Introduction
Sepsis is a dysregulated host response that can lead
to multiple organ dysfunction (1,2) and is
the primary cause of morbidity and mortality among patients in
intensive care units (ICUs) worldwide (3). The heart is one of the most
susceptible organs to sepsis-induced damage and myocardial
dysfunction is a primary cause of mortality in patients with sepsis
(4). The bacterial endotoxin
lipopolysaccharide (LPS) is the principal cause of septic heart
failure due to stimulating excessive inflammatory mediator
production, abnormal gene regulation and mitochondrial dysfunction
(5–7). At present, no effective therapeutic
strategies for sepsis-induced myocardial dysfunction have been used
in the clinic. Thus, the precise mechanism underlying LPS-induced
septic heart injury requires investigation to aid with the
development of novel therapeutic agents.
LPS stimulates inflammatory responses by activating
and releasing multiple cytokines, including IL-1β (8), which is an important contributor to
myocardial dysfunction in sepsis (9). IL-1β expression, maturation and
secretion processes are highly regulated by the NLR family pyrin
domain containing 3 (NLRP3) inflammasome (10). The NLRP3 inflammasome consists of
three essential components: NLRP3, an inflammasome sensor molecule;
apoptosis-associated speck-like protein containing A CARD (ASC), an
adaptor protein; and pro-caspase-11, an inflammatory protease
(11). The activated NLRP3
inflammasome triggers caspase-1 activation, which promotes the
cleavage and secretion of proinflammatory cytokines IL-1β and IL-18
(12). NLRP3 inflammasome
activation-induced IL-1β production involves high mobility group
box-1 protein (HMGB1) (13), which
is a key cytokine in the promotion of cellular activation and
inflammatory responses during LPS-induced myocardial injury
(14). Peroxisome
proliferator-activated receptor γ (PPARγ) is implicated in the
regulation of LPS-induced HMGB1 release in RAW 264.7 cells
(15). PPARγ activation mediates
LPS-induced inflammation and sepsis-related heart dysfunction in
mice (16). Therefore, it was
hypothesized that PPARγ activation to target HMGB1/NLRP3 signaling
may alleviate LPS-induced myocardial cell injury.
Propofol (2,6-diisopropylphenol) is widely used to
induce and maintain anesthetic effects, and sedate patients in ICUs
(17). Alongside the anesthetic
functions, propofol also displays pharmacological properties
(18,19). A clinical study demonstrated that
the antioxidant capacity of plasma is increased during propofol
anesthesia (20), and propofol
suppresses proinflammatory cytokine production in patients with
sepsis (21). In vitro and
in vivo experiments have suggested that propofol elicits
cardioprotective effects (22,23).
Intriguingly, propofol displays inhibitory effects on myocardial
ischemia-reperfusion injury in various experimental models by
reducing oxidative stress (24),
protecting mitochondrial function (25) and suppressing apoptosis (26). However, whether propofol protects
against LPS-induced myocardial cell injury is not completely
understood.
The aim of the present study was to investigate the
biological functions and the underlying mechanism of propofol in
LPS-induced cardiomyocyte injury.
Materials and methods
Neonatal rat primary cardiomyocyte
isolation and culture
The animal experimental protocol was approved by Air
Force Medical University's Institutional Animal Care and Use
Committee (approval no. 2017-01347). Healthy neonatal male
Sprague-Dawley rats (male; age, 6–24 h; weight, 5–6 g) were
obtained from the Institute of Zoology, Chinese Academy of Medical
Sciences. The rats were maintained in specific pathogen-free
conditions at 22±2°C and 60% humidity, with a 12-h light/dark cycle
and free access to food and water. The primary cardiomyocytes were
prepared from ventricles of neonatal rats according to a previously
described protocol with slight modifications (27,28).
In brief, rats were euthanized with an intraperitoneal injection of
sodium pentobarbital (200 mg/kg) under sterile conditions.
Subsequently, hearts were collected from rats and cut into pieces
in Ca2+- and Mg2+-free D-Hanks balanced salt
solution (Beyotime Institute of Biotechnology) supplemented with
0.1% trypsin. Then, 0.1% type II collagenase was added to digest
the cells four times. Cells were centrifuged at 4°C and 120 × g for
5 min. The pellets were resuspended in DMEM (Gibco; Thermo Fisher,
Scientific, Inc.) supplemented with 15% FBS (Gibco; Thermo Fisher,
Scientific, Inc.), 100 U/ml penicillin and 100 µg/ml streptomycin.
Cells were transferred into 60-mm primary culture dishes (Corning
Inc.) precoated with 1% gelatin at 37°C for 1.5 h to remove
non-cardiomyocytes. Non-adherent cardiomyocytes were plated
(1×106 cells/dish) into 60-mm gelatin-precoated primary
culture dishes. After 24 h, cardiomyocytes were washed with PBS
(Beyotime Institute of Biotechnology) thrice and cultured in
serum-free maintenance medium [80% DMEM, 20% M199 (Gibco; Thermo
Fisher, Scientific, Inc.), 100 U/ml penicillin and 100 µg/ml
streptomycin]. To validate cardiomyocytes, striated muscle-specific
sarcomeric α-actin monoclonal antibody was employed.
Cardiomyocyte transfection
Cardiomyocytes were cultured in serum-free
maintenance medium at 37°C with 5% CO2. At 80%
confluence, cells were transfected with 100 nM non-targeting small
interfering RNA (si)-negative control (NC; siNC), siHMGB1 or
siPPARγ (all purchased from Shanghai GenePharma Co., Ltd.) for 48 h
at 37°C using Lipofectamine RNAiMAX (Invitrogen; Thermo Fisher
Scientific, Inc.) according to the manufacturer's protocol.
Following 48 h of transfection, the cells were used for subsequent
experiments. The sequences of each siRNA were as follows: siNC,
5′-GCTCTGGAGCAGTTCCGATAT-3′; siHMGB1, 5′-CCATCACAGTGTTGTTAA-3′; and
siPPARγ, 5′-TAACGAATGGGATTTGTCTG-3′.
Experimental design and cell
treatments
Cardiomyocytes were divided into the following
groups: i) Control, cardiomyocytes cultured in serum-free
maintenance medium; ii) propofol, cardiomyocytes treated with
propofol (Sigma-Aldrich; Merck KGaA; 12.5, 25, 50 or 100 µM) for 6,
12, 24 or 48 h; iii) LPS, cardiomyocytes stimulated with 1 µg/ml
LPS (Escherichia coli 055:B5; Sigma-Aldrich; Merck KGaA) for
4 h; iv) propofol + LPS, cardiomyocytes treated with propofol
(12.5, 25, 50 or 100 µM) for 6, 12, 24 or 48 h then stimulated with
1 µg/ml LPS for 4 h; v) propofol + LPS + siRNA, cardiomyocytes
transfected with 100 nM siNC, siHBGB1 or siPPARγ for 24 h, treated
with propofol (50 µM) for 24 h and then stimulated with 1 µg/ml LPS
for 4 h; vi) propofol + LPS + inhibitor, cardiomyocytes pretreated
with recombinant rat HMGB1 (rHMGB1; Chimerigen Laboratories; 100
ng/ml) or GW9662 (a PPARγ activation inhibitor; MedChemExpress; 10
µM) for 30 min followed by treatment with propofol (50 µM) for 24 h
and then stimulation with 1 µg/ml LPS for 4 h. All the treatments
were incubated at 37°C in a humidified atmosphere containing 5%
CO2.
Cell viability assay
An MTT assay (Dojindo Molecular Technologies, Inc.)
was performed to measure cell viability. Cardiomyocytes were seeded
(2×104 cells/well) into a 96-well plate. Following
culture for 24 h, cells were treated as aforementioned.
Subsequently, 10 µl MTT (0.5 mg/ml) was added to each well and
incubated at 37°C for 4 h. Cell culture medium was removed and DMSO
was added to dissolve the purple formazan crystals. The absorbance
of each well was measured at a wavelength of 570 nm using a
microplate reader (Bio-Rad Laboratories, Inc.). Cell viability is
presented as a percentage of the control.
Measurement of lactate dehydrogenase
(LDH) release
Following treatment, the culture media from each
group was collected. LDH release in the culture media was measured
using the LDH assay kit (Nanjing Jiancheng Bioengineering
Institute) according to the manufacturer's protocol.
