Introduction
Malignant tumors of the central nervous system (CNS)
are among the types of cancer with the poorest prognosis, with high
levels of morbidity and mortality (1). Glioma is the most common primary CNS
tumor deriving from neuroglial stem or progenitor cells (2). Mitochondrial DNA (mtDNA) mutations
have been found to be closely associated with various types of
cancer, neurodegenerative disorders, attention-deficit
hyperactivity disorder, headache, diabetes and aging (3–8).
Therefore, a study on the effect of mtDNA heterogeneity on glioma
growth, differentiation and death is required. Mitochondria serve a
crucial and integral role in maintaining cell homeostasis,
including energy synthesis, redox regulation, cell messaging and
hematopoiesis (9,10) and they provide ~80–90% of the energy
to the cell through the respiratory chain and this percentage
reaches 95% in the heart (11,12).
Furthermore, mitochondria regulate apoptosis and cell cycle through
their role in several processes including calcium signaling,
reactive oxygen species (ROS) homeostasis, biosynthesis of heme,
biosynthesis of iron-sulfur clusters and tumorigenesis (13).
Since mtDNA mutations can compromise the
mitochondrial function and lead to human pathologies, there is a
strong and continuing interest in their role in human health and
disease. Unfortunately, the study of mtDNA mutations is not easy
due to the nuclear background diversity and the role it serves in
the modulation of mitochondrial defects (14). Fortunately, hybrid technology
provides a new strategy to study diseases associated with
mitochondrial dysfunctions. The technique is based on the use of a
cell line in which the mtDNA is depleted (ρ0 cells) and can be used
as a mitochondrial receptor when fused with platelets (15,16).
Since cytoplasmic hybrids (cybrids) have the same genetic
background, biochemical analysis is made possible. Previous studies
have demonstrated that ethidium bromide (EB), 2′,3′-dideoxycytidine
(ddC), rhodamine-6g, 1-methyl-4-phenylpyridinium, zidovudine and
stavudine, together with NT2 human teratocarcinoma, T47d human
breast cancer cells, 143B human bone osteosarcoma thymidine kinase
negative (TK-) and human mesenchymal stem cell (3a6 and KP) are
commonly used to produce ρ0 cells (13,17–19).
Therefore, in the present study, the mtDNA-depleted
cell line C6ρ0 was generated from C6 glioma cells and the
mtDNA-related deficits were determined. The results demonstrated
that C6ρ0 cells could be a useful tool in the investigation of
mitochondrial dysfunction and mtDNA mutations and their role in the
pathogenesis of glioma can be also evaluated.
Materials and methods
C6 cell culture and isolation of C6ρ0
clone cell line
The rat C6 glioma cell line was purchased from the
Institute of Culture Collection of the Chinese Academy of Sciences
and cultured in F-12 Ham medium (Gibco; Thermo Fisher Scientific,
Inc.) supplemented with 2% fetal bovine serum (Zhejiang Tianhang
Biotechnology Co., Ltd.), 15% horse serum (Tianjin Kangyuan
Biotechnology Co., Ltd.) and 1% penicillin (100 U/ml) and
streptomycin (100 µg/ml; both from Corning, Inc.). The medium was
changed every three days and sub-cultures were made when the cells
reached 80% confluence. The cells were maintained at 37°C, 5%
CO2 and 90% humidity.
The generation of C6ρ0 cells was performed by
exposing wild-type C6 glioma cells to 5 µM EB and 8 µM ddC in F-12
Ham medium supplemented with uridine (50 µg/ml) and pyruvate (0.1
mg/ml), with a daily change of the culture medium. Cells
(2×106) were incubated in the above medium at 37°C, 5%
CO2 and 90% humidity and the expression of mtDNA was
measured using a mtDNA-specific PCR for a set period of time until
mtDNA band completely disappeared.
Single cell clones were isolated and collected by
the limiting dilution method (16)
when the mtDNA band disappeared. When no mtDNA fragment was
detected by PCR, the treated cells were quickly plated into 96-well
plates at a density of one cell per well to isolate pure cell
clones derived from the heterogeneous colony. Screening and
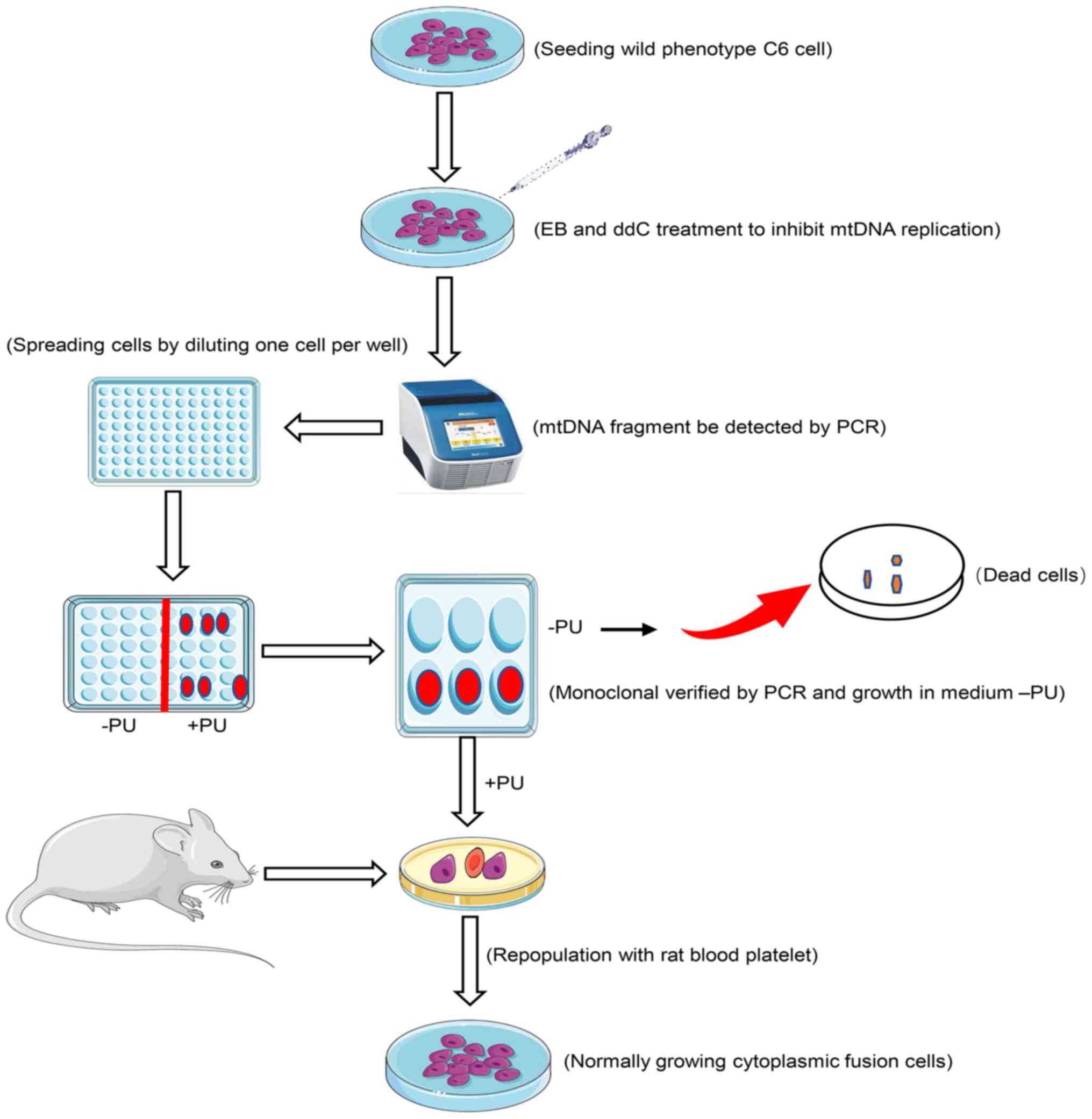
isolation of C6ρ0 cells were performed as shown in Fig. 1.
