Introduction
Pulmonary arterial hypertension (PAH) is a
pathological condition that occurs in the cardiovascular system
(1). In PAH, pulmonary vascular
resistance and pulmonary artery pressure increase, ultimately
resulting in right heart failure and even death (2,3).
Maladaptive processes, such as fibrosis, can damage or even
collapse the function of the right ventricle (RV) (4).
MicroRNAs (miRNAs/miRs) are a group of small
endogenous non-coding RNAs that can negatively regulate target gene
expression post-transcriptionally, mainly through mRNA degradation
or translational inhibition (5–8).
Alterations in the expression levels of miRNAs have been associated
with a number of pathological disease processes, such as
cardiovascular diseases. For this reason, circulating miRNAs have
been hypothesized to be potential biomarkers or therapeutic targets
for several types of disease, such as miR-29 in atrial fibrillation
and miR-133a in myocardial infarction (9,10). In
fact, several miRNAs, including miR-143, miR-124, miR-140-5p and
miR-135a, have been reported to be dysregulated in PAH animal
models or patients with PAH (11–14).
Previous studies have revealed that miR-1 was
involved in the pathogenesis of left heart failure and left
ventricle (LV) fibrosis (15,16).
Dysregulated miR-1 biogenesis was previously associated with heart
failure in aged rats, especially aged hypertensive rats (17). In addition, the expression levels of
miR-1 were upregulated in lungs from an experimental model of PAH
and in the plasma from patients with PAH, and miR-1 induced
endothelial dysfunction, suggesting a pathophysiological role for
miR-1 in PAH (18). In a previous
study, the transfection with the miR1 antagomiR downregulated the
expression levels of TGF-β and collagen hyperplasia in myocardial
infarction model mice (19).
However, to the best of our knowledge, whether miR-1 may be
involved in the regulation of PAH remains unknown.
The PI3K/AKT signaling pathway was discovered to be
involved in the regulation of cardiac fibrosis (20). A previous study revealed that
miR-132 activated the PI3K/AKT signaling pathway by downregulating
PTEN expression levels, thus inhibiting apoptosis and facilitating
cardiomyocyte proliferation and cardiac fibrosis in dilated
cardiomyopathy model rats (21).
However, whether the PI3K/AKT signaling pathway may be involved in
the regulatory effects of miR-1 on cardiac fibrosis in PAH remains
unclear.
The present study aimed to determine whether the
knockdown of miR-1 could counter PAH through attenuating RV
fibrosis in PAH model rats, and whether the PI3K/AKT signaling
pathway may be involved in the key roles of miR-1 in regulating
fibrosis in CFs.
Materials and methods
Animal studies
Experiments were performed using 78 5–6 weeks-old
male Sprague-Dawley (SD) rats (weight, 180–200 g; Beijing Vital
River Laboratory Animal Technology Co., Ltd.). All procedures were
approved by the Experimental Animal Care and Use Committee of
Nanjing Medical University (Nanjing, China; approval no. 17041015),
and were conducted in accordance with the Guide for the Care and
Use of Laboratory Animals (National Institutes of Health
publication no. 85-23, revised 1996) (22). The rats were kept in a temperature
(22±1°C) and humidity (40–60%)-controlled room under a 12-h
light/dark cycle with free access to standard chow and tap water.
The experiments were performed at the Animal Core Facility of
Nanjing Medical University.
Establishment of hypoxia rat model and
grouping
The establishment of the hypoxic condition was
performed as previously described (23). Briefly, SD rats were divided into 2
groups: i) Normoxia group (n=8), in which rats received normoxia
(21% O2) for 4 weeks; and ii) hypoxia group (n=13), in
which rats received hypoxia (10% O2) (24,25)
for 4 weeks. The expression levels of miR-1 were subsequently
determined in the RV of rats in the two groups.
In another experiment, the 5–6 weeks-old rats were
divided into the following groups: i) Normoxia + negative control
(NC) antagomiR (n=10); ii) normoxia + miR-1 antagomiR (n=10); iii)
hypoxia + NC antagomiR (n=15); and iv) hypoxia + miR-1 antagomiR
groups (n=15). Hypoxia and normoxia were administered as
aforementioned. Simultaneously, rats were injected with miR-1
antagomiR (sequence 5′-UGGAAUGUAAAGAAGUGUGUAU-3′; Guangzhou RiboBio
Co., Ltd.) or NC antagomiR (sequence 5′-CAGUACUUUUGUGUAGUACAA-3′;
Guangzhou RiboBio Co., Ltd.) via the tail vein twice a week (40
mg/kg/time). After 4 weeks, RV function and fibrosis were
determined.
Animal experiments
SD rats were anesthetized with 50 mg/kg
pentobarbital (i.p.). Using a PowerLab data acquisition system
(ADInstruments, Ltd.), a 1.4F conductance micromanometer catheter
(Millar) was inserted via the RV, across the aortic valve and into
the RV chamber to measure the right ventricular systolic pressure
(RVSP) and the mean pulmonary arterial pressure (mPAP).
Subsequently, the rats were sacrificed by cervical dislocation
following anesthesia with 3.5% isoflurane induction and 2%
isoflurane maintenance. The RV, LV and interventricular septum (S)
of the rats were separately dissected. The tibia length (TL) was
measured and weighed to calculate the ratio of RV to (LV + S) and
RV/TL, two key indicators for assessing RV hypertrophy.
Isolation and culture of cardiac
fibroblasts (CFs)
Rat CFs were isolated from 60 male and female SD
rats (age, 1–3 days old; weight, 5–8 g; Beijing Vital River
Laboratory Animal Technology Co., Ltd.), or male 9–10 weeks-old PAH
(PCFs) or normoxia (NCFs) model rats (350–400 g; n=6 for each
group). The rats were kept in a temperature (22±1°C) and humidity
(40–60%)-controlled room under a 12 h light-dark cycle with free
access to standard chow and tap water. The rats were sacrificed by
cervical dislocation following anesthesia with 3.5% isoflurane
induction for 2 min and 2% isoflurane maintenance. Death was
confirmed by the absence of a heartbeat, and corneal reflexes and
paw withdrawal response to a noxious pinch. Ventricular tissue was
subsequently dissected, washed, minced and subjected to three
digestions at 37°C for 20 min in a solution containing a mixture of
1 mg/ml collagenase A and 0.5 mg/ml hyaluronidase following an
initial digestion step in a proteinase bacterial solution (4 U/ml)
for 15 min. After each cycle of digestion, the tissue was
mechanically dissociated using a 5 ml pipette (Eppendorf), the
dissociated cells were collected and resuspended in Dulbecco's
modified Eagles medium (DMEM; Gibco; Thermo Fisher Scientific,
Inc.). CFs were separated from the cardiomyocytes by centrifugation
(1,000 × g) at 4°C for 5 min and cultured to confluence in 10-cm
cell culture dishes in DMEM supplemented with 10% FBS (Gibco;
Thermo Fisher Scientific, Inc.), 1% penicillin and 1% streptomycin,
and maintained at 37°C in a humidified atmosphere with 5%
CO2 and 95% O2. CFs from the second passage
were used for the subsequent experiments.
In the hypoxic group, CFs were exposed to 0, 3 or 5%
oxygen in an incubator connected with a chamber that was
equilibrated with a water-saturated gas mixture of 0, 3 or 5%
O2, 5% CO2 and 95, 92 or 90% N2,
respectively, at 37°C for 12, 24 or 48 h. In the normoxic group,
CFs were exposed to 5% CO2 and 95% O2.
CF transfection with miR-1
antagomiR
Negative control (NC) antagomiR and miR-1 antagomiR
were synthesized by Guangzhou RiboBio Co., Ltd. CFs were seeded
into 12-well plates at a density of 5×104 cells/ml and
transfected with 100 nM NC antagomiR or miR-1 antagomiR using
Lipofectamine 3,000 reagent (Invitrogen; Thermo Fisher Scientific,
Inc.) for 6 h. Subsequently, the CFs were treated with hypoxic or
normoxic for 24 h. The sequences of the oligonucleotides were as
follows: NC antagomiR, 5′-CAGUACUUUUGUGUAGUACAA-3′; and miR-1
antagomiR, 5′-AUACAUACUUCUUUACAUUCCA-3′.