Measurement of cardiomyocyte apoptosis
via flow cytometry
Cardiomyocyte apoptosis was determined using an
Annexin V-FITC/PI apoptosis detection kit (BD Biosciences).
Following treatment, cells were harvested, washed twice with
ice-cold PBS (pH 7.4) and centrifuged at 300 × g at 4°C for 5 min.
Cells were resuspended in 1X binding buffer to a final
concentration of 5×105 cells/ml. Subsequently, 5 µl
Annexin V-FITC and 10 µl PI were added to the cells, gently
vortexed and incubated in the dark at 37°C for 15 min. Apoptotic
cardiomyocytes were analyzed using a FACSCalibur flow cytometer (BD
Biosciences) and Cell Quest software (version 3.3; BD Biosciences).
Early (positive Annexin V-FITC staining) plus late apoptotic cells
(positive PI staining) were counted to determine the levels of
cardiomyocyte apoptosis.
Measurement of mitochondrial membrane
potential (MMP)
Cardiomyocyte MMP was measured using a
5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolocarbocyanine
iodide (JC-1) detection kit (Nanjing Jiancheng Bioengineering
Institute). JC-1 is a dual-emission MMP sensing dye that
selectively aggregates in polarized (healthy) mitochondria and
aggregated JC-1 emits red fluorescence. Mitochondrial
depolarization (loss of MMP) prevents JC-1 from entering the
mitochondria, thus monomeric JC-1 remains in the cytosol and emits
green fluorescence. Following treatment, cardiomyocytes were washed
with PBS thrice, incubated with 200 µM JC-1 at 37°C for 15 min and
washed twice with PBS. The fluorescence of aggregated JC-1 (red)
was visualized at an emission wavelength of 590 nm and the
fluorescence of monomeric JC-1 (green) was visualized at a
wavelength of 529 nm using a fluorescence microscope
(magnification, ×400; Carl Zeiss AG). The ratio of red to green
fluorescence intensity was calculated to determine the MMP.
Caspase-3 activity assay
Caspase-3 activity was detected to evaluate cell
apoptosis using a Caspase-3 activity assay kit (Beyotime Institute
of Biotechnology) according to the manufacturer's protocol.
Caspase-3 catalyzes the conversion of acetyl-Asp-Glu-Val-Asp
p-nitroanilide (Ac-DEVD-pNA) into pNA, which displays a
strong absorption peak at a wavelength of 405 nm. Following
treatment, cardiomyocytes were lysed using RIPA lysis buffer
(Beyotime Institute of Biotechnology) and centrifuged at 1,000 × g
at 4°C for 15 min. The supernatants were harvested to evaluate
caspase-3 activity. The supernatant (50 µl) was added to 2X
reaction buffer (50 µl) and Ac-DEVD-pNA (50 µl) and incubated at
37°C for 4 h. Absorbance was measured at a wavelength of 405 nm
using a microplate reader.
Reverse transcription-quantitative PCR
(qPCR)
Following treatment, total RNA was extracted from
cardiomyocytes using TRIzol® (Invitrogen; Thermo Fisher
Scientific, Inc.) according to the manufacturer's instructions.
Total RNA was reverse transcribed into cDNA using PrimeScript RT
master mix (Takara Bio, Inc.). The temperature protocol for the
reverse transcription was 5 min at 25°C, 60 min at 42°C and 15 min
at 70°C. Subsequently, qPCR was performed using SYBR-Green
Premix-Ex Tag kit (Takara Bio, Inc.) on an ABI 7500 Fast Real-time
PCR system (Applied Biosystems; Thermo Fisher Scientific, Inc.).
The following thermocycling conditions were used for the qPCR:
Initial denaturation for 10 min at 95°C; followed by 40 cycles of
15 sec at 95°C and 60 sec at 60°C. The following primers were used
for qPCR: TNF-α forward, 5′-TGGAACTGGCAGAAGAGGCACT-3′ and reverse,
5′-GTTCAGTAGACAGAAGAGCGTGGTG-3′; IL-6 forward,
5′-AGAAAAGAGTTGTGCAATGGCA-3′ and reverse,
5′-GGCAAATTTCCTGGTTATATCC-3′; IL-1β forward,
5′-CACTACAGGCTCCGAGATGAACAAC-3′ and reverse,
5′-TTGTCGTTGCTTGGTTCTCCTTGT-3′; IL-18 forward,
5′-GACTGGCTGTGACCCTATCTGTGA-3′ and reverse,
5′-TTGTGTCCTGGCACACGTTTC-3′; and GAPDH forward,
5′-TCCATGACAACTTTGGTATCG-3′ and reverse,
5′-TGTAGCCAAATTCGTTGTCA-3′. mRNA expression levels were quantified
using the 2−ΔΔCq method (29) and normalized to the internal
reference gene GAPDH.
ELISA
Following treatment, cardiomyocyte culture media was
harvested and centrifuged at 500 × g at room temperature for 5 min.
The supernatants were pooled to measure TNF-α, IL-6, IL-1β and
IL-18 levels using rat TNF-α (cat. no. 560479; BD Biosciences),
IL-6 (cat. no. 550319; BD Biosciences), IL-1β (cat. no. ab255730;
Abcam) and IL-18 (cat. no. ab213909; Abcam) ELISA kits,
respectively, according to the manufacturer's protocols.
Western blotting
Following treatment, cardiomyocytes were harvested,
lysed in RIPA lysis buffer (Beyotime Institute of Biotechnology)
and centrifuged at 1,000 × g at 4°C for 10 min. Protein
concentrations in the supernatant were quantified using a BCA assay
kit (Beyotime Institute of Biotechnology). Proteins (30 µg) were
separated via 10% SDS-PAGE and transferred onto PVDF membranes (EMD
Millipore). The membranes were blocked with 5% skimmed milk in TBS
containing 0.1% Tween-20 (TBST) for 1 h at room temperature.
Subsequently, the membranes were incubated at 4°C overnight with
primary antibodies targeted against: NLRP3 (Santa Cruz
Biotechnology, Inc.; cat. no. sc-134306; 1:1,000), ASC (Santa Cruz
Biotechnology, Inc.; cat. no. sc-514414; 1:1,000), pro-caspase-1
(Santa Cruz Biotechnology, Inc.; cat. no. sc-56036; 1:800),
caspase-1 p10 (Santa Cruz Biotechnology, Inc.; cat. no. sc-514;
1:500), pro-IL-1β (Santa Cruz Biotechnology, Inc.; cat. no.
sc-12742; 1:500), IL-1β (Santa Cruz Biotechnology, Inc.; cat. no.
sc-515598; 1:1,000), pro-IL-18 (Abcam; cat. no. ab191860; 1:100),
IL-18 (Abcam; cat. no. ab223293; 1:1,000), HMGB1 (Abcam; cat. no.
ab79823; 1:10,000), PPARγ (Abcam; cat. no. ab272718; 1:1,000) and
β-actin (Abcam; cat. no. ab6276; 1:5,000). Following primary
antibody incubation, the membranes were washed three times with
TBST and incubated with an HRP-conjugated goat anti-rabbit IgG
(Abcam; cat. no. ab6721; 1:5,000), HRP-conjugated rabbit anti-mouse
IgG (Abcam; cat. no. ab6728; 1:5,000) or HRP-conjugated goat
anti-Armenian hamster IgG (Abnova; cat. no. PAB9133; 1:3,000)
secondary antibodies for 1 h at room temperature. The membranes
were washed three times with TBST. Protein bands were visualized
using an ECL kit (Pierce; Thermo Fisher Scientific, Inc.) followed
by exposure to X-ray films. β-actin was used as the loading
control.
