Introduction
Alzheimer's disease (AD) is characterised by
behavioural impairment and cognitive dysfunction (1) and is associated with age, where the
prevalence may be as high as 50% among humans aged >95 years
(2). The pathological cause of AD
has not been fully elucidated. Previous research has indicated that
the accumulation of β-amyloid peptide (Aβ) in the brain may be an
important contributor to the development of AD (3). In patients with AD, Aβ aggregates that
form around neurons not only induce neuronal apoptosis but also
trigger a cascade of cellular damage, including τ
hyperphosphorylation and mitochondrial reactive oxygen species
(ROS) production (4). Previous
reports have indicated that symptoms of AD are attributable to the
occurrence of oxidative stress (2,5,6). In
particular, oxidative stress induces mitochondrial dysfunction and
excessive ROS accumulation, ultimately leading to neuronal cell
death (7).
Mitochondria are the primary source of intracellular
ROS and are key targets of Aβ toxicity during the AD pathological
process (8). In addition to
increasing the levels of Aβ in the brain, overaccumulation of ROS
and subsequent collapse of mitochondrial membrane potential (MMP)
lead to reductions in ATP levels (9). Therefore, the function and integrity
of the mitochondria must be maintained to protect nerve cells.
During the occurrence and development of oxidative stress, the
transcription factor nuclear factor E2-related factor 2 (Nrf2)
dissociates from Kelch-like ECH-associated protein 1 (Keap-1) to
activate numerous downstream proteins and detoxification enzymes,
including superoxide dismutase 1 (SOD 1) and haemeoxygenase-1
(HO-1) (10,11). Since activation of the Nrf2 pathway
has been documented to ameliorate oxidative stress and decreases
accumulation of Aβ (12,13), rendering Nrf2 to be a potential
therapeutic target for AD (14).
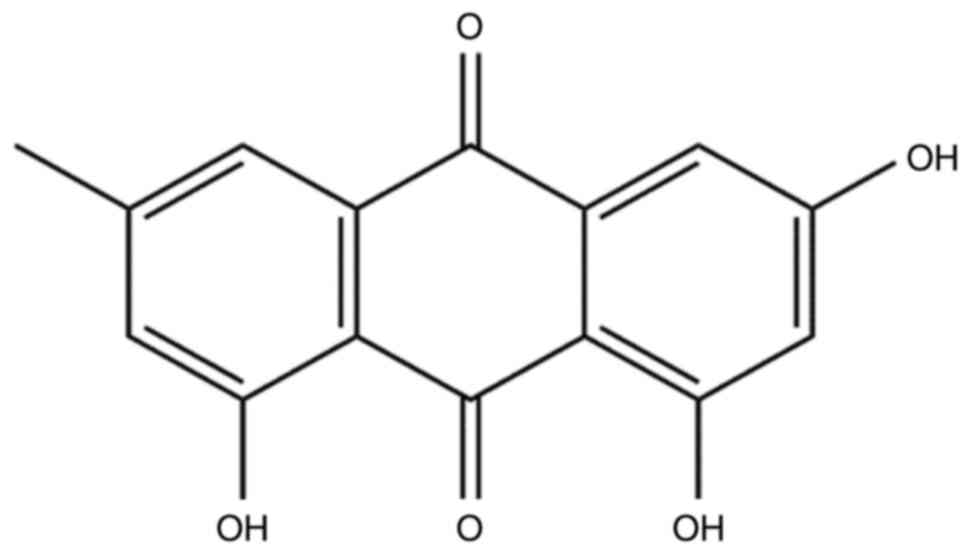
Emodin, which is structurally known as
1,3,8-trihydroxy-6-methylanthraquinone (Fig. 1), is a naturally occurring
anthraquinone derivative. This compound can be isolated from
medicinal herbs, including Rheum palmatum (15), Cassia obtusifolia (16) and Polygonum multiflorum
(17), and has been used as a
laxative in eastern Asia for 2,000 years (18). Previous studies have indicated that
emodin is multifunctional. For example, evidence suggests that
emodin enhances nerve cell survival by upregulating Bcl-2
expression levels and blocking Aβ-induced autophagy (19). In addition, emodin exhibits
antioxidative effects: In viral myocarditis, emodin alleviates
oxidative stress by increasing myocardial SOD expression levels
whilst decreasing malondialdehyde expression levels (20). Emodin has also been shown to
decrease collagen overproduction and inflammation by stimulating
the Nrf2-antioxidant signalling pathway in pulmonary fibrosis
(21), and to serve important roles
in neurological disorders (22).
For example, emodin protects neurons from Aβ25-35-induced
neurotoxicity and inhibits excess τ accumulation in cortical
neurons (19,23). To the best of our knowledge,
however, no reports have yet described the systematic protective
effects of emodin against AD or Aβ deposition in either in
vitro or in vivo mouse models.
In the present study, amyloid precursor protein
(APP)/presenilin 1 (PS1) double-transgenic mice and U251 cells
subjected to Aβ1-42-induced apoptosis were used to determine the
neuroprotective effects of emodin against AD. Emodin exerted
protective effects against Aβ toxicity in U251 cells and
ameliorated behavioural effects in the double-transgenic mouse
model. The experimental data indicated that emodin protected
neurons against neurodegeneration in AD.
Materials and methods
Cell culture
U251 cells (cat. no. KCB200965Y), a human glioma
cell line that was purchased from the Cell bank of Chinese Academy
of Sciences, were cultured in high glucose DMEM containing 10%
fetal bovine serum, 1% 100 U/ml penicillin and 100 µg/ml
streptomycin at 37°C in a 5% CO2 incubator (Thermo
Fisher Scientific, Inc.) to provide a humidified atmosphere. All
reagents were purchased from Invitrogen, Thermo Fisher Scientific,
Inc.
Measurement of cell viability
U251 cells (5×103 cells/well) cultured in
plates (96-well) were pre-treated with emodin (purity, ≥90%; CAS
no. 518-82-1; cat. no. E7881; Sigma-Aldrich; Merck KGaA) at doses
of 25 and 50 µM at 37°C for 3 h and then co-incubated with 15 µM
Aβ1-42 [cat. no. 87233; Gill Biochem (Shanghai) Co., Ltd.] or
culture medium at 37°C for a further 24 h. Non-treated cells served
as the control. Cell viability was analysed using MTT assay as
described previously (24).
Briefly, cells were exposed to 5 mg/ml (final concentration) MTT at
37°C for 4 h in the dark before the formazan precipitates were
dissolved in 100 µl DMSO (Sigma-Aldrich; Merck KGaA). Absorbance
was analysed using a Synergy™ 4 Microplate Reader at 490 nm (BioTek
Instruments, Inc.).
Measurement of lactate dehydrogenase
(LDH) levels and activity of caspases-3, −8 and −9
U251 cells (2×105 cells/well) were
cultured in plates (6-well) and treated with emodin at doses of 25
and 50 µM at 37°C for 3 h, then co-incubated with 15 µM Aβ1-42 or
culture medium at 37°C for another 24 h. Culture
medium-only-treated U251 cells served as the control group. The
intracellular levels of LDH and the activities of caspase-3, −8 and
−9 were detected using LDH (cat. no. C0016), caspase-3 (cat. no.
C1168M), −8 (cat. no. C1151) and −9 (cat. no. C1157) Activity Assay
kits (Beyotime Institute of Biotechnology) according to
manufacturer's protocols.
Flow cytometry assay
U251 cells (2×105 cells/well) were
cultured in 6-well plates with emodin at doses of 25 and 50 µM at
37°C for 3 h before being co-incubated with 15 µM Aβ1-42 or culture
medium for a further 24 h. Non-treated cells served as the control.
Cells (1×105) were collected and incubated with Annexin
V and propidium iodide (cat. no. APT750; EMD Millipore) for 15 min
at 37°C in the dark, and detected using a Muse Cell Analyzer (EMD
Millipore) and analyzed with FlowJo v10 (FlowJo LLC).
