Introduction
Hepatocellular carcinoma (HCC) has the sixth highest
incidence among all malignant tumors worldwide and the fourth
highest mortality rate worldwide (1). More than half of all patients with HCC
worldwide reside in China (2). In
recent years, the incidence of liver cancer has been on the rise,
yet <20% of patients receive timely radical surgical resection,
due to the high degree of malignancy, early metastasis and high
invasion potential of this tumor type. These properties are often
associated with the complex biochemical regulatory behavior of HCC
cells, such as excessive cell proliferation (3,4).
Although HCC treatment methods and efficiency have improved the
5-year survival rate, several methods such as radical resection,
transcatheter arterial chemoembolization, sorafenib and
chemoradiotherapy still have their respective limits (5). Radical hepatectomy is considered the
first choice for the treatment of early HCC at an early stage
(Barcelona Clinic Liver Cancer stage 0 or A). For these patients,
resection is associated with survival >60% at 5 years, with low
postoperative mortality (<3%), and as many as 70% of these
patients experience tumor recurrence at 5 years (6). Based on HCC pathogenesis, in addition
to a number of correlative studies, immunotherapy represents a
potential therapeutic option (7).
Therefore, investigating the mechanism underlying the onset of HCC
and finding a potential therapeutic target is of significance for
improving the diagnosis and treatment of HCC.
Constitutive photomorphogenesis 9 signalosome (COP9)
is a protein complex composed of eight subunits (CSN1 to CSN8) that
was initially discovered in plants as an inhibitor of
photomorphogenesis (8–12). Mammalian COP9 is involved in a
variety of biological processes, including cell cycle control,
signal transduction, transcriptional activation and tumorigenesis
(13–15). CSN subunits have a conserved
structure; for example, the Mpr1-Pad1-N-terminal (MPN) domain in
CSN5 contains a metalloproteinase motif, which has catalytic
activity and is involved in the regulation of protein demethylation
(16–18). MPN in CSN6 may serve as a structural
scaffold or play a regulatory function (16,19,20).
Previous studies have confirmed that CSN inhibits the
ubiquitin-dependent degradation of numerous tumor-related proteins
(21–23), such as P27, P53, E3
ubiquitin-protein ligase Mdm2, Smad7, Runt-related transcription
factor 3, DNA-binding protein inhibitor ID-1, S-phase
kinase-associated protein 2 and hypoxia-inducible factor 1
(14), indicating that CSN plays an
important role in tumorigenesis. In particular, CSN5 or CSN6
overexpression has been detected in numerous types of cancer, such
as myeloma, lung cancer and breast cancer (24,25).
In HCC, CSN6 overexpression promotes epithelial-mesenchymal
transition and predicts poor prognosis (26). Inhibition of CSN3 expression induced
growth arrest and apoptosis of HCC cells (27). In our preliminary study, by
analyzing the transcriptome data of patients within The Cancer
Genome Atlas, it was found that CSN1 expression was increased in
patients with HCC compared with normal controls. However, the role
and molecular mechanism of CSN1 in HCC remains to be
elucidated.
Cyclin A2 is a highly conserved cell cycle protein
encoded by the CCNA2 gene. Cyclin A2, combined with
cyclin-dependent kinase 1 (CDK1) and CDK2, controls cell cycle
progression and promotes mitosis (28) and serves a potential role in liver
tumorigenesis (29). Cyclin A2 has
been recognized as a marker for several types of cancer diagnosis
and prognosis (30), such as lung
carcinoma, breast cancer, hepatocellular carcinoma and colorectal
cancer. Moreover, the expression of cyclin A2 is increased in HCC
(31), indicating that this protein
may play an important role in the development of HCC and represent
a potential therapeutic target.
In the present study, CSN1 was upregulated in HCC
tissue samples, compared with healthy adjacent tissue. In addition,
high levels of CSN1 indicated poor prognosis in patients with HCC.
CSN1 knockdown inhibited proliferation and migration and induced
apoptosis and cell cycle arrest in HCC cells. Moreover, xenograft
tumor experiments demonstrated that CSN1 promoted HCC in
vivo. Mechanistically, cyclin A2 may be a downstream effector
of CSN1.
Materials and methods
Patients and sample collection
Primary HCC tissue samples were obtained for
retrospective analysis from 117 patients at Daping Hospital, Army
Medical University, between January 2013 and December 2014.
Matched, adjacent, non-tumor tissue samples resected 1–2 cm from
the malignant tumor were obtained from 59 of these patients.
Written informed consent was obtained from all participants. All
specimens obtained during surgery were assembled into a tissue
microarray (TMA) for further analysis. The identification of tumor
tissues and adjacent normal tissues was confirmed by pathologists.
The present study was approved by the Institutional Research Ethics
Committee of Daping Hospital, Army Medical University.
Cell culture and transfection
Human HCC cell lines (MHCC-97H, MHCC-LM3 and Hep3B)
were purchased from the Cell Bank of Academia Sinica. The
immortalized liver cell line MIHA was a kind gift from Dr Deng
Huang. The cells were cultured in DMEM (HyClone; Cytiva)
supplemented with 10% FBS (Shanghai ExCell Biology, Inc.) at 37°C
with 5% CO2.
Human CSN1 and cyclin A2 small interfering (si) RNA
molecules were synthesized by Shanghai GenePharma Co., Ltd. The
siRNA sequences were as follows: i) si-CSN1-1,
5′-CUGCCGGUUCAGGUGUUATT-3′; ii) si-CSN1-2,
5′-GAACCUUUAACGUGGACAUTT-3′; iii) si-CSN1-3,
5′-GAGACAUCAUCUUCAAAUUTT-3′; iv) si-cyclin A2,
5′-CAACCCACCAGAGACACUAAATT-3′; and v) si-negative control (NC),
5′-UUCUCCGAACGUGUCACGUTT-3′. Human CSN1 overexpression (OE) plasmid
and its corresponding empty vector (pReceiver-M02) were purchased
from GeneCopoeia, Inc. Plasmids (2 µg) or siRNA (50 nM) were
transfected using Lipofectamine® 2000 transfection
reagent (Invitrogen; Thermo Fisher Scientific, Inc.) according to
the manufacturer's instructions. Subsequent cell experiments were
conducted after transfection for 48 h.
The 293T cells were inoculated in 10-cm culture
plates at a density of 5×106 cells in 15 ml and allowed
to reach 70–80% confluence the day prior to infection. The
lentiviral plasmid GV (20 µg), and packaging plasmids pHelper 1.0
(15 µg) and pHelper 2.0 (10 µg) (Shanghai GeneChem Co., Ltd.) were
transfected into 293T cells using Genechem Transfection Reagent
(Shanghai GeneChem Co., Ltd.) for 6 h at 37°C in 5% CO2,
according to the manufacturer's protocol. Following 48–72 h,
supernatants containing lentiviral particles were harvested and
filtered through a 0.45-µm filter (EMD Millipore) to remove cell
debris. The supernatants were concentrated by ultracentrifugation
at 25,000 × rpm at 4°C for 2 h, and the lentiviral particle pellet
was resuspended in 100% FBS and stored at −80°C. The viral titers
of concentrated lentiviral particles were measured by infecting
293T cells seeded at a density of 4×104 cells/well in a
96-well plate with viral serial dilutions. Green fluorescent
protein (GFP) expression was detected 4 days later under a
fluorescence microscope and the viral titer was calculated using
the following equation: Viral titer (Tu/µl)=(% GFP+
cells × number of cells transduced)/virus volume.