Masson's trichrome staining
The rats were sacrificed by cervical dislocation
following anesthesia with 3.5% isoflurane induction and 2%
isoflurane maintenance. and the hearts were removed. The RV tissues
were fixed with 4% paraformaldehyde at 4°C for 24 h, and embedded
in paraffin. Cardiac sections (5-µm) were subsequently analyzed
using Masson's trichrome staining (Nanjing Biochannel Biotechnology
Co., Ltd.) to measure the fibrosis of cardiomyocytes. Briefly,
sections were incubated in celestine blue solution for 5 min,
washed with H2O; incubated in hemalun solution for 5
min, in H2O for 10 min, in 0.5% fuchsine acid and 1.5%
Ponceau xylidine for 5 min, washed with H2O; incubated
in 1% phosphomolybdic acid for 10 min, in 2.5% aniline blue
solution for 5 min, washed with H2O; incubated in 1%
acetic acid for 1 min, and then briefly in an ascending isopropanol
series followed by xylol. All the operations were performed at room
temperature. Then 3–5 randomly selected fields of view were
selected from each of three sections from one rat and observed
under a light microscope (magnification, ×200; Carl Zeiss AG).
Images were analyzed using Image-Pro Plus software (version 6.0;
Media Cybernetics, Inc.).
Reverse transcription-quantitative PCR
(RT-qPCR)
Total RNA was extracted from RV tissues or CFs using
TRIzol® reagent (Invitrogen; Thermo Fisher Scientific,
Inc.). Total RNA was reverse transcribed into cDNA using random
primers in a total volume of 10 µl and PrimeScript™ RT Master mix
(Takara Biotechnology Co., Ltd.), according to the manufacturer's
protocol, at 37°C for 15 min and 85°C for 5 sec. cDNA was stored at
−70°C prior to use. qPCR of miR-1, collagen I, collagen III,
α-smooth muscle actin (SMA) and connective tissue growth factor
(CTGF) expression levels were determined using SYBR Green I
fluorescence (Invitrogen; Thermo Fisher Scientific, Inc). All
samples were amplified in triplicate in 96-well plates using the
following thermocycling conditions: Initial denaturation at 95°C
for 10 min; followed by 40 cycles at 95°C for 10 sec and 60°C for 1
min. GAPDH or U6 were used as the internal controls for mRNA and
miRNA, respectively. Relative expression levels were quantified
using the 2−∆∆Cq method (26–29).
The primers used for the qPCR are shown in Table I.
 | Table I.Primers used for reverse
transcription-quantitative PCR. |
Table I.
Primers used for reverse
transcription-quantitative PCR.
| Gene | Species | Accession
number | Primer sequence
(5′→3′) |
|---|
| Collagen I | Rat | BC133728 | F:
TCAAGATGGTGGCCGTTAC |
|
|
|
| R:
CTGCGGATGTTCTCAATCTG |
| Collagen III | Rat | DN815278 | F:
CGAGATTAAAGCAAGAGGAA |
|
|
|
| R:
GAGGCTTCTTTACATACCAC |
| α-smooth muscle
actin | Rat | BC158550 | F:
GTCCCAGACATCAGGGAGTAA |
|
|
|
| R:
TCGGATACTTCAGCGTCAGGA |
| Connective tissue
growth factor | Rat | BC072503 | F:
CAGGGAGTAAGGGACACGA |
|
|
|
| R:
ACAGCAGTTAGGAACCCAGAT |
| miR-1 | Rat | NR032116 | F:
GGGGTGGAATGTAAAGAA |
|
|
|
| R:
TGCGTGTCGTGGAGTC |
| U6 | Rat | K00784 | F:
GCTTCGGCAGCACATATACTAAAAT |
|
|
|
| R:
CGCTTCACGAATTTGCGTGTCAT |
| GAPDH | Rat | AF106860 | F:
GGCACAGTCAAGGCTGAGAATG |
|
|
|
| R:
ATGGTGGTGAAGACGCCAGTA |
Western blotting
Total protein was extracted from CFs using RIPA
lysis buffer (BioChannel Biotechnology Co., Ltd.) and homogenized.
Debris that had not been homogenized was removed, and the
supernatant was obtained through centrifugation at 12,000 × g for
10 min at 4°C. Total protein was quantified by BCA (Beyotime
Institute of Biotechnology), and ~50 µg protein was separated via
8% SDS-PAGE. The separated proteins were subsequently transferred
onto PVDF membranes and blocked by 5% skimmed milk powder at room
temperature for 1 h. The membranes were then incubated with the
following primary antibodies at 4°C overnight: Anti-collagen I
(1:2,000; cat. no. ab34710; Abcam), anti-collagen III (1:5,000;
cat. no. ab7778; Abcam), anti-α-SMA (1:2,000; cat. no. ab32575;
Abcam), anti-CTGF (1:1,000; cat. no. ab6992; Abcam), anti-PI3K
(1:1,000; cat. no. 4249; Cell Signaling Technology, Inc.),
anti-phosphorylated (p)-PI3K (1:1,000; cat. no. 17366; Cell
Signaling Technology, Inc.), anti-AKT (1:1,000; cat. no. 4691; Cell
Signaling Technology, Inc.), anti-p-AKT (1:2,000; cat. no. 4060;
Cell Signaling Technology, Inc.) and anti-GAPDH (1:10,000, cat. no.
ab181602; Abcam). The horseradish peroxidase-conjugated goat
anti-rabbit secondary antibody (1:10,000, cat. no. ab7090; Abcam)
was added and incubated at room temperature for 1 h. ECL kit
(Beyotime Institute of Biotechnology) was used to visualize the
proteins. Densitometric analysis was performed using Image-Pro Plus
software (version 6.0; Media Cybernetics, Inc.).
Statistical analysis
Statistical analysis was performed using GraphPad
Prism 6.0 software (GraphPad Software, Inc.). Data are presented as
the mean ± SEM. Statistical differences between two groups were
determined using a unpaired Student's t-test, while
statistical differences between multiple groups were determined
using a one-way ANOVA followed by a Bonferroni's post hoc test. A
total of 3 experimental repeats were performed. P<0.05 was
considered to indicate a statistically significant difference.
