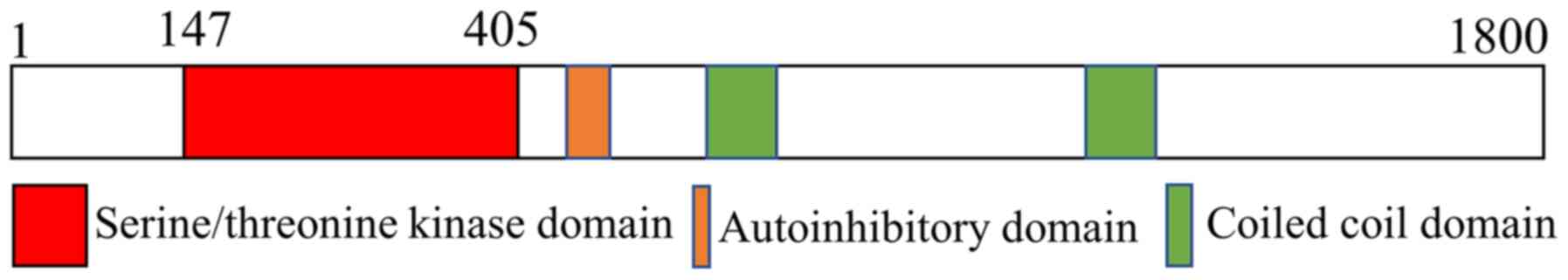
With-no-lysine kinases (WNKs) are a class of protein
kinases that were initially discovered in multicellular organisms
(1). Four WNK genes, namely WNK1,
WNK2, WNK3 and WNK4, have been identified in the human genome,
which are encoded by genes on chromosome 12, 9, X and 17,
respectively (2). A defining
characteristic of WNK family members is the absence of a catalytic
lysine residue specifically located in the N-terminal kinase domain
(3). WNKs have been reported to
play an important role in cell and body physiology (4). For example, WNK1 was found to be
expressed in a variety of tissues, including the kidney, heart and
brain (5), and two major
transcripts have been identified: One is mainly produced in the
heart, muscles and brain and is called L-WNK1, while the other
shorter transcript is primarily located in the kidney, thus is also
known as kidney-specific WNK1, as it is only expressed in distal
convoluted tubules and connecting tubules (6,7). WNK1
is widely expressed and has been reported to be involved in the
regulation of numerous cellular processes (8). For example, WNK1 in regulation of the
podocyte actin cytoskeleton, biophysical properties of glomerular
capillaries and slit diaphragm structure, all of which are
essential (9). Notably, due to the
observed association between WNK1 mutations and familial
hypertension and autonomic neuropathy, the function of WNK1 in the
kidney and nervous system has been extensively studied (10). WNK1 is self-phosphorylated on serine
residues, and mutations in the gene encoding WNK1 were found to
cause high blood pressure in humans (7). Similar to WNK1, WNK4 is also expressed
in the kidney and has been closely associated with hypertension
(11). The loss of introns in the
WNK1 and WNK4 genes has been discovered to lead to
pseudoaldosterone deficiency type II, a disease associated with
salt-sensitive hypertension and hyperkalaemia (12,13).
This may be due to the influence of WNK1 and WNK4 on the ion
reabsorption signaling pathway (14). WNK1 and WNK4 can also activate the
Na+-Cl− cotransporter (NCC) of distal
concentric tubules through the serine/threonine-protein kinase
STE20/serine/threonine kinase 39 (SPAK)/odd-skipped related
transcription factor 1 (OSR1) signaling pathway, forming the
WNK/SPAK/OSR1/NCC phosphorylation cascade, which was reported to be
involved in the regulation of renal pressure homeostasis by
regulating intracellular ions and water (15,16).
At present, to the best of our knowledge, there are
few published studies investigating WNK3 signaling in the nervous
system; however, its dysfunction in the brain has been associated
with the occurrence of several neurological diseases, including
epilepsy (29), ischemic brain
injury, intracerebral hemorrhage (27), autism (30), glioma (31), schizophrenia and autonomic nerve
pain (Table I) (32). It has been suggested that WNK3
signaling may play distinct roles in different brain diseases,
which are further discussed in more detail in the following
sections.
Autism comprises a heterogeneous range of
neurodevelopmental conditions characterized by symptoms such as
communication/language deficits, repetitive/restricted patterns of
behavior and inadequate social interactions (33,34).
Individuals with autism have difficulty with social communication
and interactions, increased rates of restricted/repetitive patterns
of behavior and increased sensory sensitivities (35). Autism is also accompanied by several
other complications, such as insomnia, intellectual disabilities,
epilepsy, self-injurious behavior, aggression, anxiety,
attention-deficit hyperactivity disorder and depression (33,36).