Measurement of MMP and ROS levels
U251 cells (2×105 cells/well) cultured in
plates (6-well) with emodin at doses of 25 and 50 µM at 37°C for 3
h and then co-incubated with 15 µM Aβ1-42 or culture medium at 37°C
for a further 24 h. Culture medium-only-treated U251 cells served
as the control group. MMP was analysed using a MMP Detection kit
(cat. no. M8650; Beijing Solarbio Science & Technology Co.,
Ltd.) according to the manufacturer's instructions. The red
fluorescence represented the higher membrane potential, and the
green fluorescence represented the damaged or lower membrane
potential. Intracellular ROS was analysed using a Reactive Oxygen
Detection kit (cat. no. S0033; Beyotime Institute of Biotechnology)
according to the manufacturer's instructions. After the cells were
washed, the fluorescence intensities were detected using a
fluorescence microscope (magnification, ×400; Eclipse TE 2000-S;
Nikon Corporation). Quantitative data analysis was performed using
ImageJ software version 1.46 (National Institutes of Health), where
the data were presented as the fluorescence intensity.
Animals and treatment
For the animal model, 8-month old male (42–49 g)
B6C3-Tg (genotype APPswe, PSEN1De9/Nju) APP/PS1 double-transgenic
mice (n=30) and wild-type (WT) littermates (n=10) were purchased
from Nanjing Biomedical Research Institute of Nanjing University
[SCXK (Su) 2015-0001; Nanjing, China]. All mice were housed in a
temperature-controlled environment with a standard 12-h light/dark
cycle at 23±1°C and 40–60% humidity, with access to food and water
ad libitum.
The present study was approved by the Animal Ethics
Committee of School of Life Sciences, Jilin University (approval
no. SY20171208; Changchun, China). APP/PS1 mice were randomly
divided into model (n=10; orally received 10 ml/Kg normal saline)
and emodin-treated groups [orally received 10 (n=10) or 20 mg/kg
(n=10) emodin once per day for 8 continuous weeks]. WT mice (n=10)
orally received normal saline once per day for 8 continuous weeks.
On the last five days of agent administration, behavioural tests
were performed on each mouse, and blood was collected from the
caudal vein of the mice under anaesthesia with isoflurane
inhalation with an initial level of 4% and a maintenance level of
1.2%. Mice were then euthanized using a small animal euthanasia
system (32×25×20 cm; cat. no. CL-1000-S2; Shanghai Yuyan Scientific
Instrument Co., Ltd.). Briefly, mice were placed in the carbon
dioxide tank for 2 min at a CO2 concentration of 30%.
The whole brain tissue was collected from each mouse for further
investigation.
Animal behaviour detection
For experimental mice, learning and memory ability
were analysed using Morris water maze test (MWM), whilst anxiety
was evaluated using the open field test as previously described
(25). For the MWM test, water was
mixed with titanium dioxide to form a white liquid to hide the
platform and the mice were placed in the water maze (cat. no.
MT-200; Techman Software Co., Ltd.) to locate a platform that was
hidden in white water. The route taken by mice was then recorded
using the watermaze software (version 2.0; Techman Software Co.,
Ltd.). In MWM, mice were trained for 7 days starting at week 7 to
allow them to learn or remember the position of platform. Open
field test was used to evaluate anxiety and performed on the day
after the MWM test. An open field experimental video analyser was
used to record the path taken by each mouse (cat. no. 1056306;
Zhong Shi Di Chuang). The box was washed after each experiment to
clear any remaining scents or traces that could affect the results
of the next test.
Immunohistochemical procedures
Brain tissue was fixed with 4% formalin solution at
25°C for 24 h, and then dehydrated with 30, 50, 70, 80, 95 and 100%
ethanol, washed in xylene, embedded in paraffin and cut into 5-µm
thick sections. All slides were gradient hydrated with 100, 95, 80,
70 and 50% ethanol and distilled water respectively (26). Following dewaxing and hydration, the
brain slides were boiled in 10 mM sodium citrate buffer (pH 6.0)
for 10 min. After cooling, the slides were incubated with 3%
hydrogen peroxide for 10 min and blocked using 10% goat serum for
30 min at 25°C. The slides were then incubated with primary
antibodies against phosphorylated (p)-τ (1:100; cat. no. SC12414;
Santa Cruz Biotechnology, Inc.), Aβ1-42 (1: 500; cat. no. ab32136;
Abcam) and 4-hydroxy-2-nonenal (4-HNE; 1:200; cat. no. ab46545;
Abcam) overnight at 4°C, followed by 1-h incubation at 25°C with
biotinylated horseradish peroxidase-conjugated secondary antibody
(anti-rabbit; 1:500; cat. no. sc-3836; Santa Cruz Biotechnology,
Inc.). After visualization using 3,3′-diaminobenzidine and Mayer's
hematoxylin and immunoperoxidase staining of Aβ1-42, p-τ and 4-HNE
at 25°C for 5 min, images were captured using light microscopy
(magnification, ×200; cat. no. IX73; Olympus Corporation).
Western blot analysis
U251 cells (2×105 cells/well) were
cultured at 37°C in 6-well plates with emodin at doses of 25 and 50
µM for 3 h, and co-incubated with 15 µM Aβ1-42 or culture medium at
37°C for a further 24 h. Culture medium only-treated U251 cells
served as the control group. Treated cells and brain tissues were
lysed using radio immunoprecipitation assay buffer (cat. no. 89900,
Thermo Fisher, Inc.) containing 1% protease inhibitor cocktail
(Sigma Aldrich; Merck KGaA). After detecting the protein
concentration using a bicinchoninic acid protein assay kit, 40 µg
protein/lane was separated by 12% SDS-PAGE and transferred onto
0.45 µm nitrocellulose membranes (Bio Basic, Inc.). The membranes
were then blocked with 5% skimmed milk at room temperature for 1 h
and incubated with primary antibodies against anti-SOD 1 (cat. no.
sc-17767), anti-HO-1 (cat. no. sc-136960), anti-CAT (cat. no.
sc-271803), anti-Nrf2 (cat. no. sc-365949), anti-Bax (cat. no.
sc-7480), anti-Bcl-2 (cat. no. sc-7382) and GAPDH (cat. no.
sc-47724; all from Santa Cruz Biotechnology, Inc.) at dilutions of
1:5,000 overnight at 4°C. The membranes were subsequently incubated
with horseradish peroxidase-conjugated mouse anti rabbit secondary
antibodies at 4°C for 4 h (dilution 1:5,000; cat. no. bs-0295G;
Bioss). ECL Detection reagent (cat. no. PE0010; Beijing Solarbio
Science & Technology Co., Ltd.) was then used to visualise the
specific bands, where the intensity was quantified by ImageJ
software (v1.46r National Institutes of Health).
Statistical analysis
Data are expressed as the mean ± SEM. Experimental
repeats were n=5 in the slide staining experiments and n=6 in the
other experiments, including apoptosis, ELISA and western blotting.
One-way ANOVA followed by Tukey's post hoc test was used to
determine statistical significance using SPSS 16.0 software (SPSS,
Inc.). P<0.05 was considered to indicate a statistically
significant difference.
