Introduction
Heart failure caused by acute myocardial infarction
(AMI) is the primary pathological cause of death and disability
worldwide (1,2). Coronary occlusion deprives
cardiomyocytes of oxygen, leading to cardiac dysfunction during AMI
(3). Cardiomyocytes are terminally
differentiated cells, thus preserving cardiomyocytes from
hypoxia-induced apoptosis has been proposed as a therapeutic
strategy for AMI (4). Previous
studies reported that hypoxia induces dissipation of mitochondrial
membrane potential and initiates mitochondrial-mediated apoptosis,
thus leading to an increase in the number of dysfunctional
mitochondria (5,6). Bcl-2 family proteins are involved in
the regulation of mitochondrial apoptosis via regulating cytochrome
C release from mitochondria (7).
Additionally, hypoxia-induced reactive oxygen species (ROS)
generation promotes the release of cytochrome C into the cytoplasm,
resulting in DNA damage, oxidative stress, apoptosis and
mitochondrial damage (8,9). Hypoxia and an altered redox balance
contribute to the mechanisms regulating the response to tissue
damage (10). For example,
excessive production of ROS initiates the intrinsic apoptosis
signaling pathway in testicular tissue cells, leading to testicular
tissue damage in individuals with varicocele (11,12).
However, the regulatory mechanisms underlying hypoxia-induced
mitochondrial-mediated apoptosis in cardiomyocytes are complex and
are not completely understood.
Activation of hypoxia-inducible factor-1 (HIF-1)
transcription factor serves a critical role in the adaptive
responses to hypoxia (13). HIF-1
is a heterodimeric transcription factor that contains two subunits,
including HIF-1α and HIF-1β/aryl hydrocarbon receptor nuclear
translocator (14). HIF-1α is
hydroxylated by prolyl hydroxylases domain (PHD) enzymes on two
conserved proline residues under normoxic conditions, which is
recognized by von Hippel-Lindau, an E3 ubiquitin ligase that
ubiquitinates HIF-1α. Under hypoxic conditions, inhibition of PHD
enzymes leads to HIF-1α accumulation and nuclear translocation
(15). Upregulation of HIF-1 target
genes is involved in oxygen transport, glycolytic metabolism,
lactate production and secretion, cell death and other processes
that can affect cell survival in hypoxia (16,17).
During hypoxia, increased glycolysis causes a surge
in lactate production (18).
Increasing evidence indicates that elevation of lactate production
is associated with myocardial infarction and volume overload
(18,19). Lactate dehydrogenase (LDH) serves a
crucial role in glucose metabolism and the Warburg effect (20). Lactate dehydrogenase A (LDHA) and
lactate dehydrogenase B (LDHB) have been reported to catalyze the
same reaction, the conversion of pyruvate to lactate at the end of
glycolysis (21). Under anaerobic
conditions, LDHA has a higher affinity for pyruvate, preferentially
converting pyruvate to lactate and NADH to NAD+. When
oxygen supplies are sufficient, LDHB has a higher affinity for
lactate, preferentially converting lactate to pyruvate and
NAD+ to NADH (22).
Moreover, hypoxia-induced elevation of LDHA expression can promote
tumor cell proliferation (23).
Human coilin interacting nuclear ATPase protein
(hCINAP) is highly conserved and ubiquitously expressed in
different eukaryotes (24).
Nucleoside-triphosphatase, a yeast homolog of hCINAP, is required
for yeast growth and 18S rRNA maturation (21,25).
hCINAP serves as a critical regulator of the 40S ribosomal protein
S14 (RPS14)/human double minute 2 (HDM2)/p53 signaling pathway by
regulating the interaction between RPS14 and HDM2 in 293T cells
(26). hCINAP knockdown can cause
defects in the formation of Cajal bodies, histone transcription and
cell viability (27). In acute
myeloid leukemia model mice, hCINAP knockdown results in increased
cell death (28). Furthermore,
hCINAP-depleted cells display increased Caspase-3 activities,
indicating that apoptosis is one of the reasons for decreased cell
viability (28). hCINAP knockdown
inhibits colorectal cancer stem cell invasion and self-renewal via
modulating the activation of LDHA (29). Collectively, the aforementioned
studies indicate that hCINAP serves a critical role in biological
processes; however, the role of hCINAP in hypoxia-induced apoptosis
of cardiomyocytes is not completely understood.
The aim of the present study was to investigated the
role of hCINAP in hypoxia-induced apoptosis and it demonstrated
that hCINAP expression was induced under hypoxic conditions in
cardiomyocytes and that hCINAP may serve as a regulator for
oxygen-dependent and lactate regulation of hypoxia responses.
Materials and methods
Cell culture
AC16 cells (a human cardiomyocyte cell line; ATCC)
were cultured in DMEM (Gibco; Thermo Fisher Scientific, Inc.)
supplemented with 10% FBS (Gibco; Thermo Fisher Scientific, Inc.)
and 1% penicillin-streptomycin at 37°C with 5% CO2. For
hypoxia treatment, cells were cultured with 1% oxygen, 5%
CO2 and 94% N2 for the indicated time points
in a hypoxia chamber (Billups-Rothenberg, Inc.).
PCR protocol
The synthesized cDNA was diluted to 15 ng/µl as cDNA
template. hCINAP cDNA was amplified by PCR conditions of initial
denaturation at 95°C for 3 min followed by 30 cycles of 95°C for 30
sec, 60°C for 20 sec, 72°C for 1 min, and final elongation at 72°C
for 5 min with Platinum II Hot-Start Green PCR Master Mix (cat. no.
14001012; Thermo Fisher Scientific, Inc.). To construct the hCINAP
promoter constructs, genomic DNA was extracted using PureLink
Genomic DNA kit (cat. no. K182002; Invitrogen; Thermo Fisher
Scientific, Inc.). Briefly, AC16 cells (4×106) were
lysed with 200 µl genomic lysis buffer (cat. no. K182302;
Invitrogen; Thermo Fisher Scientific, Inc.) supplied with 20 µl
Proteinase K (cat. no. 25530049; Invitrogen; Thermo Fisher
Scientific, Inc.) and 20 µl RNase A (cat. no. 12091021; Invitrogen;
Thermo Fisher Scientific, Inc.) at 55°C for 10 min followed by the
addition of 200 µl 95% ethanol to the lysate. The lysate was added
to a PureLink Spin Column (cat. no. K210012; Invitrogen, Thermo
Fisher Scientific, Inc.) and spun for 1 min at 10,000 × g at room
temperature. DNA fluid was obtained by centrifugation (10,000 × g
for 2 min at room temperature) using 500 µl wash buffer and 100 µl
elution buffer. hCINAP promoter (−2350 bp to +50 bp) was amplified
from genomic DNA by PCR conditions of initial denaturation at 95°C
for 3 min followed by 30 cycles of 95°C for 30 sec, 62°C for 20
sec, 72°C for 30 s, and final elongation at 72°C for 5 min with
Platinum II Hot-Start Green PCR Master Mix (cat. no. 14001012;
Thermo Fisher Scientific, Inc.). The PCR amplification conditions
for truncated hCINAP promoter constructs were the same as those
employed in amplification of full-length hCINAP promoter. A 1%
agarose gel and ethidium bromide (cat. no. 15585011; Thermo Fisher
Scientific, Inc.) were used for agarose gel electrophoresis. The
primer list for PCR is presented in Table I.
 | Table I.Sequences of primers used in the
present study. |
Table I.
Sequences of primers used in the
present study.