A single colony in 96-well was observed under an
inverted phase contrast microscope (Olympus Corporation); a single
C6ρ0 cell was marked and the wells containing >2 cells were
discarded. When the single colony was spread in the 96-well plate,
it was transferred into 48, 24, 12 and 6-well plates one after the
other after spreading the cells in each of them to gradually expand
the cell amount. To verify the absence of mtDNA, each potential
C6ρ0 cell line was divided into two halves: One half was used to
study the survival ability in pyrimidines and pyruvate-absent C6ρ0
test medium and the other half was cultured in F-12 Ham medium
containing uridine (50 µg/ml) and pyruvate (0.1 mg/ml). Trypan blue
at a dose of 0.2% was used to test and count the viable cells.
Finally, the C6ρ0 characteristic in these isolated colonies was
confirmed by mtDNA-specific PCR.
To evaluate cell growth, C6 and C6ρ0 cells were
plated in triplicate and the number of cells were counted on five
consecutive days. In order to standardize the condition of the cell
culture, C6 and C6ρ0 cells were cultured in F-12 Ham medium
supplemented with 50 µg/ml uridine and 0.1 mg/ml pyruvate to
evaluate the growth characteristics.
Production of transmitochondrial
cybrids
The fusion between C6ρ0 and rat blood platelets was
performed using the approach of standard chemical
enucleation/polyethylene glycol fusion (18). The fusion cells were placed on a
test bench for 1 min at 25°C and diluted with a mixture (10 ml) of
F-12 Ham and FBS (10%) supplemented with uridine (50 µg/ml) and
pyruvate (0.1 mg/ml) and the suspension was placed into petri
dishes (5 dishes, 2×105 cells/dish). The medium was
replaced with the selective medium (without pyruvate and uridine)
after 3 days.
PCR analysis of mtDNA
In order to detect mtDNA in C6, C6ρ0 and cybrids,
total DNA from a different number of cells was isolated as a
template using a commercial kit (Tiangen Biotech Co., Ltd.),
following the manufacturer's instructions. Oligonucleotide primers
were used to amplify the nucleotide sequences of rat mtDNA
mitochondrial D-loop region. The primers were synthesized by Sangon
Biotech Co., Ltd. mtDNA was amplified using a volume of 50 µl
containing 0.25 µl units of TakaRa Ex Taq polymerase (Takara
Biotechnology Co., Ltd.) and 0.2 µl primer pair. Primers specific
for the mtDNA D-loop region were the following: Forward
5′-CCTCCCATTCATTATCGCCGCCCTTGC-3′ and reverse
5′-GTCTGGGTCTCCTAGTAGGTCTGGGAA-3′ (235 bp). The PCR conditions
were: 35 cycles of denaturation at 98°C for 10 sec, annealing at
60°C for 30 sec and extension at 72°C for 60 sec. The products were
separated by 1% agarose gel electrophoresis and images were
detected using a 5200 Multi Luminescent image analyzer, according
to the manufacturer's protocol (Tanon Science and Technology Co.,
Ltd.). The intensity of the bands indicated the amount of
mtDNA.
Transmission electron microscopy
(TEM)
Cells for TEM were collected and plated on
coverglasses according to the method of Kukat et al
(20). Cells grown on cover slips
were fixed with a solution of 2.5% glutaraldehyde in 100 mM
cacodylate buffer, pH 7.4 for 1.5 h at 4°C, washed twice with
cacodylate buffer, followed by a fixation with 2% osmium tetroxide
in 50 mM cacodylate buffer (pH 7.4). Specimens were washed twice
with distilled water and stained over night with aqueous 0.5%
uranyl acetate at 4°C. Cells were dehydrated, embedded in Epon 812
and sectioned at 60 nm. Mitochondrial morphology was observed using
a H7000 electron microscope at 80 kV (Hitachi, Ltd.). Negatives
were digitized by scanning and processed with Adobe Photoshop CC
(Adobe Systems, Inc.).
Mitochondrial mass change and sugar
uptake
Cells (2×106 cells) were plated in 35 mm
dishes for 24 h and incubated with 100 nM Mito-Tracker Green
(Thermo Fisher Scientific, Inc.) or 10 µM 2-NBDG, a fluorescent
glucose, for 30 min at 37°C in the dark, to analyze the
mitochondrial mass and sugar intake. Cells were detached by
trypsin, collected, resuspended in saline solution and analyzed by
flow cytometry. In each measurement, fluorescence intensity data
from 2×104 single cell events were collected by an ACEA
NovoCyte2040R flow cytometer (ACEA Bioscience, Inc.; Agilent
Technologies, Inc.), using fluorescence excitation/emission (Ex/Em)
wavelengths of 490/516 nm to evaluated the mitochondrial mass and
Ex/Em of 480/525 nm to evaluate cell sugar uptake.
