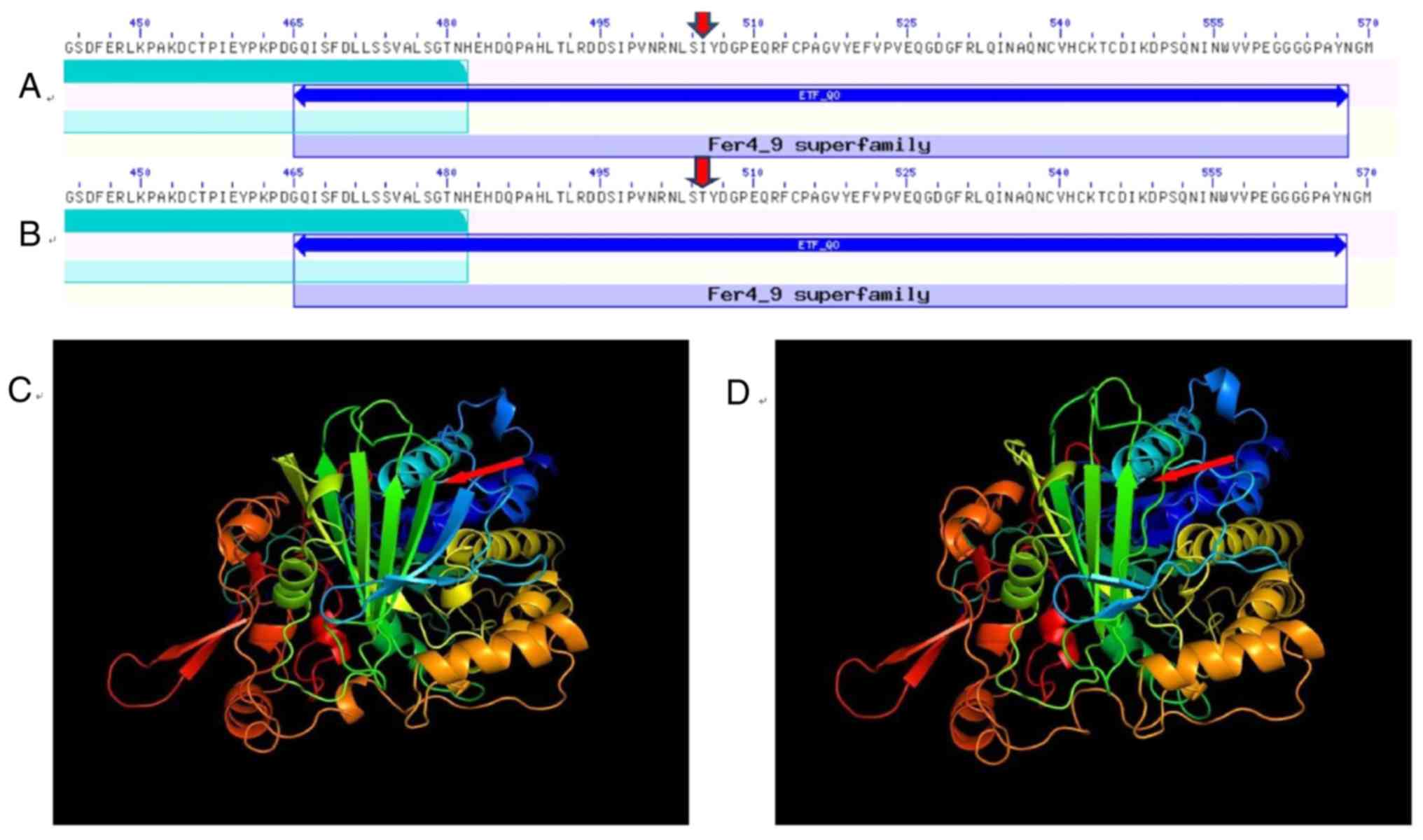
Computational mutation prediction of conserved
domain analysis of the ETFDH gene was performed using the
NCBI/Structure/Cdd. The analysis revealed a mutation, resulting in
the substitution of the 505th amino acid from isoleucine to
threonine, located in the 4Fe-4S domain of the ETFDH gene (Fig. 3). ETFDH in the inner mitochondrial
membrane accepts electrons from ETF, located in the mitochondrial
matrix, and reduces ubiquinone in the mitochondrial membrane
(7,8). The two redox centers of the protein,
flavin adenine dinucleotide (FAD) and the 4Fe-4S cluster, are
present in a 64-kDa monomer structure domain. According to the
NCBI/Structure/Cdd, a sequence-based mutation prediction for
p.I505T, indicated that this amino acid substitution could affect
the function of ETFDH.