Results
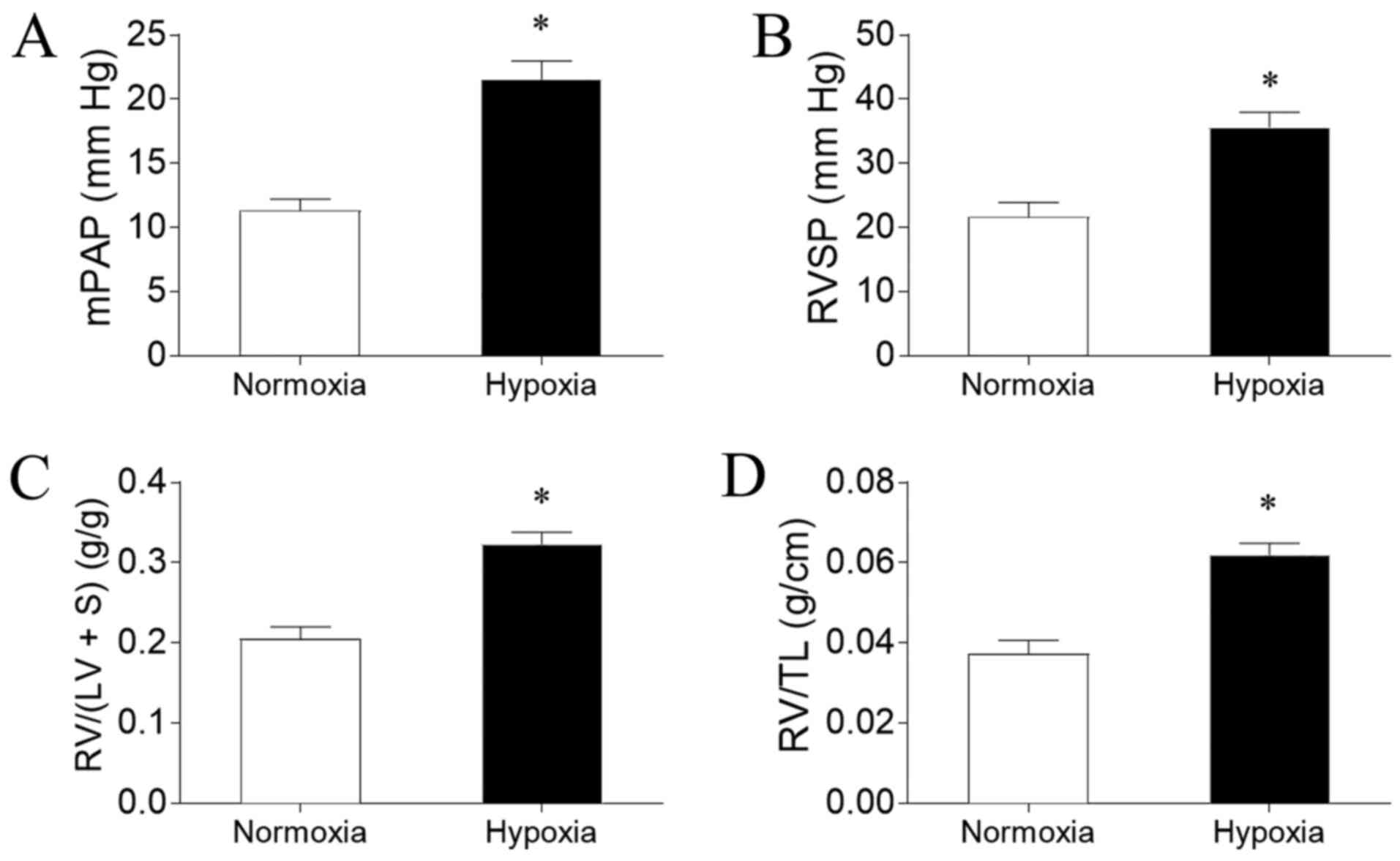
Hypoxia induces PAH in rats
PAH was successfully induced by hypoxia in the rat
model, as evidenced by an increased mPAP (Fig. 1A) and RVSP (Fig. 1B) compared with rats exposed to
normoxia. Hypoxia exposure also significantly increased RV/(LV + S)
(Fig. 1C) and RV/TL (Fig. 1D) in the rats compared with normoxia
exposure.
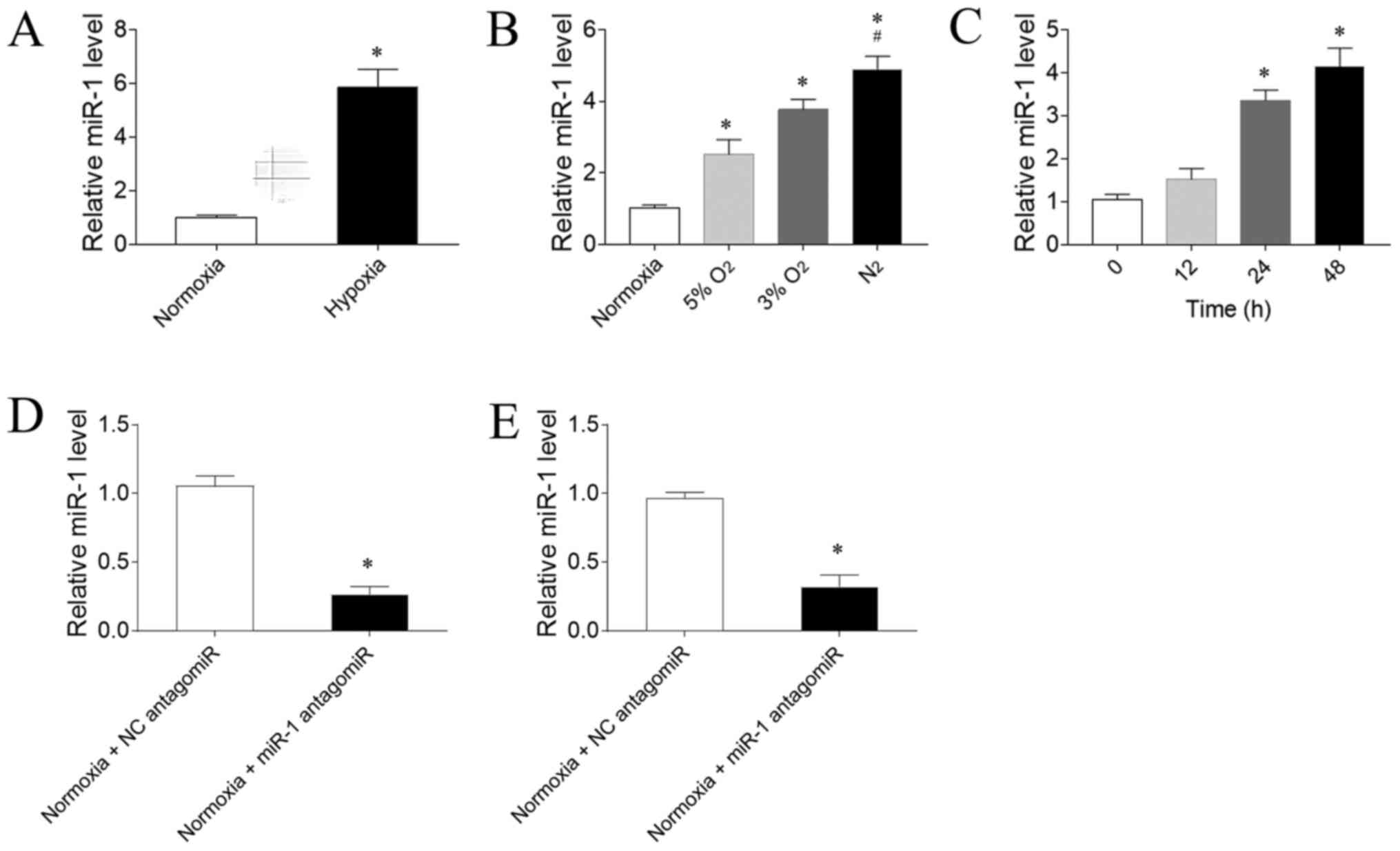
Expression levels of miR-1
The expression levels of miR-1 were significantly
increased in the RV of PAH model rats exposed to hypoxia compared
with rats exposed to normoxia (Fig.
2A). To determine the effect of hypoxia on the expression
levels of miR-1 in CFs, three gradient O2 concentrations
were used. The expression levels of miR-1 in CFs were sequentially
upregulated as the O2 concentration gradually decreased
compared with the normoxia group; the exposure to 5 or 3%
O2 significantly upregulated miR-1 expression levels
compared with exposure to normoxia. Notably, exposure to
N2 was more powerful in upregulating miR-1 expression
levels compared with 5% O2 exposure (Fig. 2B). 3% O2 was selected for
use in the following experiments. The expression levels of miR-1
were significantly upregulated following 24 h, but not 12 h, of
hypoxia exposure compared with CFs not exposed to hypoxia; however,
this upregulation was not further enhanced after 48 h of exposure
compared with 24 h (Fig. 2C). Thus,
24 h hypoxia stimulation was used in the following in vitro
experiments. miR-1 antagomiR significantly downregulated the
expression levels of miR-1 in the RV of rats compared with the NC
antagomiR (Fig. 2D). Furthermore,
the expression levels of miR-1 were significantly downregulated in
CFs transfected with miR-1 antagomiR compared with NC antagomiR
(Fig. 2E).
Effects of miR-1 antagomiR on PAH
Hypoxia-induced an increase in mPAP, which was
inhibited by miR-1 antagomiR (Fig.
3A). RVSP was increased in the rats treated with hypoxia, which
was reversed by miR antagomiR (Fig.