ATP consumption by C6 and C6ρ0
An enhanced ATP Assay kit (Beyotime Institute of
Biotechnology) was used to evaluate cellular ATP levels following
the manufacturer's instructions. The cell lysates were centrifuged
(12,000 × g at 4°C for 5 min) and the supernatant was collected and
transferred into a 96-well plate containing the detection solution.
The samples were then incubated for 30 min at 37°C and the
luminescence signal was detected. Total ATP levels were calculated
from the relationship between luminescence signals and protein
concentration.
Detection of cellular reactive oxygen
species (ROS) production
Cells were plated in 35 mm dishes (2×106
cells) for 24 h and incubated with 2,7-dichlorodihydrofluorescein
diacetate (DCFH-DA; 10 µM; Sigma-Aldrich; Merck KGaA) or 5 µM
MitoSOX Red (Thermo Fisher Scientific, Inc.) for 30 min at 37°C in
the dark to analyze total ROS and mitochondrial ROS, respectively.
Cells were detached by trypsin, collected, resuspended in saline
solution and analyzed by a NovoCyte 2040R (ACEA Biosciences Inc.)
flow cytometer. Ex/Em of 480/525 was set for the evaluation of
total ROS, while Ex/Em of 510/580 was set for the evaluation of
mitochondrial ROS. The amount of ROS produced was expressed as
fluorescence intensity relative to the one of untreated cells.
Determination of mitochondrial
membrane potential (ΔΨm)
Cells were plated in dishes (2×106 cells)
containing F-12 Ham medium for 24 h prior to the detection of ΔΨm.
The cells were then collected, washed and resuspended in
phosphate-buffered saline. Finally, 10 µM JC-1 (Beijing Solarbio
Science & Technology Co., Ltd.) stain was added into the buffer
and carbonyl cyanide m-chlorphenizonea, a potent mitochondrial
membrane disruptor, was used as the positive control. The cells
were incubated for 30 min (37°C; 5% CO2) and then
fluorescence intensity of 1×105 single cell events was
processed by a NovoCyte 2040R flow cytometer (ACEA Biosciences
Inc.) according to the manufacturer's protocol. Ex/Em of 490/530
and Ex/Em of 525/590 was used for ratio analysis. The ratio of
red/green (PE/FITC) fluorescence intensity was used to determine
ΔΨm.
Statistical analysis
Statistical analysis was performed using GraphPad
Prism software (version 6; GraphPad Software, Inc.). Results were
presented as means ± standard deviation (n=3). Comparisons among
multiple groups were analyzed using one-way ANOVA followed by
Bonferroni's post hoc test. Comparisons between two groups were
analyzed using unpaired Student's t-test. P<0.05 was considered
to indicate a statistically significant difference.
Results
Effect of EB and ddC treatment on
mtDNA content
The mtDNA content decreased with the increase of the
treatment time (Fig. 2A). After 6
weeks exposure, the amplification of mtDNA-encoded D-loop region
gene was lost in C6ρ0 cells (42 days), while the parental C6 cells
(0 day) yielded strong PCR products at the predicted size. Taken
together, the results suggested that mtDNA was specifically
depleted from C6 cells and that C6ρ0 cells survived in the culture
supplemented with uridine and pyrimidine.
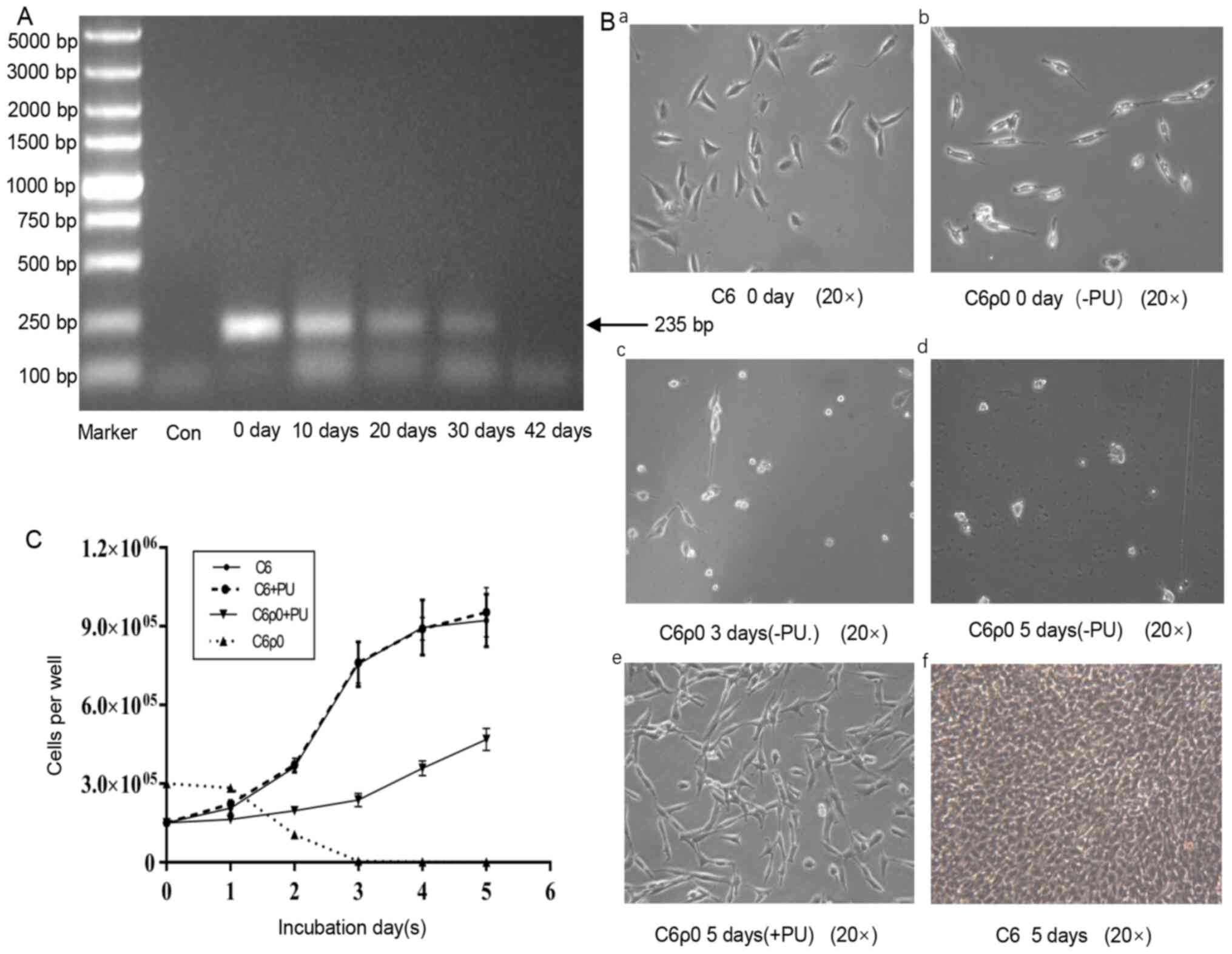 | Figure 2.Establishment of C6ρ0 cells and
growth characteristic of C6 and C6ρ0 cells in different media. (A)
Cells were treated with EB (5 µM) and ddC (8 µM) for 6 weeks. The
same amount of C6 cells (4×105) was used in the
different groups. The content of mtDNA was determined by an
mtDNA-specific PCR. The PCR products were visualized on 1% agarose
gel by EB staining. Marker: 5000 DNA ladder. (B) Representative
images of C6 and C6ρ0 clone cells growing in different media and at
a different time points. (B-a and f) C6 cells were cultured in F-12
medium at day 0 and day 5, respectively. (B-b, c and d) C6ρ0 cells
cultured in F-12 (-PU) at day 0, day 3 and day 5, respectively.