Co-immunoprecipitation (Co-IP)
Following treatment, cardiomyocytes were harvested,
lysed in RIPA lysis buffer (Beyotime Institute of Biotechnology)
and centrifuged at 1,000 × g at 4°C for 10 min. Protein
concentrations in the supernatant were quantified using a BCA assay
kit. Cell lysates (500 µg) were precleared with 20 µl of protein
A/G-agarose beads (Santa Cruz Biotechnology, Inc.), according to
the manufacturer's protocol, and incubated with anti-ASC antibody
(Santa Cruz Biotechnology, Inc. cat. no. sc-514414) or normal IgG
at 4°C overnight with gentle agitation. Subsequently, 20 µl protein
A/G-agarose beads were added to the immune complexes and incubated
for 6 h at 4°C. The resultant mixtures were centrifuged at 250 × g
at 4°C for 5 min and the supernatants were removed. The pellets
were washed three times with PBS and western blotting was performed
according to the aforementioned protocol. Proteins were separated
via SDS-PAGE and transferred onto PVDF membranes. The membranes
were incubated with primary antibodies targeted against: NLRP3, ASC
and caspase-1. Subsequently, the membranes were incubated with
HRP-conjugated secondary antibodies. Protein bands were visualized
using an ECL kit.
Statistical analysis
Data are presented as the mean ± SD from three
independent experiments. Statistical analyses were performed using
SPSS software (version 17.0; SPSS, Inc.). Comparisons among
multiple groups were analyzed using one-way ANOVA followed by
Tukey's post hoc test. P<0.05 was considered to indicate a
statistically significant difference.
Results
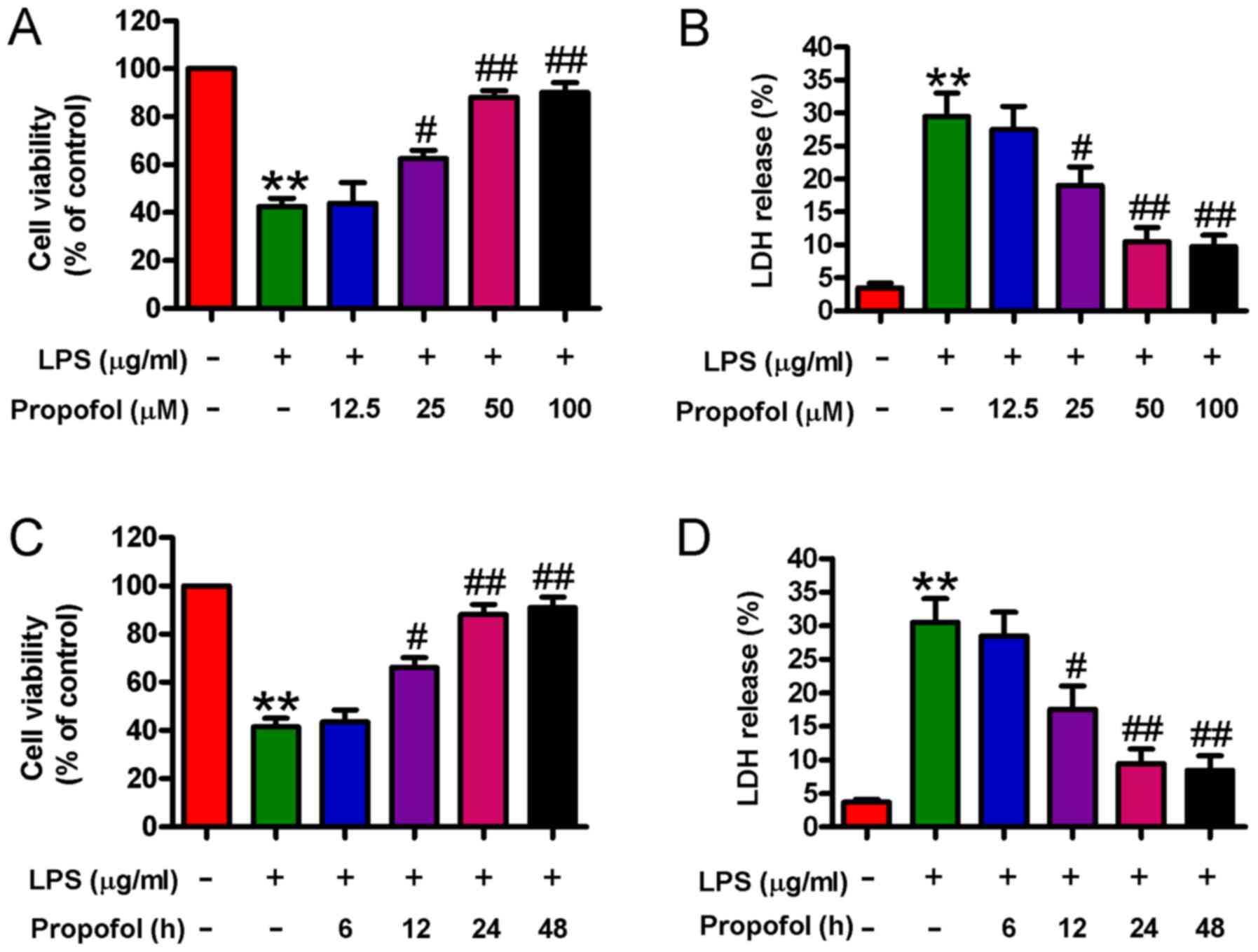
Propofol protects cardiomyocytes
against LPS-induced injury
Propofol has been reported to attenuate LPS-induced
myocardial injury (22,23). The MTT and LDH release assays were
performed to determine the effects of propofol on LPS-induced
cardiomyocyte damage. Compared with the control group, LPS
stimulation significantly reduced cardiomyocyte viability and
enhanced LDH release, whereas propofol (25, 50, or 100 µM)
pretreatment for 24 h significantly increased cell viability and
decreased LDH release in LPS-stimulated cardiomyocytes in a
concentration-dependent manner; however, there were no significant
differences observed between the 50 and 100 µM propofol groups
(Fig. 1A and B). Compared with the
LPS group, LPS-stimulated cardiomyocytes that were pretreated with
50 µM propofol for different durations (12, 24, and 48 h) displayed
significantly increased cell viability and decreased LDH release in
a time-dependent manner; however, there was no significant
difference observed between the 24 and 48 h groups (Fig. 1C and D). Therefore, 50 µM propofol
treatment for 24 h was selected for subsequent experiments due to
its protective effects against LPS-induced injury in
cardiomyocytes. The results indicated that propofol ameliorated
LPS-induced cardiomyocyte injury.
Propofol inhibits LPS-induced
cardiomyocyte apoptosis and inflammation
The effect of propofol on LPS-induced cardiomyocyte
apoptosis and inflammation was assessed. The flow cytometry results
indicated that compared with the control group, LPS significantly
increased cell apoptosis, which was significantly reduced by
pretreatment with propofol (Fig. 2A and
B). Propofol also significantly abolished LPS-induced loss of
MMP (Fig. 2C). Moreover,
LPS-induced increases in caspase-3 activity were significantly
reduced by pretreatment with propofol (Fig. 2D). The mRNA expression levels of
proinflammatory cytokines, including TNF-α and IL-6, were
significantly increased by LPS treatment compared with the control
group. However, pretreatment with propofol significantly decreased
TNF-α and IL-6 expression levels in LPS-treated cardiomyocytes
(Fig. 2E and F). Similarly,
LPS-injured cardiomyocytes that were pretreated with propofol
released significantly less TNF-α and IL-6 compared with the LPS
group (Fig. 2G and H). The results
demonstrated that propofol alleviated LPS-induced cardiomyocyte
apoptosis and inflammation.