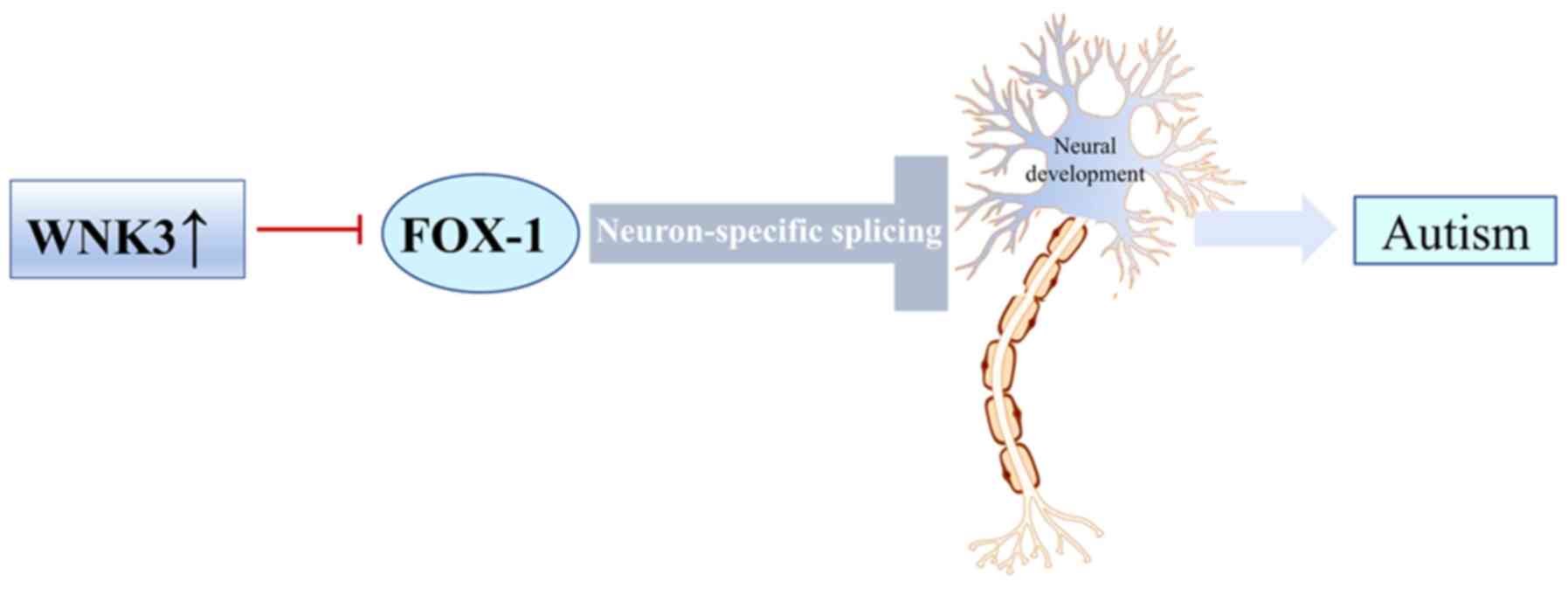
WNK3 is known to regulate the activity of the neuronal splicing
factor, RNA binding Fox-1 homolog 1 (FOX-1). FOX-1 is a
neuron-specific splicing factor which has been predicted to
regulate neuronal splicing networks that are clinically implicated
in neurodevelopmental diseases, including autism spectrum disorder
(37,38). Comparative profiling of splicing in
brains from patients with autism spectrum disorder and normal
brains revealed that FOX-1 expression was strongly associated with
autism (39). FOX-1 regulated the
excitability of neurons by specific splicing (40). Previous studies have shown that WNK3
regulated FOX-1-mediated alternative splicing and subsequently
affected autism (39,41,42).
FOX-1 and WNK3 has several intersections, which are strongly
related to the control of neuronal excitability (43). FOX-1 has been shown to regulate
alternative splicing of neuronal transcripts by binding the
sequence (U) GCAUG in introns flanking alternative exons and is
responsible for generating proper alternative splicing variants
required for normal neuronal excitability and synaptic transmission
(40). WNK3 was discovered to
affect the splicing activity of FOX-1 by affecting the subcellular
localization of FOX-1 and neuronal transcripts cannot be spliced
normally, leading to reduced neuronal excitability, which has an
important influence on the pathogenesis of autism (43). In addition, FOX-1-mediated exon
inclusion bodies were significantly downregulated after
co-expression with wild-type WNK3, while inactive WNK3 only exerted
a marginal effect (39,44,45).
Due to the role of WNK3 and FOX-1 in disorders of neuronal
development, WNK3 may represent a target for treatment of
FOX-1-induced autism (Fig. 2)
(43,46).
Epilepsy consists of a group of recurrent episodes
of abnormal neuronal firing caused by temporary central nervous
system dysfunction (47). The
typical clinical manifestations of epilepsy comprise sudden loss of
consciousness, muscle spasms, rigidity and convulsions; it can also
be accompanied by urinary incontinence, asphyxia or other symptoms
(48,49). Epilepsy is second only to stroke in
the number of years of potential life lost due to a neurological
disease. The prevalence of epilepsy is 6.4 per 1,000 individuals
and the annual incidence is 67.8 cases per 100,000 person-years
(50), which poses a major public
health burden (51–53). A previous study observed that WNK3
immunoreactivity was increased in dispersed granule neurons in
patients with epilepsy, suggesting that WNK3 may play a role in
neuronal hyperexcitability (54).
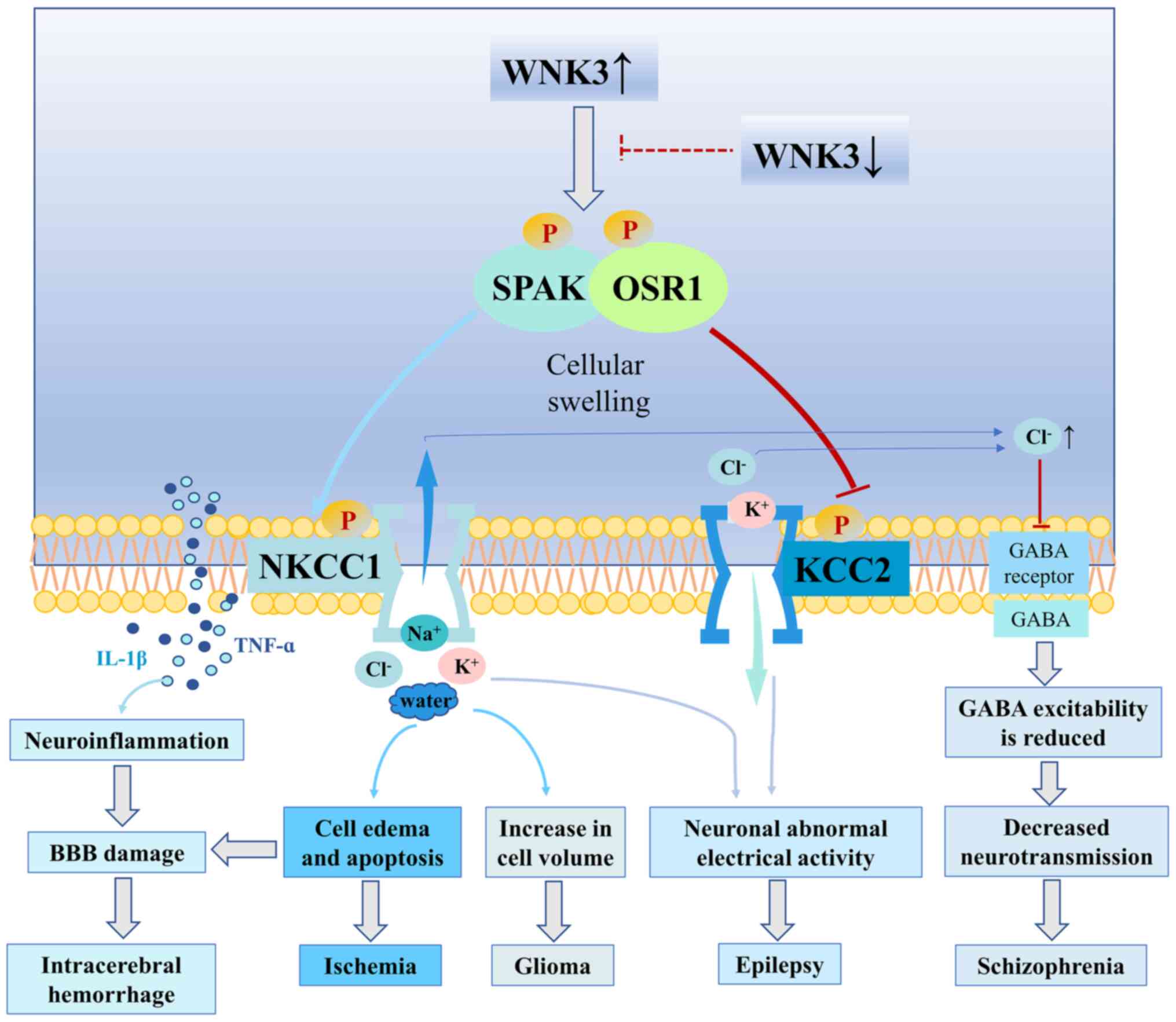
Furthermore, WNK3 phosphorylated the
Na+-K+-Cl− cotransporter (NKCC)
and inhibited the K+-Cl− cotransporter (KCC)
to regulate neuronal excitability (55). NKCC1 expression levels were
upregulated in dispersed granule cells within postmortem tissues
from patients with epilepsy (56).
In addition, previous studies have demonstrated that restoration of
KCC2 function and controlling the regulation of NKCC1 have
potential as antiepileptic therapies (57–59).
These findings suggested that the sustained activation of WNK3,
which is upstream of NKCC1 and KCC2, may contribute to the
induction of neuronal hyperexcitability and result in the
development of abnormal electrical activity within the hippocampus.
Thus, WNK3 expression may be a novel therapeutic target for
mitigating epileptogenesis (Fig. 3)
(54).
Ischemic brain damage, also known as stroke, is a
sudden cessation in cerebral blood flow. Clinical manifestations