(B-e) C6ρ0 cells cultured in F-12 (+PU) medium at day 5. (C) C6 and
C6ρ0 were cultured in media with or without pyruvate and uridine
for 5 days and cell proliferation was measured and compared. After
seeding (0 d) and growing for 5 days in various media, the number
of cells per well was counted. EB, ethidium bromide; ddC,
2′,3′-dideoxycytidine; mtDNA, mitochondrial DNA; Con, sample
without template; -PU, growth in a medium without pyruvate and
uridine; +PU, growth in a medium supplemented with pyruvate and
uridine. |
Fig. 2B-a and B-b
demonstrated that the single colony of C6ρ0 and its parent cells
were morphologically slightly different. To verify the C6ρ0 stage
of the cloned cells, the growth of C6ρ0 cells was analyzed in the
medium without uridine and pyruvate. As shown in Fig. 2B-c and B-d, the number of C6ρ0 cells
became fewer, as they were no longer able to grow in ordinary
medium, until their total disappearance. All cells could survive
for several days, but at day 5, no living C6ρ0 cells were
identified by the trypan blue exclusion method (Fig. 2B-d). By contrast, C6ρ0 cells
cultured in uridine and pyruvate and C6 cells cultured in selective
medium grew vigorously and quickly (Fig. 2B-e), thus demonstrating that the
presence or absence of uridine and pyrimidine had no effect on C6
cell growth, while their presence was essential for the growth of
C6ρ0 cells (Fig. 2C).
Repopulation with exogenous
mitochondria from rat platelets
Rat platelets were used as exogenous mitochondrial
donors to perform repopulation experiments on the C6ρ0 clone
(Fig. 3A). The cybrid cells were
analyzed to verify the presence of mtDNA using the mtDNA-specific
PCR. As shown in Fig 3B, PCR bands
corresponding to mtDNA appeared in both cybrid and wild type C6
cells, suggesting the retention of mtDNA in cybrid cells.
 | Figure 3.Construction and identification of
cytoplasmic hybrid cells. (A) C6ρ0 clone fusion with rat platelets:
Cybrid cells derived from the fusion between C6ρ0 clone and
platelets isolated from rat blood samples. (B) Presence of mtDNA in
cybrid cells and wild-type cells evaluated by mtDNA-specific PCR
after 10 days of culture. Two different amounts of cells
(2×105 cells in 1, 3, 5 lane and 4×105 cells
in 2, 4, 6 lane) were used in this experiment. The PCR products
were visualized on 1% agarose gel by EB staining. Marker, 5000 DNA
ladder. (C) TEM images of (a) C6, (b) C6ρ0 and (c) cybrid cells
(mitochondria indicated by red box), magnification ×25,000 in all
cells. (D) Representative image of cybrid cells growing normally in
media without pyruvate and uridine. mtDNA, mitochondrial DNA; EB,
ethidium bromide; Con, sample without template; Clone, monoclonal
C6ρ0; Cybrid, C6ρ0 cell fused with rat blood platelets; Wild type,
C6 cells. |
The morphology of mitochondria was significantly
different in C6 and C6ρ0 cells. Wild-type C6 and cybrid cells
displayed a typical mitochondrial morphology, with organized
cristae, electron-dense matrix and intact mitochondrial membrane
(Fig. 3C-a and C-c), while
mitochondria in C6ρ0 cells were irregularly expanded, with a
partial or almost complete dissolution of the internal cristae
(Fig. 3C-b). When the cloned C6ρ0
cells were fused with rat platelets, the cybrid cells grew well in
selective medium without pyruvate and uridine and thus, the
repopulated colonies were formed, as shown in Fig. 3D.