For lentiviral transduction, MHCC-LM3 were seeded at
1×105 cells/ml in 24-well plates, and the concentrated
lentivirus was added at MOI=20 at 37°C and 5% CO2. Seven
days after transfection, 1 µg/ml puromycin (cat. no. ST551;
Beyotime Institute of Biotechnology) was added into the cell
culture medium to select the transduced cells.
Western blot analysis
Total protein was extracted from cells using RIPA
lysis buffer (Beyotime Institute of Biotechnology), according to
the manufacturer's protocol. Total protein was quantified using a
BCA Protein Assay kit (cat. no. P0012; Beyotime Institute of
Biotechnology) and equivalent amount of 20 µg protein samples were
separated with a 4–12% Bis-Tris gel (Beyotime Institute of
Biotechnology) and transferred onto a PVDF membrane. The membrane
was blocked with 5% non-fat milk in TBS-Tween-20 for 1 h at room
temperature, incubated with the indicated primary antibodies
overnight at 4°C, and then with horseradish peroxidase-conjugated
secondary antibodies (1:5,000; ProteinTech Group, Inc.) at room
temperature for 1 h. The primary antibodies used were specific for
CSN1 (1:2,000; cat. no. 11709-1-AP; ProteinTech Group, Inc.),
cyclin A2 (1:2,000, cat. no. GTX103042; GeneTex, Inc.), CDK-2
(1:2,000; cat. no. 10122-1-AP; ProteinTech Group, Inc.), CDK-4
(1:2,000; cat. no. 11026-1-AP; ProteinTech Group, Inc.), phospho
(p)-CDK4 (1:2,000, cat. no. GTX00778; GeneTex, Inc.) or GAPDH
(1:5,000; cat. no. 10494-1-AP; ProteinTech Group, Inc.).
HRP-conjugated Affinipure goat anti-mouse IgG (H+L) (1:5,000; cat.
no. SA00001-1; ProteinTech Group, Inc.) and HRP-conjugated
AffiniPure goat anti-rabbit IgG (H+L) (1:5,000; cat. no. SA00001-2;
ProteinTech Group, Inc.) were used as secondary antibodies. Protein
signals were detected by enhanced chemiluminescence (cat. no.
KGP1121; Nanjing KeyGen Biotech Co., Ltd.). Data were
semi-quantified using ImageJ software (v1.48; National institutes
of Health). GAPDH was used as internal control.
Immunohistochemistry for TMA
TMAs were constructed from formalin-fixed,
paraffin-embedded tissue blocks stored at the Department of
Pathology, Daping Hospital. All specimens were previously fixed in
10% neutral formalin solution at room temperature for 12 h before
being paraffin-embedded and cut to 8-µm thick sections. Following
incubation at 65°C for 1 h, TMA slides were dewaxed with
dimethylbenzene and then rehydrated with graded alcohol and
distilled water. For antigen retrieval, TMA slides were incubated
in a microwave oven heated at 95°C with 10 mM citrate buffer (pH
6.0). Endogenous peroxidases were quenched with 3%
H2O2 for 20 min. The slides were treated with
normal goat serum (cat. no. C0265; Beyotime Institute of
Biotechnology) to inhibit non-specific staining at room temperature
for 15 min. Subsequently, the sections were incubated with
anti-CSN1 antibody (1:50; cat. no. 11709-1-AP; ProteinTech Group,
Inc.) overnight at 4°C and then incubated with goat anti-rabbit
antibody (1:500; cat. no. C0265; Beyotime Institute of
Biotechnology) for 1 h. Diaminobenzidine (OriGene Technologies,
Inc.) was used to produce a brown staining. Once hematoxylin
counterstaining and dehydration were completed, the sections were
sealed. Immunohistochemical staining of the TMA was assessed by
light microscopy. Three random fields of view of the IHC chips were
captured. The expression levels of CSN1 were quantified by counting
mean gray value with ImageJ (version 1.48; National Institutes of
Health). A value higher than the mean was defined as high
expression, while a value equal to or lower than the mean was
categorized as low expression in tumors.
Cell proliferation assay
HCC cells were seeded into 96-well plates at a
density of 5×103 cells/well and transfected with the
indicated siRNA or plasmids. At 48 h after transfection, cell
proliferation was determined in vitro using the EdU DNA
Proliferation Detection kit (Nanjing KeyGen Biotech Co., Ltd.)
according to the manufacturer's instructions.
Transwell cell migration and wound
healing assays
Transwell chambers (BD Biosciences) with a pore size
of 8 µm were used to determine cell migration. MHCC-97H or MHCC-LM3
cells (2×104) were seeded in the upper chamber in
serum-free DMEM (HyClone; Cytiva). Medium containing 10% FBS was
added to the lower chamber. Following incubation at 37°C for 24 h,
the cells were fixed in methanol and stained with leucocrystal
violet at room temperature for 10 min. Cells in the upper chamber
were removed, and the number of cells that migrated across the
membrane was determined by counting the number of leucocrystal
violet-stained cells. Stained cells were examined under a light
microscope and quantified by counting all the cells in the field of
view. At least three random microscopic fields were captured.
MHCC-97H or MHCC-LM3 cells (3×105
cells/well) were seeded and transfected as aforementioned in 6-well
plates. The cells were then scratched with a 10-µl pipette tip
along the center of the plate when the cells reached ~95%
confluence. The HCC cells were incubated in serum-free medium. The
distances between the cells bordering the wound were measured and
analyzed under a light microscope at 0, 24 and 48 h.
Cell cycle and cell apoptosis
analysis
For cell cycle and cell apoptosis analysis, MHCC-97H
or MHCC-LM3 cells were seeded at a density of 3×105
cells/well in a 6-well plate and transfected with the indicated
constructs. At 48 h after transfection, the cells were harvested by
centrifugation at 1,000 × g for 5 min and fixed in 70% ethanol
overnight at 4°C. The cells were then analyzed using DNA Content
Quantitation Assay (Cell Cycle) (cat. no. CA1510-50T; Beijing
Solarbio Science & Technology Co., Ltd.) according the
manufacturer's protocol.
The apoptosis rate of HCC cells was analyzed using
an Annexin V-FITC/PI apoptosis detection kit (cat.no. CA1020-50T;
Beijing Solarbio Science & Technology Co., Ltd.) following the
manufacturer's instructions. In brief, 5×105 cells were
harvested by centrifugation at 1,000 × g for 5 min and resuspended
in 200 µl binding buffer, followed by a 15-min incubation with 5 µl
Annexin V-FITC and 5 µl propidium iodide in the dark at 37°C. Data
were acquired using a FACSVerse flow cytometer (BD Biosciences) and
analyzed using FlowJo software (10.0.7; FlowJo LLC). Annexin
V+PI− cells were identified as early
apoptotic cells, whereas Annexin V+PI+ cells
were considered late apoptotic cells.