include sudden fainting, unconsciousness, sudden mouth/eye
skewness, hemiplegia and intellectual disability (60). Moreover, ischemic brain injury is
one of the most common causes of mortality and disability
worldwide, affecting 30 million people (61,62). A
previous study in mouse models revealed that inhibition of WNK3
modulation of SPAK/OSR1 and NKCC1 signaling in the brain
ameliorated both gray- and white-matter damage and promoted
neurological recovery following ischemic stroke (54). In addition, after cerebral ischemic
injury, the WNK3/SPAK/OSR1/NKCC1 signaling cascade was found to be
activated, WNK3 expression levels were upregulated, and SPAK and
OSR1 were phosphorylated, which resulted in the increased
phosphorylation of downstream NKCC1 (63). NKCC1 was also discovered to play an
important role in the pathophysiology of ischemia, where it
regulated cellular volume by regulating the entry of
Na+, K+ and Cl− into cells
(64). Under ischemic conditions,
NKCC1 induced excessive amounts of Na+, K+
and Cl− to enter into cells, leading to intracellular
ionic overload that damaged the endoplasmic reticulum and
mitochondria, and led to necrosis and apoptosis (65,66).
Hence, it was suggested that WNK3 may upregulate NKCC1 to promote
ischemic brain damage (67–69). A previous study using stroke model
mice demonstrated that WNK3-knockout mice had a reduced infarct
volume, cerebral edema and axonal demyelination following a stroke
episode compared with wild-type mice (29). These findings suggested that
cerebral ischemia may aggravate ischemic brain injury by increasing
the phosphorylation of NKCC1 through the WNK3/SPAK/OSR1 signaling
pathway. Furthermore, the knockdown of WNK3 significantly inhibited
the activity of NKCC1, suggesting that the knockdown of WNK3
expression may protect nerve cells by regulating ion and water
transport (26). WNK3/SPAK
inhibition also prevented acute cellular swelling in response to
osmotic stress and ameliorated brain swelling by simultaneously
increasing the stimulatory phosphorylation of NKCC1 and inhibiting
KCC phosphorylation (70). Notably,
in another previous study, following WNK3/SPAK inhibition, the
damage caused by cerebral ischemia was alleviated; however, the
mechanisms by which cerebral ischemia activated the
WNK3/SPAK/OSR1/NKCC1 signaling pathway remain unclear (Fig. 3) (63).
Intracerebral hemorrhage is a common type of stroke,
which is accompanied by a mortality rate of ~50%, and ~2/3 of
patients have a poor prognosis and are unable to live independently
(17,71). An intracerebral hemorrhage forms a
hematoma, which squeezes the brain tissue to cause intracranial
hypertension and pathophysiological changes in the brain, which can
lead to secondary brain injury (72). At present, reducing intracranial
pressure and blood pressure is one of the emergency treatment
methods for intracerebral hemorrhage (73,74).
Therefore, determining endogenous intervention measures is one of
the treatment approaches to improve secondary brain injury
following intracerebral hemorrhage (75,76).
Brain edema caused by blood brain barrier (BBB) damage is a common
secondary brain injury following intracerebral hemorrhage (77). After intracerebral hemorrhage, WNK3
expression levels were found to be upregulated in brain tissue,
which activated NKCC1 in microglia cells by phosphorylating SPAK
(27). The activation of the
microglia and release of the inflammatory factors, TNF-α and IL-1β
(78), leads to brain inflammation,
which further aggravates brain damage (79,80).