Mitochondrial mass change and sugar
uptake in C6 and C6ρ0
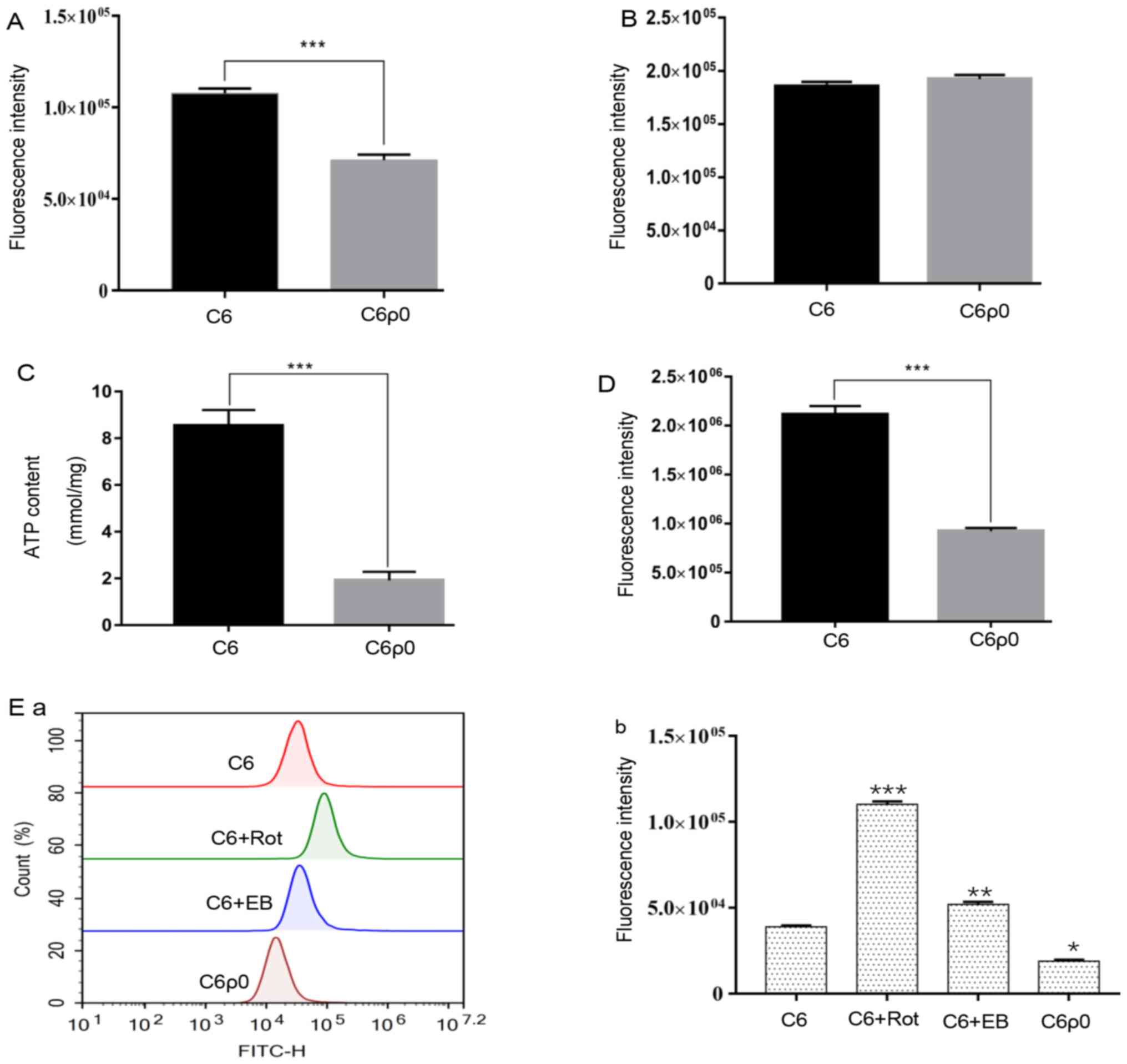
C6ρ0 cells were characterized by a notable and
significant decrease in mitochondrial mass, compared with that in
C6 cells (P<0.001; Fig. 4A).
To verify whether the decreased mitochondrial mass
had an effect on sugar uptake, the C6 and C6ρ0 cells were exposed
to 10 µM 2-NBDG for 30 min and the sugar content in the cells was
evaluated by flow cytometry. Quantitative analysis results showed
no difference in sugar uptake between C6 and C6ρ0 cells (Fig. 4B). However, the ATP assay showed
that C6ρ0 consumed less ATP compared with C6 cells (Fig. 4C).
ROS changes in C6 and C6ρ0 cells
The amount of total ROS production was significantly
reduced in C6ρ0 cells compared with its production in wild type C6
(Fig. 4D). To evaluate whether this
was due to the difference in mitochondria between the two cell
types, the source of ROS was identified. The results showed that C6
cells produced more mitochondrial ROS than C6ρ0 cells. Some
chemicals targeting mitochondria can stimulate C6 cells to produce
more mitochondrial-derived ROS; for instance, rotenone and EB
treatment can produce a large amount of mitochondrial ROS in C6
compared with in C6ρ0 (Fig. 4E).
This result confirmed that the change in ROS amount between C6 and
C6ρ0 cells was mainly due to the difference in mitochondria; the
existence and integrity of mtDNA has a great influence on the
production of ROS
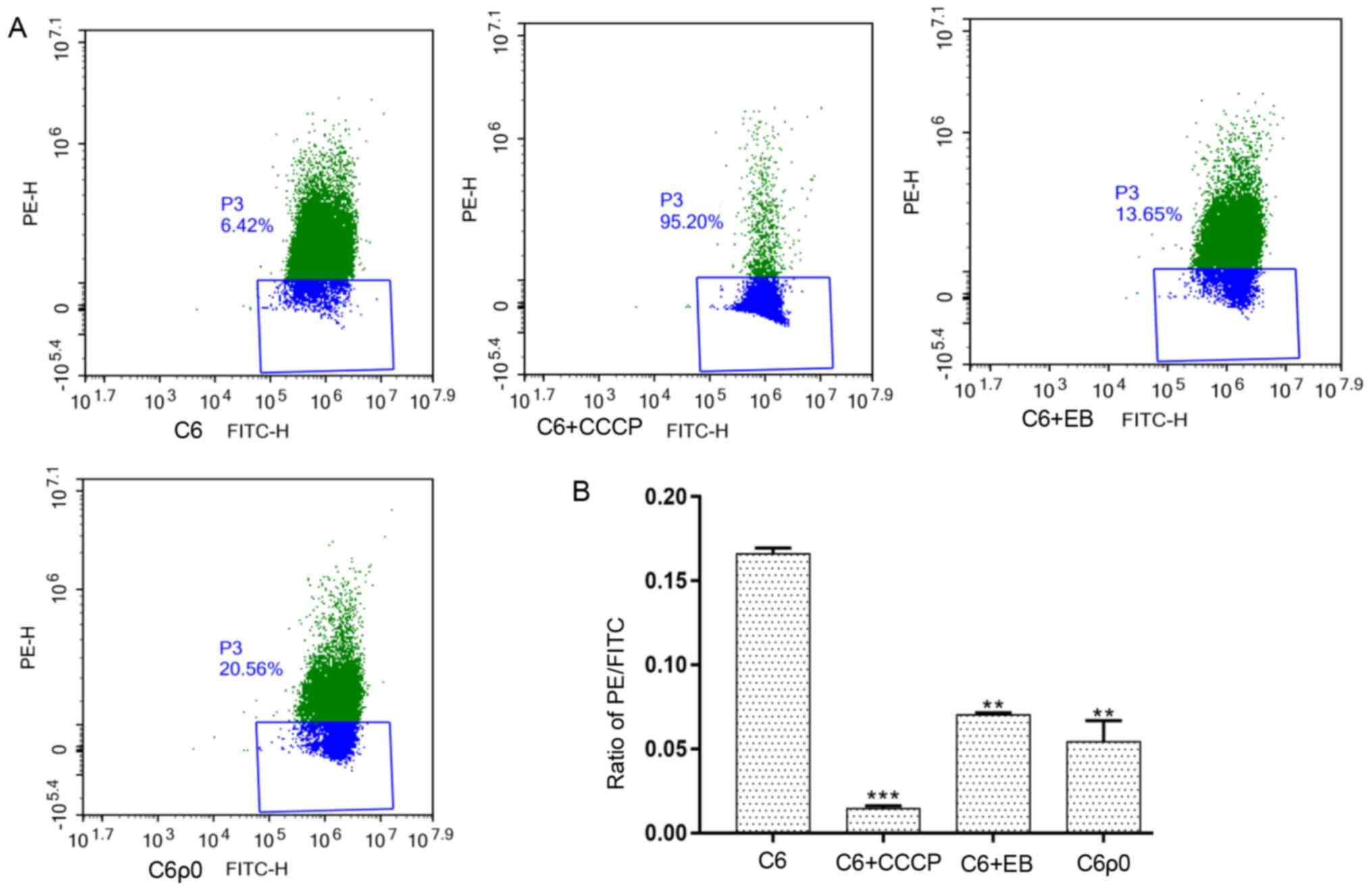
Flow cytometric analysis of ΔΨm in C6
and C6ρ0 cells
To determine the ΔΨm changes in mtDNA depleted
cells, cells were stained with JC-1 and the results of flow
cytometry showed a distinct decline in ΔΨm from untreated C6 cells,
2-day EB-treated C6 cells and C6ρ0 monoclonal cells (Fig. 5). These results suggested that the
treatment with EB induced a significant decrease in ΔΨm. Thus, the
ΔΨm detected in C6ρ0 cells was reduced by two-thirds compared with
C6 cells.