Diagnosis and treatment
Based on the mutation analysis by gene sequencing,
combined with the clinical manifestations and laboratory findings,
the patient was diagnosed with MADD. Therefore, she was immediately
treated with 80 mg vitamin B2 three times daily, 1 g L-carnitine
twice a day and 50 mg coenzyme Q10 three times daily, and her
health status was significantly improved (Tables I and II).
Discussion
MADD, also known as glutaric acidemia type II, is
characterized by elevated urine organic acid levels and is
clinically divided into three subtypes, namely the neonatal-onset
with congenital malformations MADD (type I), the neonatal-onset
without congenital malformations MADD (type II), and mild or
late-onset MADD (type III) (3,15,16).
The first two subtypes are often characterized by severe multiple
acyl-CoA dehydrogenation defects. Due to the rapid onset and severe
phenotype, the majority of children die shortly after disease
onset. Unlikely, the clinical phenotypes and age at presentation of
late-onset MADD are highly variable, ranging from early infancy to
late adulthood. This type of MADD occurs insidiously, and the
majority of patients present with intermittent muscle weakness,
paroxysmal vomiting, hypoglycemia and other clinical features,
including sensory neuropathy, tendon reflex disappearance and fatty
liver disease (15,17). Due to the lack of specific clinical
manifestations, late-onset MADD may easily be misdiagnosed as
myositis, progressive muscular dystrophy and glycogen storage
disease (5).
MSDD is caused by ETF or ETFDH deficiencies. ETF is
a heterodimer located in the mitochondrial matrix, consisting of
two subunits, namely α and β, which are encoded by the ETFα and
ETFβ gene, respectively. Furthermore, the ETFDH protein is located
in the inner mitochondrial membrane and encoded by the ETFDH gene.
ETFDH accepts electrons from ETF and transmits them to ubiquinone.
Its crystal structure is comprised of three functional domains: The
FAD, 4Fe-4S cluster and coenzyme Q10 binding domains (Fig. 3). The genes encoding ETFα, ETFβ and
ETFDH are located at 15p23-25, 19q13 and 4q33, respectively
(18,19). ETF and ETFDH link the oxidation of
fatty acids and some amino acids to the mitochondrial oxidative
respiratory chain, therefore, both proteins serve an important role
in the process of electron transfer (8). Mutations in any of the ETFα, ETFβ and
ETFDH genes may lead to functional defects of the corresponding
proteins, thus resulting in metabolism disorders of the fatty acids
and other substances, insufficient energy production and muscle
weakness accompanied by lipid deposition in muscle tissues
(20).
In the past, the diagnosis of MADD could be quickly
and easily achieved using tandem mass spectrometry of blood
samples, which could be further confirmed by gas chromatography
mass spectrometry analysis of urine organic acids (21,22).
However, in late-onset patients, the increased levels of
acylcarnitines and urinary organic acids may be mild and atypical,
or detectable only during acute episodes (5,17),
therefore, genetic analysis to detect mutations in the ETF or ETFDH
genes is considered as the key method for a definitive
diagnosis.