Results
Emodin protects U251 cells against
Aβ1-42-induced cell apoptosis
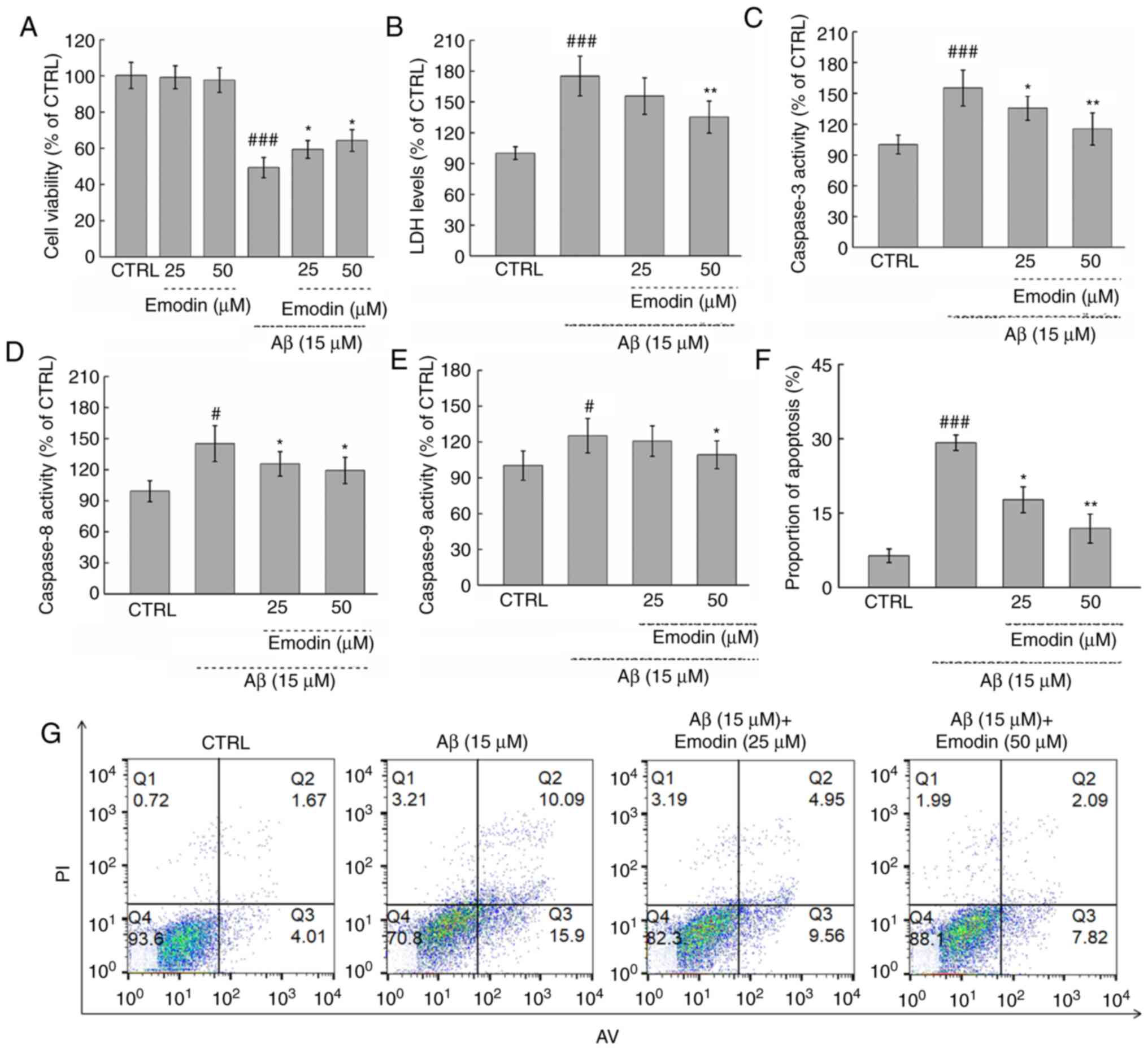
Exposure to cytotoxic Aβ1-42 (15 µM) for 24 h led to
a 50.66% decrease in the viability of U251 cells (P<0.001).
However, co-treatment with emodin at 25 and 50 µM led to 16.4 and
18.9% increases in viability, respectively (both P<0.05;
Fig. 2A). By contrast, incubation
with emodin alone for 24 h had no effects on cell viability
(Fig. 2A).
The cytotoxicity of Aβ1-42 treatment was next
evaluated by measuring the release of LDH. Notably, 3-h
pre-treatment with 50 µM emodin resulted in a 39.9% decrease of LDH
in cells exposed to Aβ1-42 for 24 h (P<0.01; Fig. 2B). Furthermore, co-treatment with
emodin at 25 µM and 50 µM suppressed Aβ1-42-induced apoptosis of
U251 cells by 11.5% (P<0.05) and 17.3% separately (P<0.01
Fig. 2F and G). The activation of
caspase −3, −8 and −9, which are classical markers of apoptosis
(27), was also subsequently
measured. It was found that 50 µM emodin treatment led to decreases
in the activity of caspase-3, −8 and −9 by 39.9% (P<0.01;
Fig. 2C), 25.9% (P<0.05;
Fig. 2D) and 15.9% (P<0.05;
Fig. 2E), respectively, in
Aβ1-42-exposed U251 cells.
Emodin ameliorates Aβ1-42-induced
mitochondrial dysfunction and activates the Nrf2 pathway
Mitochondrial injury one of the early events of cell
apoptosis (28). MMP was observed
to be significantly dissipated in Aβ1-42-exposed U251 cells.
However, co-incubation with emodin, particularly at 50 µM, restored
the MMP, as indicated by decreased green and increased red
fluorescence (Fig. 3A). The
mitochondria are the major site of ROS production in cells, such
that accumulation of ROS may cause further mitochondrial
dysfunction (29,30). In U251 cells exposed to Aβ1-42 for
24 h, 3-h pre-treatment with 50 µM emodin successfully suppressed
the overaccumulation of intracellular ROS, as indicated by the
decreased green fluorescence intensity (Fig. 3B).
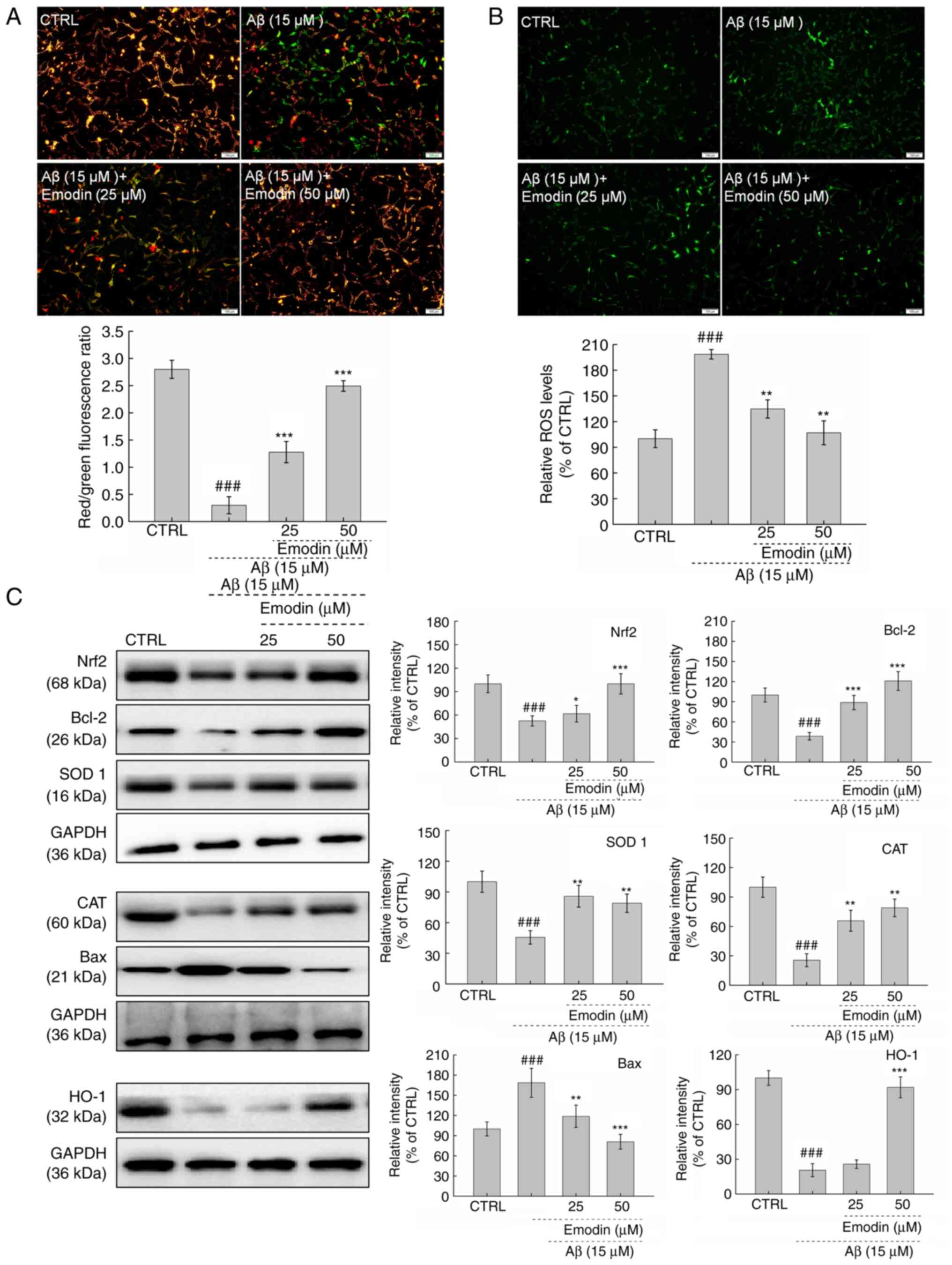 | Figure 3.Emodin suppresses oxidative stress by
increasing the expression levels of Nrf2 in U251 cells undergoing
Aβ1-42-induced apoptosis. A 3-h emodin pre-treatment (A)
ameliorated dissipation of mitochondrial membrane potential and (B)
inhibited excessive ROS accumulation in U251 cells exposed to Aβ
for 24 h (magnification, ×10; scale bar, 100 µm). (C) Levels of
oxidative stress- and apoptosis-associated proteins in
Aβ1-42-exposed U251 cells were examined by western blotting.