3B). The increase of RV/(LV+S) of rats induced by hypoxia was
alleviated by miR antagomiR administration (Fig. 3C). RV/TL was elevated in the rats
treated by hypoxia, and this increase was attenuated by
administration of miR-1 antagomiR (Fig.
3D).
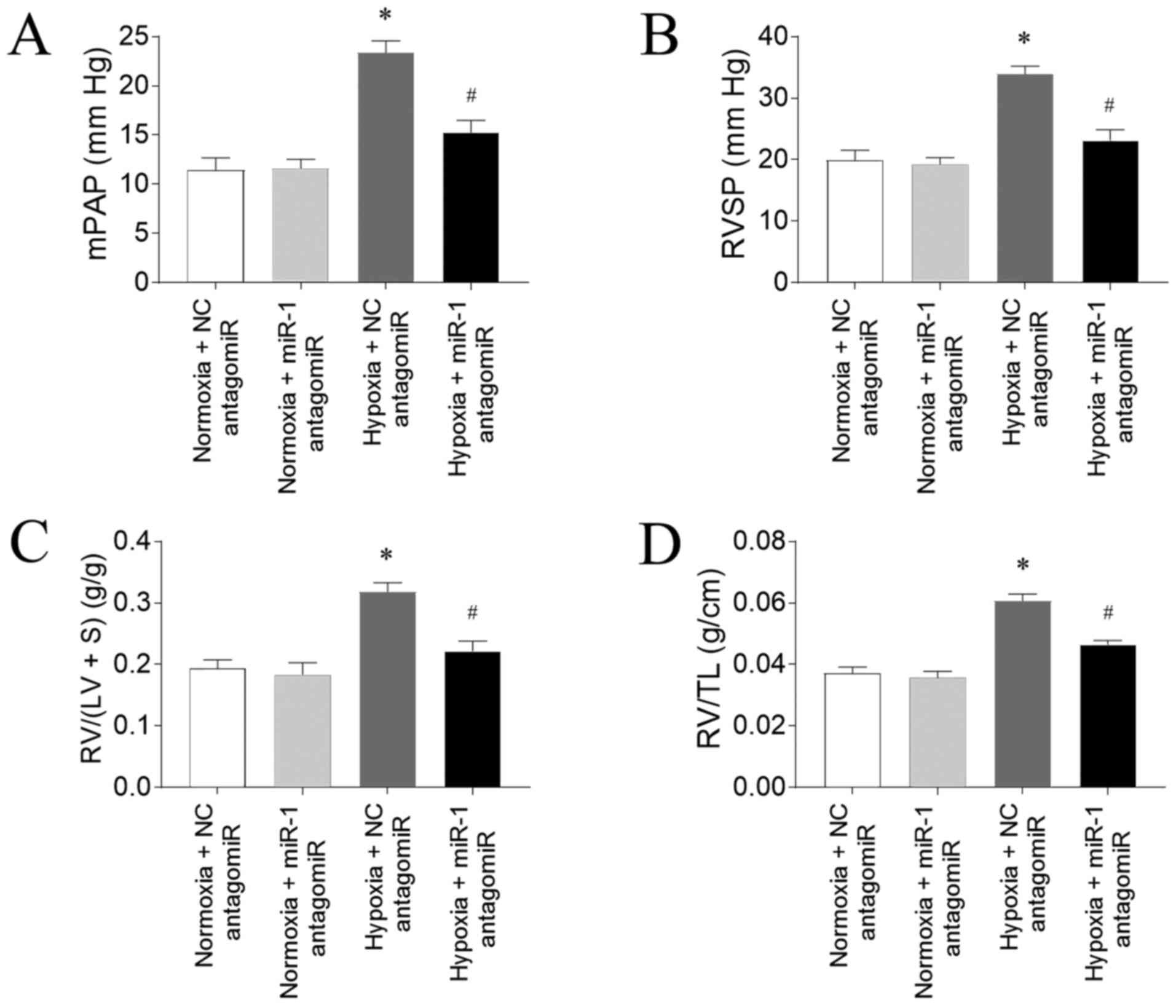 | Figure 3.miR-1 antagomiR transfection
attenuates hypoxia-induced PAH in rats. miR-1 antagomiR
transfection inhibited the increases in the (A) mPAP, (B) RVSP, (C)
RV/(LV+S) and (D) RV/TL in PAH model rats exposed to hypoxia. The
results are presented as the mean ± SEM. n=10 in normoxia + NC
antagomiR and normoxia + miR-1 antagomiR groups and n=15 in hypoxia
+ NC antagomiR and hypoxia + miR-1 antagomiR groups. *P<0.05 vs.
normoxia + NC antagomiR; #P<0.05 vs. hypoxia + NC
antagomiR. miR, microRNA; PAH, pulmonary arterial hypertension;
mPAP, mean pulmonary arterial pressure; RVSP, right ventricle
systolic pressure; RV, right ventricle; LV, left ventricle; S,
interventricular septum; TL, tibia length; NC, negative
control. |
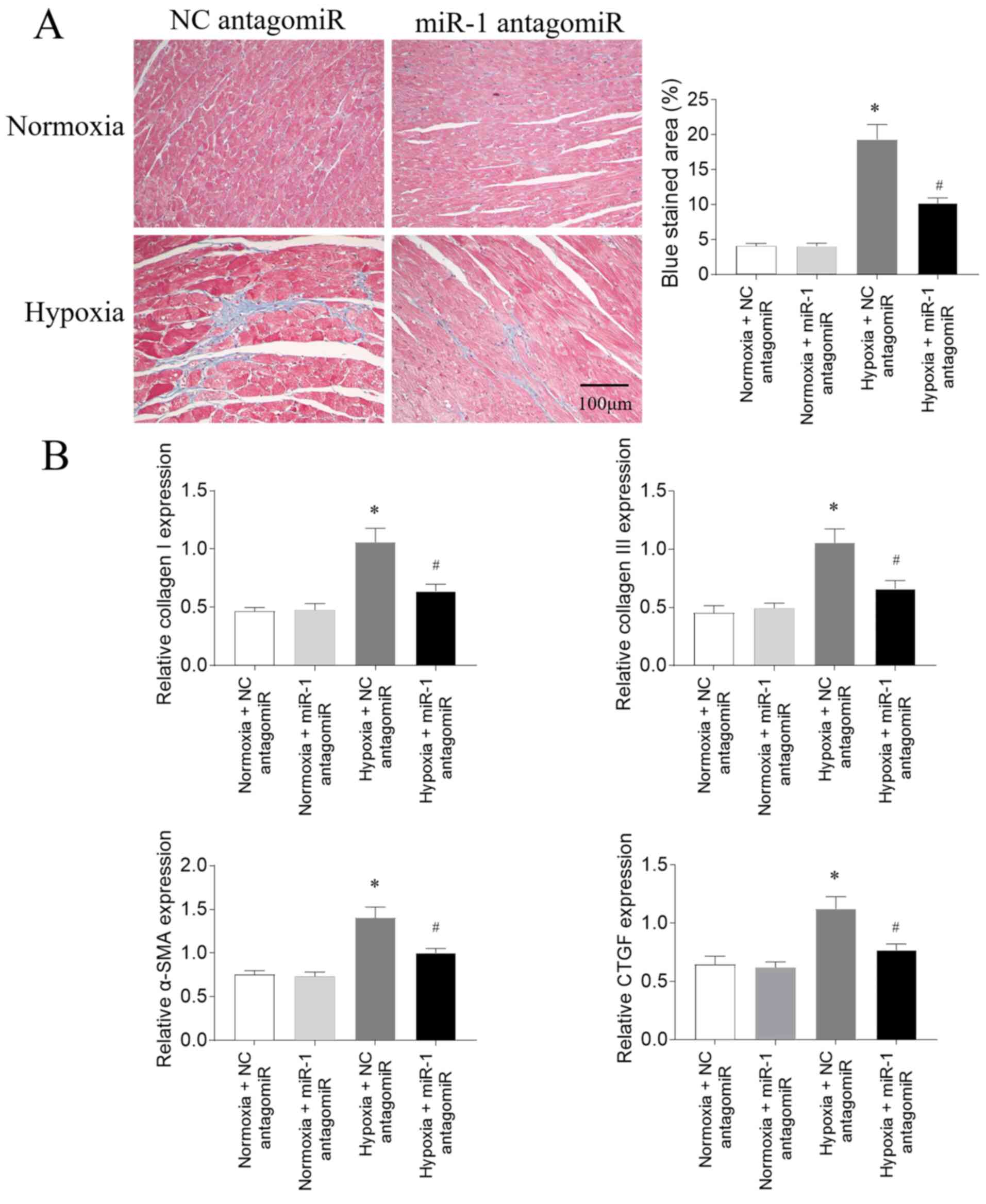
Effects of miR-1 antagomiR on fibrosis
in PAH model rats
According to the results of Masson's trichrome
staining, the RV fibrosis was increased following hypoxia
treatment; this increase was subsequently partially reversed
following miR-1 antagomiR transfection (Fig. 4A). The mRNA expression levels of
collagen I, collagen III, α-SMA and CTGF in the RV of PAH model
rats exposed to hypoxia were significantly upregulated; these
increases were partially inhibited following miR-1 antagomiR
transfection (Fig. 4B).