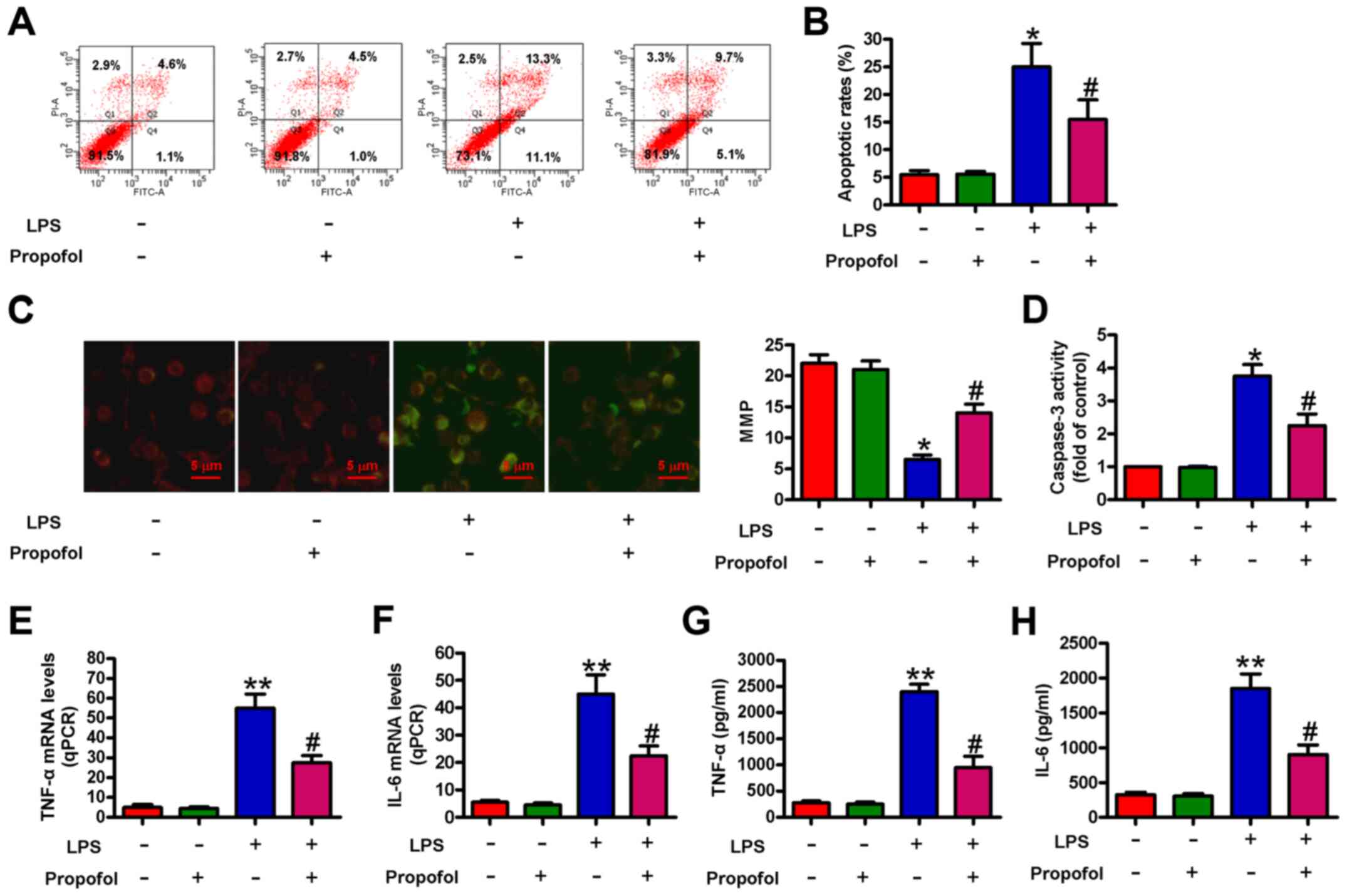 | Figure 2.Propofol reduces LPS-induced
cardiomyocyte apoptosis and inflammation. Cardiomyocytes were
pretreated with 50 µM propofol for 24 h followed by stimulation
with LPS (1 µg/ml) for 4 h. Cardiomyocyte apoptosis was (A)
determined by flow cytometry and (B) quantified. (C)
5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolocarbocyanine
iodide staining was performed to assess the MMP. Scale bar, 5-µm.
(D) Caspase-3 activity was measured to evaluate cell apoptosis. (E)
TNF-α and (F) IL-6 mRNA expression levels were measured via reverse
transcription-quantitative PCR. (G) TNF-α and (H) IL-6 levels in
the supernatants were measured by performing ELISAs. Data are from
three independent experiments. *P<0.05 and **P<0.01 vs.
control; #P<0.05 vs. LPS. LPS, lipopolysaccharide;
MMP, mitochondrial membrane potential; qPCR, quantitative PCR. |
Propofol prevents LPS-induced NLRP3
inflammasome activation in cardiomyocytes
The NLRP3 inflammasome is a prominent and early
mediator of inflammatory responses in myocardial injury (30). NLRP3 inflammasome activation serves
an important role in high glucose-induced H9c2 cell toxicity
(31). Therefore, whether propofol
prevented NLRP3 inflammasome activation in LPS-induced
cardiomyocytes was investigated. Propofol markedly reduced
LPS-induced increases in the protein expression levels of NLRP3,
ASC, caspase-1 p10, IL-1β and IL-18 in cardiomyocytes (Fig. 3A). The Co-IP assay results
demonstrated that the formation of the NLRP3-ASC-pro-caspase-1
complex was notably inhibited by pretreatment with propofol in
LPS-injured cardiomyocytes (Fig.
3B). LPS-induced upregulation of IL-1β and IL-18 mRNA
expression levels was significantly decreased by pretreatment with
propofol (Fig. 3C and D). Propofol
also significantly inhibited IL-1β and IL-18 release in LPS-injured
cardiomyocytes (Fig. 3E and F).
Collectively, the results suggested that propofol inhibited
LPS-induced NLRP3 inflammasome activation in cardiomyocytes.
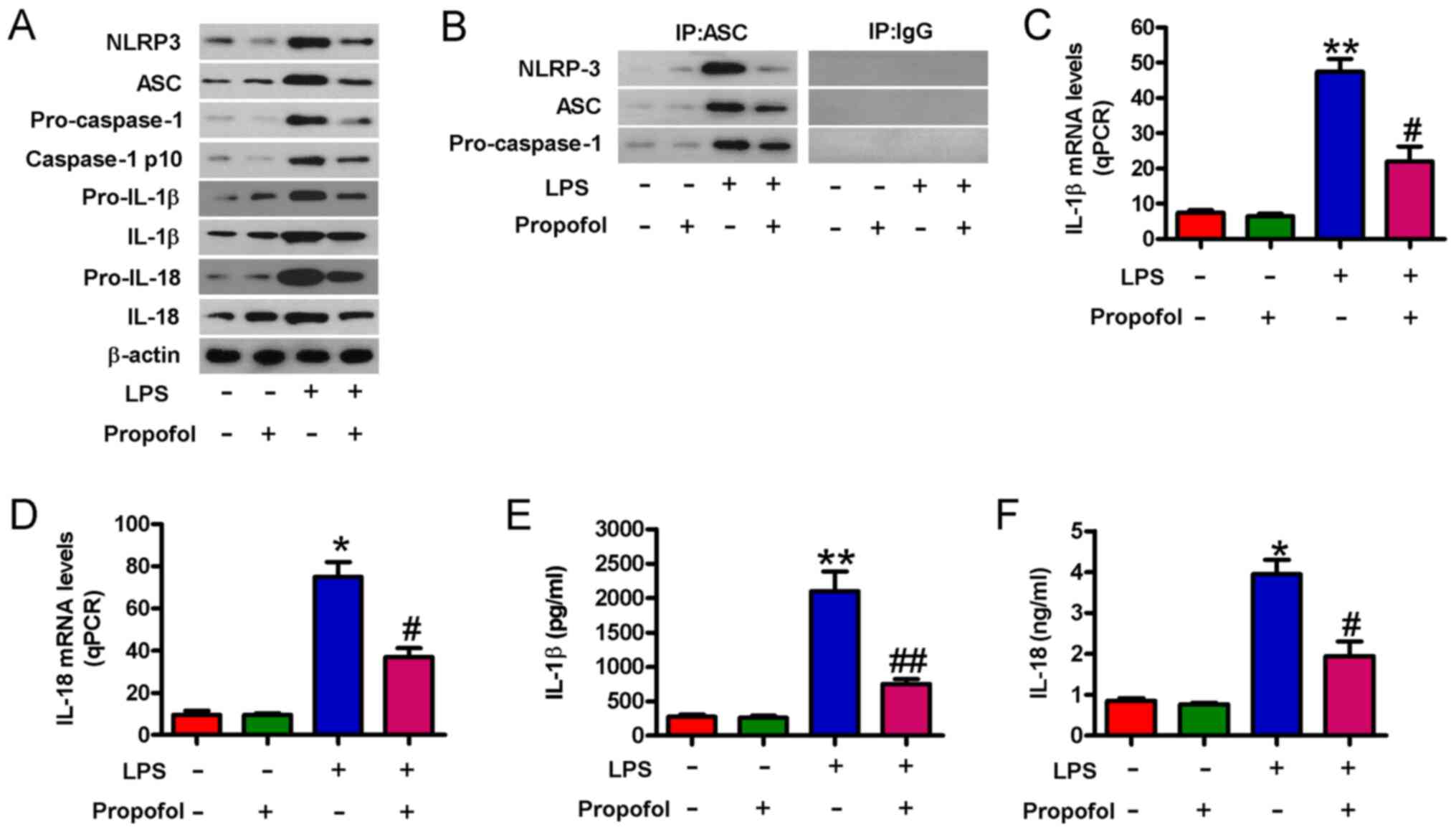 | Figure 3.Propofol suppresses NLRP3
inflammasome activation in LPS-injured cardiomyocytes.