| Primer | Sequence
(5′→3) | Primer purpose |
|---|
| GAPDH | F:
TCCTGGTATGACAACGAAT | RT-qPCR |
|
| R:
GGTCTCTCTCTTCCTCTTG |
|
| hCINAP | F:
GTTGGTTCAGTTACTAGCAGACC | RT-qPCR |
|
| R:
CCTTTGACCTGTGAGGGACATG |
|
| hCI-pm | F:
CTCGAGAATCTTGGCCCCTTTCCTCT | Luciferase reporter
assay |
|
| R:
GGTACCGCCCTTCGCTTGCGCC |
|
| hCI-pm1 | F:
CTCGAGAATCTTGGCCCCTTTCCTCTATA | Luciferase reporter
assay |
|
| R:
GGTACCGGATTTGCGAGCTCAACCC |
|
| hCI-pm2 | F:
CTCGAGCGTTTCAAAAGGTATACAGGTGG | Luciferase reporter
assay |
|
| R:
GGTACCGCCCTTCGCTTGCGCCGA |
|
| hCI-pm3 | F:
CTCGAGCGTTTCAAAAGGTATACAGGTGG | Luciferase reporter
assay |
|
| R:
GGTACCGGATTTGCGAGCTCAACCC |
|
| hCI-pm-4 | F:
CTCGAGCGTTTCAAAAGGTATACAGGTGG | Luciferase reporter
assay |
|
| R:
GGTACCTTGTTTTGAGACGGAGTCTCAC |
|
| hCI-pm-5 | F:
CTCGAGGGTGAAACCCCGTCTCTATTAA | Luciferase reporter
assay |
|
| R:
GGTACCGGATTTGCGAGCTCAACCC |
|
| hCINAP | F:
CGTTTCAAAAGGTATACAGGTGG | ChIP-qPCR |
|
| R:
TTAATAGAGACGGGGTTTCACC |
|
Plasmids and reagents
The cDNA of human hCINAP was amplified by PCR and
cloned into the pB513B-Flag empty vector (cat. no. 5619; Biovector
NTCC, Inc.) to generate the pB513B-hCINAP-Flag vector. The coding
sequence of HIF-1α was cloned in-frame with an ATG start codon into
the pcDNA 6.0 expression vector (cat. no. 3791; Biovector NTCC,
Inc.) to generate the pcDNA-HIF-1α vector. For LDHA inhibition,
cells were treated with 10 µM LDHA inhibitor FX11 (MedChemExpress)
and incubated at 37°C for 24 h. For lactate treatment, cells were
treated with 20 mM lactate (Sigma-Aldrich; Merck KGaA) and
incubated at 37°C for 24 h. Cytoplasmic and mitochondrial protein
fractions were prepared using the Cytoplasmic and Mitochondrial
Protein Extraction kit (cat. no. G007-1-1; Nanjing Jiancheng
Bioengineering Institute). Caspase-9 and Caspase-3 activities were
measured using Caspase-9 Activity Assay kit (cat. no. C1157;
Beyotime Institute of Biotechnology) and Caspase-3 Activity Assay
kit (cat. no. C1115; Beyotime Institute of Biotechnology) according
to the manufacturer's protocol. Lactate production was measured
using a Lactate Assay kit (cat. no. BC2230; Beijing Solarbio
Science & Technology Co., Ltd.), according to the
manufacturer's protocol. LDHA enzyme activity was measured using a
LDHA Assay kit (cat. no. BC0680; Beijing Solarbio Science &
Technology Co., Ltd.), according to the manufacturer's
protocol.
Chromatin-immunoprecipitation
(ChIP)
ChIP assays were performed using a Simple ChIP
PlusEnzymatic Chromatin IP kit (Agarose Beads; cat. no. 9004; Cell
Signaling Technology, Inc.) according to the manufacturer's
protocol. Briefly, AC16 cells (4×106) were seeded into
10-cm dishes and allowed to adhere overnight. Following treatment,
cells were fixed with 1% formaldehyde at room temperature for 10
min, followed by incubating with ChIP lysis buffer plus protease
inhibitors (PIC) on ice for 10 min. Next the lysates were incubated
at 37°C for 20 min with micrococcal nuclease (cat. no. 10011; Cell
Signaling Technology, Inc.) to digest the chromatin DNA.
Immunoprecipitation was performed using an IgG (1 µg; cat. no.
2729; Cell Signaling Technology, Inc.) or HIF-1α antibody (1 µg;
cat. no. NB100-105; Novus Biologicals, LLC) and ChIP-Grade Protein
G Agarose Beads (cat. no. 9007; Cell Signaling Technology, Inc.)
incubated at 4°C for 2 h. Subsequently, chromatin complexes were
eluted from the beads using ChIP elution buffer as described in
manufacturer's instructions. ChIP DNA was purified with a DNA
purification columns (cat. no. 10010; Cell Signaling Technology,
Inc.). Immunoprecipitation was used as templates for Chip-qPCR
using appropriate primers. Primer sequences for ChIP-qPCR are
listed in Table I. ChIP-qPCR were
performed as previously described (30).
Western blotting
Total protein was extracted from cells using RIPA
buffer (Beijing Solarbio Science & Technology Co., Ltd.) with
protease inhibitor cocktail and protein phosphatase inhibitor
(Beijing Solarbio Science & Technology Co., Ltd.). Protein
concentration was determined by Pierce BCA Protein Assay kit (cat.
no. 23227; Thermo Fisher Scientific, Inc.). Total Proteins (30 µg)
were separated via SDS-PAGE (8%) and transferred onto PVDF
membranes. After blocking with 5% BSA (cat. no. A8010; Beijing
Solarbio Science & Technology Co., Ltd.) at room temperature
for 1 h and the membranes were incubated at 4°C overnight with the
following primary antibodies in 5% BSA: HIF-1α (1:2,000; cat. no.
NB100-105; Novus Biologicals, LLC), hCINAP (1:1,000; cat. no.
ab192653; Abcam), β-actin (1:2,000; cat. no. 3700S; Cell Signaling
Technology, Inc.), β-tubulin (1:3,000; cat. no. 2146S; Cell
Signaling Technology, Inc.), phosphorylated LDHA (1:500; cat. no.
8176S; Cell Signaling Technology, Inc.), Flag-tag (1:3,000; cat.
no. 14793S; Cell Signaling Technology, Inc.), LDHA (1:3,000; cat.
no. 2012S; Cell Signaling Technology, Inc.), cytochrome C (1:2,000;
cat. no. 4280S; Cell Signaling Technology, Inc.), hsp60 (1:1,000;
cat. no. 611563; BD Pharmingen; BD Biosciences), monocarboxylate
transporter 1 (MCT1; 1:2,000; cat. no. 20139-1-AP; ProteinTech
Group, Inc.). Subsequently, the membranes were incubated with
HRP-linked anti-mouse IgG (1:8,000; cat. no. 7076P2; Cell Signaling
Technology, Inc.) and HRP-linked anti-rabbit IgG (1:5,000; cat. no.
7074P2; Cell Signaling Technology, Inc.) secondary antibodies at
room temperature for 1 h. To visualize the images, ECL reagent
(cat. no. WBKLS0050; EMD Millipore) was applied and the images were
captured by CCD camera (LAS-4000; Fujifilm Life Science). Protein
expression was semi-quantified using ImageLab software version
5.2.1. (Bio-Rad Laboratories, Inc.) with β-actin or β-tubulin as
the loading control.
RT-qPCR
Total RNA was extracted and purified from AC16 cells
using TRIzol® reagent (Thermo Fisher Scientific, Inc.)
according to the manufacturer's instructions. Total RNA was reverse
transcribed into cDNA at 37°C for 15 min using PrimeScript RT
Master Mix cDNA synthesis system (Bio-Rad Laboratories, Inc.).
Subsequently, qPCR was performed using SYBR Green (Bio-Rad
Laboratories, Inc.). The following thermocycling conditions were
used for qPCR: Preheated at 95°C for 2 min; followed by 34 cycles
of 95°C for 10 sec, 60°C for 10 sec and 72°C for 15 sec. The
primers used for qPCR are presented in Table I. mRNA expression levels were
quantified using the 2−∆∆Cq method (31) and normalized to the internal
reference gene GAPDH.
In vitro cell viability assay
Cell viability was determined by performing a Cell
Counting Kit-8 (CCK-8) assay (Beijing Solarbio Science &
Technology Co., Ltd.) according to the manufacturer's protocol.
Briefly, AC16 cells were seeded into 96-well plates and grown to
75% confluence. Following transfection, CCK-8 reagent was added to
each well and incubated for 1 h at 37°C. The absorbance of each
well was measured at a wavelength of 450 nm on a microplate reader
(Thermo Fisher Scientific, Inc.). For clonogenic assays, AC16 cells
were plated into 6-well plates at a density of 4,000–8,000
cells/well. After 24 h later, cells were cultured in a hypoxia
chamber (Billups-Rothenberg, Inc.) at 37°C for the indicated time
points. Cells were fixed with 10% formalin at room temperature for
10 min, followed by staining with 0.02% crystal violet at room
temperature for 10 min. Cell numbers were counted under a
dissection microscope.
Mitochondrial membrane potential
analysis
Mitochondrial membrane potential was measured via
flow cytometry using rhodamine-123 (Beijing Solarbio Science &
Technology Co., Ltd.). Following treatment, AC16 cells
(105 cells/ml) were incubated with 5 µM rhodamine-123 at
37°C for 30 min. Cells were washed twice with PBS and re-suspended
in 300 µl PBS. Subsequently, cells were analyzed via FACSCanto II
flow cytometer (BD Biosciences) and data analyzed with FlowJo v10
(FlowJo, LLC).
Transfection and
immunoprecipitation
To knockdown hCINAP or HIF-1α in AC16 cells, the
following siRNAs (all purchased from Sangon Biotech Co., Ltd.) were
used: siRNA-hCINAP (5′-GAGAGAAGGUGGAGUUAUU-3′), siRNA-HIF-1α
(5′-AAGCAUUUCUCUCAUUUCCUCAUGG-3′) and siRNA-ctrl (scrambled siRNAs)
(5′-GACUACUGGUCGUUGAACU-3′). AC16 cells (1×105
cells/well) were transfected with 30 nM siRNA-ctrl, 30 nM
siRNA-hCINAP, 1 µg empty vector or 1 µg hCINAP-Flag vector at room
temperature using Effectene Transfection Reagent (Qiagen GmbH)
according to the manufacturer's protocol and flow experiments were
performed 48 h after transfection. To induce LDHA activation, AC16
cells were transfected with 50 or 100 ng vector of hCINAP-Flag at
room temperature using Effectene Transfection Reagent (Qiagen GmbH)
according to the manufacturer's protocol.