Reverse transcription-quantitative PCR
(RT-qPCR)
Total RNA was isolated from cells using the TRIpure
total RNA extraction kit (cat. no. RP1001; BioTeke Corporation). RT
was performed using the PrimeScript™ RT Reagent kit, and qPCR was
performed using SYBR Premix Ex Taq II (both from Takara Bio, Inc.)
according to the manufacturer's instructions. RT was carried out at
37°C for 15 min then 85°C for 15 sec. qPCR was performed at 95°C
for 30 sec, followed by 40 cycles of 5 sec at 95°C and 30 sec at
60°C. GAPDH was used as reference gene. The following primer pairs
were used: GAPDH forward, 5′-ACTCCTCCACCTTTGACGC-3′ and reverse,
5′-GCTGTAGCCAAATTCGTTGTC-3′; cyclin A2 forward,
5′-CCAGGAGAATATCAACCCGGA-3′ and reverse, 5′-GTGCAACCCGTCTCGTC-3′
The relative expression levels of each target were quantified using
the 2−ΔΔCq method (32).
Protein half-life assay
To examine the half-life of cyclin A2 protein,
cycloheximide pulse-chase experiments were performed as previously
described (33). MHCC-LM3 cells
were seeded at 3×105 cells/well in 6-well plates.
Twenty-four hours later, the cells were treated with 75 µg/ml
cycloheximide (CHX; Sigma-Aldrich; Merck KGaA) to block the
endogenous protein synthesis for 0, 1, 2, 4, 6 and 8 h, and were
subsequently lysed for western blotting. The bands were scanned,
and the image semi-quantified using ImageJ software (v1.48;
National institutes of Health). The relative concentration at 0 h
was defined as 100%. The fraction of remaining cyclin A2 at each
indicated time point was normalized by comparing the relative
concentration with that at time 0 h.
Protein synthesis inhibition
assay
CHX or actinomycin D were used to block the
endogenous protein synthesis. MHCC-LM3 cells were seeded at
3×105 cells/well in a 6-well plate and transfected with
the indicated constructs. At 48 h after transfection, the cells
were treated with 0.1% DMSO (Beijing Solarbio Science &
Technology Co., Ltd.), 75 µg/ml CHX (Sigma-Aldrich; Merck KGaA) and
50 µg/ml actinomycin D (Selleck, Inc.) respectively for 6 h. The
cells were then lysed for western blotting. The bands were scanned,
and the image semi-quantified using ImageJ software (v1.48;
National institutes of Health).
Xenograft growth assay
BALB/cA-nu nude male mice (4-week-old) were obtained
from the Animal Laboratory of Daping Hospital, Army Medical
University. The mice were maintained under standard animal housing
conditions (24°C, 60% humidity, 12-h light/dark cycle, free access
to food and purified water) and were divided into two groups (n=5
each). A total of 5×106 MHCC-LM3 cells from CSN1
lentivirus-transfected cells and negative control cells were
injected subcutaneously into the mice. At 8 days post-injection,
tumor appearance was examined in the mice. The subcutaneous tumor
size was calculated and recorded every 4 days using a Vernier
caliper. The tumor volume (mm3) was calculated as
[length × (width)2]/2. At 28 days post-cellular
injection, the mice were anesthetized with pentobarbital sodium
(100 mg/kg body weight) and sacrificed by cervical dislocation.
Subsequently, the tumors were excised, and tumor weights were
recorded for analysis. All animal experiments were conducted in
accordance with the Guide for the Care and Use of Laboratory
Animals by the National Institutes of Health. The experimental
study design was approved by the Laboratory Animal Welfare and
Ethics Committee of the Army Medical University.
Statistical analysis
All statistical analyses were performed using SPSS
20.0 (IBM Corp.) and GraphPad Prism 8.0 (GraphPad Software, Inc.).
All results are presented as the mean ± SD of three experiments.
Differences between two groups were analyzed using the unpaired
Student's t-test. Differences among >2 groups were analyzed
using one-way ANOVA followed by Tukey's post hoc test. Wilcoxon
signed-rank test (paired data) and Mann-Whitney's U test (unpaired
data) were used to analyze the difference in CSN1 expression
between HCC and adjacent tissues. Survival analysis was evaluated
using the Kaplan-Meier method and the log-rank test. P<0.05 was
considered to indicate a statistically significant difference.
Results
Elevated CSN1 expression indicates
poor prognosis in patients with HCC
To characterize CSN1 expression in HCC, CSN1
immunohistochemical staining was conducted on a tissue microarray,
which included 117 HCC tissue samples and 58 matched adjacent
paracancerous tissue samples. Higher levels of CSN1 were observed
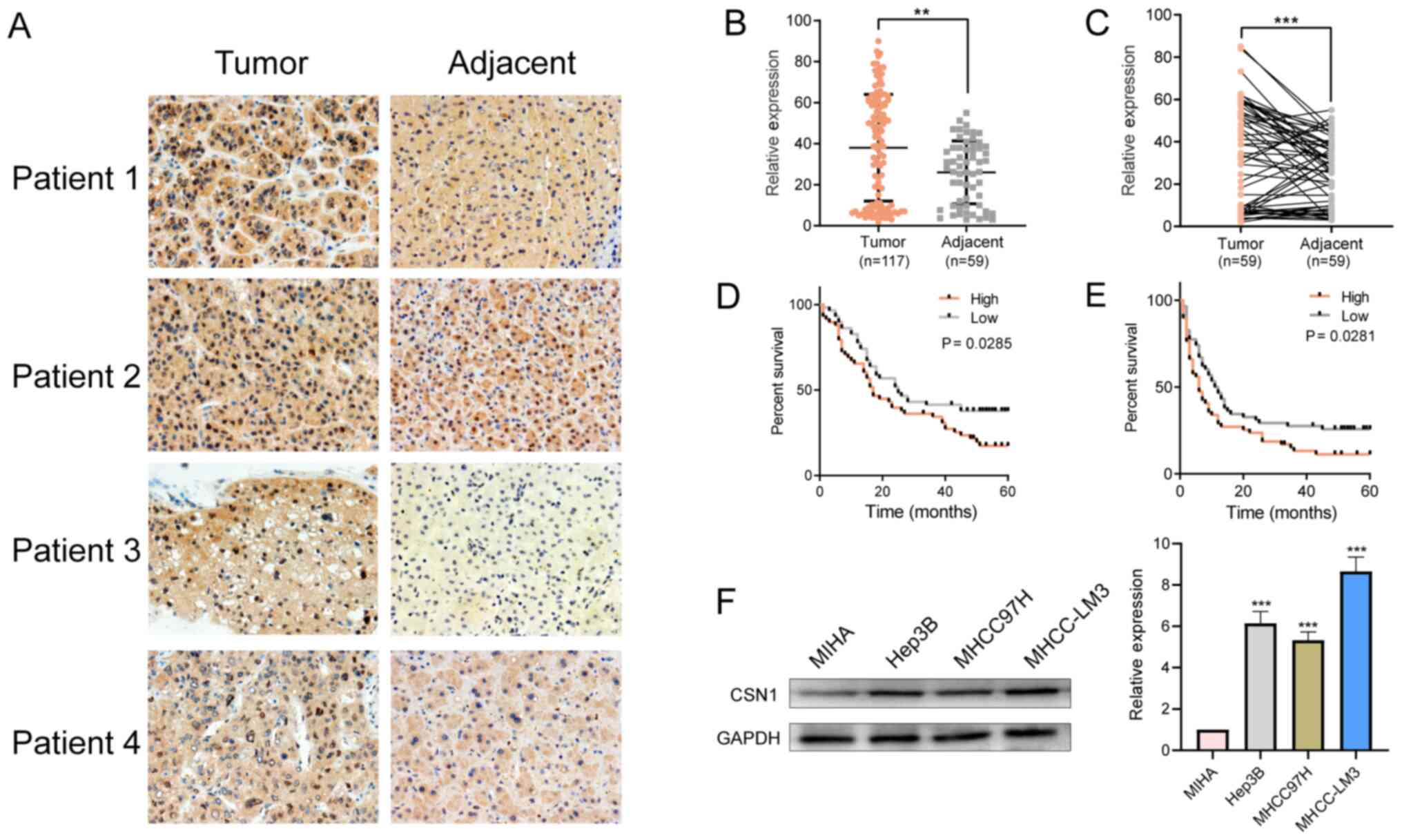
in HCC tissue, compared with the matched adjacent tissues (Fig. 1A-C). Subsequently, the effects of
CSN1 expression on the pathological characteristics and prognosis
of patients with HCC were analyzed. CSN1 expression levels
correlated with tumor size, lymph node metastasis, vascular
invasion, intrahepatic metastasis and tumor stage (Table I). Moreover, the overall survival
(17.38 vs. 37.78%; P=0.0285) and the disease-free survival rates
(11.30 vs. 25.74%; P=0.0281) of HCC patients with high CSN1
expression were significantly lower compared with patients with low
expression (Fig. 1D and E). In
addition, CSN1 expression was significantly higher in the HCC cell
lines MHCC-LM3, Hep3B and MHCC-97H compared with the normal liver
cell line MIHA (Fig. 1F). These
results demonstrated that elevated CSN1 levels were associated with
poor prognosis.