The overexpression of WNK3 could also increase the phosphorylation
of NKCC1, and phosphorylated NKCC1 stimulated the microglia to
secrete inflammatory factors, which simultaneously expanded the
cell volume, destroyed the tight connection of cells, accelerated
the diffusion of inflammatory factors, destroyed the BBB, led to
neuronal apoptosis and aggravated brain edema (27). Conversely, knocking out WNK3
expression had the opposite effects. These findings indicated that
inhibiting the WNK3 signaling pathway may play a role in brain
protection, and WNK3 may improve secondary brain injury caused by
intracerebral hemorrhage (Fig. 3)
(27).
Brain tumors originate from glial cells, and gliomas
are among the most problematic primary cancers to treat (81). Furthermore, gliomas are the most
common type of tumor of the central nervous system (82). Gliomas invade by diffusing into the
surrounding brain parenchyma, thereby making surgical resection
difficult (83). Glioma cells
change in morphology and volume to migrate to adjacent brain
parenchyma, and the transport of Na+, Cl− and
water was discovered to play a crucial role in this process
(84,85). Compared with those in the normal
brain, the expression levels of WNK and SPAK/OSR1 were found to be
upregulated in the brain tissues of patients with glioma (31). In addition, WNK3 was discovered to
be involved in the regulation of cellular volume, which is an
important parameter for the migration of glioma (31). Previous studies have shown that WNK3
facilitates intracellular entry of Cl− and water,
resulting in changed cell volume; cell volume serves a significant
role in tumor migration (86).
Changes in cellular volume have also been increasingly recognized
as an important requirement for cancer cell invasion and metastasis
(87). During the migration of
tumor cells, ions must accumulate in cells to allow them to flow
along an electrochemical gradient (88). NKCC1 was demonstrated to regulate
the entry of ions into cells and played an indispensable role in
regulating intracellular pressure and tumor cell migration
(89). In addition, a previous
study revealed that NKCC1 co-transported water and ions, making it
ideally suited to transporting salt and water across the plasma
membrane during cytoplasmic cell-volume regulation (90,91). A
previous study by Haas et al (31) showed that the knockdown of WNK3 with
small interfering RNA promoted loss of NKCC1 function, which
alleviated cell-volume changes associated with cellular invasion.
These findings suggested that WNK3 may influence glioma migration
through its regulation of NKCC1. Thus, it has been hypothesized
that SPAK/OSR1 may participate in the regulation of WNK3 over NKCC1
to inhibit the signaling pathway, which may help reduce
intracellular ionic influx and prevent cellular volumes from being
too large to inhibit the metastasis and invasion of gliomas
(31,86). Therefore, understanding the role of
WNK3 in facilitating glioma migration and invasion may provide
evidence to suggest the potential of WNK3 as a therapeutic target
for glioma (Fig. 3) (28,30).
At the beginning of the 20th century, psychiatrists
considered blunted affect and emotional withdrawal as key symptoms
of schizophrenia (92,93). The main clinical manifestations of
patients with schizophrenia comprise anhedonia, loneliness,
avolition, emotional immaturity and asociality (94). These emotional and cognitive
deficits are caused by neural-network dysfunctions, which may, at
least partly, be due to abnormal neurotransmission of
γ-aminobutyric acid (GABA) in the dorsolateral prefrontal cortex
(95,96). According to previous studies, the
upregulated expression levels of WNK3 and oxidative stress
responsive kinase 1 (OXSR1) modulated the activity of
Cl− transporters, which led to a change in the
concentration of Cl− ions inside and outside of neurons
that affected GABAergic neurotransmission in patients with
schizophrenia (55,97). WNK3 was also found to effectively
activate NKCC1, which is co-expressed in neurons, and was found to
have a OXSR1/STK39 binding motif (97). These findings suggested that WNK3
may regulate NKCC1 expression through OXSR1, thereby regulating the
flow of Cl− ions and increasing intracellular
Cl− concentrations to inhibit GABA receptors, which
ultimately reduces the excitability and hyperpolarization of GABA
(98). Upregulated expression
levels of WNK3 were found to be accompanied by upregulated OXSR1
expression levels and enhanced NKCC1 activity, which promoted the
flow of Cl− ions and led to abnormal GABAergic
transmission (99,100). Previous studies have also reported
that WNK signaling not only affected downstream NKCC1, but it also
influenced the flow of Cl− ions inside and outside the
cell by regulating the expression of members of the KCC family
(101,102). It is well established that WNK3 is
highly expressed in the brain. Several previous studies
investigating the WNK kinase family have reported that WNK3 could
simultaneously activate NKCC1 and inhibit KCC2 (55, 97,103). The
regulatory effect of WNK3 on NKCC1 and KCC2 altered the flow of
Cl− ions, facilitating Cl− accumulation in
cells to produce higher concentrations of intracellular
Cl−, thus affecting the excitability of GABA receptors
(97). In addition, the expression
levels and kinase activities of WNK3 and OXSR1 were upregulated in
patients with schizophrenia, which further increased the activity
of NKCC1 and reduced the activity of KCC2, resulting in a high
concentration of intracellular Cl− and increased
depolarization of the postsynaptic membrane; these effects
ultimately altered the function of the GABA receptors (97,104).
A small clinical trial involving 42 patients with schizophrenia and
42 matched healthy subjects revealed that OXSR1 and WNK3 expression
levels were markedly upregulated in patients with schizophrenia
compared with healthy subjects (1,97). In
schizophrenia, upregulated WNK3 and OXSR1 expression levels led to
increased phosphorylation and consequently increased NKCC1 activity
and decreased KCC2 activity, thereby increasing the intracellular
Cl− concentration (97).
Thus, when GABA receptors are activated, Cl− influx is
reduced and the GABAergic neurotransmission is altered. At present,
although there are relatively few studies reporting the association
between WNK3 and schizophrenia, WNK3 is expected to represent a
promising target for alleviating and/or treating schizophrenia
(Fig. 3).
WNK3 expression levels have been reported to be
upregulated in the brain, and among the members of the WNK family,
the expression levels of WNK3 are the highest in the brain
(105). This distribution pattern
suggests that WNK3 may play an important role in brain diseases.