Effects of miR-1 antagomiR on fibrosis
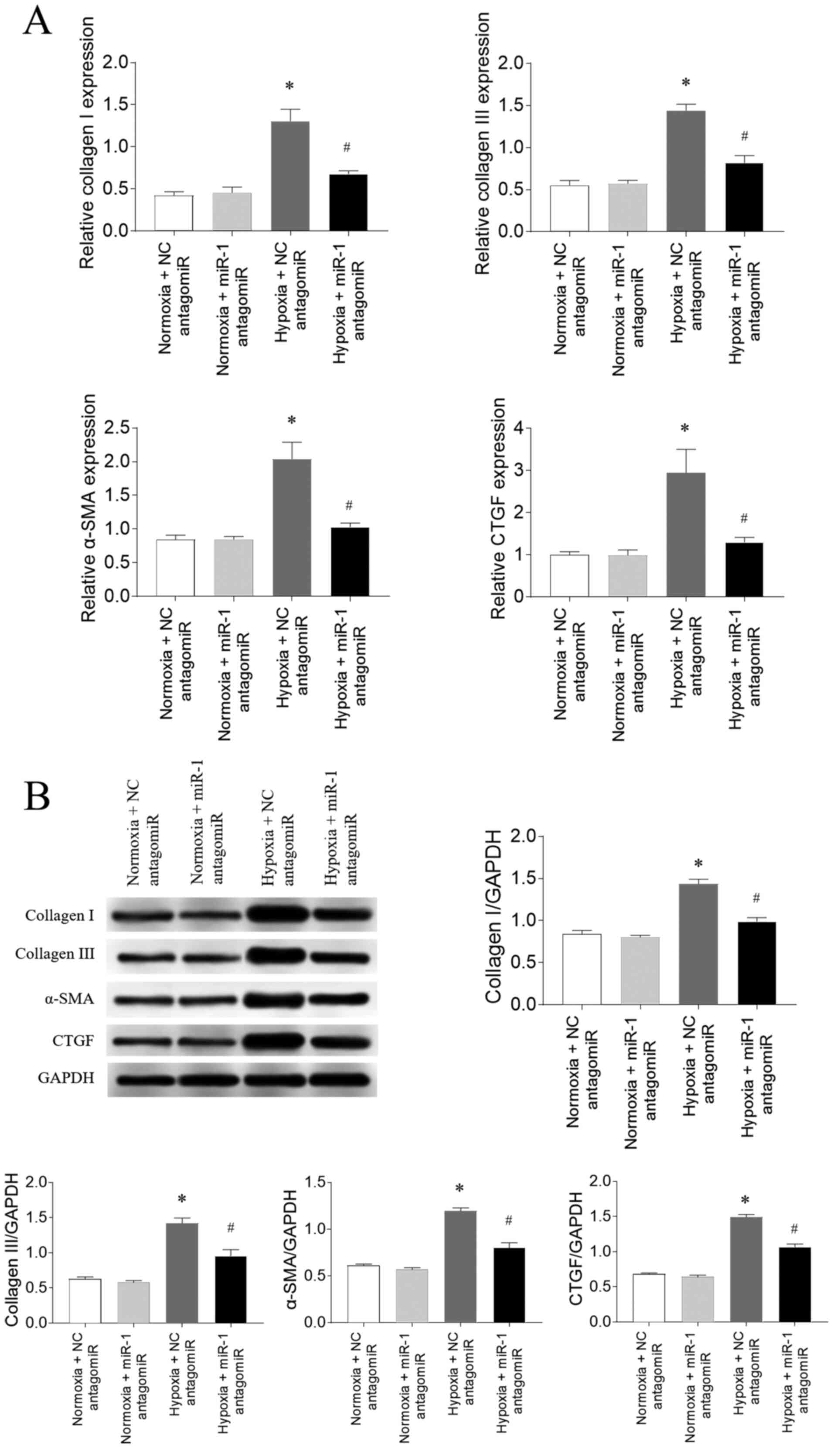
in CFs
Following 3% O2 exposure (hypoxia), the
mRNA expression levels of collagen I, collagen III, α-SMA and CTGF
were significantly upregulated in CFs compared with the normoxia
group, which were all subsequently attenuated following miR-1
antagomiR transfection (Fig. 5A).
The protein expression levels of collagen I, collagen III, α-SMA
and CTGF were also significantly upregulated in CFs exposed to
hypoxia compared with the normoxia group, and these increases were
partially inhibited by miR-1 antagomiR transfection (Fig. 5B).
Effects of miR-1 antagomiR on fibrosis
in CFs
The expression levels of collagen I in CFs isolated
from PAH model rats (PCFs) were significantly upregulated compared
with CFs isolated from normoxia rats (NCFs), while the subsequent
transfection with miR-1 antagomiR inhibited this upregulation
(Fig. 6). Collagen III expression
levels were also significantly upregulated in PAFs compared with
NCFs, and were also attenuated by miR-1 antagomiR transfection.
Similarly, the expression levels of α-SMA and CTGF in PCFs treated
with NC antagomiR were upregulated compared with in NCFs treated
with NC antagomiR, and the increases in α-SMA and CTGF expression
levels in PCFs were inhibited by miR-1antagomiR transfection.