 | Table I.Relationship between the expression
of CSN1 and clinicopathological characteristics of patients with
hepatocellular carcinoma. |
Table I.
Relationship between the expression
of CSN1 and clinicopathological characteristics of patients with
hepatocellular carcinoma.
| Clinicopathological
characteristics | Cases, n (%) | CSN1 expression,
mean ± SEM | t-value | P-value |
|---|
| Sex |
|
Male | 106 (90.6) | 39.12±2.50 | 1.414 | 0.160 |
|
Female | 11 (9.4) | 27.52±8.48 |
|
|
| Age, years |
|
≥50 | 74 (63.3) | 36.71±3.03 | 0.712 | 0.475 |
|
<50 | 43 (36.7) | 40.29±3.98 |
|
|
| HBsAg |
|
Positive | 106 (90.6) | 37.57±2.57 | 0.591 | 0.556 |
|
Negative | 11 (9.4) | 42.45±6.45 |
|
|
| AFP, ng/ml |
|
≥400 | 65 (55.6) | 40.02±3.27 | 0.923 | 0.358 |
|
<400 | 52 (44.4) | 35.54±3.55 |
|
|
| Tumor size, cm |
| ≥5 | 74 (63.2) | 42.42±2.96 | 2.448 | 0.016 |
|
<5 | 43 (36.8) | 30.47±3.90 |
|
|
|
Differentiation |
|
High/moderate | 93 (79.5) | 37.44±2.71 | 0.476 | 0.635 |
|
Low | 24 (20.5) | 40.29±5.31 |
|
|
| TNM
stagea |
|
I/II | 64 (54.7) | 31.21±3.33 | 3.241 | 0.002 |
|
III/IV | 53 (45.3) | 46.26±3.15 |
|
|
| BCLC
stageb |
|
A/B | 63 (53.8) | 30.83±3.36 | 3.373 | 0.001 |
|
C/D | 54 (46.2) | 46.43±3.10 |
|
|
| Vascular
invasion |
|
Yes | 38 (32.5) | 46.95±3.60 | 2.636 | 0.010 |
| No | 79 (67.5) | 33.74±3.01 |
|
|
| Intrahepatic
metastasis |
|
Yes | 40 (34.2) | 46.25±3.86 | 2.519 | 0.013 |
| No | 77 (65.8) | 33.76±2.96 |
|
|
| Lymph node
metastasis |
|
Yes | 12 (10.3) | 54.00±7.96 | 2.285 | 0.024 |
| No | 105 (89.7) | 36.20±2.47 |
|
|
CSN1 knockdown inhibits HCC cell
proliferation, migration and invasion
To investigate the role of CSN1 in HCC cells, three
siRNA candidates were designed and transfected into MHCC-LM3, Hep3B
and MHCC-97H cells, and the most efficient siRNA was screened for
using western blotting (Fig. 2A).
Subsequently, functional analyses were conducted in MHCC-LM3 and
Hep3B cells following CSN1 siRNA knockdown. CSN1 knockdown
significantly reduced cell proliferation, compared with NC
(Fig. 2B). Moreover, cell migration
also significantly decreased after CSN1 knockdown (Fig. 2C and D). Lastly, CSN1 knockdown
increased the apoptosis rate (Fig.
2E) and affected cell cycle distribution. The proportion of
cells in the G0/G1 phase significantly
increased, while the proportion of cells in S and G2/M
phases significantly decreased after CSN1 knockdown (Fig. 2F). In conclusion, CSN1 knockdown
inhibited HCC cell proliferation, migration and invasion.
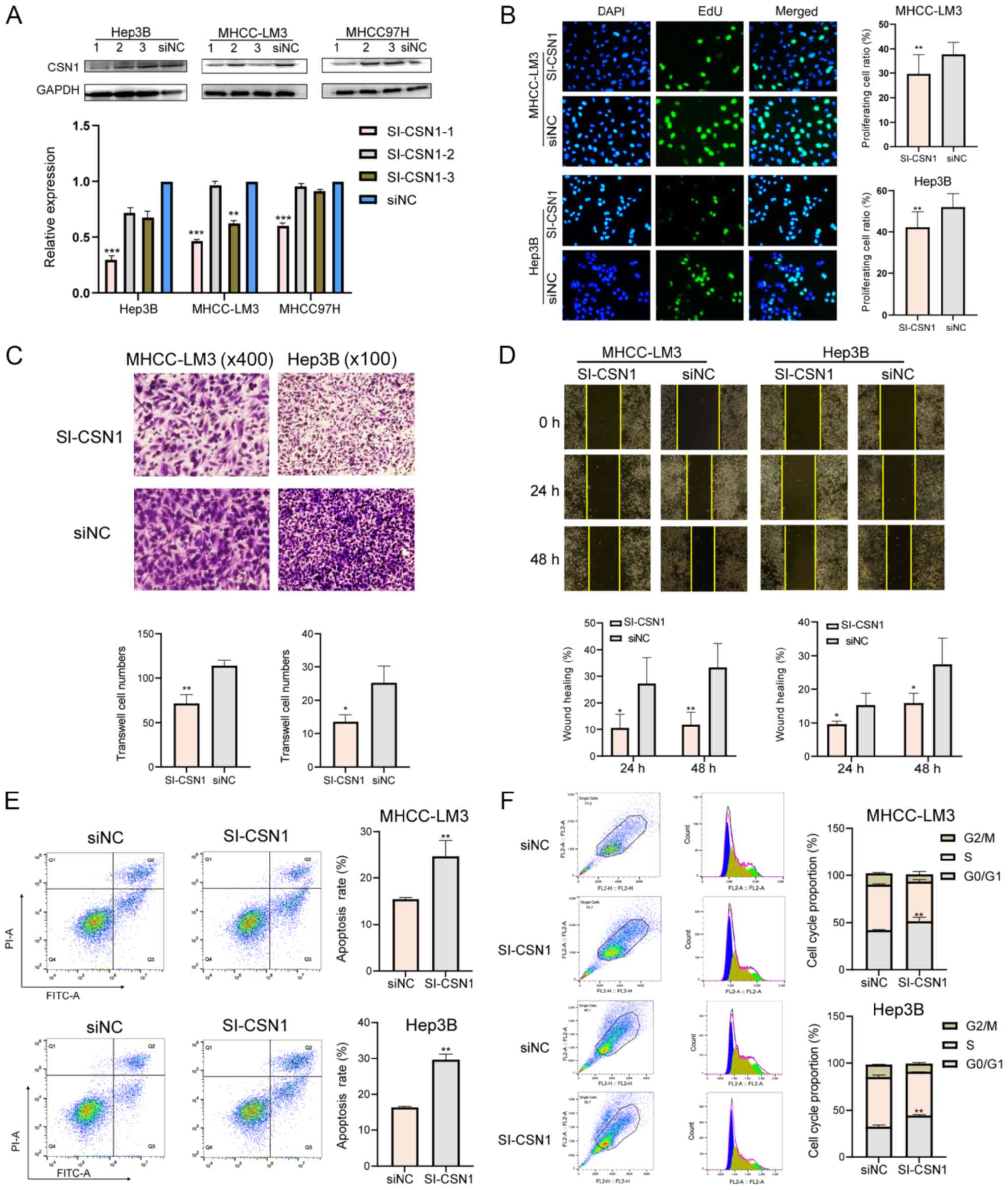 | Figure 2.Effects of CSN1 on hepatocellular
carcinoma cell proliferation, cell cycle and apoptosis. (A) CSN1
expression levels in MHCC-LM3, Hep3B and MHCC-97H cells transfected
with si-CSN1-1, −2 and −3 or siNC. (B) Proliferation of MHCC-LM3
and Hep3B cells following CSN1 knockdown. Magnification, ×200. (C)
Representative images (upper panel) and quantification (lower
panel) of the Transwell assays for MHCC-LM3 and Hep3B cells
transfected with si-CSN1 or si-NC. Magnifications, ×100 and ×400.