Discussion
Mitochondria are the main source of cellular energy
and serve a role in cell proliferation, growth and differentiation
through continuous fusion and division (21). The mitochondrial genome is essential
for a normal cellular function and it encodes important subunits of
the mitochondrial respiratory chain and F0F1-ATPase, which are
required for oxidative phosphorylation. Depletion of mtDNA results
in the inhibition of the mitochondrial respiratory chain and the
loss of mtDNA-encoded mitochondrial enzyme activity (16,22–24).
In the present study, persistent mtDNA damage of C6
cells by EB and ddC successfully induced the C6ρ0 cell phenotype.
EB is known for its insertion in mammalian mtDNA, thus inhibiting
mtDNA replication (25,26), and ddC is used as an antiretroviral
drug that can reduce the level of mtDNA by inhibiting mitochondrial
DNA polymerase γ (27). Therefore,
these two compounds have the ability to reduce the amount of mtDNA
by interfering with mtDNA replication or inhibiting polymerase
γ.
Indeed, in the present study C6ρ0 monoclonal cells
were successfully obtained through the treatment with the mixture
of EB and ddC as reported by the literature. As a result, enzymes
involved in pyrimidine biosynthesis cannot be activated, resulting
in the inability of C6ρ0 cells to remain alive without uridine and
pyruvate (28). The morphology of
mitochondria in C6 and C6ρ0 cells observed by TEM revealed the
presence of elongated mitochondria with parallel and normal
electron density in the wild-type cells, while disordered swollen
mitochondria were found in C6ρ0 cells. The changes in C6ρ0 cells
were due to the loss of the mitochondrial genome and in agreement
with the results of previous studies (17,20,29).
However, not all cell types treated with EB and ddC
can result in pure ρ0 cells. Indeed, in the present study HepG2 and
Huh7 cells did not lose all their mtDNA following the above
treatment (data not shown). This is because not all cells are
sensitive to drugs that destroy mtDNA. Thus far, only several cell
types can form pure ρ0 cells that can be used for further research
(17,20,26,30).
Since mtDNA-depleted cells provide a cytoplasmic
hybrid model for studying mtDNA single nucleotide polymorphisms
with the same nuclear DNA background, they are considered an
effective tool for studying mitochondrial disorders (15). mtDNA and nuclear DNA can control
mitochondrial functions and the cybrids allow researchers to
evaluate whether the mtDNA or nuclear DNA function is involved in a
mitochondrial defect (31). In the
present study, a practical and reliable technique was developed to
produce C6ρ0 monoclonal cells and cybrids generated with rat
platelets. The experiments on the growth characteristics of C6 and
C6ρ0 cells showed that the growth rate of C6ρ0 was significantly
reduced compared with the parental cells and that uridine and
pyruvate were necessary for their growth. Nevertheless, cybrids
grew as much as the wild-type cells and did not need the help of
uridine and pyruvate. However, it has been reported that the
T47Dρ0, MOLT-4ρ0, HeLaρ0, 143BTK−ρ0 cells grow slower
than their parent cells (17,20,29,32).
It is noteworthy that the hepatoma Hepal1-6ρ0 and HepG2ρ0 cells
proliferate several times more than the wild-type cells, suggesting
that different types of cells may exhibit different sensitivities
to the intracellular changes caused by mtDNA and not all cell types
proliferate less after mtDNA depletion (33).
The number of mitochondria is closely associated
with the energy cell metabolism (34). In the present study, the
mitochondrial mass of C6ρ0 cells was notably decreased compared
with the mitochondrial mass in C6 cells. The hypothesis was that
the decrease of mtDNA content in C6ρ0 cells led to mitochondrial
dysfunction and that changes in energy metabolism and ATP content
may destroy unconstrained tumor cell proliferation and invasion
(35). A series of cellular changes
and diseases are closely associated with mtDNA mutations (36). By contrast, some studies have
identified that the increasing mtDNA content is a self-protection
mechanism that prevents apoptosis and increases tumor cell
sensitivity to chemotherapeutic drugs (37,38).
The mitochondrial content in the cell determines the apoptotic fate
and modulates the time of death, since cells with higher
mitochondrial content are more prone to die (39). In fact, the amount of all apoptotic
proteins is modulated by the mitochondrial content (40). It is also hypothesized that mtDNA
destruction can enhance the apoptosis of certain cells in
vivo and inhibit the tumorigenicity of certain tumor cells
(41).
Mitochondria are the major source of ROS, which
serve key roles in both physiological and pathological processes
and act as mediators in a number of cellular signaling pathways in
the cell (42). An increase in ROS
levels can trigger a protective response by activating antioxidant
response elements (43). However,
the increase of ROS damages proteins, fats and nucleic acids,
triggering other stress responses or cell death (44,45).