Quantitative protein expression levels were normalised to those of
GAPDH. Data are presented as the mean ± SEM (n=6 experiments).
###P<0.001 vs. CTRL. *P<0.05, **P<0.01 and
***P<0.001 vs. Aβ1-42 only. Nrf2, nuclear factor E2-related
factor 2; Aβ, β-amyloid peptide; ROS, reactive oxygen species;
CTRL, control; SOD, superoxide dismutase; CAT, catalase; HO, heme
oxygenase. |
Both anti- and pro-apoptotic members of the Bcl-2
protein family serve key roles in modulating mitochondrial cell
apoptosis (31). Compared with U251
cells exposed to Aβ1-42 alone, those co-treated with emodin
exhibited significantly increased Bcl-2 (P<0.001) and decreased
Bax expression levels (P<0.01; Fig.
3C).
Nrf2 is activated to regulate the functions of
mitochondria during the initiation and progression of oxidative
stress (32). Compared with
Aβ1-42-exposed U251 cells, emodin-treated cells exhibited increased
expression levels of Nrf2 (P<0.05), SOD 1 (P<0.01), HO-1
(P<0.001) and the antioxidant enzyme CAT (P<0.01; Fig. 3C). Taken together, these results
suggest that emodin restored MMP and decreased ROS in cells whilst
activating the Nrf2 pathway.
Emodin improves AD-like behaviour and
suppresses Aβ1-42, p-τ and 4-HNE deposition in APP/PS1 mice
Cognitive impairment and anxiety are the primary
clinical manifestations of AD (33). Therefore, MWM and open field tests
were used to evaluate these manifestations in APP/PS1 mice.
Compared with untreated mice, those treated with emodin at 20 mg/kg
exhibited a 14.3% decrease in time required to locate the platform
during the MWM test, indicating that emodin enhanced the spatial
memory and learning ability of the mice (P<0.05; Fig. 4A). In the open field test, mice
treated with emodin at 20 mg/kg spent significantly less time in
the centre of the field, which indicated that this agent
ameliorated anxiety in the mouse model (P<0.01; Fig. 4B).
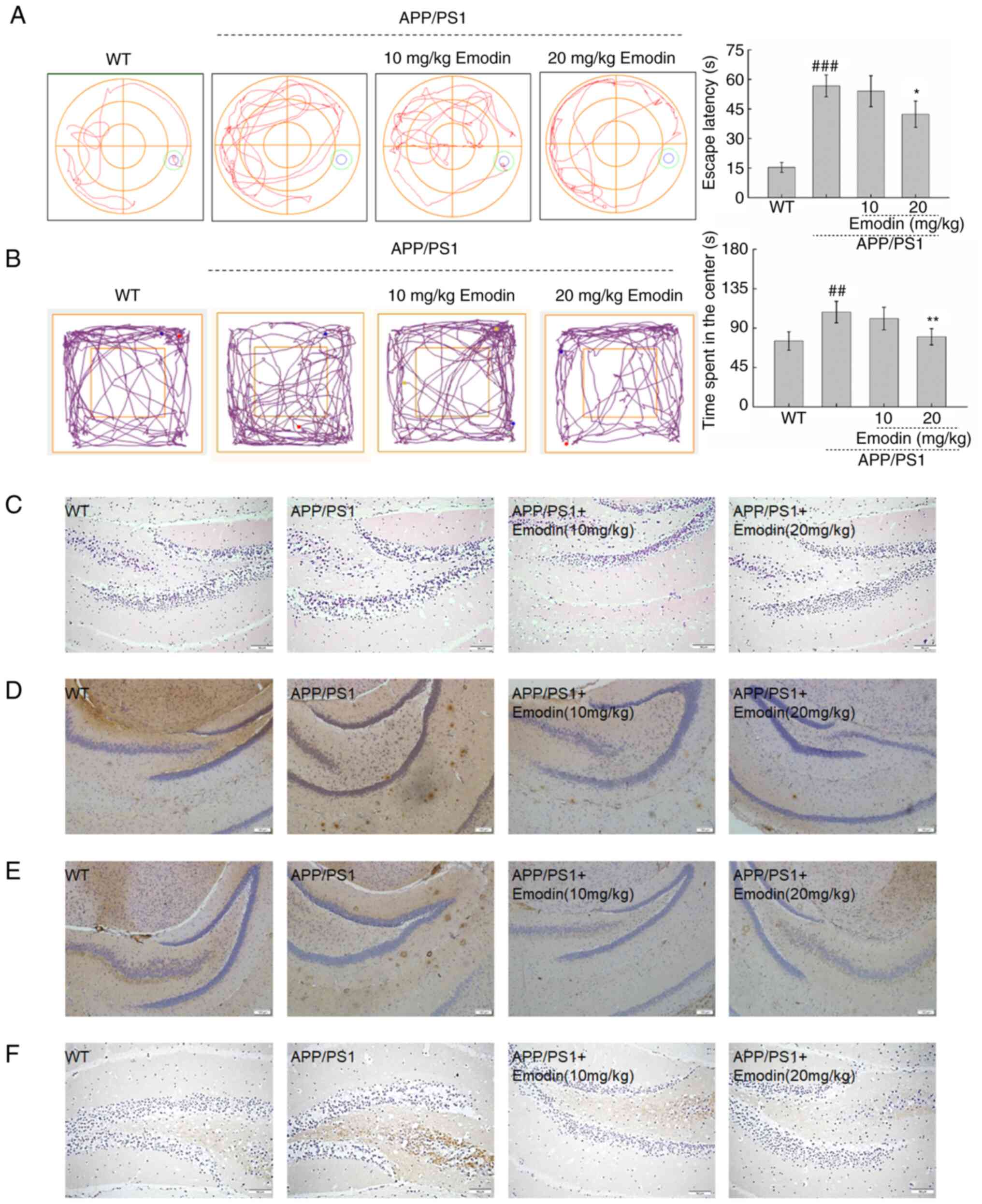 | Figure 4.Emodin improves behavioural
performance of APP/PS1 mice by decreasing deposition of Aβ1-42, p-τ
and 4-HNE. (A) Emodin decreased the escape latency time of APP/PS1
mice during the Morris water maze test. (B) Emodin decreased the
time spent by APP/PS1 mice in the central area during the open
field test. Data are presented as the mean ± SEM (n=10).