For co-immunoprecipitation experiments, total
extracts of AC16 cells were lysed using IP lysis buffer (150 mM
NaCl, 25 mM Tris-HCl, pH 7.4, 5% glycerol, 1% NP-40, 1 mM EDTA).
Lysate (1 ml) was incubated overnight at 4°C with 1 µg hCINAP
antibody. Protein A/G-Sepharose (60 µl) was added to samples and
incubated at room temperature for 4 h on a rotary shaker (300 × g).
The pellets were washed three times with 1 ml lysis buffer and
eluted by boiling with 200 µl SDS-PAGE loading buffer. Samples were
analyzed via western blotting as described above.
Annexin V/PI staining
Cells were incubated with Annexin V-FITC Apoptosis
Detection kit (Beijing Solarbio Science & Technology Co., Ltd.)
according to the manufacturer's protocol. Briefly, cells were
stained with 5 µl of Annexin V-FITC and 5 µl PI at room temperature
for 15 min. Following centrifugation at 6,00 × g for 3 min at room
temperature. The population of early apoptotic cells (Annexin
V+/PI−) and late-apoptotic/necrotic cells
(Annexin V+/PI+) was evaluated using a
FACSCanto II flow cytometer (BD Biosciences) and both Annexin
V+ cell populations were considered to be apoptotic
cells. Data was measured by FlowJo v10 (FlowJo, LLC).
Luciferase reporter assay
JASPAR (jaspar.genereg.net) was used to predict whether HIF-1α
bound to the promoter of hCINAP. Promoter regions were defined
using Eukaryotic Promoter Database (epd.vital-it.ch). To construct
pGL3-hCINAP promoter-luciferase constructs, hCINAP promoter (−2350
to +50 bp) was amplified from genomic DNA and assembled into the
pGL3-Basic vector (cat. no. E1751; Promega Corporation). To
construct the truncated hCINAP promoter constructs,
hCINAP-promoter-1 (−2350 to −270 bp), hCINAP-promoter-2 (−901 to
+50 bp), hCINAP-promoter-3 (−901 to −270 bp), hCINAP-promoter-4
(−901 to −549 bp) and hCINAP-promoter-5 (−715 to −270 bp) were
amplified from the pGL3-hCINAP promoter-luciferase vector and
assembled into the pGL3-Basic vector. The primers used for the
establishment of promoter constructs are presented in Table I. AC16 cells were plated
(3×104 cells/well) into 24-well plates and
co-transfected with 150 ng pGL3-hCINAP promoter-luciferase, 15 ng
pcDNA-HIF-1α or empty vector and 15 ng pRL-TK vector (cat. no.
E2241; Promega Corporation) using Effectene Transfection Reagent
(Qiagen GmbH). At 36 h post-transfection, luciferase activities
were measured using a Dual-luciferase reporter kit (Promega
Corporation). Firefly luciferase activities were normalized to
Renilla luciferase activities.
Intracellular immunostaining
AC16 cells (2×105) grown on glass
coverslips were fixed with 4% paraformaldehyde at 4°C for 30 min
and permeabilized with 0.5% Triton X-100 for 15 min at room
temperature. Subsequently, cells were blocked with 5% BSA at room
temperature for 1 h, followed by incubation with cytochrome C
antibody (1:500) overnight at 4°C. Following washing with PBS,
cells were incubated with fluorescein-conjugated goat anti-mouse
IgG (1:1,000; cat. no. A-21202; Thermo Fisher Scientific, Inc.) at
room temperature for 1 h. Subsequently, cells were counterstained
at room temperature for 5 min with Hoechst 33258 (Beijing Solarbio
Science & Technology Co., Ltd.) and observed using an inverted
fluorescence microscope (magnification, ×200).
Statistical analysis
Data are presented the mean ± SEM of at least three
independent experiments. Comparisons among multiple groups were
analyzed using one-way ANOVA followed by Tukey's post hoc test.
Statistical analyses were performed using SPSS software (version
22.0; IBM Corp.). P<0.05 was considered to indicate a
statistically significant difference.
Results
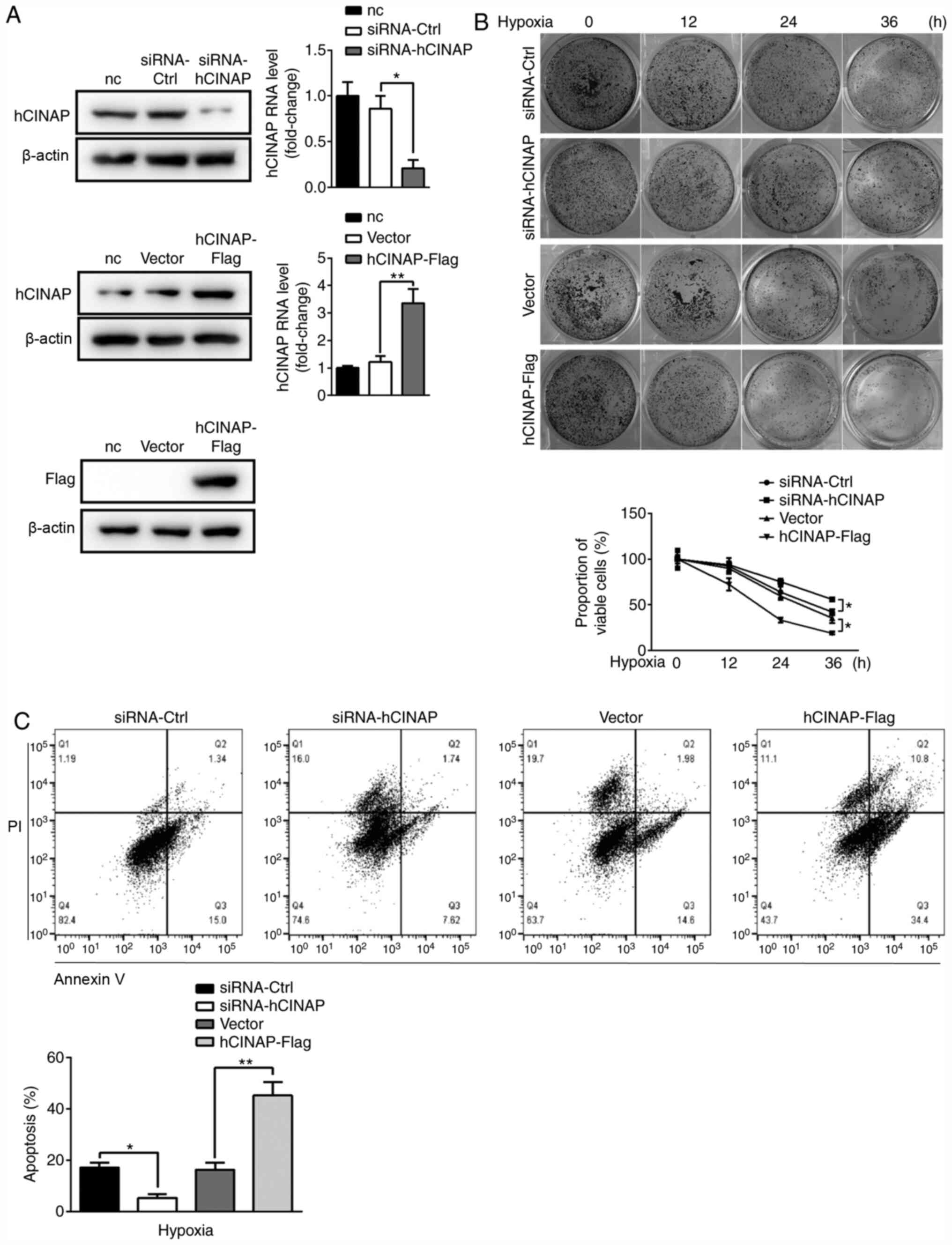
hCINAP knockdown increases cell
survival in response to hypoxia
To identify the role of hCINAP in hypoxia, gain- and
loss-of-function experiments were performed using hCINAP-Flag
vector and siRNA-hCINAP, respectively. Compared with siRNA-Ctrl,
siRNA-hCINAP decreased hCINAP mRNA and protein expression levels,
whereas compared with vector, hCINAP-Flag markedly elevated hCINAP
expression levels (Fig. 1A).