(D) Measurement of the migration rate after CSN1 knockdown in
MHCC-LM3 and Hep3B cells. Magnification, ×100. (E) Apoptosis and
(F) cell cycle measurement following CSN1 silencing in MHCC-LM3 and
Hep3B cells. *P<0.05, **P<0.01 and ***P<0.001. CSN1,
constitutive photomorphogenesis 9 signalosome subunit 1; si, small
interfering RNA; NC, negative control. |
CSN1 affects HCC cell proliferation
and migration via regulating cyclin A2 expression
Cyclin A2 is reported to regulate tumor growth and
metastasis (34,35). To investigate the mechanisms by
which CSN1 affects HCC proliferation, the expression levels of
several cell cycle-related proteins were assessed following CSN1
overexpression. Cyclin A2 and p-CDK4 expressions were upregulated
by CSN1 overexpression (Fig.
3A).
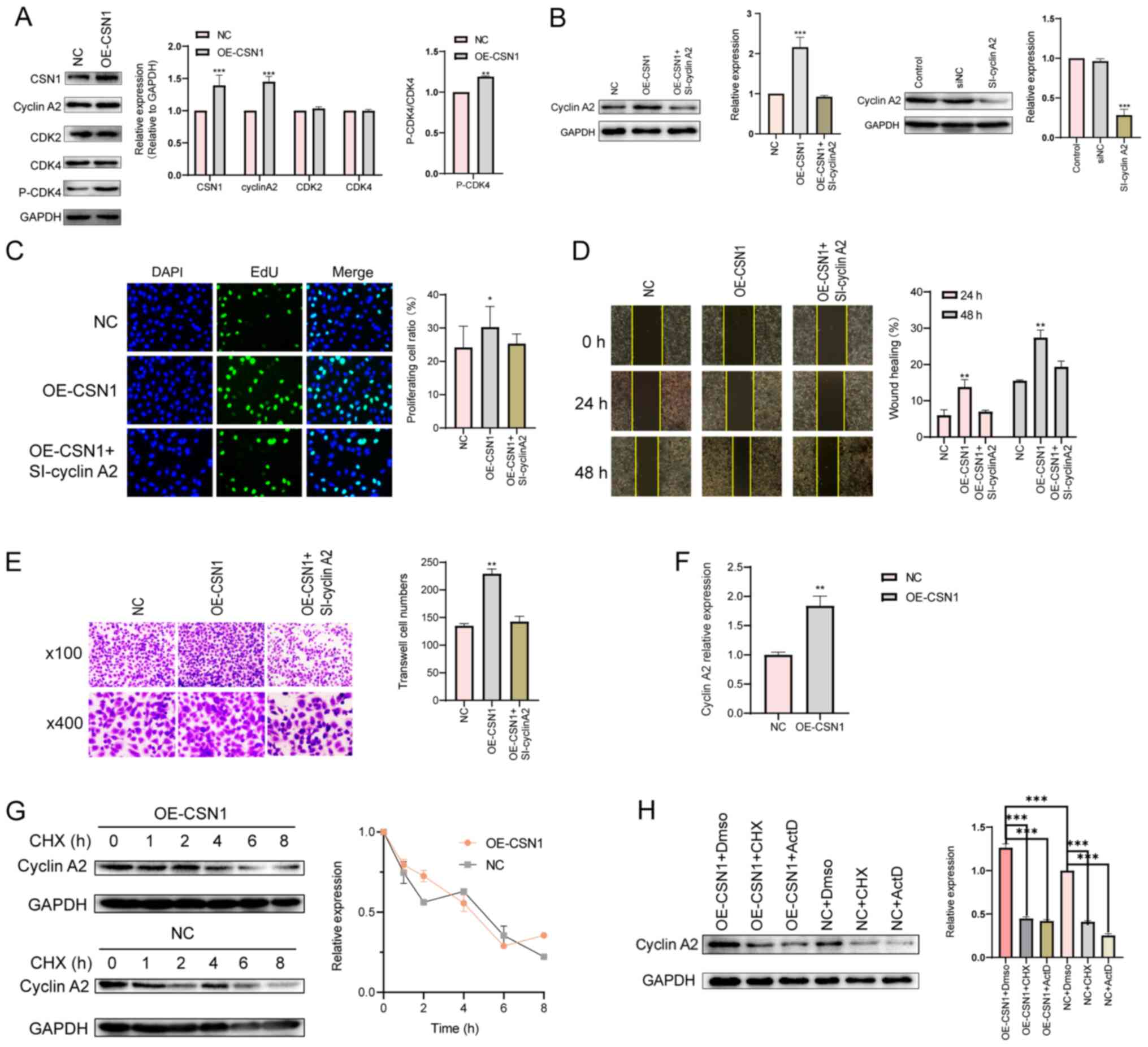 | Figure 3.CSN1 interacts with cyclin A2. (A)
Representative western blots of CSN1, cyclin A2 and cell
cycle-related proteins following CSN1 overexpression in MHCC-LM3
cells. Cells transfected with empty vectors were used as NC group.
(B) Western blots of cyclin A2 protein levels in MHCC-LM3 cells
transfected with NC or OE-CSN1 plasmids, with or without si-cyclin
A2. (C) Proliferation (magnification, ×200), (D) wound healing
(magnification, ×100) (E) and Transwell assay (magnification, ×100
and ×400) of MHCC-LM3 cells transfected with NC or OE-CSN1
plasmids, with or without si-cyclin A2. (F) Effect of OE-CSN1 on
cyclin A2 mRNA levels. (G) Representative western blots of cyclin
A2 turnover rate in OE-CSN1 MHCC-LM3 cells following CXH treatment.