##P<0.01 and ###P<0.001 vs. WT mice.
*P<0.05 and **P<0.01 vs. untreated APP/PS1 mice. (C)
Haematoxylin and eosin staining of brain tissue (scale bar, 150 µm;
n=5 experiments). (D) Emodin markedly suppressed deposition of
Aβ1-42. (E) Overaccumulation of p-τ in the brain of APP/PS1 mice
(scale bar, 1100 µm; n=5 experiments). (F) High expression levels
of 4-HNE (scale bar, 150 µm; n=5 experiments) in the brain of
APP/PS1 mice detected by immunohistochemistry. APP, amyloid
precursor protein; PS1, presenilin-1; Aβ, β-amyloid peptide; p-,
phosphorylated; 4-HNE, 4-hydroxy-2-nonenal; WT, wild-type. |
H&E staining was used to detect neuronal damage
in brain tissue. However, there was no obvious pathological
reaction in the brain (Fig. 4C).
The presence of extracellular Aβ and intracellular neurofibrillary
tangles in the brain are two defining aetiological characteristics
of AD (34). Compared with
untreated APP/PS1 mice, emodin-treated mice, particularly those
that received a dose of 20 mg/kg, exhibited lower expression levels
of Aβ1-42 (Fig. 4D) and p-τ
(Fig. 4E) in the brain. Tissue was
also stained for 4-HNE, a biomarker of oxidative stress, to further
elucidate the function of emodin in this process. Notably, emodin
decreased expression levels of 4-HNE in the brain compared with
those in untreated APP/PS1 mice (Fig.
4F).
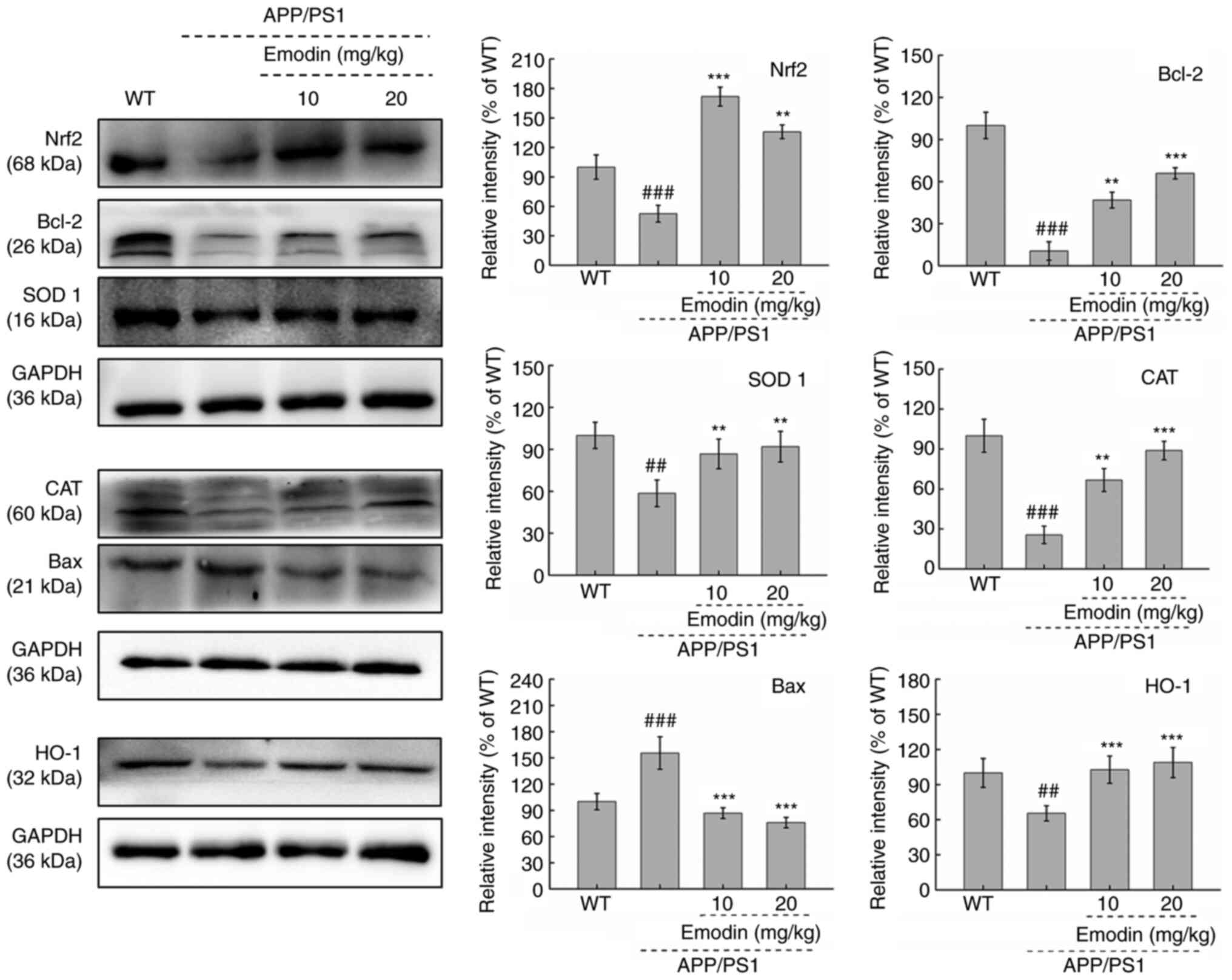
Emodin regulates Nrf2 signalling in the brain of
mice. To further investigate the possible mechanism by which emodin
exerts its effects in APP/PS1 mice, the expression levels of
proteins associated with the Bcl-2 family and the Nrf2 pathway, in
addition to the antioxidant enzyme CAT, were evaluated. Compared
with those in untreated APP/PS1 mice, 20 mg/kg emodin exhibited
significantly increased expression levels of Bcl-2 (P<0.01) and
significantly decreased expression levels of Bax in the brain
tissue of mice (P<0.001; Fig.
5). Furthermore, an 8-week course of 20 mg/kg emodin
administration led to significant increases in Nrf2 (P<0.01),
SOD 1 (P<0.01) and HO-1 expression (P<0.001) and a
significant decrease in CAT expression in the brain tissue, even at
an emodin dose of 10 mg/kg (P<0.01; Fig. 5). These results demonstrated the
ability of emodin to regulate Nrf2 pathway activity in APP/PS1 mice
and its potential as an antioxidant.
Discussion
The majority of symptoms of AD can be attributed to
oxidative stress (35,36). Therefore, the elimination of excess
ROS or induction of endogenous antioxidant activity may be a
potential strategy for the treatment of AD (37). Emodin is a biologically active
compound that can be found in a number of herbal laxatives and
exhibits pharmacological activity due to its strong antioxidative
effects (38). The present study
confirmed the neuroprotective effects of emodin against AD by using
U251 cells subjected to Aβ1-42-induced apoptosis and in APP/PS1
double-transgenic mice.
The U251 human astrocyte cell line has previously
been used to investigate AD (39).
In the present study, U251 cells were treated with Aβ1-42 to induce
cell injury. Aβ, a peptide containing 39–43 amino acids, has been
shown to exert numerous toxic effects both in vitro and
in vivo (40). For example,
Aβ accumulation is neurotoxic and can depolarise the cell membrane,
decrease mitochondrial potential and increase the production of
ROS, induce synaptic dysfunction and cause oxidative stress to
mediate mitochondrial damage (41).
The present study demonstrated that emodin improved cell viability
and suppressed apoptosis in cells exposed to Aβ1-42. Apoptotic
neuronal death causes fatal injury to the brain tissue (42). Caspases-3, −8 and −9 are major
components of the classical apoptosis pathway (43). Caspase-8 is located primarily in the
mitochondria and is activated to form the apoptosome, which
recruits pro-caspase-9 and induces pro-caspase-3 cleavage,
ultimately leading to mitochondrial apoptosis (44).