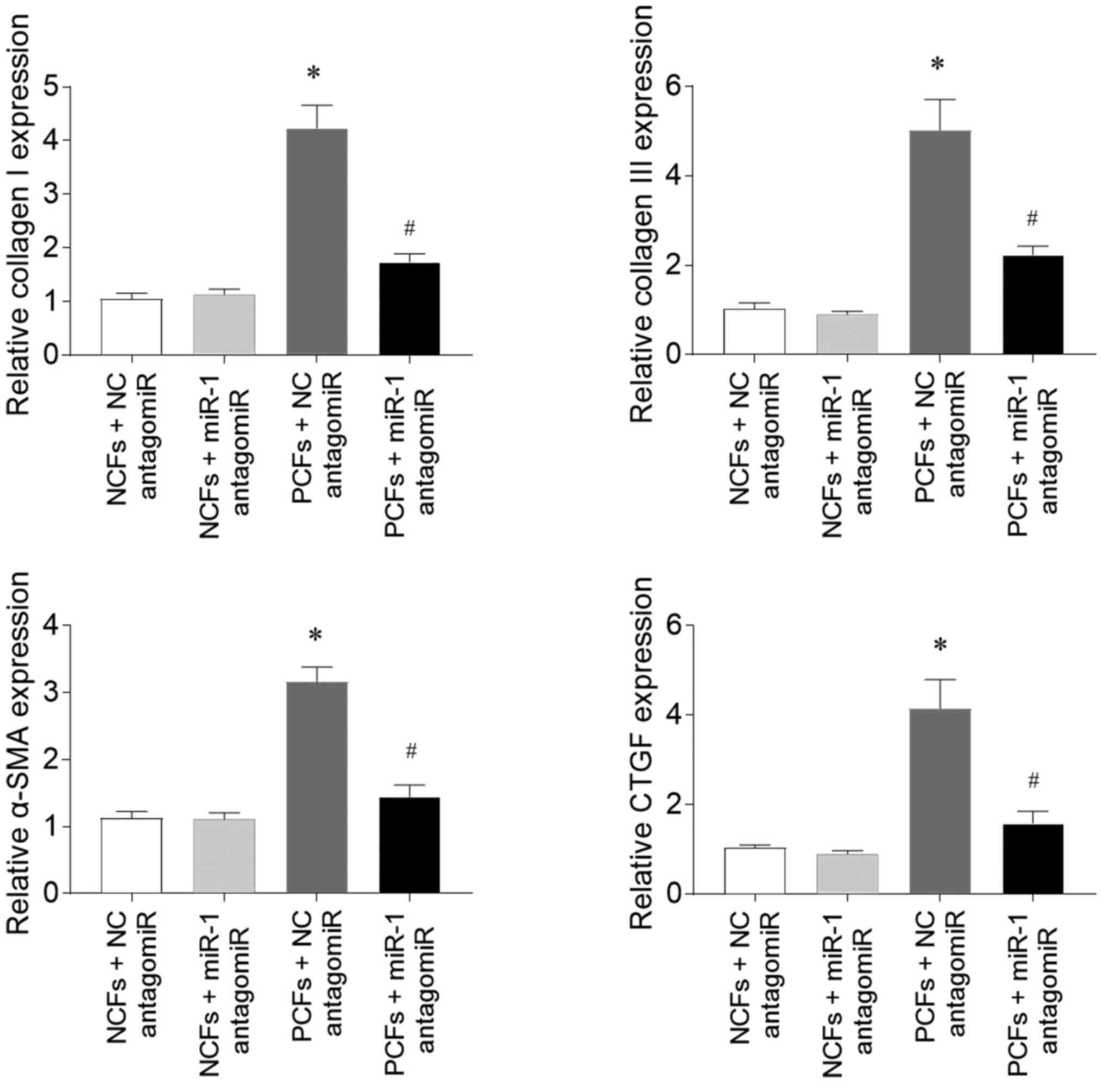 | Figure 6.miR-1 antagomiR attenuates the
fibrosis in CFs isolated from PAH model rats. The expression levels
of collagen I, collagen III, α-SMA and CTGF in PCFs treated with NC
antagomiR were upregulated compared with NCFs treated with NC
antagomiR. These increases were subsequently inhibited by
transfection with the miR-1 antagomiR. The results are presented as
the mean ± SEM. *P<0.05 vs. normoxia + NC antagomiR;
#P<0.05 vs. hypoxia + NC antagomiR. miR, microRNA;
CFs, cardiac fibroblasts; PAH, pulmonary arterial hypertension;
SMA, smooth muscle actin; CTGF, connective tissue growth factor;
NC, negative control; PCFs, CFs isolated from PAH model rats; NCFs,
CFS isolated from normoxia rats. |
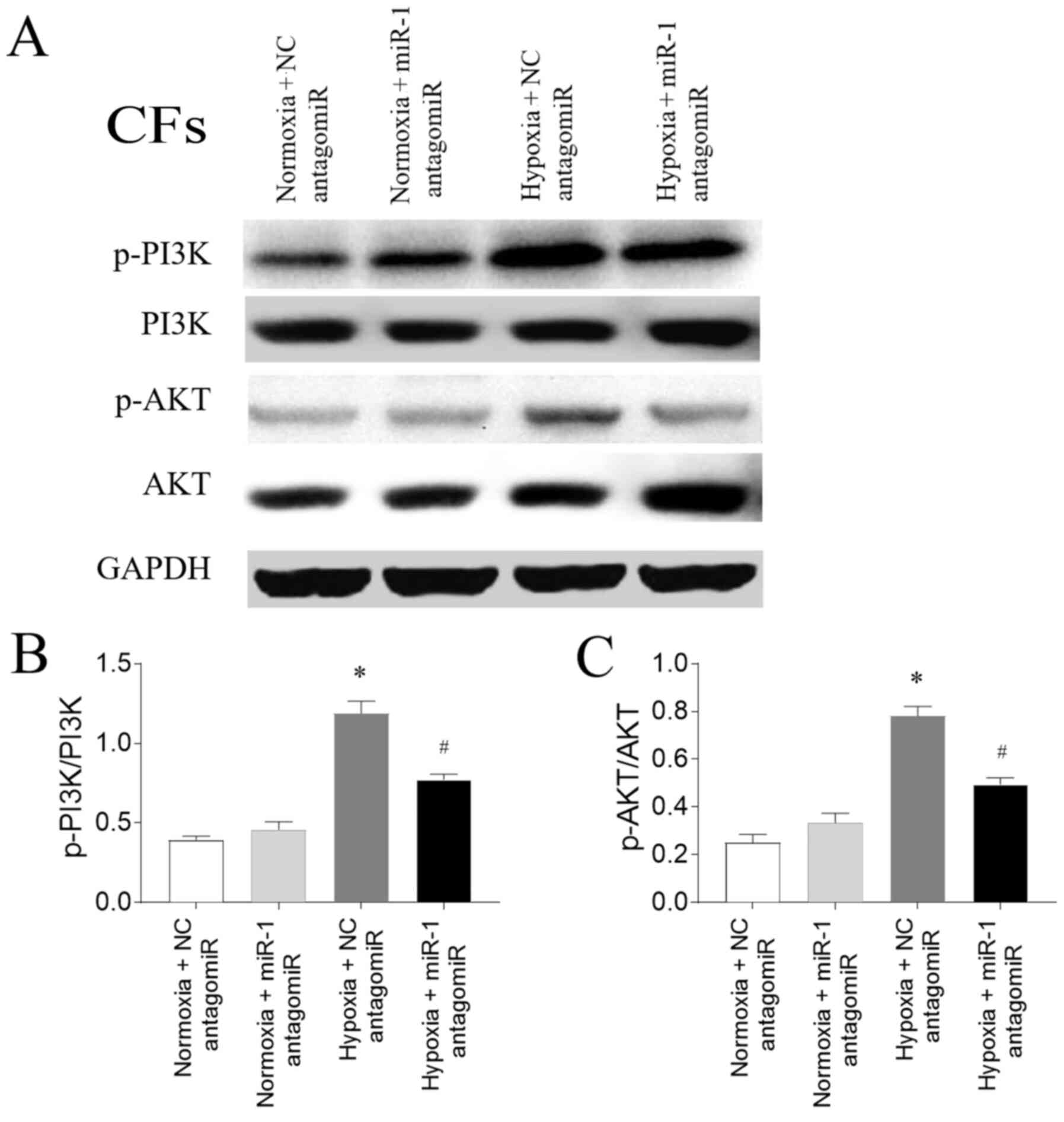
Involvement of the PI3K/AKT signaling
pathway in PAH
The expression levels of p-PI3K/PI3K were
upregulated in CFs exposed to hypoxia, and this increase was
subsequently inhibited by miR-1 antagomiR transfection.
Furthermore, the expression levels of p-AKT were also upregulated
in CFs exposed to hypoxia compared with the normoxia CFs, and this
increase was partially reversed following miR-1 antagomiR
transfection (Fig. 7).
Discussion
Hypoxia plays an initiating role in the pathogenesis
of PAH. Numerous miRs have been found to be dysregulated in the
lung and heart of PAH model rats under chronic hypoxic and
monocrotaline (MCT) environments (30–32).
The results of the present study demonstrated that knocking down
miR-1 expression attenuated PAH and RV fibrosis in PAH model rats,
a process that was suggested to involve the PI3K/AKT signaling
pathway.