The present review summarized the current roles and applications of
WNK3 in the nervous system, and discovered that the WNK3 signaling
pathway may be involved in the regulation of a number of
neurological diseases. Thus, WNK3 is suggested to play an important
role in nervous system diseases both physiological and pathological
processes. Briefly, the aforementioned studies indicated that WNK3
may increase the activity of proapoptotic pathways in central
nervous system diseases, partially accelerate the progression of
numerous diseases, and worsen the poor prognosis of nervous system
diseases and secondary brain damage (27,31).
The mechanism through which the WNK3 signaling
pathway may regulate the pathology of brain diseases remains
complex and at present, the understanding of the role of WNK3 in
nervous system diseases is not complete. WNK3 has been shown to
play different roles in the pathological processes of nervous
system diseases by regulating multiple different signaling
pathways. The most common mechanism identified to date is that WNK3
may regulate downstream ionic transport by phosphorylating
SPAK/OSR1 (27). Increases in
intracellular ionic concentrations via NKCC1 inhibit KCC activity,
which regulates ion influx/efflux across the plasma membrane,
increases the number of ions and water molecules inside the cell,
alters the cellular structure/volume, and destroys cytoskeletal
structures within glia and endothelial cells (97). These processes were discovered to
contribute to brain edema and trigger apoptosis, which compromises
the normal physiological functioning of the brain. The regulation
of this signaling pathway was demonstrated to serve an important
role in glioma, intracerebral hemorrhage and ischemic brain injury
(29,31,63).
In addition, WNK3 is involved in regulating the flow of
Cl− ions and promoting the accumulation of
Cl− in cells, thereby affecting the excitability of GABA
receptors, which may be related to schizophrenia (97). WNK3 was also reported to lead to the
increase in intracellular Cl− concentrations by
regulating NKCC1 and KCC expression, which promoted abnormal
electrical activity in the hippocampus and thereby induced epilepsy
(55). In addition, WNK3 was found
to be closely associated with autism by inhibiting the shearing
activity of FOX-1, which ultimately affected neural development
(43). Previous studies have also
shown that WNK3 was associated with neuropathic pain (104), spasticity (1) and other related diseases of the
nervous system. However, to the best of our knowledge, currently,
the underlying mechanisms of how nervous system diseases may induce
WNK3 activation remain unclear.
Although the potential of WNK3 as a novel
therapeutic target for brain diseases has been discussed in the
present study, whether therapeutic strategies that target WNK3
signaling will be successful in the clinic remains unknown.
However, since compounds that inhibit WNK3 have been discovered or
developed, such as WNK463 (106),
WNK3 inhibitors may represent promising novel targets for the
treatment of nervous system diseases.
Not applicable.
The present study was supported by the Zhangjiagang
Science and Technology Project (grant no. ZKS1914) and Zhangjiagang
Health Youth Science and Technology Project (grant no.
ZJGQNKJ202030).
GC and BQD conceived and designed the study. YTG and
MYW wrote the manuscript. JCS created the figures. JFT and JL
assisted in the literature search and analyzing the literature. RG
reviewed and revised the manuscript. BQD and GC confirm the
authenticity of all the raw data. All authors read and approved the
final manuscript.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
|
1
|
Siew K and O'Shaughnessy KM: Extrarenal
roles of the with-no-lysine[K] kinases (WNKs). Clin Exp Pharmacol
Physiol. 40:885–894. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Xu B, English JM, Wilsbacher JL, Stippec
S, Goldsmith EJ and Cobb MH: WNK1, a novel mammalian
serine/threonine protein kinase lacking the catalytic lysine in
subdomain II. J Biol Chem. 275:16795–16801. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Akella R, Drozdz MA, Humphreys JM, Jiou J,
Durbacz MZ, Mohammed ZJ, He H, Liwocha J, Sekulski K and Goldsmith
EJ: A phosphorylated intermediate in the activation of WNK kinases.
Biochemistry. 59:1747–1755. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Thomson MN, Cuevas CA, Bewarder TM,
Dittmayer C, Miller LN, Si J, Cornelius RJ, Su XT, Yang CL,
McCormick JA, et al: WNK bodies cluster WNK4 and SPAK/OSR1 to
promote NCC activation in hypokalemia. Am J Physiol Renal Physiol.
318:F216–F228. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Gao JL, Peng K, Shen MW, Hou YH, Qian XB,
Meng XW, Ji FH, Wang LN and Yang JP: Suppression of WNK1-SPAK/OSR1
attenuates bone cancer pain by regulating NKCC1 and KCC2. J Pain.
20:1416–1428. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Bergaya S, Vidal-Petiot E, Jeunemaitre X
and Hadchouel J: Pathogenesis of pseudohypoaldosteronism type 2 by
WNK1 mutations. Curr Opin Nephrol Hypertens. 21:39–45. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Naray-Fejes-Toth A, Snyder PM and
Fejes-Toth G: The kidney-specific WNK1 isoform is induced by
aldosterone and stimulates epithelial sodium channel-mediated
Na+ transport. Proc Natl Acad Sci USA. 101:17434–17439.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Huang CL, Jian X and Yuh CH:
Wnk1-Osr1/spak kinase cascade is important for angiogenesis. Trans
Am Clin Climatol Assoc. 131:140–146. 2020.PubMed/NCBI
|
|
9
|
Liu Z, Yoon J, Wichaidit C, Jaykumar AB,
Dbouk HA, Embry AE, Liu L, Henderson JM, Chang AN, Cobb MH and
Miller RT: Control of podocyte and glomerular capillary wall
structure and elasticity by WNK1 kinase. Front Cell Dev Biol.
8:6188982020. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Chi RA, Wang T, Huang CL, Wu SP, Young SL,
Lydon JP and DeMayo FJ: WNK1 regulates uterine homeostasis and its
ability to support pregnancy. JCI Insight. 5:e1418322020.