The mitochondria are energy-producing organelles
that serve as the primary source of ROS (45). Aβ accumulation directly exhibits
negative effects on mitochondrial energy metabolism, specifically
on α-ketoglutarate dehydrogenase and pyruvate dehydrogenase
activity, which ultimately results in cell apoptosis (46). The Bcl-2 family member of proteins,
which include the pro-apoptotic protein Bax and the anti-apoptotic
protein Bcl-2, have also been reported to be involved in
mitochondrial injury (47). The
pro-apoptotic protein Bax is activated in response to apoptotic
stimuli (48), which leads to
mitochondrial oxidative respiratory chain damage and decreased MMP
(49).
In the present study, emodin also enhanced the
activity of the antioxidant enzyme CAT and activated the
transcription factor Nrf2. The latter is known to regulate a number
of cytoprotective and detoxification genes. For example, Nrf2 binds
with Keap-1 in the cytoplasm, which represses Nrf2 nuclear
translocation (50). However, this
interaction is disrupted by oxidative stress, which allows Nrf2 to
translocate into the nucleus t interact with the antioxidant
response element, thus activating an effector cascade involving
HO-1 and SOD1 (10). Both HO-1 and
SOD1 are powerful antioxidative reagents that can neutralize toxic
superoxide radicals produced in AD (51,52).
In addition to oxidative stress, Nrf2 controls the expression of
nuclear genes that encode mitochondrial proteins, which affect
mitochondrial biological function. For example, Nrf2 deficiency
affects mitochondrial electron transport chain activity, fatty acid
oxidation and availability of substrates (NADH and FADH2/succinate)
for respiration, and ATP synthesis (53), furthermore, it can also exacerbate
APP and τ pathology (54). In the
present study, emodin strongly decreased activity of casapase-3, −8
and −9, improved mitochondrial function, decreased ROS
accumulation, enhanced the Bcl-2/Bax ratio and activated the Nrf2
pathway in both U251 cells subjected to Aβ1-42-induced apoptosis
and APP/PS1 mice, suggesting that emodin exerts both antioxidant
and neuroprotective effects.
In the present study, double transgenic APP/PS1 mice
were used to generate a model of AD with severe pathology. In this
transgenic model, overexpression of the gene encoding APP and a
mutant form of PS1 have previously been demonstrated to impair the
processing of amyloid proteins and increase levels of Aβ (55). Accumulation of Aβ exacerbates
cognitive impairment and induces anxiety in humans with AD
(56). Therefore, the
manifestations in APP/PS1 mice were similar to AD-like symptoms
(57). The present study used a MWM
test to evaluate the spatial learning ability (58) of mice and the open field test to
evaluate anxiety (59). In neurons,
aggregation of Aβ affects the activity of kinases and phosphatases,
leading to the hyperphosphorylation of τ protein and formation of
neurofibrillary tangles (60). An
increase in the number of neuroinflammatory plaques caused by
excess p-τ is another pathological feature of AD that has
previously been observed in the brain of APP/PS1 mice (61). In turn, excess p-τ inhibits
kinesin-dependent transportation and blocks APP transport into
axons and dendrites, which increases accumulation of Aβ in the
neurons (62). In the present
study, emodin treatment led to significant improvements in AD-like
behaviour of APP/PS1 mice and decreased the levels of aggregated
Aβ, pathogenic τ and the peroxidation product 4-HNE.
Increases in the production of
H2O2 and other oxidative products lead to
accumulation of Aβ and thus contribute to the initiation and
progression of AD (35,63,64).
An effective antioxidant therapy is necessary to relieve the
symptoms of AD. The present study demonstrated that emodin improved
AD-associated behaviour in APP/PS1 mice, ameliorated severe
oxidative stress and activated the Nrf2 pathway both in vivo
and in vitro.
In conclusion, emodin was demonstrated to exert
protective effects against Aβ1-42-induced cell apoptosis in
vitro and Aβ deposition in vivo in APP/PS1
double-transgenic mice. These effects are likely due to the
Nrf2-mediated anti-oxidative activity of emodin and suggest a
potential role for this agent in AD protection.
Acknowledgements
Not applicable.
Funding
The present study was supported by the Special
Projects of the Cooperation between Jilin University and Jilin
Province (grant no. SXGJXX2017-1), the Projects from the Science
and Technology Department of Jilin Province in China (grant nos.
20200708091YY and 20200708068YY) and the Department of Finance of
Jilin Province, China (grant no. JLSCZD2019-012).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
YZ and XF designed the experiments, drafted and
revised the manuscript. ZL, HB and HJ performed the experiments and
analyzed the data. JS and QM revised the manuscript critically for
important intellectual content. All authors read and approved the
final manuscript.
Ethics approval and consent to
participate
The experiments were approved by Animal Ethics
Committee of School of Life Sciences, Jilin University (approval
no. SY20171208).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
APP
|
amyloid precursor protein
|
|
PS1
|
presenilin-1
|
|
LDH
|
lactate dehydrogenase
|
|
ROS
|
reactive oxygen species
|
|
AD
|
Alzheimer's disease
|
|
MMP
|
mitochondrial membrane potential
|
|
Keap-1
|
Kelch-like ECH-associated
protein-1
|
|
4-HNE
|
4-hydroxy-2-nonenal
|
|
SOD
|
superoxide dismutase
|
|
CAT
|
catalase
|
|
HO-1
|
heme oxygenase-1
|
|
MWM
|
Morris water maze
|
References
|
1
|
Rygiel K and Rygiel K: Novel strategies
for Alzheimer's disease treatment: An overview of anti-amyloid beta
monoclonal antibodies. Indian J Pharmacol. 48:629–636. 2016.
View Article : Google Scholar
|
|
2
|
Viña J, Lloret A, Giraldo E, Badia MC and
Alonso MD: Antioxidant pathways in Alzheimers disease:
Possibilities of intervention. Curr Pharm Des. 17:3861–3864. 2011.
View Article : Google Scholar
|
|
3
|
Evans DA, Beckett LA, Field TS, Feng L,
Albert MS, Bennett DA, Tycko B and Mayeux R: Apolipoprotein E
epsilon4 and incidence of Alzheimer disease in a community
population of older persons. JAMA. 277:822–824. 1997. View Article : Google Scholar
|
|
4
|
Faizi M, Seydi E, Abarghuyi S, Salimi A,
Nasoohi S and Pourahmad J: A Search for Mitochondrial Damage in
Alzheimer's Disease Using Isolated Rat Brain Mitochondria. Iran J
Pharm Res. 15 (Suppl):185–195. 2016.
|
|
5
|
Chen Z and Zhong C: Oxidative stress in
Alzheimer's disease. Neurosci Bull. 30:271–281. 2014. View Article : Google Scholar
|
|
6
|
Ahmad W, Ijaz B, Shabbiri K, Ahmed F and
Rehman S: Oxidative toxicity in diabetes and Alzheimer's disease:
Mechanisms behind ROS/ RNS generation. J Biomed Sci. 24:762017.
View Article : Google Scholar
|
|
7
|
Angelova PR and Abramov AY: Role of
mitochondrial ROS in the brain: From physiology to
neurodegeneration. FEBS Lett. 592:692–702. 2018. View Article : Google Scholar
|
|
8
|
Nesi G, Sestito S, Digiacomo M and
Rapposelli S: Oxidative Stress, Mitochondrial Abnormalities and
Proteins Deposition: Multitarget Approaches in Alzheimer's Disease.
Curr Top Med Chem. 17:3062–3079. 2017.
|
|
9
|
Hardy J: Alzheimer's disease: The amyloid
cascade hypothesis: an update and reappraisal. J Alzheimers Dis. 9
(Suppl. 3):151–153. 2006. View Article : Google Scholar
|
|
10
|
Kang MI, Kobayashi A, Wakabayashi N, Kim
SG and Yamamoto M: Scaffolding of Keap1 to the actin cytoskeleton
controls the function of Nrf2 as key regulator of cytoprotective
phase 2 genes. Proc Natl Acad Sci USA. 101:2046–2051. 2004.