PAH is refractory and devastating; however, there
are currently no effective treatments available. miRNAs have
emerged as novel targets for PAH treatment and numerous miRNAs play
a role in the development of PAH (33). For example, one previous study
reported that miR-204 expression levels were downregulated in lung
tissue from humans, mice and rats with PAH, and knocking down
miR-204 expression increased the proliferation and decreased the
apoptosis of pulmonary artery smooth muscle cells in patients with
PAH (34). The expression levels of
miRNAs show model-derived differences. For example, MCT and hypoxia
induced consistent changes in miR-30c and miR-451, yet regulated
miR-22 and miR-21 differently, suggesting that hypoxia- and
MCT-induced PAH share some common elements relating to miRs
regulation and differential regulation on miRs (31). The results of the present study
revealed that the expression levels of miR-1 in the RV were
upregulated in rats with hypoxia-induced PAH. In PAH model rats,
mPAP and RVSP were also increased, while knocking down miR-1
expression with an antagomiR reversed these increases in PAH model
rats. These results suggested that the expression levels of miR-1
may be dysregulated in the RV of PAH model rats, and that knocking
down miR-1 expression may significantly attenuate PAH.
PAH exerts significant pressure on the RV, usually
resulting in RV remodeling (35).
Pathological hypertrophy is a feature of RV remodeling (36). Restoring the expression of miR-223
in the lungs of rats with MCT-induced PAH provided beneficial
effects on RV hypertrophy and vascular remodeling in a previous
study (37). In the present study,
RV hypertrophy was increased in PAH model rats exposed to hypoxia,
as indicated by the increases in the RV/(LV+S) and RV/TL.
Transfection with the miR-1 antagomiR reversed these increases,
indicating that knocking down miR-1 may control RV hypertrophy in
PAH model rats.
RV fibrosis is another feature of PAH-induced RV
remodeling, which is consistently observed in patients with PAH
(38,39) and animal models (40,41).
The present study found that the expression levels of collagen I,
collagen III, α-SMA and CTGF were upregulated in the RV of PAH
model rats exposed to normoxia, and these increases were inhibited
following miR-1 antagomiR transfection. miR-1 antagomiR also
attenuated the increases in the expression levels of collagen I,
collagen III, α-SMA and CTGF in CFs stimulated with hypoxia.
Similarly, the expression levels of collagen I, collagen III, α-SMA
and CTGF in CFs from PAH model rats were upregulated, which were
downregulated by miR-1 antagomiR transfection. These results
suggested that knocking down miR-1 expression may reverse the
fibrosis of RV in PAH model rats.
The PI3K/AKT signaling pathway plays a key role in
the fibrosis of the heart (42).
Cardiac fibroblast proliferation and migration following myocardial
infarction were found to be regulated by the PTEN/PI3K/AKT/mTOR
signaling pathway (20). PI3K/AKT
signaling was also demonstrated to be necessary for hypoxia-induced
CF differentiation and extracellular matrix synthesis (43). The results of the present study
reported that the expression levels of p-PI3K were upregulated in
CFs exposed with hypoxia, while the transfection with the miR-1
antagomiR partially inhibited this increase. Furthermore, the
expression levels of p-AKT were also upregulated in CFs exposed
with hypoxia, and these expression levels were also reversed
following miR-1 antagomiR transfection. These results indicated
that the PI3K/AKT signaling pathway may be involved in the
regulation of miR-1 in the cardiac fibrosis of PAH.
In conclusion, the findings of the present study
suggested that knocking down miR-1 expression may control PAH, and
attenuate RV hypertrophy and fibrosis induced by PAH. The results
indicated that these effects may occur via a regulatory mechanism
that may involve the PI3K/AKT signaling pathway. Future studies
should aim to analyze the expression levels of miR-1 in patients
with PAH to determine whether miR-1 expression is dysregulated. The
present results suggested that miR-1 may be a potential novel
target for the treatment of PAH.
Acknowledgements
Not applicable
Funding
The present study was supported by grants from the
Priority Academic Program Development of Jiangsu Higher Education
Institutions (PAPD).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
Yu L and Yo L made substantial contributions to
conception and design, and acquisition of data. JL and XZ made
substantial contributions to analysis of data. QC and WX made
substantial contributions to interpretation of data. HW made
substantial contributions to design, and drafting the manuscript
and revising it. Yu L and HW were responsible for confirming the
authenticity of raw data. All authors reviewed and approved the
final manuscript.
Ethics approval and consent to
participate
All procedures were approved by the Experimental
Animal Care and Use Committee of Nanjing Medical University
(Nanjing, China; approval no. 17041015), and were conducted in
accordance with the Guide for the Care and Use of Laboratory
Animals (National Institutes of Health publication no. 85-23,
revised 1996) (22).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Kovacs G, Dumitrescu D, Barner A, Greiner
S, Grünig E, Hager A, Köhler T, Kozlik-Feldmann R, Kruck I, Lammers
AE, Mereles D, et al: Definition, clinical classification and
initial diagnosis of pulmonary hypertension: Updated
recommendations from the cologne consensus conference 2018. Int J
Cardiol. 272S:11–19. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Lan NSH, Massam BD, Kulkarni SS and Lang
CC: Pulmonary arterial hypertension: Pathophysiology and treatment.
Diseases. 6:382018. View Article : Google Scholar
|
|
3
|
Guiot J, Parzibut G, Weber T, Davin L,
Dulgheru R, Lancellotti P, Louis R and Vachiery JL: Pulmonary
arterial hypertension. Rev Med Liege. 74:139–45. 2019.(In French).
PubMed/NCBI
|
|
4
|
Thenappan T, Ormiston ML, Ryan JJ and
Archer SL: Pulmonary arterial hypertension: Pathogenesis and
clinical management. BMJ. 360:j54922018. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Bartel DP: MicroRNAs: Genomics,
biogenesis, mechanism, and function. Cell. 116:281–297. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Javadian M, Gharibi T, Shekari N,
Abdollahpour-Alitappeh M, Mohammadi A, Hossieni A, Mohammadi H and
Kazemi T: The role of microRNAs regulating the expression of matrix
metalloproteinases (MMPs) in breast cancer development,
progression, and metastasis. J Cell Physiol. 234:5399–5412. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
van Rooij E: The art of microRNA research.
Circ Res. 108:219–234. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Mohr AM and Mott JL: Overview of microRNA
biology. Semin Liver Dis. 35:3–11. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Dawson K, Wakili R, Ordog B, Clauss S,
Chen Y, Iwasaki Y, Voigt N, Qi XY, Sinner MF, Dobrev D, et al:
MicroRNA29: A mechanistic contributor and potential biomarker in
atrial fibrillation. Circulation. 127:1466–1475, 1475e1-28. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Eitel I, Adams V, Dieterich P, Fuernau G,
de Waha S, Desch S, Schuler G and Thiele H: Relation of circulating
MicroRNA-133a concentrations with myocardial damage and clinical
prognosis in ST-elevation myocardial infarction. Am Heart J.
164:706–714. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Deng L, Blanco FJ, Stevens H, Lu R,
Caudrillier A, McBride M, McClure JD, Grant J, Thomas M, Frid M, et
al: MicroRNA-143 activation regulates smooth muscle and endothelial
cell crosstalk in pulmonary arterial hypertension. Circ Res.
117:870–883. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Caruso P, Dunmore BJ, Schlosser K, Schoors
S, Dos Santos C, Perez-Iratxeta C, Lavoie JR, Zhang H, Long L,
Flockton AR, et al: Identification of MicroRNA-124 as a major
regulator of enhanced endothelial cell glycolysis in pulmonary
arterial hypertension via PTBP1 (Polypyrimidine Tract Binding
Protein) and pyruvate kinase M2. Circulation. 136:2451–2467. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Rothman AM, Arnold ND, Pickworth JA,
Iremonger J, Ciuclan L, Allen RM, Guth-Gundel S, Southwood M,
Morrell NW, Thomas M, et al: MicroRNA-140-5p and SMURF1 regulate
pulmonary arterial hypertension. J Clin Invest. 126:2495–2508.