The conserved domain analysis of the ETFDH gene
revealed that the c.1514T>C mutation, resulting in the
substitution of isoleucine with threonine, was located in the
4Fe-4S domain of the ETFDH gene. Wen et al (3) and Wang et al (23) demonstrated that the vast majority
of ETFDH mutations were concentrated in the FAD-binding domain,
followed by the coenzyme Q10 binding domain. However, mutations
within the 4Fe-4S cluster structural domain were rare. Although the
4Fe-4S cluster binding domain does not directly bind and transfer
electrons, it is involved in electron transfer by acting as a
cofactor. Usselman et al (24) revealed that amino acid variations
in the 4Fe-4S cluster could attenuate the activity of ubiquinone
reductase and slow down the electron transfer rate to the oxidative
respiratory chain. A search in the HGMDpro database and the
retrieval of several major public databases revealed that the
c.1514T>C mutation had not been previously reported, suggesting
that this mutation was extremely rare in the general population.
The novel mutation site was located in the 4Fe-4S cluster domain,
which combined with the previously reported mutations, broadened
the ETFDH gene mutation spectrum.
Previous studies have reported that some patients
with MADD are sensitive to riboflavin, also known as
riboflavin-responsive MADD (RR-MADD), which is more common in the
late-onset type (3,5,25).
However, there is no specific treatment for RR-MADD, and patients
are usually treated with riboflavin accompanied with L-carnitine
and coenzyme Q10 administration as adjuvant therapy. In this case,
when treatment is accompanied by a low fat, low protein and high
carbohydrate diet, the effects are significant for the patient's
health. Therefore, the symptoms of muscle weakness are
significantly improved, and the abnormal laboratory parameters are
also gradually corrected. However, the exact mechanism underlying
the effect of riboflavin in the treatment of MADD remains unclear.
It has been suggested that FAD may act as a molecular chaperone
that mediates the proper folding of certain flavoproteins.
Therefore, the elevated FAD concentration within mitochondria may
compensate for the decreased folding capacity of the mutated
proteins. As a precursor of the coenzyme FAD, riboflavin therapy
may enhance the stability of mutant flavoproteins and the activity
of the ETFDH proteins (23,25).
As there are only a few previous reports regarding mutations in the
4Fe-4S domain, it is therefore difficult to determine the
pathophysiological mechanisms of riboflavin treatment on such
mutations. We speculate that mutations in the 4Fe-4S domain may not
only lead to reduced structural stability of the ETFDH protein, but
may also affect the activity of ubiquitin reductase. Therefore,
increasing FAD concentration may partially compensate for these
defects. Besides, it has been demonstrated that in recessive
genetic diseases, the phenotype of the patient is often determined
by the milder genetic variant when different mutations are present
in the two alleles (25). The
present study indicated that the c.1514T>C mutation was a mild
ETFDH gene mutation, which could also partially explain why this
patient was very sensitive to riboflavin treatment.
The association between the genotype and clinical
phenotype in patients with MADD has been extensively studied.
Although some patients carry the same mutation, the onset time and
clinical manifestations vary among them (5), indicating that the environment may
have an impact on the phenotype. However, further studies are still
needed to elucidate these differences. In the present study the
patient suffered from late-onset MADD and only showed intermittent
muscle weakness, and no other clinical manifestations such as
myalgia, hypoglycemia or severe fatty liver disease, which further
illustrated the high heterogeneity of the disease.
In conclusion, a case of late-onset MADD with limb
weakness and exercise intolerance as the first symptoms was
reported. Genetic analysis revealed that the c.1514T>C was a
pathogenic mutation, which broadened the spectrum of the ETFDH gene
mutations and provided a valuable reference for early diagnosis and
treatment. Special consideration should be given to adult patients
with unexplained myasthenia and elevated muscle enzyme levels to
the possibility of late-onset MADD. When muscle biopsy indicates
lipid storage myopathy, genetic testing should be performed to
further confirm the first diagnosis. Nevertheless, early diagnosis
and reasonable treatment may effectively improve the prognosis of
patients with MADD.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
HL conceived the present study, participated in its
methodology design, drafted the manuscript and interpreted the
data. CW conceptualized the study, obtained molecular genetic
literature, drafted the manuscript and interpreted data. CW and YM
acquired the data. Data analysis was performed by XX and YM. YM
obtained molecular genetic literature and coordinated the study.