Subsequently, whether altered hCINAP expression affected AC16 cell
viability under hypoxia was investigated. The results indicated
that AC16 cell viability of control group (siRNA-ctrl group and
vector group) was gradually decreased by ~50% over the 36 h of
hypoxia. Following hypoxia for 36 h, cell viability was
significantly higher in hCINAP-knockdown cells compared with the
siRNA-Ctrl group, whereas cell viability was significantly
decreased in hCINAP-overexpression cells compared with the vector
group. Similar results were obtained for the clonogenic assay
(Fig. 1B). The flow cytometry
results indicated that the percentage of apoptotic cells in the
siRNA-hCINAP group was significantly decreased compared with the
siRNA-Ctrl group, whereas hCINAP overexpression displayed the
opposite effect compared with the vector group (Fig. 1C), indicating that hCINAP knockdown
inhibited apoptosis under hypoxic conditions. Collectively, the
aforementioned results suggested that hCINAP knockdown promoted
AC16 cell survival.
hCINAP overexpression elevates
hypoxia-induced lactate production via facilitating LDHA
phosphorylation
It has been reported that hCINAP binds to LDHA and
promotes the phosphorylation of LDHA in colorectal cancer stem
cells (25). Therefore, it was
hypothesized that hCINAP interacted with LDHA in AC16 cells and
co-immunoprecipitation assays were performed. The results indicated
that exogenous hCINAP interacted with LDHA in AC16 cells (Fig. 2A). Compared with the siRNA-Ctrl
group, hCINAP knockdown markedly decreased LDHA phosphorylation,
whereas compared with the vector group, hCINAP overexpression
notably increased LDHA phosphorylation (Fig. 2B). Alterations to hCINAP expression
displayed no obvious effects on LDHA expression, which was
consistent with a previous study (29). Accordingly, LDHA activity was
significantly decreased by hCINAP knockdown compared with the
siRNA-Ctrl group. By contrast, hCINAP overexpression significantly
increased LDHA activity compared with the vector group (Fig. 2C). Lactate serves as an energy
substrate and gluconeogenic precursor, which is essential for
cardiomyocyte survival under hypoxic conditions (32). In the present study, lactate
accumulation was increased in AC16 cells following hypoxia exposure
for 12 h compared with the normoxia group, but lactate accumulation
in the hypoxia and normoxia groups began to fall at 36 h (Fig. 2D). In AC16 cells, hCINAP knockdown
significantly decreased lactate production compared with the
siRNA-Ctrl group, whereas hCINAP overexpression significantly
increased lactate production compared with the vector group
(Fig. 2E). Furthermore, compared
with the vector + hypoxia group, hCINAP overexpression markedly
increased hypoxia-induced LDHA phosphorylation in a dose-dependent
manner (Fig. 2F). The results
indicated that hCINAP promoted hypoxia-induced lactate production
via binding to LDHA and modulating LDHA activity.
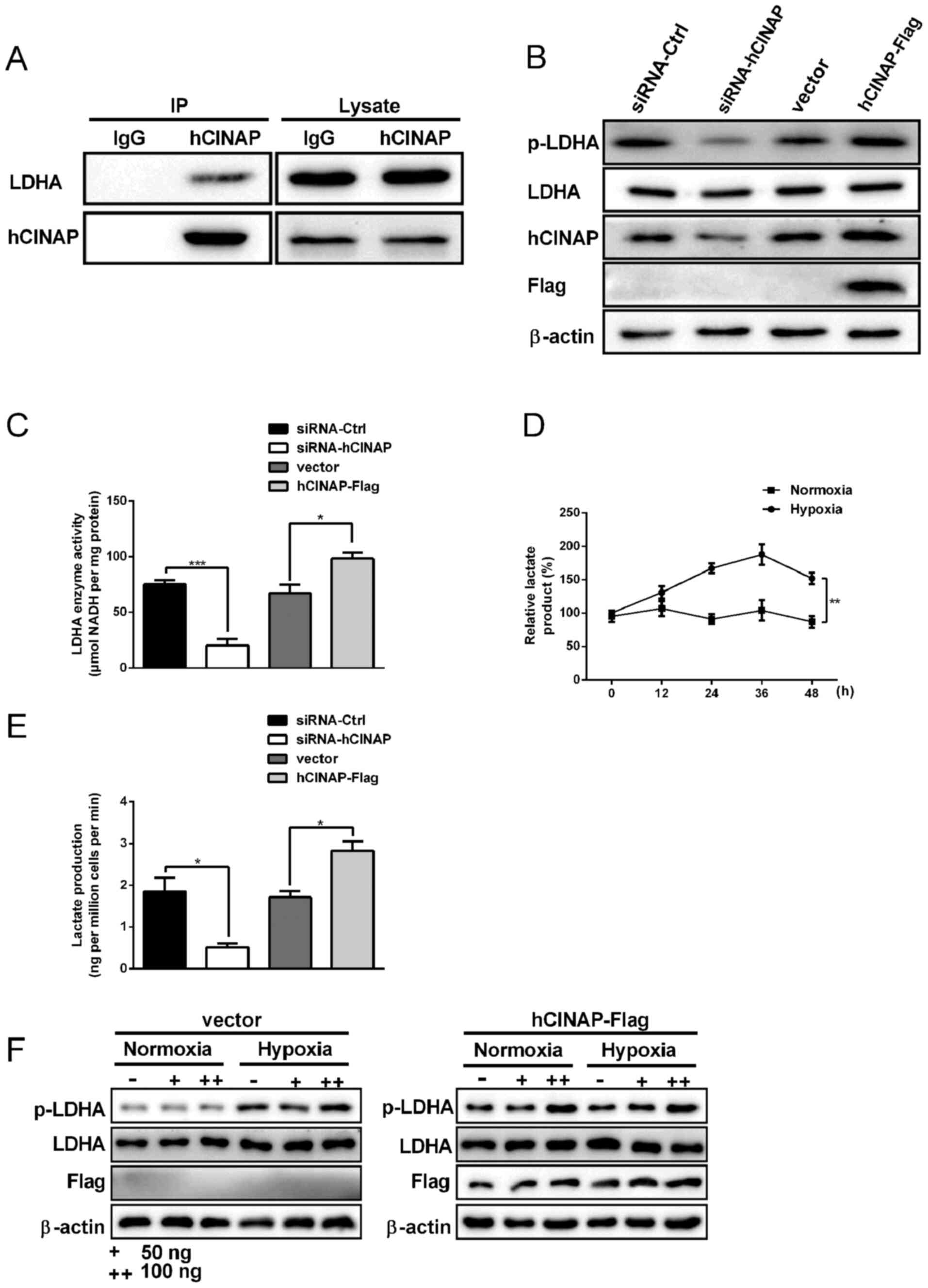 | Figure 2.hCINAP facilitates LDHA
phosphorylation and lactate production. (A) AC16 cells were
transfected with or without hCINAP-Flag. Subsequently,
immunoprecipitation was performed using anti-hCINAP and then
western blotting was performed. (B) AC16 cells were transfected
with siRNA-Ctrl, siRNA-hCINAP, vector or hCINAP-Flag. Subsequently,
western blotting was performed to measure p-LDHA, LDHA, hCINAP and
Flag protein expression levels. (C) LDHA enzyme activities. Lactate
levels in AC16 cells (D) under normoxic and hypoxic conditions or
(E) following transfection with siRNA-Ctrl, siRNA-hCINAP, vector or
hCINAP-Flag. (F) AC16 cells were transfected with 50 or 100 ng
vector of hCINAP-Flag. Subsequently western blotting was performed
to measure p-LDHA, LDHA and Flag protein expression levels under
normoxic and hypoxic conditions. *P<0.05, **P<0.01 and
***P<0.001. hCINAP, human coilin interacting nuclear ATPase
protein; LDHA, lactate dehydrogenase A; siRNA, small interfering
RNA; Ctrl, control; p, phosphorylated; IP, immunoprecipitation. |
HIF-α is recruited to the hCINAP
promoter in hypoxia
To further investigate the role of hCINAP in
hypoxia, whether hCINAP protein expression was increased under
hypoxia was investigated. HIF-1α expression gradually increased in
a time-dependent manner in response to hypoxia. Similarly, hCINAP
protein expression was also increased in a time-dependent manner in
response to hypoxia (Fig. 3A). To
assess whether HIF-1α regulated hypoxia-induced hCINAP, AC16 cells
were transfected with siRNAs targeted against HIF-1α. The western
blotting results demonstrated that hCINAP protein expression levels
were markedly reduced in the siRNA-HIF-1α group compared with the
siRNA-Ctrl group under normoxic and hypoxic conditions (Fig. 3B), suggesting that HIF-1α expression
was required for hCINAP expression. The appropriate hCINAP promoter
sequence ranging from −2,350 bp upstream to +50 bp downstream of
the transcription start site was retrieved from the Eukaryotic
Promoter Database (epd.vital-it.ch). Subsequently, the hCINAP
promoter was screened for the presence of HIF-1α binding sites
using JASPAR (jaspar.genereg.net). Luciferase reporter assays were
performed to identify HIF-1α binding sites within the hCINAP
promoter. Compared with the vector + HIF-1α group, HIF-1α
overexpression significantly increased the luciferase activity of
the hCINAP promoter (−2,350 to +50 bp), suggesting that HIF-1α
bound to the hCINAP promoter and promoted its transcription.
Subsequently, two different regions of the hCINAP promoter were
designed (hCINAP-promoter-1, −2,350 to −270 bp; hCINAP-promoter-2,
−901 to +50 bp) and analyzed by performing luciferase reporter
assays. Compared with the vector + HIF-1α group, HIF-1α
overexpression significantly increased the luciferase activity of
hCINAP-promoter-1 and hCINAP-promoter-2, indicating that the
overlap region of the two promoter regions was required for HIF-1α
binding. Subsequently, the overlap region (−901 to −270 bp) with
the hCINAP promoter was constructed as hCINAP-promoter-3 and
further segmented into hCINAP-promoter-4 (−901 to −549 bp) and
hCINAP-promoter-5 (−715 to −270 bp). Compared with the vector +
HIF-1α group, HIF-1α overexpression significantly enhanced the
luciferase activity of hCINAP-promoter-3 and hCINAP-promoter-4, but
failed to significantly alter the activity of hCINAP-promoter-5,
suggesting that the specific region (−901 to −715 bp) of
hCINAP-promoter-4 was required for HIF-1α binding (Fig. 3C). Furthermore, this key region of
the hCINAP promoter for HIF-1α binding was confirmed by performing
a ChIP-qPCR assay under hypoxic conditions (Fig. 3D). Collectively, the results
demonstrated that hCINAP was a direct target of HIF-1α.