2016. View
Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lee HW and Park SH: Elevated microRNA-135a
is associated with pulmonary arterial hypertension in experimental
mouse model. Oncotarget. 8:35609–35618. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Karakikes I, Chaanine AH, Kang S, Mukete
BN, Jeong D, Zhang S, Hajjar RJ and Lebeche D: Therapeutic
cardiac-targeted delivery of miR-1 reverses pressure
overload-induced cardiac hypertrophy and attenuates pathological
remodeling. J Am Heart Assoc. 2:e0000782013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yin H, Zhao L, Zhang S, Zhang Y and Lei S:
MicroRNA1 suppresses cardiac hypertrophy by targeting nuclear
factor of activated T cells cytoplasmic 3. Mol Med Rep.
12:8282–8288. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Lapikova-Bryhinska T, Zhukovska A, Nagibin
V, Tumanovska L, Portnichenko G, Goncharov S, Portnychenko A and
Dosenko V: Altered biogenesis of microRNA-1 is associated with
cardiac dysfunction in aging of spontaneously hypertensive rats.
Mol Cell Biochem. 459:73–82. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Mondejar-Parreno G, Callejo M, Barreira B,
Morales-Cano D, Esquivel-Ruiz S, Filice M, Moreno L, Cogolludo A
and Perez-Vizcaino F: miR-1 induces endothelial dysfunction in rat
pulmonary arteries. J Physiol Biochem. 75:519–529. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wei L, Zhang Y, Qi X, Sun X, Li Y and Xu
Y: Ubiquitinproteasomes are the dominant mediators of the
regulatory effect of microRNA1 on cardiac remodeling after
myocardial infarction. Int J Mol Med. 44:1899–1907. 2019.PubMed/NCBI
|
|
20
|
Yang W, Wu Z, Yang K, Han Y, Chen Y, Zhao
W, Huang F, Jin Y and Jin W: BMI1 promotes cardiac fibrosis in
ischemia-induced heart failure via the PTEN-PI3K/Akt-mTOR signaling
pathway. Am J Physiol Heart Circ Physiol. 316:H61–H69. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zhang CJ, Huang Y, Lu JD, Lin J, Ge ZR and
Huang H: Upregulated microRNA-132 rescues cardiac fibrosis and
restores cardiocyte proliferation in dilated cardiomyopathy through
the phosphatase and tensin homolog-mediated PI3K/Akt signal
transduction pathway. J Cell Biochem. Sep 14–2018.(Epub ahead of
print). doi: 10.1002/jcb.27081.
|
|
22
|
National Research Council: Guide for the
Care and Use of Laboratory Animals. National Academies Press;
Washington, DC: 1996
|
|
23
|
Luo H, Liu B, Zhao L, He J, Li T, Zha L,
Li X, Qi Q, Liu Y and Yu Z: Galectin-3 mediates pulmonary vascular
remodeling in hypoxia-induced pulmonary arterial hypertension. J Am
Soc Hypertens. 11:673–683.e3. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wang Y, Pandey RN, York AJ, Mallela J,
Nichols WC, Hu YC, Molkentin JD, Wikenheiser-Brokamp KA and Hegde
RS: The EYA3 tyrosine phosphatase activity promotes pulmonary
vascular remodeling in pulmonary arterial hypertension. Nat Commun.
10:41432019. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Luo Y, Teng X, Zhang L, Chen J, Liu Z,
Chen X, Zhao S, Yang S, Feng J and Yan X: CD146-HIF-1alpha hypoxic
reprogramming drives vascular remodeling and pulmonary arterial
hypertension. Nat Commun. 10:35512019. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zhao H, Ma TF, Lin J, Liu LL, Sun WJ, Guo
LX, Wang SQ, Otecko NO and Zhang YP: Identification of valid
reference genes for mRNA and microRNA normalisation in prostate
cancer cell lines. Sci Rep. 8:19492018. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Peltier HJ and Latham GJ: Normalization of
microRNA expression levels in quantitative RT-PCR assays:
Identification of suitable reference RNA targets in normal and
cancerous human solid tissues. Rna. 14:844–852. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Inada K, Okoshi Y, Cho-Isoda Y, Ishiguro
S, Suzuki H, Oki A, Tamaki Y, Shimazui T, Saito H, Hori M, et al:
Endogenous reference RNAs for microRNA quantitation in
formalin-fixed, paraffin-embedded lymph node tissue. Sci Rep.
8:59182018. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Zhu Z, Fang Z, Hu X and Zhou S: MicroRNAs
and mesenchymal stem cells: Hope for pulmonary hypertension. Rev
Bras Cir Cardiovasc. 30:380–385. 2015.PubMed/NCBI
|
|
31
|
Caruso P, MacLean MR, Khanin R, McClure J,
Soon E, Southgate M, MacDonald RA, Greig JA, Robertson KE, Masson
R, et al: Dynamic changes in lung microRNA profiles during the
development of pulmonary hypertension due to chronic hypoxia and
monocrotaline. Arterioscler Thromb Vasc Biol. 30:716–723. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Zhou SS, Jin JP, Wang JQ, Zhang ZG,
Freedman JH, Zheng Y and Cai L: miRNAS in cardiovascular diseases:
Potential biomarkers, therapeutic targets and challenges. Acta
Pharmacol Sin. 39:1073–1084. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zhou G, Chen T and Raj JU: MicroRNAs in
pulmonary arterial hypertension. Am J Respir Cell Mol Biol.
52:139–151. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Courboulin A, Paulin R, Giguere NJ,
Saksouk N, Perreault T, Meloche J, Paquet ER, Biardel S, Provencher
S, Côté J, et al: Role for miR-204 in human pulmonary arterial
hypertension. J Exp Med. 208:535–548. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Tadic M, Cuspidi C, Bombelli M and Grassi
G: Right heart remodeling induced by arterial hypertension: Could
strain assessment be helpful? J Clin Hypertens (Greenwich).
20:400–407. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Maron BA and Loscalzo J: Pulmonary
hypertension: Pathophysiology and signaling pathways. Handb Exp
Pharmacol. 218:31–58. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Meloche J, Le Guen M, Potus F, Vinck J,
Ranchoux B, Johnson I, Antigny F, Tremblay E, Breuils-Bonnet S,
Perros F, et al: miR-223 reverses experimental pulmonary arterial
hypertension. Am J Physiol Cell Physiol. 309:C363–C372. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
McCann GP, Gan CT, Beek AM, Niessen HW,
Vonk Noordegraaf A and van Rossum AC: Extent of MRI delayed
enhancement of myocardial mass is related to right ventricular
dysfunction in pulmonary artery hypertension. AJR Am J Roentgenol.
188:349–355. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Shehata ML, Lossnitzer D, Skrok J, Boyce
D, Lechtzin N, Mathai SC, Girgis RE, Osman N, Lima JA, Bluemke DA,
et al: Myocardial delayed enhancement in pulmonary hypertension:
Pulmonary hemodynamics, right ventricular function, and remodeling.
AJR Am J Roentgenol. 196:87–94. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Hessel MH, Steendijk P, den Adel B,
Schutte CI and van der Laarse A: Characterization of right
ventricular function after monocrotaline-induced pulmonary
hypertension in the intact rat. Am J Physiol Heart Circ Physiol.
291:H2424–H2430. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Drake JI, Bogaard HJ, Mizuno S, Clifton B,
Xie B, Gao Y, Dumur CI, Fawcett P, Voelkel NF and Natarajan R:
Molecular signature of a right heart failure program in chronic
severe pulmonary hypertension. Am J Respir Cell Mol Biol.
45:1239–1247. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
MacLean J and Pasumarthi KB: Signaling
mechanisms regulating fibroblast activation, phenoconversion and
fibrosis in the heart. Indian J Biochem Biophys. 51:476–482.
2014.PubMed/NCBI
|
|
43
|
Zhang J, Fan G, Zhao H, Wang Z, Li F,
Zhang P, Zhang J, Wang X and Wang W: Targeted inhibition of focal
adhesion kinase attenuates cardiac fibrosis and preserves heart
function in adverse cardiac remodeling. Sci Rep. 7:431462017.
View Article : Google Scholar : PubMed/NCBI
|