View Article : Google Scholar
|
|
11
|
Zhao X, Lai G, Tu J, Liu S and Zhao Y:
Crosstalk between phosphorylation and ubiquitination is involved in
high salt-induced WNK4 expression. Exp Ther Med. 21:1332021.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Sie ZL, Li RY, Sampurna BP, Hsu PJ, Liu
SC, Wang HD, Huang CL and Yuh CH: WNK1 kinase stimulates
angiogenesis to promote tumor growth and metastasis. Cancers
(Basel). 12:5752020. View Article : Google Scholar
|
|
13
|
Rafael C, Chavez-Canales M and Hadchouel
J: New perspective on the role of WNK1 and WNK4 in the regulation
of NaCl reabsorption and K(+) secretion by the distal nephron. Med
Sci (Paris). 32:274–280. 2016.(In French). View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Delaloy C, Lu J, Houot AM, Disse-Nicodeme
S, Gasc JM, Corvol P and Jeunemaitre X: Multiple promoters in the
WNK1 gene: One controls expression of a kidney-specific
kinase-defective isoform. Mol Cell Biol. 23:9208–9221. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Furusho T, Uchida S and Sohara E: The WNK
signaling pathway and salt-sensitive hypertension. Hypertens Res.
43:733–743. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Anderegg MA, Albano G, Hanke D, Deisl C,
Uehlinger DE, Brandt S, Bhardwaj R, Hediger MA and Fuster DG: The
sodium/proton exchanger NHA2 regulates blood pressure through a
WNK4-NCC dependent pathway in the kidney. Kidney Int. 99:350–363.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Klebe D, Iniaghe L, Burchell S, Reis C,
Akyol O, Tang J and Zhang JH: Intracerebral hemorrhage in mice.
Methods Mol Biol. 1717:83–91. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Rinehart J, Vazquez N, Kahle KT, Hodson
CA, Ring AM, Gulcicek EE, Louvi A, Bobadilla NA, Gamba G and Lifton
RP: WNK2 kinase is a novel regulator of essential neuronal
cation-chloride cotransporters. J Biol Chem. 286:30171–30180. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Costa AM, Pinto F, Martinho O, Oliveira
MJ, Jordan P and Reis RM: Silencing of the tumor suppressor gene
WNK2 is associated with upregulation of MMP2 and JNK in gliomas.
Oncotarget. 6:1422–1434. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Alves ALV, Costa AM, Martinho O, da Silva
VD, Jordan P, Silva VAO and Reis RM: WNK2 inhibits autophagic flux
in human glioblastoma cell line. Cells. 9:4852020. View Article : Google Scholar
|
|
21
|
Moniz S, Martinho O, Pinto F, Sousa B,
Loureiro C, Oliveira MJ, Moita LF, Honavar M, Pinheiro C, Pires M,
et al: Loss of WNK2 expression by promoter gene methylation occurs
in adult gliomas and triggers Rac1-mediated tumour cell
invasiveness. Hum Mol Genet. 22:84–95. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Holden S, Cox J and Raymond FL: Cloning,
genomic organization, alternative splicing and expression analysis
of the human gene WNK3 (PRKWNK3). Gene. 335:109–119. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Moniz S and Jordan P: Emerging roles for
WNK kinases in cancer. Cell Mol Life Sci. 67:1265–1276. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kahle KT, Ring AM and Lifton RP: Molecular
physiology of the WNK kinases. Annu Rev Physiol. 70:329–355. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Verissimo F, Silva E, Morris JD, Pepperkok
R and Jordan P: Protein kinase WNK3 increases cell survival in a
caspase-3-dependent pathway. Oncogene. 25:4172–4182. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
de Los Heros P, Pacheco-Alvarez D and
Gamba G: Role of WNK kinases in the modulation of cell volume. Curr
Top Membr. 81:207–235. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Wu D, Lai N, Deng R, Liang T, Pan P, Yuan
G, Li X, Li H, Shen H, Wang Z and Chen G: Activated WNK3 induced by
intracerebral hemorrhage deteriorates brain injury maybe via
WNK3/SPAK/NKCC1 pathway. Exp Neurol. 332:1133862020. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Pacheco-Alvarez D and Gamba G: WNK3 is a
putative chloride-sensing kinase. Cell Physiol Biochem.
28:1123–1134. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Begum G, Yuan H, Kahle KT, Li L, Wang S,
Shi Y, Shmukler BE, Yang SS, Lin SH, Alper SL and Sun D: Inhibition
of WNK3 kinase signaling reduces brain damage and accelerates
neurological recovery after stroke. Stroke. 46:1956–1965. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Tang BL: (WNK)ing at death: With-no-lysine
(Wnk) kinases in neuropathies and neuronal survival. Brain Res
Bull. 125:92–98. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Haas BR, Cuddapah VA, Watkins S, Rohn KJ,
Dy TE and Sontheimer H: With-no-lysine kinase 3 (WNK3) stimulates
glioma invasion by regulating cell volume. Am J Physiol Cell
Physiol. 301:C1150–C1160. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Shekarabi M, Zhang J, Khanna AR, Ellison
DH, Delpire E and Kahle KT: WNK kinase signaling in ion homeostasis
and human disease. Cell Metab. 25:285–299. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Schreck KA and Richdale AL: Sleep
problems, behavior, and psychopathology in autism:
inter-relationships across the lifespan. Curr Opin Psychol.