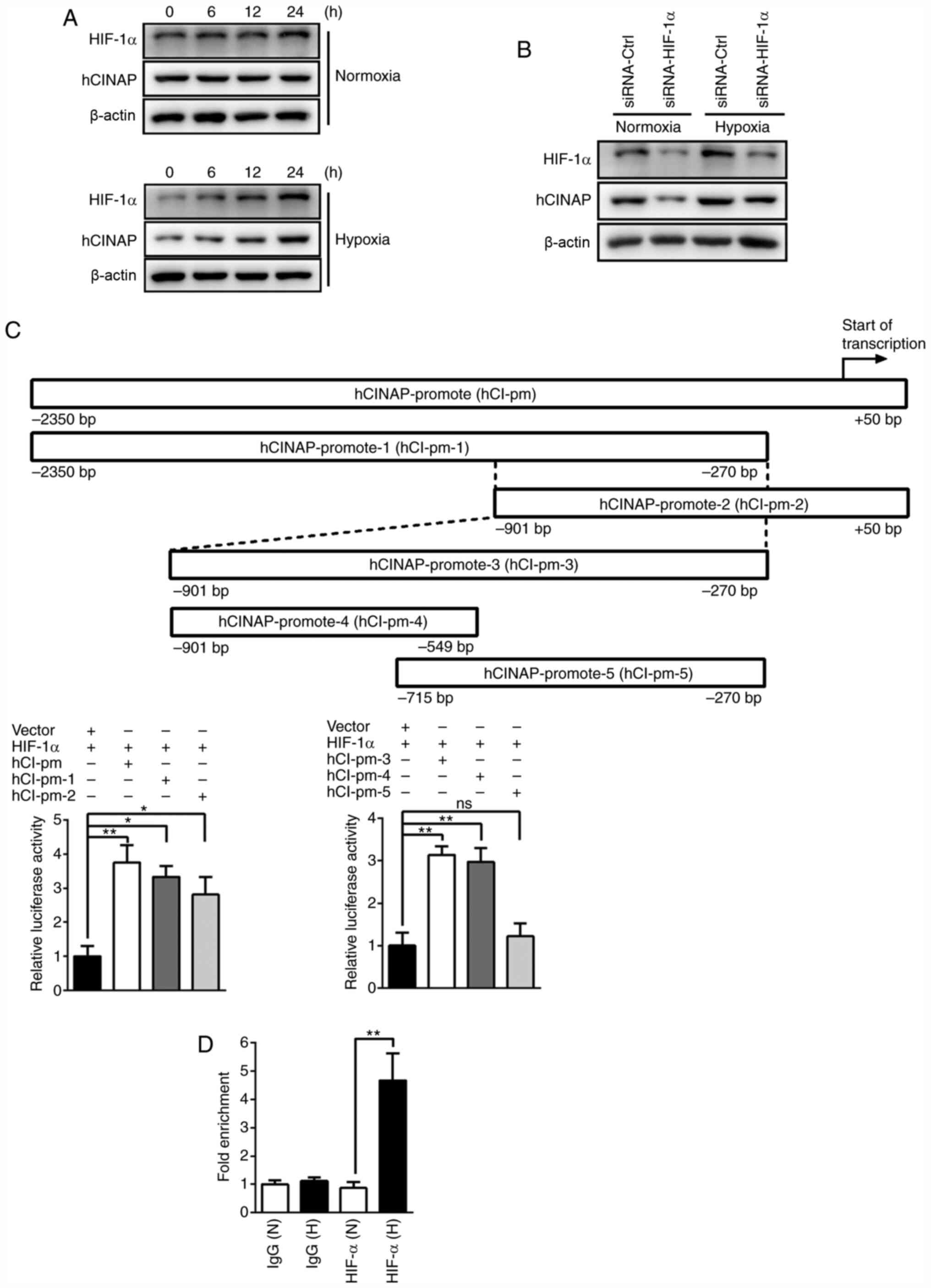 | Figure 3.HIF-1α promotes hCINAP expression by
directly binding to the hCINAP promoter. (A) Western blotting was
performed to measure HIF-1α and hCINAP protein expression levels in
AC16 cells under normoxic or hypoxic conditions. (B) Following
transfection with siRNA-Ctrl or siRNA-HIF-1α, western blotting was
performed to measure HIF-1α and hCINAP protein expression levels in
AC16 cells under normoxic or hypoxic conditions. (C) Different
regions of the hCINAP promoter (hCI-pm-1, −2, −3 −4 and −5) were
constructed into a luciferase reporter vector, and luciferase
reporter assays were performed to determine the effects of HIF-1α
on different partial regions of hCINAP promoter activity. (D)
Chromatin-immunoprecipitation-quantitative PCR was performed to
verify hCINAP as a direct binding target of HIF-1α. *P<0.05 and
**P<0.01. HIF-1α, hypoxia-inducible factor-1α; hCINAP, human
coilin interacting nuclear ATPase protein; siRNA, small interfering
RNA; Ctrl, control; ns, not significant; N, normoxia; H,
hypoxia. |
hCINAP knockdown attenuates
hypoxia-induced apoptosis via the mitochondrial-mediated signaling
pathway
Emerging evidence has indicated that hypoxia-induced
apoptosis involving activation of Caspases can occur in a
cytochrome C-dependent manner (27). Subsequently, the role of hCINAP in
cytochrome C release and Caspase activation was examined.
Immunofluorescence staining indicated that under hypoxic
conditions, cytochrome C was released into the cytoplasm in
siRNA-Ctrl-transfected cells, whereas hCINAP knockdown inhibited
cytochrome C (Fig. 4A).
Consistently, the western blotting results indicated that hypoxia
led to cytochrome C release from the mitochondria into the
cytoplasm in siRNA-Ctrl-transfected cells, but this effect was not
observed in hCINAP-knockdown cells (Fig. 4B), suggesting that hCINAP was
involved in the mitochondrial-mediated apoptotic signaling pathway.
Furthermore, compared with the siRNA-Ctrl group, hCINAP knockdown
significantly decreased hypoxia-induced activation of Caspase-3 and
−9, whereas hCINAP overexpression displayed the opposite effect on
Caspase activation compared with the vector group. Moreover, hCINAP
displayed no significant effect on Caspase activation under
normoxic conditions (Fig. 4C). In
addition, compared with the hypoxia + siRNA-Ctrl group, hCINAP
knockdown significantly inhibited hypoxia-induced depletion of the
mitochondrial membrane potential (Fig.
4D). Therefore, the results indicated that hCINAP knockdown
protected AC16 cells against hypoxia-induced apoptosis via the
mitochondrial-mediated apoptotic signaling pathway.