Conversely, appropriate levels of ROS can reduce cancer, diabetes
and delay aging (46). In the
present study, the increase in ROS content of rotenone-treated C6
cells was more than twice that of non-treated C6 cells, while ROS
levels in monoclonal C6ρ0 clearly decreased. These results
demonstrated that drugs targeting the complex I, III and V of the
respiratory chain (11,47) can cause the increase of ROS, but
they have a weak effect on mitochondrial DNA depleted cells.
Mitochondria are key regulators of cell death
through alterations in the ΔΨm to respond to various unfavorable
situations. The results of the present study showed that the ΔΨm in
C6 cells was reduced following EB treatment, while the reduction of
ΔΨm in C6ρ0 cells was one third of that of the parent cells. In
addition, the C6ρ0 cells remained alive at relatively low ΔΨm.
These results indicated that the complete depletion of the mtDNA in
C6ρ0 cells was not associated to a complete disappearance of the
ΔΨm. Some studies have demonstrated that mitochondria with a
dysfunctional respiratory chain can still generate ΔΨm by the use
of glycolytic ATP (18,48). A previous study suggested that the
change in the membrane status leads to changes in external
oxidative stress tolerance and has an influence on the efficacy of
cancer therapy (32).
Overall, the method of the present study proved
efficient and reliable; the defects in the mitochondrial structure
and function in C6ρ0 cells can lead to impairments in cell
proliferation in vitro. Therefore, C6ρ0 cell lines with
different nuclear backgrounds could be used as a model in future
experiments to study glioma associated with mitochondria.
Acknowledgements
Not applicable.
Funding
The present study was supported by grants from the
National Science Foundation of China (grant no. 31870333), Qinghai
Provincial Science Foundation (grant no. 2019-ZJ-7023), Qinghai
Province International Cooperation Project (grant no. 2018-HZ-812),
the Taishan Scholar Program of Shandong Province (grant no.
tshw201502046) and the Shuangbai Project of Yantai and Youth
Innovation Promotion Association, CAS.
Availability of data and materials
The datasets used and/or analyzed in the current
study are available from the corresponding author upon reasonable
request.
Authors' contributions
HW and ZW conceived the study. JL and BB designed
the experiments. YJ and GL performed the experiments. YJ and GL
analyzed the data. YJ wrote the manuscript. All authors read and
approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Ostrom QT, Gittleman H, Fulop J, Liu M,
Blanda R, Kromer C, Wolinsky Y, Kruchko C and Barnholtz-Sloan JS:
CBTRUS statistical report: Primary brain and central nervous system
tumors diagnosed in the united states in 2008–2012. Neuro Oncol. 17
(Suppl 4):iv1–iv62. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Liu CA, Chang CY, Hsueh KW, Su HL, Chiou
TW, Lin SZ and Harn HJ: Migration/invasion of malignant gliomas and
implications for therapeutic treatment. Int J Mol Sci. 19:11152018.
View Article : Google Scholar
|
|
3
|
Chatterjee A, Mambo E and Sidransky D:
Mitochondrial DNA mutations in human cancer. Oncogene.
25:4663–4674. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Rocha EM, De Miranda B and Sanders LH:
Alpha-synuclein: Pathology, mitochondrial dysfunction and
neuroinflammation in Parkinson's disease. Neurobiol Dis.
109:249–257. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Verma P, Singh A, Nthenge-Ngumbau DN,
Rajamma U, Sinha S, Mukhopadhyay K and Mohanakumar KP: Attention
deficit-hyperactivity disorder suffers from mitochondrial
dysfunction. BBA Clin. 6:153–158. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kraya T, Deschauer M, Joshi PR, Zierz S
and Gaul C: Prevalence of headache in patients with mitochondrial
disease: A cross-sectional study. Headache. 58:45–52. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Szendroedi J, Phielix E and Roden M: The
role of mitochondria in insulin resistance and type 2 diabetes
mellitus. Nat Rev Endocrinol. 8:92–103. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Theurey P and Pizzo P: The aging
mitochondria. Genes (Basel). 9:222018. View Article : Google Scholar
|
|
9
|
Schapira AH: Mitochondrial disease.
Lancet. 368:70–82. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Sharma P and Sampath H: Mitochondrial DNA
integrity: Role in health and disease. Cells. 8:1002019. View Article : Google Scholar
|
|
11
|
Dias N and Bailly C: Drugs targeting
mitochondrial functions to control tumor cell growth. Biochem
Pharmacol. 70:1–12. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Govindaraj P, Rani B, Sundaravadivel P,
Vanniarajan A, Indumathi KP, Khan NA, Dhandapany PS, Rani DS,
Tamang R, Bahl A, et al: Mitochondrial genome variations in
idiopathic dilated cardiomyopathy. Mitochondrion. 48:51–59. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Spadafora D, Kozhukhar N, Chouljenko VN,
Kousoulas KG and Alexeyev MF: Methods for efficient elimination of
mitochondrial DNA from cultured cells. PLoS One. 11:e01546842016.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Yang L, Long Q, Liu J, Tang H, Li Y, Bao
F, Qin D, Pei D and Liu X: Mitochondrial fusion provides an
‘initial metabolic complementation’ controlled by mtDNA. Cell Mol
Life Sci. 72:2585–2598. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Chomyn A: Platelet-mediated transformation
of human mitochondrial DNA-less cells. Methods Enzymol.
264:334–339. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yoon YG, Oh YJ and Yoo YH: Rapid isolation
of mitochondrial DNA-depleted mammalian cells by ethidium bromide
and dideoxycytidine treatments. J App Biol Chem. 57:259–265. 2014.
View Article : Google Scholar
|
|
17
|
Yu M, Shi Y, Wei X, Yang Y, Zhou Y, Hao X,
Zhang N and Niu R: Depletion of mitochondrial DNA by ethidium
bromide treatment inhibits the proliferation and tumorigenesis of
T47D human breast cancer cells. Toxicol Lett. 170:83–93. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Fernández-Moreno M, Hermida-Gómez T,
Gallardo ME, Dalmao-Fernández A, Rego-Pérez I, Garesse R and Blanco
FJ: Generating rho-0 cells using mesenchymal stem cell lines. PLoS
One. 11:e01641992016. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Binder DR, Dunn WH Jr and Swerdlow RH:
Molecular characterization of mtDNA depleted and repleted NT2 cell
lines. Mitochondrion. 5:255–265. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kukat A, Kukat C, Brocher J, Schäfer I,
Krohne G, Trounce IA, Villani G and Seibel P: Generation of rho0
cells utilizing a mitochondrially targeted restriction endonuclease
and comparative analyses. Nucleic Acids Res. 36:e442008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Holmuhamedova E, Jahangira A,
Bienengraebera M, Lewisb LD and Terzic A: Deletion of mtDNA
disrupts mitochondrial function and structure, but not biogenesis.