Resources and interpretation of data were provided and performed by
CW, XX and QL. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
This study was conducted with approval from the
Ethics Committee of The First Hospital of Lanzhou University and in
accordance with the Declaration of Helsinki. Written informed
consent was obtained from the patient and her parents for
participation in the study.
Patient consent for publication
Written informed consent was obtained from the
patient and her parents for publication of case details.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
MADD
|
multiple acyl-CoA dehydrogenase
deficiency
|
|
RR-MADD
|
riboflavin-responsive MADD
|
|
CoA
|
Coenzyme A
|
|
ETF
|
electron transfer flavoprotein
|
|
ETFDH
|
ETF dehydrogenase
|
|
FAD
|
flavin adenine dinucleotide
|
References
|
1
|
Frerman FE and Goodman SI: Deficiency of
electron transfer flavoprotein or electron transfer flavoprotein:
Ubiquinone oxidoreductase in glutaric acidemia type II fibroblasts.
Proc Natl Acad Sci USA. 82:4517–4520. 1985. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Vergani L, Barile M, Angelini C, Burlina
AB, Nijtmans L, Freda MP, Brizio C, Zerbetto E and Dabbeni-Sala F:
Riboflavin therapy. Biochemical heterogeneity in two adult lipid
storage myopathies. Brain. 122:2401–2411. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Wen B, Dai T, Li W, Zhao Y, Liu S, Zhang
C, Li H, Wu J, Li D and Yan C: Riboflavin-responsive lipid-storage
myopathy caused by ETFDH gene mutations. J Neurol Neurosurg
Psychiatry. 81:231–236. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Nilipour Y, Fatehi F, Sanatinia S,
Bradshaw A, Duff J, Lochmüller H, Horvath R and Nafissi S: Multiple
acyl-coenzyme A dehydrogenase deficiency shows a possible founder
effect and is the most frequent cause of lipid storage myopathy in
Iran. J Neurol Sci. 411:1167072020. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Fu HX, Liu XY, Wang ZQ, Jin M, Wang DN, He
JJ, Lin MT and Wang N: Significant clinical heterogeneity with
similar ETFDH genotype in three Chinese patients with late-onset
multiple acyl-CoA dehydrogenase deficiency. Neurol Sci.
37:1099–1105. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Przyrembel H, Wendel U, Becker K, Bremer
HJ, Bruinvis L, Ketting D and Wadman SK: Glutaric aciduria type II:
Report on a previously undescribed metabolic disorder. Clin Chim
Acta. 66:227–239. 1976. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Chautard R, Laroche-Raynaud C, Lia AS,
Chazelas P, Derouault P, Sturtz F, Baaj Y, Veauville-Merllié A,
Acquaviva C, Favreau F and Faye PA: A case report of a mild form of
multiple acyl-CoA dehydrogenase deficiency due to compound
heterozygous mutations in the ETFA gene. BMC Med Genom. 13:122020.
View Article : Google Scholar
|
|
8
|
Lucas TG, Henriques BJ and Gomes CM:
Conformational analysis of the riboflavin-responsive
ETF:QO-p.Pro456Leu variant associated with mild multiple acyl-CoA
dehydrogenase deficiency. Biochim Biophys Acta Proteins Proteom.
1868:1403932020. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Chen S, Zhou Y, Chen Y and Gu J: fastp: An
ultra-fast all-in-one FASTQ preprocessor. Bioinformatics.