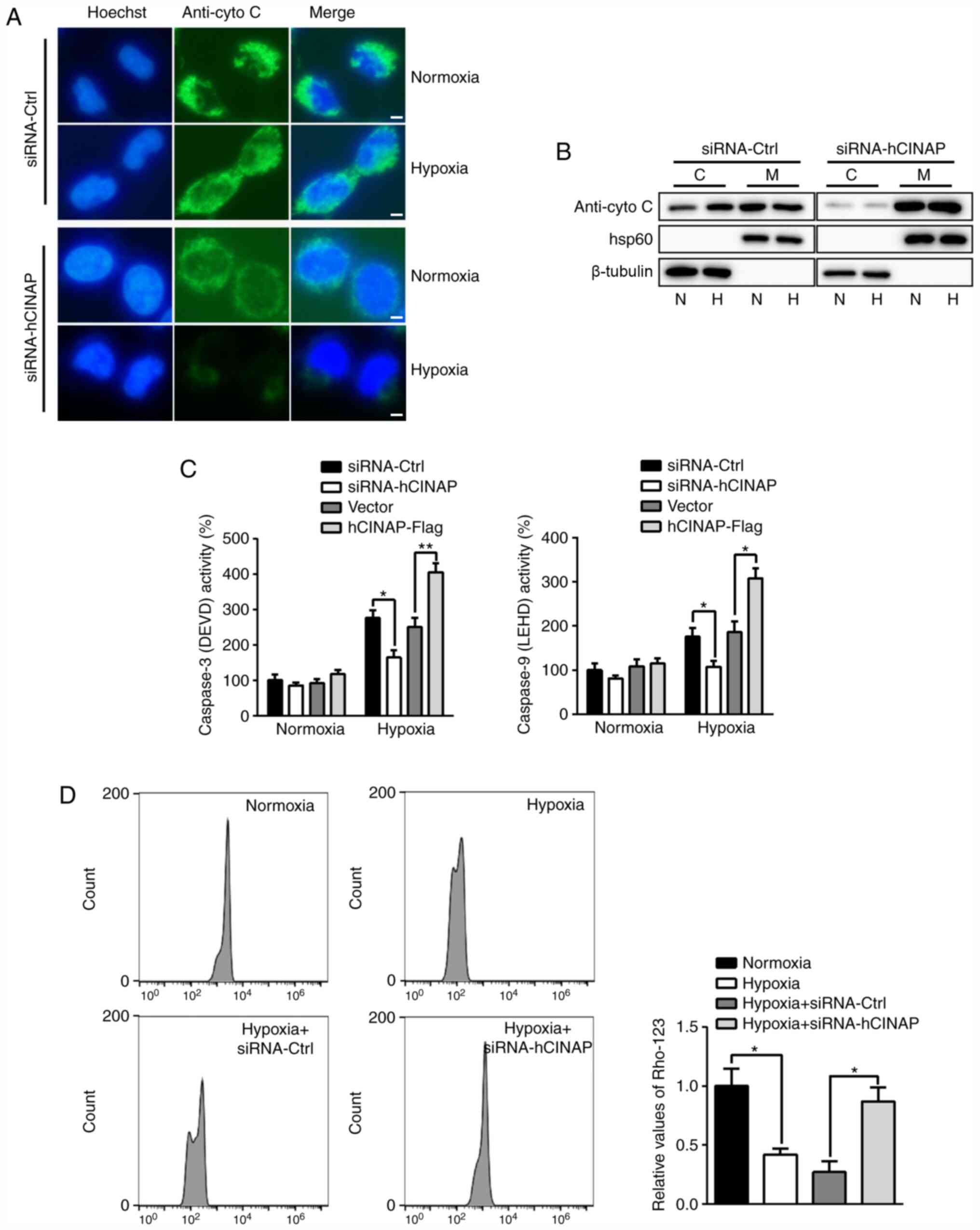 | Figure 4.hCINAP regulates
mitochondrial-mediated apoptosis in hypoxia. Following transfection
with siRNA-Ctrl, siRNA-hCINAP, vector or hCINAP-Flag, AC16 cells
were exposed to normoxia or hypoxia. (A) Cells were harvested and
stained with anti-cytochrome C and Hoechst 33258. Scar bar, 20 µm.
(B) Western blotting was performed to measure the protein
expression levels of cytochrome C in cytosolic and mitochondrial
fractions. (C) Caspase activities were measured using luminogenic
substrate of Ac-DEVD-AMC (Caspase-3) and Z-LEHD-aminoluciferin
(Caspase-9). (D) Mitochondrial membrane potentials were assessed by
staining with rhodamine-123 followed by flow cytometry. *P<0.05
and **P<0.01. hCINAP, human coilin interacting nuclear ATPase
protein; siRNA, small interfering RNA; Ctrl, control; C, cytosolic;
M, mitochondrial; N, normoxia; H, hypoxia; Rho-123, rhodamine-123;
cyto C, cytochrome C. |
hCINAP knockdown inhibits
lactate-induced cardiomyocyte apoptosis
The aforementioned results indicated that hCINAP
promoted LDHA phosphorylation and hypoxia-induced apoptosis.
Therefore, whether hCINAP was involved in lactate
accumulation-induced cell damage was investigated. Following
treatment with 20 mM lactate for 24 h, AC16 cells displayed
significantly increased apoptosis compared with control cells,
which was significantly reduced by transfection with siRNA-hCINAP
compared with siRNA-Ctrl (Fig. 5A),
indicating that hCINAP knockdown blocked lactate-induced apoptosis.
To assess the role of hCINAP in the regulation of lactate-induced
apoptosis via LDHA, an LDHA inhibitor, FX11, was used to
downregulate LDHA activity. Compared with the vector group, hCINAP
overexpression markedly increased Caspase-3 and −9 activation in
response to lactate treatment (Fig.
5B). However, LDHA inhibition by FX11 significantly attenuated
the effect of hCINAP overexpression on Caspase-3 and −9 activation,
suggesting that hCINAP regulated Caspase activation via LDHA.
Furthermore, blocking of LDHA significantly decreased hCINAP
overexpression-induced excessive Caspase-3 and −9 activation under
hypoxic conditions (Fig. 5C). In
addition, compared with the corresponding control groups, hCINAP
knockdown and overexpression displayed no notable effects on MCT1
expression, which facilitates lactate entry into or efflux out of
cells depending on their metabolic state (33) (Fig.
5D). Collectively, the results suggested that hCINAP knockdown
mitigated lactate accumulation-induced apoptosis.
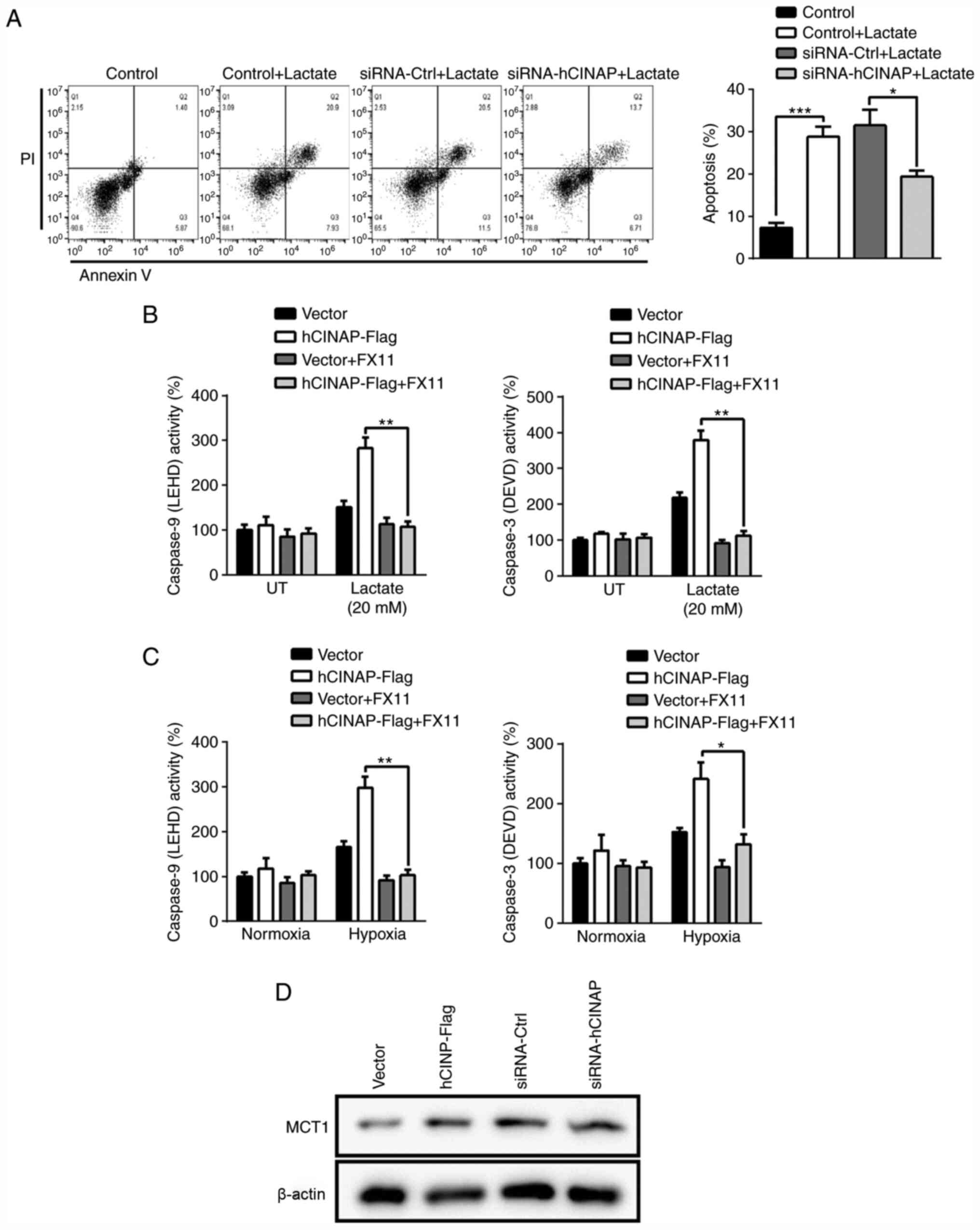 | Figure 5.hCINAP knockdown prevents
lactate-induced apoptosis. (A) Following transfection with
siRNA-Ctrl or siRNA-hCINAP, AC16 cells were treated with lactate
for 12 h. Cell apoptosis was examined by performing Annexin V/PI
staining followed by flow cytometry. (B) Following transfection
with vector or hCINAP-Flag, AC16 cells were treated with or without
LDHA inhibitor FX11 (10 µM), then treated with or without lactate
(20 mM) for 12 h. Caspase activities were measured using
luminogenic substrate of Ac-DEVD-AMC (Caspase-3) and
Z-LEHD-aminoluciferin (Caspase-9). (C) Following transfection with
vector or hCINAP-Flag, AC16 cells were treated with or without LDHA
inhibitor FX11 (10 µM), then exposed to normoxia or hypoxia for 24
h. Caspase activities were measured using luminogenic substrate of
Ac-DEVD-AMC (Caspase-3) and Z-LEHD-aminoluciferin (Caspase-9). (D)
Following transfection with siRNA-Ctrl, siRNA-hCINAP, vector or
hCINAP-Flag, western blotting was performed to measure MCT1 protein
expression levels in AC16 cells. *P<0.05, **P<0.01 and
***P<0.001. hCINAP, human coilin interacting nuclear ATPase
protein; siRNA, small interfering RNA; Ctrl, control; LDHA, lactate
dehydrogenase A; MCT1, monocarboxylate transporter 1; UT,
untreated. |
Discussion
Oxygen deprivation of cardiomyocytes causes a
notable alteration in gene expression and characterizes major
pathological processes, such as myocardial ischemia and myocardial
infarction (3). Cardiomyocyte
apoptosis is a major cellular injury that occurs during AMI
(34). Therefore, understanding the
potential molecular mechanisms underlying hypoxia-induced apoptosis
could aid with the development of therapeutic strategies to prevent
and treat AMI. In the present study, the results indicated that
hCINAP mediated lactate production via LDHA. Compared with normoxia
conditions, hCINAP expression was increased under hypoxic
conditions, leading to LDHA activation. hCINAP knockdown attenuated
mitochondrial-mediated apoptosis signaling in response to
hypoxia.