Cardiomyocytes were pretreated with 50 µM propofol for 24 h
followed by stimulation with LPS (1 µg/ml) for 4 h. (A) Western
blotting was performed to measure the protein expression levels of
NLRP3, ASC, pro-caspase-1, caspase-1 p10, pro-IL-1β, IL-1β, IL-18
and pro-IL-18. (B) The co-immunoprecipitation assay was performed
to assess the relationship among NLPR3, ASC and pro-caspase-1. (C)
IL-1β and (D) IL-18 mRNA expression levels were measured via
reverse transcription-quantitative PCR. (E) IL-1β and (F) IL-18
levels in the supernatants were evaluated by performing ELISAs.
Data are from three independent experiments. *P<0.05 and
**P<0.01 vs. control; #P<0.05 and
##P<0.01 vs. LPS. NLRP3, NLR family pyrin domain
containing 3; LPS, lipopolysaccharide; ASC, apoptosis-associated
speck-like protein containing A CARD; qPCR, quantitative PCR; IP,
immunoprecipitation. |
Propofol-mediated inactivation of the
NLRP3 inflammasome is dependent on HMGB1 downregulation in
LPS-injured cardiomyocytes
HMGB1 produced by cardiomyocytes contributes to
LPS-induced myocardial dysfunction (14). To assess whether propofol reduced
LPS-induced HMGB1 expression in cardiomyocytes, western blotting
was performed. Compared with the control group, LPS notably
increased HMGB1 expression levels, which were obviously reduced by
pretreatment with propofol (Fig.
4A). HMGB1 activates the NLRP3 inflammasome in vascular smooth
muscle cells (13). Subsequently,
whether propofol-mediated inactivation of the NLRP3 inflammasome
was HMGB1-dependent was investigated. siHMGB1 transfection markedly
decreased HMGB1 protein expression levels in control and
LPS-stimulated cardiomyocytes compared with siNC transfection
(Fig. 4B). In LPS-stimulated
cardiomyocytes, propofol-mediated downregulation of NLRP3, ASC,
pro-caspase-1 and caspase-1 p10 protein expression levels were
obviously counteracted by rHMGB1 pretreatment, but notably enhanced
by siHMGB1 transfection (Fig. 4C).
Similarly, rHMGB1 pretreatment significantly attenuated
propofol-mediated inhibition of IL-1β and IL-18 release in
LPS-stimulated cardiomyocytes, whereas HMGB1 knockdown enhanced the
inhibitory effects of propofol on IL-1β and IL-18 release (Fig. 4D and E). The results suggested that
propofol inhibited NLRP3 inflammasome activation by reducing HMGB1
expression in LPS-injured cardiomyocytes.
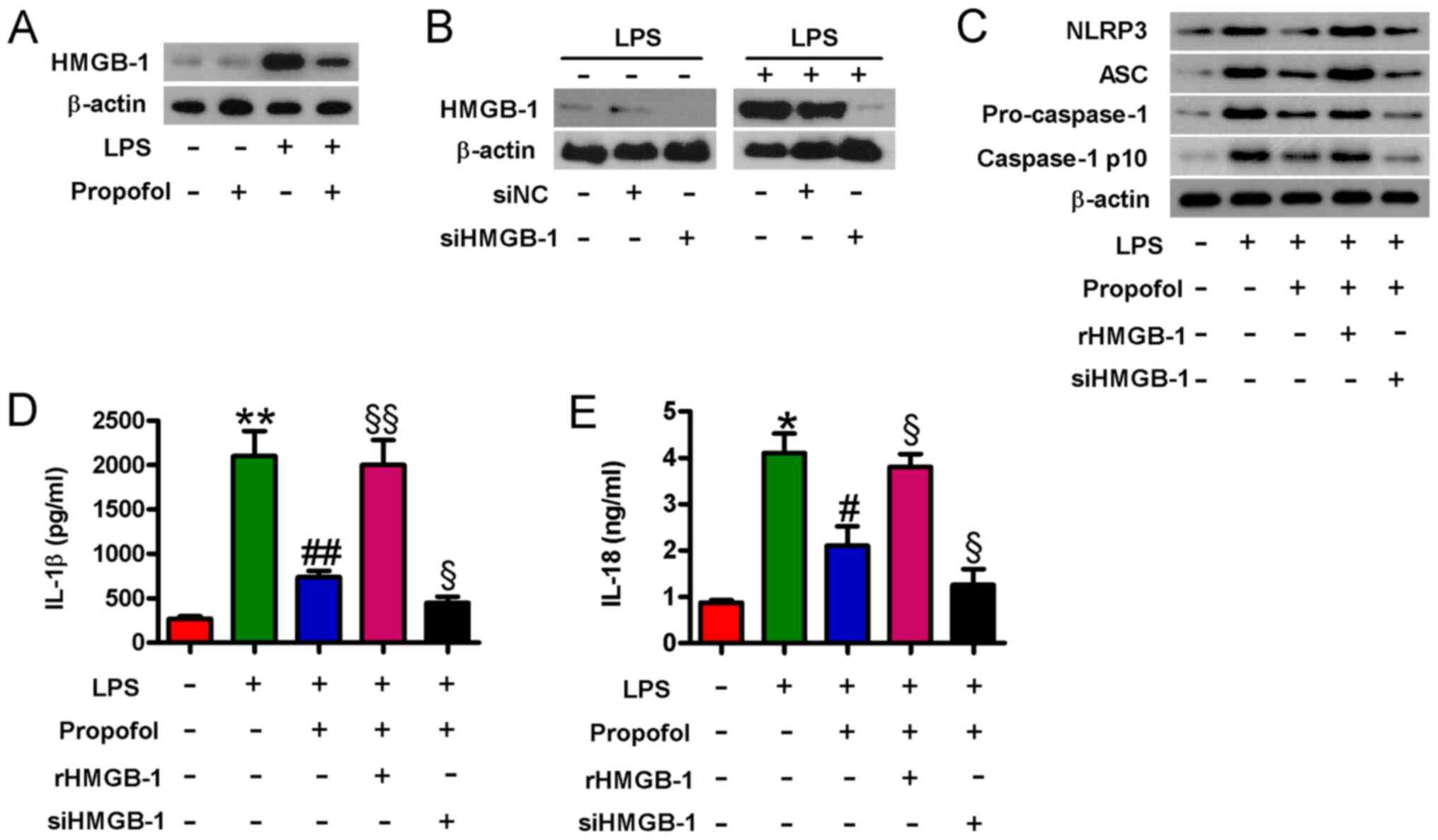 | Figure 4.Propofol inactivates the NLRP3
inflammasome by downregulating HMGB1 in LPS-injured cardiomyocytes.
(A) Cardiomyocytes were pretreated with 50 µM propofol for 24 h
followed by stimulation with LPS (1 µg/ml) for 4 h. Western
blotting was performed to measure HMGB1 protein expression levels.
(B) Cardiomyocytes were transfected with 100 µM siHMGB1 or siNC for
48 h followed by stimulation with LPS (1 µg/ml) for 4 h. Western
blotting was performed to measure HMGB1 protein expression levels.
Cardiomyocytes were pretreated with rHMGB1 (100 ng/ml) for 30 min
or transfected with 100 µM siHMGB1 or siNC for 24 h, followed by
treatment with propofol (50 µM) for 24 h then stimulation with LPS
(1 µg/ml) for 4 h. (C) NLRP3, ASC, pro-caspase-1 and caspase-1 p10
protein expression levels were measured via western blotting. (D)
IL-1β and (E) IL-18 levels in the supernatants were evaluated by
performing ELISAs. Data are from three independent experiments.