34:i884–i890. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
McKenna A, Hanna M, Banks E, Sivachenko A,
Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly
M and DePristo MA: The Genome Analysis Toolkit: A MapReduce
framework for analyzing next-generation DNA sequencing data. Genome
Res. 20:1297–1303. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Jian X, Boerwinkle E and Liu X: In silico
prediction of splice-altering single nucleotide variants in the
human genome. Nucleic Acids Res. 42:13534–13544. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Paternostro-Sluga T, Grim-Stieger M, Posch
M, Schuhfried O, Vacariu G, Mittermaier C, Bittner C and
Fialka-Moser V: Reliability and validity of the Medical Research
Council (MRC) scale and a modified scale for testing muscle
strength in patients with radial palsy. J Rehabil Med. 40:665–671.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Koenig M, Beggs AH, Moyer M, Scherpf S,
Heindrich K, Bettecken T, Meng G, Müller CR, Lindlöf M, Kaariainen
H, et al: The molecular basis for Duchenne versus Becker Muscular
Dystrophy: Correlation of severity with type of deletion. Am J Hum
Genet. 45:498–506. 1989.PubMed/NCBI
|
|
14
|
Moser H: Duchenne muscular dystrophy:
Pathogenetic aspects and genetic prevention. Human Genetics.
66:17–40. 1984. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Goodman SI and Frerman FE: Glutaric
acidaemia type II (multiple Acyl-Coa dehydrogenation deficiency). J
Inherit Metab Dis. 7:33–37. 1984. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Gordon N: Glutaric aciduria types I and
II. Brain Dev. 28:136–140. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
al-Essa MA, Rashed MS, Bakheet SM, Patay
ZJ and Ozand PT: Glutaric aciduria type II: Observations in seven
patients with neonatal- and late-onset disease. J Perinatol.
20:120–128. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Schiff M, Froissart R, Olsen RK, Acquaviva
C and Vianey-Saban C: Electron transfer flavoprotein deficiency:
Functional and molecular aspects. Mol Genet Metab. 88:153–158.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhang J, Frerman FE and Kim JJ: Structure
of electron transfer flavoprotein-ubiquinone oxidoreductase and
electron transfer to the mitochondrial ubiquinone pool. Proc Natl
Acad Sci USA. 103:16212–16217. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Angelini C, Tavian D and Missaglia S:
Heterogeneous phenotypes in lipid storage myopathy Due to ETFDH
gene mutations. JIMD Rep. 38:33–40. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Champion M: An approach to the diagnosis
of inherited metabolic disease. Arch Dis Child Educ Pract Ed.
95:40–46. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Li Q, Wei S, Wu D, Wen C and Zhou J:
Urinary metabolomics study of patients with gout using gas
chromatography-mass spectrometry. Biomed Res Int. 2018:34615722018.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wang ZQ, Chen XJ, Murong SX, Wang N and Wu
ZY: Molecular analysis of 51 unrelated pedigrees with late-onset
multiple acyl-CoA dehydrogenation deficiency (MADD) in southern
China confirmed the most common ETFDH mutation and high carrier
frequency of c.250G>A. J Mol Med (Berl). 89:569–576. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Usselman RJ, Fielding AJ, Frerman FE,
Watmough NJ, Eaton GR and Eaton SS: Impact of mutations on the
midpoint potential of the [4Fe-4S]+1,+2 cluster and on catalytic
activity in electron transfer flavoprotein-ubiquinone
oxidoreductase (ETF-QO). Biochemistry. 47:92–100. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Olsen RKJ, Olpin SE, Andresen BS,
Miedzybrodzka ZH, Pourfarzam M, Merinero B, Frerman FE, Beresford
MW, Dean JC, Cornelius N, et al: ETFDH mutations as a major cause
of riboflavin-responsive multiple acyl-CoA dehydrogenation
deficiency. Brain. 130:2045–2054. 2007. View Article : Google Scholar : PubMed/NCBI
|