Homozygous hCINAP knockout mice display embryonic
lethality as ATPase hCINAP regulates 18S rRNA processing, and is
essential for embryogenesis and tumor growth (35). However, the specific role of hCINAP
in cardiomyocytes is not completely understood. The results of the
present study indicated that compared with the vector group, hCINAP
overexpression decreased cardiomyocyte viability, and compared with
the siRNA-Ctrl group, hCINAP knockdown reduced apoptosis under
hypoxic conditions. Conversely, a previous study reported that
hCINAP knockdown increased apoptosis in colorectal cancer stem
cells, whereas colon organoids displayed low sensitivity to hCINAP
knockdown-induced alterations to apoptosis (29), indicating that hCINAP serves
different roles in cancer cells compared with healthy cells. Cancer
cells can acquire necessary nutrients from the malnourished
environment, which are then used to maintain viability and build
new biomass (36). A major
characteristic of tumor cells is the generation of ATP via
glycolysis followed by lactate production, instead of oxidative
phosphorylation, even when sufficient oxygen is present, a
phenomenon that is known as the Warburg effect or aerobic
glycolysis (37). It has been
suggested that hCINAP enhanced aerobic glycolysis via the positive
regulation of LDHA activity, leading to the generation of
extracellular lactate to provide a favorable microenvironment for
CRCSC proliferation and invasion (29). In the present study, the results
demonstrated that hCINAP interacted with LDHA, and hCINAP was
required for the phosphorylation of LDHA in cardiomyocytes.
Furthermore, compared with the siRNA-Ctrl group, hCINAP knockdown
impaired LDHA activity, suggesting that LDHA phosphorylation was
responsible for its enzymatic activity. Lactate is formed by a lack
of oxygen and is utilized under fully aerobic conditions in
mitochondria. The anaerobic metabolism causes overproduction of
lactate and metabolic acidosis, which results in cardiomyocyte
damage (38). Consistent with
previous reports, the present study demonstrated that
cardiomyocytes exposed to hypoxia for a short duration displayed
increased lactate production, whereas hypoxia exposure for a long
duration decreased lactate levels (18,19).
Moreover, compared with the siRNA-Ctrl group, hCINAP knockdown
suppressed hypoxia-induced lactate production. Collectively, the
aforementioned results suggested that hCINAP knockdown protected
against hypoxia-induced damage by regulating the lactate
pathway.
Transcriptional induction of LDHA is caused by
hypoxia, with both HIF-1α and HIF-2α binding to LDHA at 89 bp under
hypoxic conditions (23). In the
present study, HIF-1α bound 901–715 bp upstream of the hCINAP
transcription start site, suggesting that hypoxia-induced
expression of hCINAP was dependent on HIF-1α. Alterations to hCINAP
expression levels did not affect the total expression levels of
LDHA, indicating that hCINAP was involved in the
post-transcriptional regulation of LDHA.
Under normal conditions, ROS display beneficial
effects as they regulate several important physiological reactions
via redox-responsive signaling pathways (39). Additionally, ROS regulate cellular
proliferation, differentiation, vascular tone and adhesion
(40). Tissue hypoxia elevates
intracellular ROS levels, leading to deregulation of cellular ion
homeostasis, cell death and tissue inflammation (10). Moreover, Bcl-2 has been reported to
modulate antioxidant defenses and lipid peroxidation without
altering intracellular ROS levels (41).
Lactate accumulation-induced cytosolic acidification
leads to mitochondrial-dependent apoptosis, including dissipation
of mitochondrial membrane potential, release of cytochrome C and
activation of Caspases (42). The
results of the present study suggested that hCINAP promoted
hypoxia-induced apoptosis by increasing cytochrome C release. In
addition, compared with the siRNA-Ctrl group, hCINAP knockdown
attenuated hypoxia-induced mitochondrial membrane potential
dissipation and impaired the activity of Caspase-3 and −9,
indicating that hCINAP aggravated hypoxia-induced cell death
involving Caspases. However, alterations to hCINAP expression did
not significantly affect Caspase activity under normoxic
conditions. It is likely that low levels of lactate are not
sufficient for inducing Caspase activity under normoxic conditions.
Therefore, the results indicated that hCINAP may participate in the
regulation of mitochondrial-mediated apoptosis under hypoxic
conditions. A previous study also emphasized the oncogene function
of hCINAP in acute myeloid leukemia model mice (28). hCINAP knockdown results in higher
levels of DNA damage and renders cells more sensitive to
chemotherapeutic drugs (28). MCT1
is abundantly expressed in heart tissue localized in mitochondrial
membranes, where it facilitates transport of lactate into
mitochondria for oxidative metabolism (33). Under pathological conditions, such
as hypoxia and ischemia, increased MCT1 expression mitigates
intracellular acidification (19).
In the right atrial appendage of patients with atrial fibrillation,
cytoplasmic lactate concentration and mitochondrial MCT1 expression
were elevated, which suggested that highly expressed MCT1 is
closely related to cardiovascular diseases (43). Previous study reported that MCT1
protein expression was significantly increased in
lactate-stimulated cardiomyocytes when compared with unstimulated
cells (44), whereas hCINAP
knockdown did not notably affect MCT1 expression levels compared
with the siRNA-Ctrl group, indicating that hCINAP regulated lactate
signaling via specifically binding to LDHA. Furthermore, hCINAP
serves as a negative regulator in NF-κB signaling by recruiting
serine/threonine-protein phosphatase 1 to deactivate IKK (45); therefore, the role of hCINAP in
hypoxia-induced inflammatory signaling requires further
investigation.
Collectively, the results of the present study
demonstrated that hCINAP-knockdown cardiomyocytes displayed higher
cell viability and lower apoptosis compared with the siRNA-Ctrl
group, and hCINAP expression was increased in a HIF-1α-dependent
manner under hypoxic conditions. The results also indicated that
hCINAP was involved in mitochondrial-mediated apoptosis, including
the release of cytochrome C and activation of Caspases.
Furthermore, compared with the siRNA-Ctrl group, hCINAP knockdown
suppressed lactate accumulation via inhibiting LDHA activity, which
might contribute to the protective effects of hCINAP in
hypoxia-induced cardiomyocytes. Based on the important role of
hCINAP in regulating hypoxia-induced cell death, understanding the
transcriptional network that regulates hCINAP expression will aid
with the identification of genes that regulate hCINAP expression
under hypoxic conditions. Unfortunately, at present, there are no
drugs or inhibitors that specifically target hCINAP, although it is
a promising candidate for cancer treatment (46). The protective effect of hCINAP
knockdown against cardiomyocyte apoptosis identified in the present
study suggested that selective small-molecule inhibitors and
monoclonal antibodies targeting hCINAP may serve as therapeutic
strategies for hypoxia-related diseases, such as chronic
obstructive pulmonary disease, chronic mountain sickness and
tumorigenesis.
Acknowledgements
Not applicable.
Funding
The present study was supported by the National
Nature Science Foundation of China (grant no. 81470247).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
HX and GX designed the experiments. HX, ZY and GX
analyzed the data. ZY and YG wrote the manuscript. All authors
contributed to data interpretation and critically revised the
manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Benjamin EJ, Virani SS, Callaway CW,
Chamberlain AM, Chang AR, Cheng S, Chiuve SE, Cushman M, Delling
FN, Deo R, et al: Heart disease and stroke statistics-2018 update:
A report from the American heart association. Circulation.
137:e67–e492. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Halestrap AP and Wilson MC: The
monocarboxylate transporter family-role and regulation. IUBMB Life.
64:109–119. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
3
|
Anderson JL and Morrow DA: Acute
myocardial infarction. N Engl J Med. 376:2053–2064. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Nakada Y, Canseco DC, Thet S, Abdisalaam
S, Asaithamby A, Santos CX, Shah AM, Zhang H, Faber JE, Kinter MT,
et al: Hypoxia induces heart regeneration in adult mice. Nature.