(H) Cyclin A2 protein levels after OE-CSN1 transfection, and CHX or
ActD treatment in MHCC-LM3 cells. *P<0.05, **P<0.01 and
***P<0.001. CHX, cycloheximide; CSN1, constitutive
photomorphogenesis 9 signalosome subunit 1; OE, overexpression;
ActD, actinomycin D; CDK, cyclin-dependent kinase; si, small
interfering; NC, negative control. |
Moreover, co-transfection of si-cyclin A2 and
OE-CSN1 were carried out. The efficiency of cyclin A2 siRNA
transfections was confirmed by western blotting (Fig. 3B). Co-transfection of si-cyclin A2
with OE-CSN1 significantly reduced the effects of OE-CSN1 on cyclin
A2 levels, proliferation and migration, compared with cells
transduced with OE-CSN1 alone. Thus, cyclin A2 may be a downstream
regulator of CSN1 (Fig. 3B-E).
Since CSN subunits may inhibit the ubiquitination
and degradation of several protein substrates (22), the effect of CSN1 on cyclin A2
degradation was evaluated. Cyclin A2 mRNA levels were elevated by
CSN1 overexpression (Fig. 3F), and
the half-life of cyclin A2 protein was not affected by CSN1
overexpression (Fig. 3G). Moreover,
inhibition of mRNA transcription or translation by actinomycin D
and cycloheximide, respectively, reduced cyclin A2 protein levels
in CSN1-overexpressing cells (Fig.
3H), indicating that CSN1 may regulate cyclin A2 expression at
the translational level or in an indirect manner. In conclusion,
these results indicated that CSN1 promoted HCC cell proliferation
and migration by upregulating cyclin A2 expression.
CSN1 facilitates HCC growth in
vivo
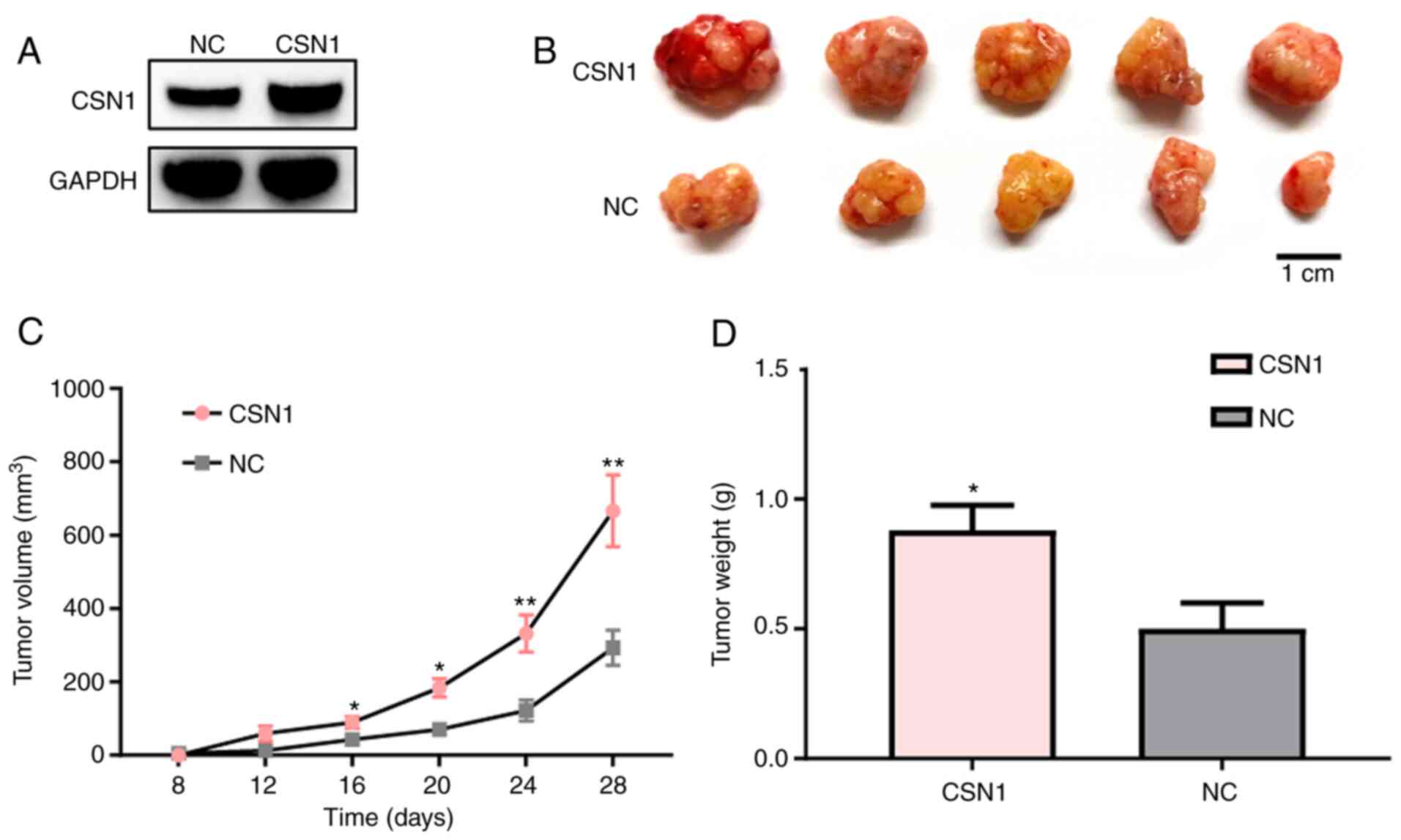
To further determine the effects of CSN1 on the
tumor growth of HCC cells in vivo, a xenograft tumor assay
was performed with stably infected OE-CSN1 and NC MHCC-LM3 cells
(Fig. 4A). CSN1 overexpression
significantly increased the tumor volumes and weights of the
MHCC-LM3 cells compared with the negative control group (Fig. 4B). Based on the tumor volumes, CSN1
overexpression was associated with higher tumor growth rate and
tumor weights (Fig. 4C and D).
These results suggested that CSN1 overexpression can facilitate
tumor growth in vivo.
Discussion
Hepatocellular carcinoma (HCC) is the most common
primary malignant tumor of the liver, accounting for ~85% of all
cases (36). The high mortality
rate of liver cancer is mainly associated with tumor invasion,
metastasis and tumor recurrence after surgical resection (6,37). In
recent years, a number of studies have confirmed that the CSN
superfamily is widely involved in the regulation of several
important intracellular pathways, including cell proliferation,
migration, invasion and signal transduction, and plays an important
role in tumor development (13,38–40).
For example, CSN5 and CSN6 are highly expressed in myeloma, lung,
colon, breast cancer, malignant glioma and leukemia clinical
tissues (14). However, the
expression pattern and role of CSN1 in HCC have yet to be
determined.