*P<0.05 and **P<0.01 vs. control; #P<0.05 and
##P<0.01 vs. LPS; §P<0.05 and
§§P<0.01 vs. LPS + propofol. NLRP3, NLR family pyrin
domain containing 3; HMGB1, high mobility group box-1; LPS,
lipopolysaccharide; si, small interfering RNA; NC, negative
control; ASC, apoptosis-associated speck-like protein containing A
CARD; r, recombinant; ns, no significance. |
PPARγ contributes to propofol-mediated
inactivation of the HMGB1-dependent NLRP3 inflammasome in
LPS-injured cardiomyocytes
PPARγ inhibits LPS-induced HMGB1 release in RAW
264.7 cells (15). Previous studies
have revealed that propofol represses inflammation by activating
PPARγ in the renal tissues of sepsis model mice (32) and by upregulating PPARγ expression
in THP-1 macrophage-derived foam cells (33). Compared with the control group,
PPARγ expression was notably downregulated in LPS-injured
cardiomyocytes, which was reversed by pretreatment with propofol
(Fig. 5A). Subsequently, whether
PPARγ was involved in propofol-mediated inactivation of the
HMGB1-dependent NLRP3 inflammasome in LPS-injured cardiomyocytes
was assessed. In control and LPS-stimulated cardiomyocytes, PPARγ
protein expression levels were obviously lower in
siPPARγ-transfected cardiomyocytes compared with control or
siNC-transfected cardiomyocytes (Fig.
5B). siPPARγ transfection or GW9662 treatment reversed
propofol-mediated downregulation of HMGB1, NLRP3, ASC,
pro-caspase-1 and caspase-1 p10 protein expression levels in
LPS-injured cardiomyocytes (Fig.
5C). Moreover, propofol-mediated inhibition of IL-1β and IL-18
release was significantly reversed by siPPARγ transfection or
GW9662 treatment in LPS-injured cardiomyocytes (Fig. 5D and E). The results indicated that
PPARγ was involved in propofol-mediated inactivation of the
HMGB1-dependent NLRP3 inflammasome in LPS-injured
cardiomyocytes.
 | Figure 5.Propofol inhibits HMGB1-dependent
NLRP3 inflammasome activation by activating PPARγ in LPS-injured
cardiomyocytes. (A) Cardiomyocytes were pretreated with 50 µM
propofol for 24 h followed by stimulation with LPS (1 µg/ml) for 4
h. Western blotting was performed to measure PPARγ protein
expression levels. (B) Cardiomyocytes were transfected with 100 µM
siPPARγ or siNC for 48 h followed by stimulation with LPS (1 µg/ml)
for 4 h. Western blotting was performed to measure PPARγ protein
expression levels. Cardiomyocytes were pretreated with GW9662 (10
µM) for 30 min or transfected with 100 µM siPPARγ or siNC for 24 h,
followed by treatment with propofol (50 µM) for 24 h and then
stimulation with LPS (1 µg/ml) for 4 h. (C) HMGB1, NLRP3, ASC,
pro-caspase-1 and caspase-1 p10 protein expression levels were
measured via western blotting. (D) IL-1β and (E) IL-18 levels in
the supernatants were evaluated by performing ELISAs. Data are from
three independent experiments. *P<0.05 and **P<0.01 vs.
control; #P<0.05 and ##P<0.01 vs. LPS;
§P<0.05 and §§P<0.01 vs. LPS +
propofol. HMGB1, high mobility group box-1; NLRP3, NLR family pyrin
domain containing 3; PPARγ, peroxisome proliferator-activated
receptor γ; LPS, lipopolysaccharide; si, small interfering RNA; NC,
negative control; ASC, apoptosis-associated speck-like protein
containing A CARD. |
Discussion
In the present study, the effects and mechanisms
underlying propofol in LPS-induced cardiomyocyte injury were
investigated. Propofol pretreatment significantly enhanced cell
viability and decreased LDH release in LPS-stimulated
cardiomyocytes. Propofol also protected against LPS-induced
cardiomyocyte apoptosis and inflammation. Moreover, LPS-induced
NLRP3 inflammasome activation in cardiomyocytes was inhibited by
pretreatment with propofol, which occurred via downregulation of
HMGB1. In addition, PPARγ participated in propofol-mediated
inactivation of the HMGB1-dependent NLRP3 inflammasome in
LPS-injured cardiomyocytes. Overall, the results indicated that the
cardioprotective role of propofol was partly reliant on its ability
to inhibit activation of the HMGB1-dependent NLRP3 signaling
pathway via activating PPARγ.
Myocardial injury is a distinctive characteristic of
sepsis (4). LPS, the primary
component of the outer membrane of Gram-negative bacteria, is one
of the major causes of sepsis (34). LPS is commonly utilized to induce
cardiomyocyte lesions (35–37). In the present study, cardiomyocyte
viability was significantly decreased and LDH release was
significantly increased in LPS-stimulated cardiomyocytes compared
with control cells, suggesting that LPS induced cardiomyocyte
damage. In LPS-induced myocardial injury, the excessive generation
of inflammatory mediators is considered as one of the principal
underlying mechanisms (38).
Inflammatory mediators can result in loss of the MMP and caspase-3
activation, which ultimately leads to cardiomyocyte apoptosis
(35). Cardiomyocyte apoptosis has
been implicated in sepsis-induced myocardial dysfunction (39). In LPS-challenged mice, the
myocardial levels of TNF-α and IL-1β are increased (40). In the present study, the levels of
proinflammatory factors, such as TNF-α, IL-6, IL-1β and IL-18, were
increased, and cell apoptosis, loss of the MMP and caspase-3
activation were enhanced in LPS-injured cardiomyocytes compared
with control cells. Propofol is a widely used anesthetic agent that
displays cardioprotective effects in various injury models
(22–26). Propofol reduces the production of
IL-1, IL-6 and TNF-α during LPS-induced myocardial injury in
vitro and in vivo (22,23).
Propofol attenuates H2O2-induced oxidative
stress and apoptosis via the mitochondrial-mediated signaling
pathway in neonatal rat cardiomyocytes (41). In the present study, the results
indicated that propofol pretreatment protected cardiomyocytes
against LPS-induced inflammation and apoptosis, as evidenced by
decreased inflammatory cytokines, reduced apoptosis, alleviation of
loss of the MMP and decreased caspase-3 activation in
propofol-pretreated LPS-injured cardiomyocytes compared with
LPS-injured cardiomyocytes. The results suggested that propofol may
serve as a therapeutic agent for septic myocardial dysfunction due
to its anti-inflammatory and anti-apoptotic properties.
NLRP3 inflammasome activation is a major contributor
to myocardial injury (42).
Activation of the NLRP3 inflammasome is a multi-step process that
includes priming and activating (43). A priming stimulus increases the
intracellular levels of pro-IL-1β and NLRP3, and the activation
stimulus results in NLRP3 interacting with procaspase-1 via the
adaptor protein ASC (44). The
assembly of the NLRP3 inflammasome initiates caspase-1 activation
via the cleavage of pro-caspase-1. The active form of caspase-1
causes enzymatic cleavage of pro-IL-1β and pro-IL-18, leading to
IL-1β and IL-18 maturation and release (45). The present study indicated that
propofol pretreatment markedly reduced LPS-induced increases in
NLRP3, ASC, pro-caspase-1, caspase-1 p10, IL-1β and IL-18
expression levels, and IL-1β and IL-18 production in
cardiomyocytes. Propofol pretreatment also prevented the
association of NLRP3, ASC and pro-caspase-1 in LPS-injured
cardiomyocytes. The results indicated that propofol pretreatment
suppressed the priming and activation of NLRP3 inflammasome in
LPS-injured cardiomyocytes.
HMGB1 can increase the expression of NLRP3
inflammasome components (NLRP3, ASC and caspase-1) (13). HMGB1 was originally described as a
nuclear DNA-binding protein that is present in the nuclei and the
cytoplasm, and is involved in maintaining the nucleosome structure
and regulating gene transcription (46). HMGB1 is derived from stimulated
macrophages or monocytes, and can be released from injured or
necrotic cardiomyocytes (14,47).