View Article : Google Scholar
|
|
11
|
Kaur SJ, McKeown SR and Rashid S: Mutant
SOD1 mediated pathogenesis of Amyotrophic Lateral Sclerosis. Gene.
577:109–118. 2016. View Article : Google Scholar
|
|
12
|
Kensler TW, Wakabayashi N and Biswal S:
Cell survival responses to environmental stresses via the
Keap1-Nrf2-ARE pathway. Annu Rev Pharmacol Toxicol. 47:89–116.
2007. View Article : Google Scholar
|
|
13
|
Kärkkäinen V, Pomeshchik Y, Savchenko E,
Dhungana H, Kurronen A, Lehtonen S, Naumenko N, Tavi P, Levonen AL,
Yamamoto M, et al: Nrf2 regulates neurogenesis and protects neural
progenitor cells against Aβ toxicity. Stem Cells. 32:1904–1916.
2014. View Article : Google Scholar
|
|
14
|
Calkins MJ, Johnson DA, Townsend JA,
Vargas MR, Dowell JA, Williamson TP, Kraft AD, Lee JM, Li J and
Johnson JA: The Nrf2/ARE pathway as a potential therapeutic target
in neurodegenerative disease. Antioxid Redox Signal. 11:497–508.
2009. View Article : Google Scholar
|
|
15
|
Wang JB, Zhao HP, Zhao YL, Jin C, Liu DJ,
Kong WJ, Fang F, Zhang L, Wang HJ and Xiao XH: Hepatotoxicity or
hepatoprotection? Pattern recognition for the paradoxical effect of
the Chinese herb Rheum palmatum L. in treating rat liver
injury. PLoS One. 6:e244982011. View Article : Google Scholar
|
|
16
|
Yang YC, Lim M-Y and Lee H-S: Emodin
isolated from Cassia obtusifolia (Leguminosae) seed shows
larvicidal activity against three mosquito species. J Agric Food
Chem. 51:7629–7631. 2003. View Article : Google Scholar
|
|
17
|
Lee MH, Kao L and Lin C-C: Comparison of
the antioxidant and transmembrane permeative activities of the
different Polygonum cuspidatum extracts in
phospholipid-based microemulsions. J Agric Food Chem. 59:9135–9141.
2011. View Article : Google Scholar
|
|
18
|
Dong X, Fu J, Yin X, Cao S, Li X, Lin L,
Ni J and Emodin: A Review of its Pharmacology, Toxicity and
Pharmacokinetics. Phytother Res. 30:1207–1218. 2016. View Article : Google Scholar
|
|
19
|
Liu T, Jin H, Sun QR, Xu JH and Hu HT:
Neuroprotective effects of emodin in rat cortical neurons against
beta-amyloid-induced neurotoxicity. Brain Res. 1347:149–160. 2010.
View Article : Google Scholar
|
|
20
|
Lin J, Ma C and Lin HH: Emodin Alleviates
Viral Myocarditis in BALB/c Mice and Underlying Mechanisms. Lat Am
J Pharm. 38:1979–1984. 2019.
|
|
21
|
Park SY, Jin ML, Ko MJ, Park G and Choi
YW: Anti-neuroinflammatory Effect of Emodin in LPS-Stimulated
Microglia: Involvement of AMPK/Nrf2 Activation. Neurochem Res.
41:2981–2992. 2016. View Article : Google Scholar
|
|
22
|
Tian SL, Yang Y, Liu XL and Xu QB: Emodin
Attenuates Bleomycin-Induced Pulmonary Fibrosis via
Anti-Inflammatory and Anti-Oxidative Activities in Rats. Med Sci
Monit. 24:1–10. 2018. View Article : Google Scholar
|
|
23
|
Mizuno M, Kawamura H, Takei N and Nawa H:
The anthraquinone derivative Emodin ameliorates neurobehavioral
deficits of a rodent model for schizophrenia. J Neural Transm
(Vienna). 115:521–530. 2008. View Article : Google Scholar
|
|
24
|
Li Z, Chen X, Zhang Y, Liu X, Wang C, Teng
L and Wang D: Protective roles of Amanita caesarea polysaccharides
against Alzheimer's disease via Nrf2 pathway. Int J Biol Macromol.
121:29–37. 2019. View Article : Google Scholar
|
|
25
|
Han Y, Nan S, Fan J, Chen Q and Zhang Y:
Inonotus obliquus polysaccharides protect against Alzheimer's
disease by regulating Nrf2 signaling and exerting antioxidative and
antiapoptotic effects. Int J Biol Macromol. 131:769–778. 2019.
View Article : Google Scholar
|
|
26
|
Zhang Y, Wang J, Wang C, Li Z, Liu X,
Zhang J, Lu J and Wang D: Pharmacological Basis for the Use of
Evodiamine in Alzheimer's Disease: Antioxidation and Antiapoptosis.
Int J Mol Sci. 19:192018.
|
|
27
|
Sharifi AM, Eslami H, Larijani B and
Davoodi J: Involvement of caspase-8, −9, and −3 in high
glucose-induced apoptosis in PC12 cells. Neurosci Lett. 459:47–51.
2009. View Article : Google Scholar
|
|
28
|
Berry BJ, Trewin AJ, Amitrano AM, Kim M
and Wojtovich AP: Use the Protonmotive Force: Mitochondrial
Uncoupling and Reactive Oxygen Species. J Mol Biol. 430:3873–3891.
2018. View Article : Google Scholar
|
|
29
|
Tan S, Sagara Y, Liu Y, Maher P and
Schubert D: The regulation of reactive oxygen species production
during programmed cell death. J Cell Biol. 141:1423–1432. 1998.
View Article : Google Scholar
|
|
30
|
Grivennikova VG and Vinogradov AD:
Generation of superoxide by the mitochondrial Complex I. Biochim
Biophys Acta. 1757:553–561. 2006. View Article : Google Scholar
|
|
31
|
Reed JC, Jurgensmeier JM and Matsuyama S:
Bcl-2 family proteins and mitochondria. Biochim Biophys Acta.
1366:127–137. 1998. View Article : Google Scholar
|
|
32
|
McMahon M, Itoh K, Yamamoto M, Chanas SA,
Henderson CJ, McLellan LI, Wolf CR, Cavin C and Hayes JD: The
Cap'n'Collar basic leucine zipper transcription factor Nrf2 (NF-E2
p45-related factor 2) controls both constitutive and inducible
expression of intestinal detoxification and glutathione
biosynthetic enzymes. Cancer Res. 61:3299–3307. 2001.
|
|
33
|
Benedict C: Candidate mechanisms
underlying the association between poor sleep and obesity.
Endocrine Abstracts. 49:pp. S282017, simplehttps://doi.org/10.1530/endoabs.49.S28.1
|
|
34
|
Choi ML and Gandhi S: Crucial role of
protein oligomerization in the pathogenesis of Alzheimer's and
Parkinson's diseases. FEBS J. 285:3631–3644. 2018. View Article : Google Scholar
|
|
35
|
Ansari MA and Scheff SW: Oxidative stress
in the progression of Alzheimer disease in the frontal cortex. J
Neuropathol Exp Neurol. 69:155–167. 2010. View Article : Google Scholar
|
|
36
|
Selkoe DJ: Alzheimer's disease: Genes,
proteins, and therapy. Physiol Rev. 81:741–766. 2001. View Article : Google Scholar
|
|
37
|
Saxena G, Singh SP, Agrawal R and Nath C:
Effect of donepezil and tacrine on oxidative stress in
intracerebral streptozotocin-induced model of dementia in mice. Eur
J Pharmacol. 581:283–289. 2008. View Article : Google Scholar
|
|
38
|
Monisha BA, Kumar N and Tiku AB: Emodin
and Its Role in Chronic Diseases. Adv Exp Med Biol. 928:47–73.