34:105–111. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Fakhoury M: Autistic spectrum disorders: A
review of clinical features, theories and diagnosis. Int J Dev
Neurosci. 43:70–77. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Richdale AL and Schreck KA: Sleep problems
in autism spectrum disorders: Prevalence, nature, and possible
biopsychosocial aetiologies. Sleep Med Rev. 13:403–411. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Horvath GA, Stowe RM, Ferreira CR and Blau
N: Clinical and biochemical footprints of inherited metabolic
diseases. III. Psychiatric presentations. Mol Genet Metab. 130:1–6.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Fogel BL, Wexler E, Wahnich A, Friedrich
T, Vijayendran C, Gao F, Parikshak N, Konopka G and Geschwind DH:
RBFOX1 regulates both splicing and transcriptional networks in
human neuronal development. Hum Mol Genet. 21:4171–4186. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Wen M, Yan Y, Yan N, Chen XS, Liu SY and
Feng ZH: Upregulation of RBFOX1 in the malformed cortex of patients
with intractable epilepsy and in cultured rat neurons. Int J Mol
Med. 35:597–606. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Voineagu I, Wang X, Johnston P, Lowe JK,
Tian Y, Horvath S, Mill J, Cantor RM, Blencowe BJ and Geschwind DH:
Transcriptomic analysis of autistic brain reveals convergent
molecular pathology. Nature. 474:380–384. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Sebat J, Lakshmi B, Malhotra D, Troge J,
Lese-Martin C, Walsh T, Yamrom B, Yoon S, Krasnitz A, Kendall J, et
al: Strong association of de novo copy number mutations with
autism. Science. 316:445–449. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Qiao Y, Liu X, Harvard C, Hildebrand MJ,
Rajcan-Separovic E, Holden JJ and Lewis ME: Autism-associated
familial microdeletion of Xp11.22. Clin Genet. 74:134–144. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Edens AC, Lyons MJ, Duron RM, Dupont BR
and Holden KR: Autism in two females with duplications involving
Xp11.22-p11.23. Dev Med Child Neurol. 53:463–466. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Lee AY, Chen W, Stippec S, Self J, Yang F,
Ding X, Chen S, Juang YC and Cobb MH: Protein kinase WNK3 regulates
the neuronal splicing factor Fox-1. Proc Natl Acad Sci USA.
109:16841–16846. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Chung BH, Drmic I, Marshall CR,
Grafodatskaya D, Carter M, Fernandez BA, Weksberg R, Roberts W and
Scherer SW: Phenotypic spectrum associated with duplication of
Xp11.22-p11.23 includes autism spectrum disorder. Eur J Med Genet.
54:e516–e520. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Gehman LT, Stoilov P, Maguire J, Damianov
A, Lin CH, Shiue L, Ares M Jr, Mody I and Black DL: The splicing
regulator Rbfox1 (A2BP1) controls neuronal excitation in the
mammalian brain. Nat Genet. 43:706–711. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Piton A, Gauthier J, Hamdan FF, Lafrenière
RG, Yang Y, Henrion E, Laurent S, Noreau A, Thibodeau P, Karemera
L, et al: Systematic resequencing of X-chromosome synaptic genes in
autism spectrum disorder and schizophrenia. Mol Psychiatry.
16:867–880. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Navidhamidi M, Ghasemi M and Mehranfard N:
Epilepsy- associated alterations in hippocampal excitability. Rev
Neurosci. 28:307–334. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Zhou Y, Liu M and Liang WN: Progress on
the epidemiological study of epilepsy. Zhonghua Liu Xing Bing Xue
Za Zhi. 28:92–94. 2007.(In Chinese). PubMed/NCBI
|
|
49
|
Thurman DJ, Hesdorffer DC and French JA:
Sudden unexpected death in epilepsy: Assessing the public health
burden. Epilepsia. 55:1479–1485. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Devinsky O, Vezzani A, O'Brien TJ, Jette
N, Scheffer IE, de Curtis M and Perucca P: Epilepsy. Nat Rev Dis
Primers. 4:180242018. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Chen Z, Brodie MJ, Liew D and Kwan P:
Treatment outcomes in patients with newly diagnosed epilepsy
treated with established and new antiepileptic drugs: A 30-year
longitudinal cohort study. JAMA Neurol. 75:279–286. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Shima A, Nitta N, Suzuki F, Laharie AM,
Nozaki K and Depaulis A: Activation of mTOR signaling pathway is
secondary to neuronal excitability in a mouse model of
mesio-temporal lobe epilepsy. Eur J Neurosci. 41:976–988. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Schmeiser B, Zentner J, Prinz M, Brandt A
and Freiman TM: Extent of mossy fiber sprouting in patients with
mesiotemporal lobe epilepsy correlates with neuronal cell loss and
granule cell dispersion. Epilepsy Res. 129:51–58. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Jeong KH, Kim SH, Choi YH, Cho I and Kim
WJ: Increased expression of WNK3 in dispersed granule cells in
hippocampal sclerosis of mesial temporal lobe epilepsy patients.
Epilepsy Res. 147:58–61. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Kahle KT, Rinehart J, de Los Heros P,
Louvi A, Meade P, Vazquez N, Hebert SC, Gamba G, Gimenez I and
Lifton RP: WNK3 modulates transport of Cl- in and out of cells:
Implications for control of cell volume and neuronal excitability.
Proc Natl Acad Sci USA. 102:16783–16788. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Huberfeld G, Blauwblomme T and Miles R:
Hippocampus and epilepsy: Findings from human tissues. Rev Neurol
(Paris). 171:236–251. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Eftekhari S, Mehvari Habibabadi J, Najafi
Ziarani M, Hashemi Fesharaki SS, Gharakhani M, Mostafavi H,
Joghataei MT, Beladimoghadam N, Rahimian E and Hadjighassem MR:
Bumetanide reduces seizure frequency in patients with temporal lobe
epilepsy. Epilepsia. 54:e9–e12. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Loscher W, Puskarjov M and Kaila K:
Cation-chloride cotransporters NKCC1 and KCC2 as potential targets
for novel antiepileptic and antiepileptogenic treatments.
Neuropharmacology. 69:62–74. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Silayeva L, Deeb TZ, Hines RM, Kelley MR,
Munoz MB, Lee HH, Brandon NJ, Dunlop J, Maguire J, Davies PA and
Moss SJ: KCC2 activity is critical in limiting the onset and
severity of status epilepticus. Proc Natl Acad Sci USA.
112:3523–3528. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Chen Y, Zhou H, Jin T, Ye T and Xie W:
Clinical observation of the phased acupuncture for ischemic stroke
hemiplegia. Zhongguo Zhen Jiu. 38:1027–1034. 2018.(In Chinese).
PubMed/NCBI
|
|
61
|
Hu YY, Li L, Xian XH, Zhang M, Sun XC, Li
SQ, Cui X, Qi J and Li WB: GLT-1 upregulation as a potential
therapeutic target for ischemic brain injury. Curr Pharm Des.
23:5045–5055. 2017.PubMed/NCBI
|
|
62
|
Tuttolomondo A, Puleo MG, Velardo MC,
Corpora F, Daidone M and Pinto A: Molecular biology of
atherosclerotic ischemic strokes. Int J Mol Sci. 21:93722020.