HMGB1 is an alarmin cytokine implicated in inflammation and cell
injury during myocardial dysfunction (14). HMGB1 is an early mediator of
inflammation and necrosis, and a late mediator of lethal sepsis
(48,49). Chen et al (47) reported that LPS augments HMGB1
expression and facilitates its translocation from the nucleus to
the cytoplasmic compartment in cardiomyocytes. HMGB1 antibody
administration effectively decreases the mortality of LPS-induced
sepsis model animals (50). In the
present study, propofol pretreatment obviously decreased
LPS-induced HMGB1 expression in cardiomyocytes. Propofol-induced
reductions in the expression levels of NLRP3, ASC, pro-caspase-1
and caspase-1, and production of IL-1β and IL-18 were reversed by
rHMGB1 pretreatment, but enhanced by siHMGB1 transfection in
LPS-injured cardiomyocytes. The results suggested that
propofol-mediated inactivation of the NLRP3 inflammasome was
HMGB1-dependent in LPS-injured cardiomyocytes.
PPARγ, a ligand-inducible nuclear receptor belonging
to the nuclear transcription factor PPAR superfamily, is expressed
in a wide range of tissues and cells (51). PPARγ can protect against
ischemia-reperfusion cardiomyocyte injury by regulating oxidative
stress, inflammatory responses, glucose and lipid metabolism, and
apoptosis (52). PPARγ activator
inhibits LPS-induced TNF-α expression in neonatal rat
cardiomyocytes (53). PPARγ agonist
prevents LPS-mediated reductions in the number of mitochondria and
improves cardiac dysfunction in wild-type mice, and a high survival
rate is observed in PPARγ-transgenic mice (16). Hwang et al (15) reported that PPARγ ligand
rosiglitazone ablates LPS-stimulated HMGB1 release in RAW 264.7
cells, whereas siPPARγ transfection or GW9662 treatment abolishes
the effect of rosiglitazone on HMGB1 release. Previous studies
demonstrated the inhibitory effects of PPARγ on NLRP3 activation in
monosodium urate-treated HK-2 cells and
LPS/H2O2-challenged Kupffer cells (54,55).
Propofol can upregulate PPARγ expression in THP-1
macrophage-derived foam cells (33). Consistently, the present study
demonstrated that propofol enhanced PPARγ expression in LPS-injured
cardiomyocytes, and GW9662 treatment or siPPARγ transfection
alleviated propofol-mediated downregulation of LPS-induced
increases in HMGB1, NLRP3, ASC, pro-caspase-1 and caspase-1
expression levels, and IL-1β and IL-18 production. Therefore, the
results suggested that the potential beneficial effects of propofol
against septic myocardial injury were dependent on PPARγ activation
to suppress HMGB1-dependent NLRP3 inflammasome activation.
In summary, the present study suggested that the
protective effects of propofol on LPS-induced cardiomyocyte injury
were partially reliant on PPARγ activation, which inhibited
activation of the NLRP3 inflammasome by downregulating HMGB1
expression during myocardial injury. Moreover, propofol reduced
LPS-induced cardiomyocyte inflammation and apoptosis. Collectively,
the present study suggested that propofol may serve as a potential
therapeutic agent for septic myocardial dysfunction.
Acknowledgements
Not applicable.
Funding
The present study was supported by the National
Science Foundation for Young Scientists of China (grant no.
81101367).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
HC conceived and designed the study. HZ performed
the experiments and drafted the manuscript. YG analyzed the data.
HZ and HC wrote the manuscript. All authors confirm the
authenticity of all the raw data, and read and approved the final
manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Angus DC and van der Poll T: Severe sepsis
and septic shock. N Engl J Med. 369:840–851. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Cohen J, Vincent JL, Adhikari NK, Machado
FR, Angus DC, Calandra T, Jaton K, Giulieri S, Delaloye J, Opal S,
et al: Sepsis: A roadmap for future research. Lancet Infect Dis.
15:581–614. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Delano MJ and Ward PA: Sepsis-induced
immune dysfunction: Can immune therapies reduce mortality? J Clin
Invest. 126:23–31. 2016. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Romero-Bermejo FJ, Ruiz-Bailen M,
Gil-Cebrian J and Huertos-Ranchal MJ: Sepsis-induced
cardiomyopathy. Curr Cardiol Rev. 7:163–183. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Li F, Lang F, Zhang H, Xu L, Wang Y and
Hao E: Role of TFEB mediated autophagy, oxidative stress,
inflammation, and cell death in endotoxin induced myocardial
toxicity of young and aged mice. Oxid Med Cell Longev.
2016:53803192016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Yasuda S and Lew WY: Lipopolysaccharide
depresses cardiac contractility and beta-adrenergic contractile
response by decreasing myofilament response to Ca2+ in
cardiac myocytes. Circ Res. 81:1011–1020. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Suliman HB, Welty-Wolf KE, Carraway M,
Tatro L and Piantadosi CA: Lipopolysaccharide induces oxidative
cardiac mitochondrial damage and biogenesis. Cardiovasc Res.
64:279–288. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Cavaillon JM: Exotoxins and endotoxins:
Inducers of inflammatory cytokines. Toxicon. 149:45–53. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Fallach R, Shainberg A, Avlas O, Fainblut
M, Chepurko Y, Porat E and Hochhauser E: Cardiomyocyte Toll-like
receptor 4 is involved in heart dysfunction following septic shock
or myocardial ischemia. J Mol Cell Cardiol. 48:1236–1244. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Latz E, Xiao TS and Stutz A: Activation
and regulation of the inflammasomes. Nat Rev Immunol. 13:397–411.
2013. View
Article : Google Scholar : PubMed/NCBI
|
|
11
|
Schroder K and Tschopp J: The
inflammasomes. Cell. 140:821–832. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Rathinam VA, Vanaja SK and Fitzgerald KA:
Regulation of inflammasome signaling. Nat Immunol. 13:333–342.
2012. View
Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kim EJ, Park SY, Baek SE, Jang MA, Lee WS,
Bae SS, Kim K and Kim CD: HMGB1 increases IL-1β production in
vascular smooth muscle cells via NLRP3 inflammasome. Front Physiol.
9:3132018. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Xu H, Su Z, Wu J, Yang M, Penninger JM,
Martin CM, Kvietys PR and Rui T: The alarmin cytokine, high
mobility group box 1, is produced by viable cardiomyocytes and
mediates the lipopolysaccharide-induced myocardial dysfunction via
a TLR4/phosphatidylinositol 3-kinase gamma pathway. J Immunol.
184:1492–1498. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Hwang JS, Kang ES, Ham SA, Yoo T, Lee H,
Paek KS, Park C, Kim JH, Lim DS and Seo HG: Activation of
peroxisome proliferator-activated receptor gamma by rosiglitazone
inhibits lipopolysaccharide-induced release of high mobility group
box 1. Mediators Inflamm. 2012:3528072012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Drosatos K, Khan RS, Trent CM, Jiang H,
Son NH, Blaner WS, Homma S, Schulze PC and Goldberg IJ: Peroxisome
proliferator-activated receptor-g activation prevents
sepsis-related cardiac dysfunction and mortality in mice. Circ
Heart Fail. 6:550–562. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Green TR, Bennett SR and Nelson VM:
Specificity and properties of propofol as an antioxidant free
radical scavenger. Toxicol Appl Pharmacol. 129:163–169. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Marik PE: Propofol: An immunomodulating
agent. Pharmacotherapy. 25:28S–33S. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Vasileiou I, Xanthos T, Koudouna E, Perrea
D, Klonaris C, Katsargyris A and Papadimitriou L: Propofol: A
review of its non-anaesthetic effects. Eur J Pharmacol. 605:1–8.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Hans P, Deby-Dupont G, Deby C, Pieron F,
Verbesselt R, Franssen C and Lamy M: Increase in antioxidant
capacity of plasma during propofol anesthesia. J Neurosurg
Anesthesiol. 9:234–236. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Vanlersberghe C and Camu F: Propofol.
Handb Exp Pharmacol. 227–252. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Tang J, Hu JJ, Lu CH, Liang JN, Xiao JF,
Liu YT, Lin CS and Qin ZS: Propofol inhibits
lipopolysaccharide-induced tumor necrosis factor-alpha expression
and myocardial depression through decreasing the generation of
superoxide anion in cardiomyocytes. Oxid Med Cell Longev.