Mitochondrion. 3:13–19. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Alston CL, Rocha MC, Lax NZ, Turnbull DM
and Taylor RW: The genetics and pathology of mitochondrial disease.
J Pathol. 241:236–250. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Meiliana A, Dewi NM and Wijaya A:
Mitochondria in health and disease. Indonesian Biomedical J.
11:1–15. 2019. View Article : Google Scholar
|
|
24
|
Miller SW, Trimmer PA, Davis Parker W Jr
and Davis RE: Creation and characterization of mitochondrial
DNA-depleted cell lines with ‘neuronal-like’ properties. J
Neurochem. 67:1897–1907. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Desjardins P, Frost E and Morais R:
Ethidium bromide-induced loss of mitochondrial DNA from primary
chicken embryo fibroblasts. Mol Cell Biol. 5:1163–1169. 1985.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Armand R, Channon JY, Kintner J, White KA,
Miselis KA, Perez RP and Lewis LD: The effects of ethidium bromide
induced loss of mitochondrial DNA on mitochondrial phenotype and
ultrastructure in a human leukemia T-cell line (MOLT-4 cells).
Toxicol Appl Pharmacol. 196:68–79. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Nelson I, Hanna MG, Wood NW and Harding
AE: Depletion of mitochondrial DNA by ddC in untransformed human
cell lines. Somat Cell Mol Genet. 23:287–290. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
King MP and Attardi G: Isolation of human
cell lines lacking mitochondrial DNA. Methods Enzymol. 264:304–313.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Wochna A, Niemczyk E, Kurono C, Masaoka M,
Majczak A, Kedzior J, Slominska E, Lipinski M and Wakabayashi T:
Role of mitochondria in the switch mechanism of the cell death mode
from apoptosis to necrosis-studies on rho0 cells. J Electron
Microsc (Tokyo). 54:127–138. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Schauen M, Spitkovsky D, Schubert J,
Fischer JH, Hayashi J and Wiesner RJ: Respiratory chain deficiency
slows down cell-cycle progression via reduced ROS generation and is
associated with a reduction of p21CIP1/WAF1. J Cell Physiol.
209:103–112. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Inoue K, Takai D, Hosaka H, Ito S, Shitara
H, Isobe K, LePecq JB, Segal-Bendirdjian E and Hayashi J: Isolation
and characterization of mitochondrial DNA-less lines from various
mammalian cell lines by application of an anticancer drug,
ditercalinium. Biochem Biophys Res Commun. 239:257–260. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Tomita K, Kuwahara Y, Takashi Y, Tsukahara
T, Kurimasa A, Fukumoto M, Nishitani Y and Sato T: Sensitivity of
mitochondrial DNA depleted ρ0 cells to H2O2
depends on the plasma membrane status. Biochem Biophys Res Commun.
490:330–335. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Boland ML, Chourasia AH and Macleod KF:
Mitochondrial dysfunction in cancer. Front Oncol. 3:2922013.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Vyas S, Zaganjor E and Haigis MC:
Mitochondria and Cancer. Cell. 166:555–566. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Penrose HM, Heller S, Cable C, Nakhoul H,
Ungerleider N, Baddoo M, Pursell ZF, Flemington EK, Crawford SE and
Savkovic SD: In colonic ρ° (rho0) cells reduced mitochondrial
function. Oncoscience. 4:189–198. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Pessôa LVF, Bressan FF, Chiaratti MR,
Pires PR, Perecin F, Smith LC and Meirelles FV: Mitochondrial DNA
dynamics during in vitro culture and pluripotency induction of a
bovine Rho0 cell line. Genet Mol Res. 14:14093–14104. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Mei H, Sun S, Bai Y, Chen Y, Chai R and Li
H: Reduced mtDNA copy number increases the sensitivity of tumor
cells to chemotherapeutic drugs. Cell Death Dis. 6:e17102015.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Zong WX, Rabinowitz JD and White E:
Mitochondria and cancer. Mol Cell. 61:667–676. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Grady JP, Pickett SJ, Ng YS, Alston CL,
Blakely EL, Hardy SA, Feeney CL, Bright AA, Schaefer AM, Gorman GS,
et al: mtDNA heteroplasmy level and copy number indicate disease
burden in m.3243A>G mitochondrial disease. EMBO Mol Med.
10:e82622018. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Márquez-Jurado S, Díaz-Colunga J, das
Neves RP, Martinez-Lorente A, Almazán F, Guantes R and Iborra FJ:
Mitochondrial levels determine variability in cell death by
modulating apoptotic gene expression. Nat Commun. 9:3892018.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Papal S and Skulachev VP: Reactive oxygen
species, mitochondria, apoptosis and aging. Mol Cell Biochem.
174:305–319. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Frazier AE, Thorburn DR and Compton AG:
Mitochondrial energy generation disorders: Genes, mechanisms, and
clues to pathology. J Biol Chem. 294:5386–5395. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Shadel GS and Horvath TL: Mitochondrial
ROS signaling in organismal homeostasis. Cell. 163:560–569. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Nunnari J and Suomalainen A: Mitochondria:
In sickness and in health. Cell. 148:1145–1159. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Meyer JN, Hartman JH and Mello DF:
Mitochondrial toxicity. Toxicol Sci. 162:15–23. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Ristow M, Zarse K, Oberbach A, Klöting N,
Birringer M, Kiehntopf M, Stumvoll M, Kahn CR and Blüher M:
Antioxidants prevent health-promoting effects of physical exercise
in humans. Proc Natl Acad Sci USA. 106:8665–8670. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Zorov DB, Juhaszova M and Sollott SJ:
Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS
release. Physiol Rev. 94:909–950. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Buchet K and Godinot C: Functional
F1-ATPase essential in maintaining growth and membrane potential of
human mitochondrial DNA-depleted rho degrees cells. J Biol Chem.
273:22983–22989. 1998. View Article : Google Scholar : PubMed/NCBI
|