The present study reported that CSN1 expression
levels were increased in HCC tissues and HCC cells compared to
normal controls. Of note, because of the tumor heterogeneity, 30.5%
of the HCC tissues displayed a decrease or no difference in CSN1
expression compared to adjacent tissues. High CSN1 levels indicated
poor prognosis in patients. In addition, in vitro assays
indicated that CSN1 knockdown inhibited the proliferation and
migration of HCC cells, suggesting that CSN1 may promote the
occurrence and development of HCC by enhancing proliferation and
inhibiting apoptosis. Moreover, the xenograft growth assay revealed
that CSN1 facilitated HCC growth in vivo. Mechanically, CSN1
can affect the expression of cyclin A2 and thus participate in the
regulation of the cell cycle, cell proliferation and migration.
Cyclin A2 is a key regulator of cell cycle transition and can
regulate cell growth through its pro-mitotic effects. Cyclin A2
modulates epithelial-mesenchymal transition via β-catenin and
phospholipase C pathways (41). The
present study supported the notion that CSN1 played a role in
carcinogenesis and cancer progression.
The role of the CSN complex in ubiquitin-mediated
protein degradation is well-characterized. Several CSN subunits
exert de-ubiquitination activity. For example, CSN5 promoted the
proliferation of non-small cell lung cancer cells by inhibiting the
ubiquitination-mediated degradation of survivin (17). Du et al (42) demonstrated that CSN6 promoted the
occurrence of gastric cancer via the ubiquitin-independent
proteasomal degradation of p16INK4a. In addition, CSN is also
involved in ubiquitin-like modifications. For example, the CSN
complex de-neddylates cullins and destabilizes E3 ligase complexes
(43). De-neddylation by the CSN
complex inactivates a cullin1 complex that ubiquitinates capicua
following its phosphorylation by MAP kinase in response to EGFR
signaling (44). The
CUL4A-RBX1-DDB1-DDB2 complex (CRL4ADDB2) is inactive in
the absence of damaged DNA and requires CSN to regulate the repair
process (45).
However, the present results indicated that although
CSN1 can increase the expression of cyclin A2 mRNA, the regulatory
effects of CSN1 on cyclin A2 may not occur at the
post-translational level, since the protein half-time of cyclin A2
was not affected by CSN1. There are several possible reasons for
this unexpected result. First, CSN1 may not interact with cyclin A2
directly, and there may be other mediators involved in the
CSN1-cyclin A2 axis. Secondly, cyclin A2 may not be a direct
substrate in CSN-mediated ubiquitination. Moreover, CSN1 might not
be the subunit in the CSN complex that mediates the ubiquitination
of its substrates. It has been suggested that the MPN domain in
CSN6 and CSN5 may be responsible for regulating ubiquitination and
cullin de-neddylation (14,46,47).
Since CSN1 lacks an MPN domain, there is still no evidence that
CSN1 is directly involved in ubiquitination.
There are several limitations in the present study.
The exact molecular mechanism of CSN1 in HCC tumorigenesis needs
further investigation, especially regarding how CSN1 affects cyclin
A2 to regulate HCC cell proliferation and migration. Moreover, how
CSN1 and other CSN components interplay to regulate HCC
tumorigenesis should also be evaluated in the future.
Taken together, the present findings indicated that
high expression levels of CSN1 may contribute to HCC progression.
CSN1 knockdown inhibited HCC cell proliferation and induced
apoptosis and migration by affecting cyclin A2 expression. The
present study determined the function of CSN1 and may provide new
insights and therapeutic strategies for HCC prevention and
treatment.
Acknowledgements
The authors would like to thank the Hepatobiliary
Surgery laboratory of Xinan Hospital, Army Medical University for
providing laboratory resources.
Funding
The present study was supported by the National
Natural Science Foundation of China (grant no. 81270523).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
PC and JC designed the experiments. HF and YZ
performed most of the experiments and drafted the manuscript. YC
conducted the experiments on the clinical samples. All authors read
and approved the final manuscript.
Ethics approval and consent to
participate
Informed consent was obtained from all participants.
Experiments involving clinical samples conformed to the principles
of the Declaration of Helsinki and were approved by The Ethics
Committee of Daping Hospital, Army Medical University. All
procedures involving mice and the corresponding experimental
protocols were approved by The Laboratory Animal Welfare and Ethics
Committee of the Army Medical University.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
AFP
|
α-fetoprotein
|
|
CSN1
|
constitutive photo morphogenesis 9
signalosome subunit 1
|
|
HCC
|
hepatocellular carcinoma
|
|
BCLC
|
Barcelona Clinic Liver Cancer
|
|
HBsAg
|
hepatitis B surface antigen
|
References
|
1
|
Bray F, Ferlay J, Soerjomataram I, Siegel
RL, Torre LA and Jemal A: Global cancer statistics 2018: GLOBOCAN
estimates of incidence and mortality worldwide for 36 cancers in
185 countries. CA Cancer J Clin. 68:394–424. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Chen W, Zheng R, Baade PD, Zhang S, Zeng
H, Bray F, Jemal A, Yu XQ and He J: Cancer statistics in China,
2015. CA Cancer J Clin. 66:115–132. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Juarez-Hernandez E, Motola-Kuba D,
Chavez-Tapia NC, Uribe M and Barbero Becerra V: Biomarkers in
hepatocellular carcinoma: An overview. Expert Rev Gastroenterol
Hepatol. 11:549–558. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Yan X, Shao C, Chen C, Chen J, Gu S, Huang
L, Fu X, Zhao H and Qiu Y: Mutation detection of fibroblast growth
factor receptor 3 for infiltrative hepatocellular carcinoma by
whole-exome sequencing. Dig Dis Sci. 62:407–417. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Zhu Q, Li N, Zeng X, Han Q, Li F, Yang C,
Lv Y, Zhou Z and Liu Z: Hepatocellular carcinoma in a large medical
center of China over a 10-year period: Evolving therapeutic option
and improving survival. Oncotarget. 6:4440–4450. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kumari R, Sahu MK, Tripathy A, Uthansingh
K and Behera M: Hepatocellular carcinoma treatment: Hurdles,
advances and prospects. Hepatic Oncol. 5:Hep082018. View Article : Google Scholar
|
|
7
|
Heinrich B, Czauderna C and Marquardt JU:
Immunotherapy of hepatocellular carcinoma. Oncol Res Treat.
41:292–297. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Chamovitz DA, Wei N, Osterlund MT, von
Arnim AG, Staub JM, Matsui M and Deng XW: The COP9 complex, a novel
multisubunit nuclear regulator involved in light control of a plant
developmental switch. Cell. 86:115–121. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wei N and Deng XW: COP9: A new genetic
locus involved in light-regulated development and gene expression
in arabidopsis. Plant Cell. 4:1507–1518. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wei N and Deng XW: Making sense of the
COP9 signalosome. A regulatory protein complex conserved from
Arabidopsis to human. Trends Genet. 15:98–103. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wang H, Ma LG, Li JM, Zhao HY and Deng XW:
Direct interaction of Arabidopsis cryptochromes with COP1 in light
control development. Science. 294:154–158. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Schwechheimer C and Deng XW: The
COP/DET/FUS proteins-regulators of eukaryotic growth and
development. Semin Cell Dev Biol. 11:495–503. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Richardson KS and Zundel W: The emerging
role of the COP9 signalosome in cancer. Mol Cancer Res. 3:645–653.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lee MH, Zhao R, Phan L and Yeung SC: Roles
of COP9 signalosome in cancer. Cell Cycle. 10:3057–3066. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wei N and Deng XW: The COP9 Signalosome.