2016. View Article : Google Scholar
|
|
39
|
Handattu SP, Monroe CE, Nayyar G,
Palgunachari MN, Kadish I, van Groen T, Anantharamaiah GM and
Garber DW: In vivo and in vitro effects of an apolipoprotein E
mimetic peptide on amyloid-β pathology. J Alzheimers Dis.
36:335–347. 2013. View Article : Google Scholar
|
|
40
|
Carrillo-Mora P, Luna R and Colín-Barenque
L: Amyloid beta: Multiple mechanisms of toxicity and only some
protective effects? Oxid Med Cell Longev. 2014:7953752014.
View Article : Google Scholar
|
|
41
|
Anandatheerthavarada HK, Biswas G, Robin
M-A and Avadhani NG: Mitochondrial targeting and a novel
transmembrane arrest of Alzheimer's amyloid precursor protein
impairs mitochondrial function in neuronal cells. J Cell Biol.
161:41–54. 2003. View Article : Google Scholar
|
|
42
|
Pluta R, Ułamek-Kozioł M and Czuczwar SJ:
Neuroprotective and Neurological/Cognitive Enhancement Effects of
Curcumin after Brain Ischemia Injury with Alzheimer's Disease
Phenotype. Int J Mol Sci. 19:40022018. View Article : Google Scholar
|
|
43
|
Viswanath V, Wu Y, Boonplueang R, Chen S,
Stevenson FF, Yantiri F, Yang L, Beal MF and Andersen JK: Caspase-9
activation results in downstream caspase-8 activation and bid
cleavage in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced
Parkinson's disease. J Neurosci. 21:9519–9528. 2001. View Article : Google Scholar
|
|
44
|
Cain K, Bratton SB, Langlais C, Walker G,
Brown DG, Sun XM and Cohen GM: Apaf-1 oligomerizes into
biologically active approximately 700-kDa and inactive
approximately 1.4-MDa apoptosome complexes. J Biol Chem.
275:6067–6070. 2000. View Article : Google Scholar
|
|
45
|
Luca M, Luca A and Calandra C: The Role of
Oxidative Damage in the Pathogenesis and Progression of Alzheimer's
Disease and Vascular Dementia. Oxid Med Cell Longev.
2015:5046782015. View Article : Google Scholar
|
|
46
|
Casley CS, Canevari L, Land JM, Clark JB
and Sharpe MA: Beta-amyloid inhibits integrated mitochondrial
respiration and key enzyme activities. J Neurochem. 80:91–100.
2002. View Article : Google Scholar
|
|
47
|
Gross A, McDonnell JM and Korsmeyer SJ:
BCL-2 family members and the mitochondria in apoptosis. Genes Dev.
13:1899–1911. 1999. View Article : Google Scholar
|
|
48
|
Zhu W, Cowie A, Wasfy GW, Penn LZ, Leber B
and Andrews DW: Bcl-2 mutants with restricted subcellular location
reveal spatially distinct pathways for apoptosis in different cell
types. EMBO J. 15:4130–4141. 1996. View Article : Google Scholar
|
|
49
|
Hsu YT and Youle RJ: Nonionic detergents
induce dimerization among members of the Bcl-2 family. J Biol Chem.
272:13829–13834. 1997. View Article : Google Scholar
|
|
50
|
Itoh K, Wakabayashi N, Katoh Y, Ishii T,
Igarashi K, Engel JD and Yamamoto M: Keap1 represses nuclear
activation of antioxidant responsive elements by Nrf2 through
binding to the amino-terminal Neh2 domain. Genes Dev. 13:76–86.
1999. View Article : Google Scholar
|
|
51
|
Zhang Y, Unnikrishnan A, Deepa SS, Liu Y,
Li Y, Ikeno Y, Sosnowska D, Van Remmen H and Richardson A: A new
role for oxidative stress in aging: The accelerated aging phenotype
in Sod1−/− mice is correlated to increased cellular
senescence. Redox Biol. 11:30–37. 2017. View Article : Google Scholar
|
|
52
|
Kansanen E, Kuosmanen SM, Leinonen H and
Levonen AL: The Keap1-Nrf2 pathway: Mechanisms of activation and
dysregulation in cancer. Redox Biol. 1:45–49. 2013. View Article : Google Scholar
|
|
53
|
Dinkova-Kostova AT and Abramov AY: The
emerging role of Nrf2 in mitochondrial function. Free Radic Biol
Med. 88B:B179–B188. 2015. View Article : Google Scholar
|
|
54
|
Rojo AI, Pajares M, Rada P, Nuñez A,
Nevado-Holgado AJ, Killik R, Van Leuven F, Ribe E, Lovestone S,
Yamamoto M, et al: NRF2 deficiency replicates transcriptomic
changes in Alzheimer's patients and worsens APP and TAU pathology.
Redox Biol. 13:444–451. 2017. View Article : Google Scholar
|
|
55
|
Hu D, Serrano F, Oury TD, Klann E and Hu
D: Aging-dependent alterations in synaptic plasticity and memory in
mice that overexpress extracellular superoxide dismutase. J
Neurosci. 26:3933–3941. 2006. View Article : Google Scholar
|
|
56
|
Pietrzak RH, Lim YY, Neumeister A, Ames D,
Ellis KA, Harrington K, Lautenschlager NT, Restrepo C, Martins RN,
Masters CL, et al Australian Imaging, Biomarkers, Lifestyle
Research Group, : Amyloid-β, anxiety, and cognitive decline in
preclinical Alzheimer disease: A multicenter, prospective cohort
study. JAMA Psychiatry. 72:284–291. 2015. View Article : Google Scholar
|
|
57
|
Delatour B, Guégan M, Volk A and Dhenain
M: In vivo MRI and histological evaluation of brain atrophy in
APP/PS1 transgenic mice. Neurobiol Aging. 27:835–847. 2006.
View Article : Google Scholar
|
|
58
|
Mehta MA: Morris Water Maze. Encyclopedia
of Psychopharmacology. Stolerman IP: Springer; Berlin, Heidelberg:
2010, simplehttps://doi.org/10.1007/978-3-540-68706-1_1638
|
|
59
|
Prut L and Belzung C: The open field as a
paradigm to measure the effects of drugs on anxiety-like behaviors:
A review. Eur J Pharmacol. 463:3–33. 2003. View Article : Google Scholar
|
|
60
|
Kamat PK, Kalani A, Rai S, Swarnkar S,
Tota S, Nath C and Tyagi N: Mechanism of Oxidative Stress and
Synapse Dysfunction in the Pathogenesis of Alzheimer's Disease:
Understanding the Therapeutics Strategies. Mol Neurobiol.
53:648–661. 2016. View Article : Google Scholar
|
|
61
|
Leroy K, Ando K, Laporte V, Dedecker R,
Suain V, Authelet M, Héraud C, Pierrot N, Yilmaz Z, Octave JN, et
al: Lack of tau proteins rescues neuronal cell death and decreases
amyloidogenic processing of APP in APP/PS1 mice. Am J Pathol.
181:1928–1940. 2012. View Article : Google Scholar
|
|
62
|
Stamer K, Vogel R, Thies E, Mandelkow E
and Mandelkow EM: Tau blocks traffic of organelles, neurofilaments,
and APP vesicles in neurons and enhances oxidative stress. J Cell
Biol. 156:1051–1063. 2002. View Article : Google Scholar
|
|
63
|
Smith MA, Hirai K, Hsiao K, Pappolla MA,
Harris PL, Siedlak SL, Tabaton M and Perry G: Amyloid-beta
deposition in Alzheimer transgenic mice is associated with
oxidative stress. J Neurochem. 70:2212–2215. 1998. View Article : Google Scholar
|
|
64
|
Wirths O, Multhaup G, Czech C, Feldmann N,
Blanchard V, Tremp G, Beyreuther K, Pradier L and Bayer TA:
Intraneuronal APP/A beta trafficking and plaque formation in
beta-amyloid precursor protein and presenilin-1 transgenic mice.
Brain Pathol. 12:275–286. 2002. View Article : Google Scholar
|