541:222–227. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Yin J, Ni B, Liao WG and Gao YQ:
Hypoxia-induced apoptosis of mouse spermatocytes is mediated by
HIF-1α through a death receptor pathway and a mitochondrial
pathway. J Cell Physiol. 233:1146–1155. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zhang H, Liu B, Li T, Zhu Y, Luo G, Jiang
Y, Tang F, Jian Z and Xiao Y: AMPK activation serves a critical
role in mitochondria quality control via modulating mitophagy in
the heart under chronic hypoxia. Int J Mol Med. 41:69–76.
2018.PubMed/NCBI
|
|
7
|
Kim R, Emi M and Tanabe K: Role of
mitochondria as the gardens of cell death. Cancer Chemother
Pharmacol. 57:545–553. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Balaban RS, Nemoto S and Finkel T:
Mitochondria, oxidants, and aging. Cell. 120:483–495. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Soltani M, Moghimian M, Abtahi-Eivari SH,
Shoorei H, Khaki A and Shokoohi M: Protective effects of matricaria
chamomilla extract on torsion/detorsion-induced tissue damage and
oxidative stress in adult rat testis. Int J Fertil Steril.
12:242–248. 2018.PubMed/NCBI
|
|
10
|
Lokmic Z, Musyoka J, Hewitson TD and Darby
IA: Hypoxia and hypoxia signaling in tissue repair and fibrosis.
Int Rev Cell Mol Biol. 296:139–185. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Shokoohi M, Khaki A, Shoorei H, Khaki AA,
Moghimian M and Abtahi-Eivary SH: Hesperidin attenuated
apoptotic-related genes in testicle of a male rat model of
varicocoele. Andrology. 8:249–258. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ameli M, Hashemi MS, Moghimian M and
Shokoohi M: Protective effect of tadalafil and verapamil on
testicular function and oxidative stress after torsion/detorsion in
adult male rat. Andrologia. 50:e130682018. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Denko NC: Hypoxia, HIF1 and glucose
metabolism in the solid tumour. Nat Rev Cancer. 8:705–713. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Mandl M and Depping R: Hypoxia-inducible
aryl hydrocarbon receptor nuclear translocator (ARNT) (HIF-1β): Is
it a rare exception? Mol Med. 20:215–220. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Depping R, Jelkmann W and Kosyna FK:
Nuclear-cytoplasmatic shuttling of proteins in control of cellular
oxygen sensing. J Mol Med (Berl). 93:599–608. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Brahimi-Horn MC, Bellot G and Pouyssegur
J: Hypoxia and energetic tumour metabolism. Curr Opin Genet Dev.
21:67–72. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Loor G and Schumacker PT: Role of
hypoxia-inducible factor in cell survival during myocardial
ischemia-reperfusion. Cell Death Differ. 15:686–690. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Evans RK, Schwartz DD and Gladden LB:
Effect of myocardial volume overload and heart failure on lactate
transport into isolated cardiac myocytes. J Appl Physiol (1985).
94:1169–1176. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Johannsson E, Lunde PK, Heddle C, Sjaastad
I, Thomas MJ, Bergersen L, Halestrap AP, Blackstad TW, Ottersen OP
and Sejersted OM: Upregulation of the cardiac monocarboxylate
transporter MCT1 in a rat model of congestive heart failure.
Circulation. 104:729–734. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Fiume L, Manerba M, Vettraino M and Di
Stefano G: Inhibition of lactate dehydrogenase activity as an
approach to cancer therapy. Future Med Chem. 6:429–445. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ždralević M, Brand A, Di Ianni L, Dettmer
K, Reinders J, Singer K, Peter K, Schnell A, Bruss C, Decking SM,
et al: Double genetic disruption of lactate dehydrogenases A and B
is required to ablate the ‘Warburg effect’ restricting tumor growth
to oxidative metabolism. J Biol Chem. 293:15947–15961. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Urbanska K and Orzechowski A:
Unappreciated role of LDHA and LDHB to control apoptosis and
autophagy in tumor cells. Int J Mol Sci. 20:20852019. View Article : Google Scholar
|
|
23
|
Cui XG, Han ZT, He SH, Wu XD, Chen TR,
Shao CH, Chen DL, Su N, Chen YM, Wang T, et al: HIF1/2α mediates
hypoxia-induced LDHA expression in human pancreatic cancer cells.
Oncotarget. 8:24840–24852. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Santama N, Ogg SC, Malekkou A, Zographos
SE, Weis K and Lamond AI: Characterization of hCINAP, a novel
coilin-interacting protein encoded by a transcript from the
transcription factor TAFIID32 locus. J Biol Chem. 280:36429–36441.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Granneman S, Nandineni MR and Baserga SJ:
The putative NTPase Fap7 mediates cytoplasmic 20S pre-rRNA
processing through a direct interaction with Rps14. Mol Cell Biol.
25:10352–10364. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zhang J, Bai D, Ma X, Guan J and Zheng X:
hCINAP is a novel regulator of ribosomal protein-HDM2-p53 pathway
by controlling NEDDylation of ribosomal protein S14. Oncogene.
33:246–254. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhang J, Zhang F and Zheng X: Depletion of
hCINAP by RNA interference causes defects in Cajal body formation,
histone transcription, and cell viability. Cell Mol Life Sci.
67:1907–1918. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Xu R, Yu S, Zhu D, Huang X, Xu Y, Lao Y,
Tian Y, Zhang J, Tang Z, Zhang Z, et al: hCINAP regulates the
DNA-damage response and mediates the resistance of acute myelocytic
leukemia cells to therapy. Nat Commun. 10:38122019. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Ji Y, Yang C, Tang Z, Yang Y, Tian Y, Yao
H, Zhu X, Zhang Z, Ji J and Zheng X: Adenylate kinase hCINAP
determines self-renewal of colorectal cancer stem cells by
facilitating LDHA phosphorylation. Nat Commun. 8:153082017.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Iida A, Iwagawa T, Kuribayashi H, Satoh S,
Mochizuki Y, Baba Y, Nakauchi H, Furukawa T, Koseki H, Murakami A
and Watanabe S: Histone demethylase Jmjd3 is required for the
development of subsets of retinal bipolar cells. Proc Natl Acad Sci
USA. 111:3751–3756. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Brooks GA: Energy flux, lactate shuttling,
mitochondrial dynamics, and hypoxia. Adv Exp Med Biol. 903:439–455.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Bonen A: The expression of lactate
transporters (MCT1 and MCT4) in heart and muscle. Eur J Appl
Physiol. 86:6–11. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Prech M, Marszalek A, Schröder J, Filas V,
Lesiak M, Jemielity M, Araszkiewicz A and Grajek S: Apoptosis as a
mechanism for the elimination of cardiomyocytes after acute
myocardial infarction. Am J Cardiol. 105:1240–1245. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Bai D, Zhang J, Li T, Hang R, Liu Y, Tian
Y, Huang D, Qu L, Cao X, Ji J and Zheng X: The ATPase hCINAP
regulates 18S rRNA processing and is essential for embryogenesis
and tumour growth. Nat Commun. 7:123102016. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Leong SP, Aktipis A and Maley C: Cancer
initiation and progression within the cancer microenvironment. Clin
Exp Metastasis. 35:361–367. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Warburg O: On the origin of cancer cells.
Science. 123:309–314. 1956. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Saddik M and Lopaschuk GD: Myocardial
triglyceride turnover and contribution to energy substrate
utilization in isolated working rat hearts. J Biol Chem.
266:8162–8170. 1991.PubMed/NCBI
|
|
39
|
Liochev SI: Reactive oxygen species and
the free radical theory of aging. Free Radic Biol Med. 60:1–4.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Martinon F: Signaling by ROS drives
inflammasome activation. Eur J Immunol. 40:616–619. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Hockenbery DM, Oltvai ZN, Yin XM, Milliman
CL and Korsmeyer SJ: Bcl-2 functions in an antioxidant pathway to
prevent apoptosis. Cell. 75:241–251. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Jeong D, Kim TS, Lee JW, Kim KT, Kim HJ,
Kim IH and Kim IY: Blocking of acidosis-mediated apoptosis by a
reduction of lactate dehydrogenase activity through antisense mRNA
expression. Biochem Biophys Res Commun. 289:1141–1149. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Xu J, Xu X, Si L, Xue L, Zhang S, Qin J,
Wu Y, Shao Y, Chen Y and Wang X: Intracellular lactate signaling
cascade in atrial remodeling of mitral valvular patients with
atrial fibrillation. J Cardiothorac Surg. 8:342013. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Gao C, Wang F, Wang Z, Zhang J and Yang X:
Asiatic acid inhibits lactate-induced cardiomyocyte apoptosis
through the regulation of the lactate signaling cascade. Int J Mol
Med. 38:1823–1830. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Qu L, Ji Y, Zhu X and Zheng X: hCINAP
negatively regulates NF-κB signaling by recruiting the phosphatase
PP1 to deactivate IKK complex. J Mol Cell Biol. 7:529–542. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Yan Y, Yuan X, Xue C and He Y: Human
coilin interacting nuclear ATPase protein in cancer: Uncovering new
insights into pathogenesis and therapy. Am J Transl Res.
12:4051–4058. 2020.PubMed/NCBI
|