Annu Rev Cell Dev Biol. 19:261–286. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Gusmaroli G, Figueroa P, Serino G and Deng
XW: Role of the MPN subunits in COP9 signalosome assembly and
activity, and their regulatory interaction with Arabidopsis
Cullin3-based E3 ligases. Plant Cell. 19:564–581. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Li J, Li Y, Wang B, Ma Y and Chen P:
CSN5/Jab1 facilitates non-small cell lung cancer cell growth
through stabilizing survivin. Biochem Biophys Res Commun.
500:132–138. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Liu H, Hu J, Pan H, Luo D, Huang M and Xu
W: CSN5 promotes hepatocellular carcinoma progression by SCARA5
inhibition through suppressing β-catenin ubiquitination. Dig Dis
Sci. 63:155–165. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Mao Z, Sang MM, Chen C, Zhu WT, Gong YS
and Pei DS: CSN6 promotes the migration and invasion of cervical
cancer cells by inhibiting autophagic degradation of cathepsin L.
Int J Biol Sci. 15:1310–1324. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Shi J, Guan X, Zhan F, Liu C, Li Z, Yao Y,
Wang B, Lou C and Zhang Y: CSN6 expression is associated with
pancreatic cancer progression and predicts poor prognosis. Cancer
Biol Ther. 20:1290–1299. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Suisse A, Bekes M, Huang TT and Treisman
JE: The COP9 signalosome inhibits Cullin-RING E3 ubiquitin ligases
independently of its deneddylase activity. Fly (Austin).
12:118–126. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Schwechheimer C and Deng XW: COP9
signalosome revisited: A novel mediator of protein degradation.
Trends Cell Biol. 11:420–426. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Bech-Otschir D, Seeger M and Dubiel W: The
COP9 signalosome: At the interface between signal transduction and
ubiquitin-dependent proteolysis. J Cell Sci. 115:467–473.
2002.PubMed/NCBI
|
|
24
|
Rhodes DR, Kalyana-Sundaram S, Mahavisno
V, Varambally R, Yu J, Briggs BB, Barrette TR, Anstet MJ,
Kincead-Beal C, Kulkarni P, et al: Oncomine 3.0: Genes, pathways,
and networks in a collection of 18,000 cancer gene expression
profiles. Neoplasia. 9:166–180. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Rhodes DR, Yu J, Shanker K, Deshpande N,
Varambally R, Ghosh D, Barrette T, Pandey A and Chinnaiyan AM:
ONCOMINE: A cancer microarray database and integrated data-mining
platform. Neoplasia. 6:1–6. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Xu M, Zhen L, Lin L, Wu K, Wang Y and Cai
X: Overexpression of CSN6 promotes the epithelial-mesenchymal
transition and predicts poor prognosis in hepatocellular carcinoma.
Clin Res Hepatol Gastroenterol. 44:340–348. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Yu YS, Tang ZH, Pan QC, Chen XH, Liu XN
and Zang GQ: Inhibition of Csn3 expression induces growth arrest
and apoptosis of hepatocellular carcinoma cells. Cancer Chemother
Pharmacol. 69:1173–1180. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Deng H, Cheng Y, Guo Z, Zhang F, Lu X,
Feng L, Wang X and Xu Z: Overexpression of CyclinA2 ameliorates
hypoxia-impaired proliferation of cardiomyocytes. Exp Ther Med.
8:1513–1517. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Gopinathan L, Tan SL, Padmakumar VC,
Coppola V, Tessarollo L and Kaldis P: Loss of Cdk2 and cyclin A2
impairs cell proliferation and tumorigenesis. Cancer Res.
74:3870–3879. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Yasmeen A, Berdel WE, Serve H and
Müller-Tidow C: E- and A-type cyclins as markers for cancer
diagnosis and prognosis. Expert Rev Mol Diagn. 3:617–633. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Ohashi R, Gao C, Miyazaki M, Hamazaki K,
Tsuji T, Inoue Y, Uemura T, Hirai R, Shimizu N and Namba M:
Enhanced expression of cyclin E and cyclin A in human
hepatocellular carcinomas. Anticancer Res. 21:657–662.
2001.PubMed/NCBI
|
|
32
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Fu H, Zhang Y, Chen J, Zhou B, Chen G and
Chen P: Tmub1 suppresses hepatocellular carcinoma by promoting the
Ubiquitination of ΔNp63 isoforms. Mol Ther Oncolytics. 18:126–136.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Blanchard JM: Cyclin A2 transcriptional
regulation: Modulation of cell cycle control at the G1/S transition
by peripheral cues. Biochem Pharmacol. 60:1179–1184. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Bendris N, Cheung CT, Leong HS, Lewis JD,
Chambers AF, Blanchard JM and Lemmers B: Cyclin A2, a novel
regulator of EMT. Cell Mol Life Sci. 71:4881–4894. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Neureiter D, Stintzing S, Kiesslich T and
Ocker M: Hepatocellular carcinoma: Therapeutic advances in
signaling, epigenetic and immune targets. World J Gastroenterol.
25:3136–3150. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Chen CP: Role of radiotherapy in the
treatment of hepatocellular carcinoma. J Clin Transl Hepatol.
7:183–190. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Wicker CA and Izumi T: Analysis of RNA
expression of normal and cancer tissues reveals high correlation of
COP9 gene expression with respiratory chain complex components. BMC
Genomics. 17:9832016. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Wang L, Zheng JN and Pei DS: The emerging
roles of Jab1/CSN5 in cancer. Med Oncol. 33:902016. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Mao Z, Chen C and Pei DS: The emerging
role of CSN6 in biological behavior and cancer progress. Anticancer
Agents Med Chem. 19:1198–1204. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Cheung CT, Bendris N, Paul C, Hamieh A,
Anouar Y, Hahne M, Blanchard JM and Lemmers B: Cyclin A2 modulates
EMT via β-catenin and phospholipase C pathways. Carcinogenesis.
36:914–924. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Du W, Liu Z, Zhu W, Li T, Zhu Z, Wei L,
Song J and Pei D: CSN6 promotes tumorigenesis of gastric cancer by
ubiquitin-independent proteasomal degradation of
p16INK4a. Cancer Biol Med. 16:514–529. 2019.PubMed/NCBI
|
|
43
|
Schwechheimer C: NEDD8-its role in the
regulation of Cullin-RING ligases. Curr Opin Plant Biol.
45:112–119. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Su H, Huang W and Wang X: The COP9
signalosome negatively regulates proteasome proteolytic function
and is essential to transcription. Int J Biochem Cell Biol.
41:615–624. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Cavadini S, Fischer ES, Bunker RD, Potenza
A, Lingaraju GM, Goldie KN, Mohamed WI, Faty M, Petzold G, Beckwith
RE, et al: Cullin-RING ubiquitin E3 ligase regulation by the COP9
signalosome. Nature. 531:598–603. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Lim SO, Li CW, Xia W, Cha JH, Chan LC, Wu
Y, Chang SS, Lin WC, Hsu JM, Hsu YH, et al: Deubiquitination and
stabilization of PD-L1 by CSN5. Cancer Cell. 30:925–939. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Hou J, Deng Q, Zhou J, Zou J, Zhang Y, Tan
P, Zhang W and Cui H: CSN6 controls the proliferation and
metastasis of glioblastoma by CHIP-mediated degradation of EGFR.
Oncogene. 36:1134–1144. 2017. View Article : Google Scholar : PubMed/NCBI
|