2014:1573762014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Yang Z, Cheng F, Yan G, Xiong L and Liu H:
Propofol protects against endotoxin-induced myocardial injury by
inhibiting NF-kB-mediated inflammation. Exp Ther Med. 15:2032–2036.
2018.PubMed/NCBI
|
|
24
|
Shinjo T, Tanaka T, Okuda H, Kawaguchi AT,
Oh-Hashi K, Terada Y, Isonishi A, Morita-Takemura S, Tatsumi K,
Kawaguchi M and Wanaka A: Propofol induces nuclear localization of
Nrf2 under conditions of oxidative stress in cardiac H9c2 cells.
PLoS One. 13:e01961912018. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Javadov SA, Lim KH, Kerr PM, Suleiman MS,
Angelini GD and Halestrap AP: Protection of hearts from reperfusion
injury by propofol is associated with inhibition of the
mitochondrial permeability transition. Cardiovasc Res. 45:360–369.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Jin YC, Kim W, Ha YM, Shin IW, Sohn JT,
Kim HJ, Seo HG, Lee JH and Chang KC: Propofol limits rat myocardial
ischemia and reperfusion injury with an associated reduction in
apoptotic cell death in vivo. Vascul Pharmacol. 50:71–77. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Huang B, You J, Qiao Y, Wu Z, Liu D, Yin
D, He H and He M: Tetramethylpyrazine attenuates
lipopolysaccharide-induced cardiomyocyte injury via improving
mitochondrial function mediated by 14-3-3g. Eur J Pharmacol.
832:67–74. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Jiang H and Qu P: Effects of Ginkgo biloba
leaf extract on local renin-angiotensin system through TLR4/NF-κB
pathway in cardiac myocyte. Exp Ther Med. 14:5857–5862.
2017.PubMed/NCBI
|
|
29
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Toldo S and Abbate A: The NLRP3
inflammasome in acute myocardial infarction. Nat Rev Cardiol.
15:203–214. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Huang Z, Zhuang X, Xie C, Hu X, Dong X,
Guo Y, Li S and Liao X: Exogenous hydrogen sulfide attenuates high
glucose-induced cardiotoxicity by inhibiting NLRP3 inflammasome
activation by suppressing TLR4/NF-κB pathway in H9c2 cells. Cell
Physiol Biochem. 40:1578–1590. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Hsing CH, Chou W, Wang JJ, Chen HW and Yeh
CH: Propofol increases bone morphogenetic protein-7 and decreases
oxidative stress in sepsis-induced acute kidney injury. Nephrol
Dial Transplant. 26:1162–1172. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Ma X, Li SF, Qin ZS, Ye J, Zhao ZL, Fang
HH, Yao ZW, Gu MN and Hu YW: Propofol up-regulates expression of
ABCA1, ABCG1, and SR-B1 through the PPARγ/LXRα signaling pathway in
THP-1 macrophage-derived foam cells. Cardiovasc Pathol. 24:230–235.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Munford RS: Endotoxemia-menace, marker, or
mistake? J Leukoc Biol. 100:687–698. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Turdi S, Han X, Huff AF, Roe ND, Hu N, Gao
F and Ren J: Cardiac-specific overexpression of catalase attenuates
lipopolysaccharide-induced myocardial contractile dysfunction: Role
of autophagy. Free Radic Biol Med. 53:1327–1338. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Liu D, Yi B, Liao Z, Tang L, Yin D, Zeng
S, Yao J and He M: 14-3-3γ protein attenuates
lipopolysaccharide-induced cardiomyocytes injury through the Bcl-2
family/mitochondria pathway. Int Immunopharmacol. 21:509–515. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Xianchu L, Lan PZ, Qiufang L, Yi L,
Xiangcheng R, Wenqi H and Yang D: Naringin protects against
lipopolysaccharide-induced cardiac injury in mice. Environ Toxicol
Pharmacol. 48:1–6. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Du J, An J, Wei N, Guan T, Pritchard KA Jr
and Shi Y: Increased resistance to LPS-induced myocardial
dysfunction in the brown norway rats versus Dahl S rats: Roles of
inflammatory cytokines and nuclear factor kappaB pathway. Shock.
33:332–336. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Matsuno K, Iwata K, Matsumoto M, Katsuyama
M, Cui W, Murata A, Nakamura H, Ibi M, Ikami K, Zhang J, et al:
NOX1/NADPH oxidase is involved in endotoxin-induced cardiomyocyte
apoptosis. Free Radic Biol Med. 53:1718–1728. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Essandoh K, Yang L, Wang X, Huang W, Qin
D, Hao J, Wang Y, Zingarelli B, Peng T and Fan GC: Blockade of
exosome generation with GW4869 dampens the sepsis-induced
inflammation and cardiac dysfunction. Biochim Biophys Acta.
1852:2362–2371. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Liu XR, Cao L, Li T, Chen LL, Yu YY, Huang
WJ, Liu L and Tan XQ: Propofol attenuates
H2O2-induced oxidative stress and apoptosis
via the mitochondria- and ER-medicated pathways in neonatal rat
cardiomyocytes. Apoptosis. 22:639–646. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Toldo S, Mezzaroma E, Mauro AG, Salloum F,
Van Tassell BW and Abbate A: The inflammasome in myocardial injury
and cardiac remodeling. Antioxid Redox Signal. 22:1146–1161. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Guo H, Callaway JB and Ting JP:
Inflammasomes: Mechanism of action, role in disease, and
therapeutics. Nat Med. 21:677–687. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Ulland TK, Ferguson PJ and Sutterwala FS:
Evasion of inflammasome activation by microbial pathogens. J Clin
Invest. 125:469–477. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Haneklaus M and O'Neill LA: NLRP3 at the
interface of metabolism and inflammation. Immunol Rev. 265:53–62.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Klune JR, Dhupar R, Cardinal J, Billiar TR
and Tsung A: HMGB1: Endogenous danger signaling. Mol Med.
14:476–484. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Chen HM, Liou SF, Hsu JH, Chen TJ, Cheng
TL, Chiu CC and Yeh JL: Baicalein inhibits HMGB1 release and
MMP-2/-9 expression in lipopolysaccharide-induced cardiac
hypertrophy. Am J Chin Med. 42:785–797. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Ding HS and Yang J: High mobility group
box-1 and cardiovascular diseases. Saudi Med J. 31:486–489.
2010.PubMed/NCBI
|
|
49
|
Qin S, Wang H, Yuan R, Li H, Ochani M,
Ochani K, Rosas-Ballina M, Czura CJ, Huston JM, Miller E, et al:
Role of HMGB1 in apoptosis-mediated sepsis lethality. J Exp Med.
203:1637–1642. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Wang H, Ward MF and Sama AE: Novel
HMGB1-inhibiting therapeutic agents for experimental sepsis. Shock.
32:348–357. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Ricote M and Glass CK: PPARs and molecular
mechanisms of transrepression. Biochim Biophys Acta. 1771:926–935.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Zhong CB, Chen X, Zhou XY and Wang XB: The
role of peroxisome proliferator-activated receptor g in mediating
cardioprotection against ischemia/reperfusion injury. J Cardiovasc
Pharmacol Ther. 23:46–56. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Takano H, Nagai T, Asakawa M, Toyozaki T,
Oka T, Komuro I, Saito T and Masuda Y: Peroxisome
proliferator-activated receptor activators inhibit
lipopolysaccharide-induced tumor necrosis factor-alpha expression
in neonatal rat cardiac myocytes. Circ Res. 87:596–602. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Hong W, Hu S, Zou J, Xiao J, Zhang X, Fu
C, Feng X and Ye Z: Peroxisome proliferator-activated receptor
gamma prevents the production of NOD-like receptor family, pyrin
domain containing 3 inflammasome and interleukin 1β in HK-2 renal
tubular epithelial cells stimulated by monosodium urate crystals.
Mol Med Rep. 12:6221–6226. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Xu Y, Yao J, Zou C, Zhang H, Zhang S, Liu
J, Ma G, Jiang P and Zhang W: Asiatic acid protects against hepatic
ischemia/reperfusion injury by inactivation of Kupffer cells via
PPARγ/NLRP3 inflammasome signaling pathway. Oncotarget.
8:86339–86355. 2017. View Article : Google Scholar : PubMed/NCBI
|