View Article : Google Scholar
|
|
63
|
Zhao H, Nepomuceno R, Gao X, Foley LM,
Wang S, Begum G, Zhu W, Pigott VM, Falgoust LM, Kahle KT, et al:
Deletion of the WNK3-SPAK kinase complex in mice improves
radiographic and clinical outcomes in malignant cerebral edema
after ischemic stroke. J Cereb Blood Flow Metab. 37:550–563. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Demian WL, Persaud A, Jiang C, Coyaud É,
Liu S, Kapus A, Kafri R, Raught B and Rotin D: The ion transporter
NKCC1 links cell volume to cell mass regulation by suppressing
mTORC1. Cell Rep. 27:1886–1896.e6. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Yan Y, Dempsey RJ, Flemmer A, Forbush B
and Sun D: Inhibition of Na(+)-K(+)-Cl(−) cotransporter during
focal cerebral ischemia decreases edema and neuronal damage. Brain
Res. 961:22–31. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Chen H, Luo J, Kintner DB, Shull GE and
Sun D: Na(+)-dependent chloride transporter (NKCC1)-null mice
exhibit less gray and white matter damage after focal cerebral
ischemia. J Cereb Blood Flow Metab. 25:54–66. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Krueger M, Hartig W, Reichenbach A,
Bechmann I and Michalski D: Blood-brain barrier breakdown after
embolic stroke in rats occurs without ultrastructural evidence for
disrupting tight junctions. PLoS One. 8:e564192013. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Chen H, Kintner DB, Jones M, Matsuda T,
Baba A, Kiedrowski L and Sun D: AMPA-mediated excitotoxicity in
oligodendrocytes: Role for Na(+)-K(+)-Cl(−) co-transport and
reversal of Na(+)/Ca(2+) exchanger. J Neurochem. 102:1783–1795.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Hossain Khan MZ, Sohara E, Ohta A, Chiga
M, Inoue Y, Isobe K, Wakabayashi M, Oi K, Rai T, Sasaki S and
Uchida S: Phosphorylation of Na-Cl cotransporter by OSR1 and SPAK
kinases regulates its ubiquitination. Biochem Biophys Res Commun.
425:456–461. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Zhang J, Gao G, Begum G, Wang J, Khanna
AR, Shmukler BE, Daubner GM, de Los Heros P, Davies P, Varghese J,
et al: Functional kinomics establishes a critical node of
volume-sensitive cation-Cl-cotransporter regulation in the
mammalian brain. Sci Rep. 6:359862016. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Zhang P, Wang T, Zhang D, Zhang Z, Yuan S,
Zhang J, Cao J, Li H, Li X, Shen H and Chen G: Exploration of
MST1-mediated secondary brain injury induced by intracerebral
hemorrhage in rats via hippo signaling pathway. Transl Stroke Res.
10:729–743. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Kamel H and Hemphill JC III:
Characteristics and sequelae of intracranial hypertension after
intracerebral hemorrhage. Neurocrit Care. 17:172–176. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Honner SK, Singh A, Cheung PT, Alter HJ,
Dutaret CG, Patel AK and Acharya A: Emergency department control of
blood pressure in intracerebral hemorrhage. J Emerg Med.
41:355–361. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Zheng H, Chen C, Zhang J and Hu Z:
Mechanism and therapy of brain edema after intracerebral
hemorrhage. Cerebrovasc Dis. 42:155–169. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Dang G, Yang Y, Wu G, Hua Y, Keep RF and
Xi G: Early erythrolysis in the hematoma after experimental
intracerebral hemorrhage. Transl Stroke Res. 8:174–182. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Hemphill JC III, Greenberg SM, Anderson
CS, Becker K, Bendok BR, Cushman M, Fung GL, Goldstein JN,
Macdonald RL, Mitchell PH, et al: Guidelines for the management of
spontaneous intracerebral hemorrhage: A guideline for healthcare
professionals from the American Heart Association/American Stroke
Association. Stroke. 46:2032–2060. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Zhang X, Gu Y, Li P, Jiang A, Sheng X, Jin
X, Shi Y and Li G: Matrix metalloproteases-mediated cleavage on
β-dystroglycan may play a key role in the blood-brain barrier after
intracerebral hemorrhage in rats. Med Sci Monit. 25:794–800. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Mracsko E and Veltkamp R:
Neuroinflammation after intracerebral hemorrhage. Front Cell
Neurosci. 8:3882014. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Wu X, Fu S, Liu Y, Luo H, Li F, Wang Y,
Gao M, Cheng Y and Xie Z: NDP-MSH binding melanocortin-1 receptor
ameliorates neuroinflammation and BBB disruption through
CREB/Nr4a1/NF-κB pathway after intracerebral hemorrhage in mice. J
Neuroinflammation. 16:1922019. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Tian Y, Guo SX, Li JR, Du HG, Wang CH,
Zhang JM and Wu Q: Topiramate attenuates early brain injury
following subarachnoid haemorrhage in rats via duplex protection
against inflammation and neuronal cell death. Brain Res.
1622:174–185. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Digregorio M, Lombard A, Lumapat PN,
Scholtes F, Rogister B and Coppieters N: Relevance of translation
initiation in diffuse glioma biology and its therapeutic potential.
Cells. 8:15422019. View Article : Google Scholar
|
|
82
|
Giese A and Westphal M: Glioma invasion in
the central nervous system. Neurosurgery. 39:232–250. 1996.
|
|
83
|
de Paula LB, Primo FL and Tedesco AC:
Nanomedicine associated with photodynamic therapy for glioblastoma
treatment. Biophys Rev. 9:761–773. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Sontheimer H: Ion channels and amino acid
transporters support the growth and invasion of primary brain
tumors. Mol Neurobiol. 29:61–71. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Sontheimer H: An unexpected role for ion
channels in brain tumor metastasis. Exp Biol Med (Maywood).
233:779–791. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Garzon-Muvdi T, Schiapparelli P, ap Rhys
C, Guerrero-Cazares H, Smith C, Kim DH, Kone L, Farber H, Lee DY,
An SS, et al: Regulation of brain tumor dispersal by NKCC1 through
a novel role in focal adhesion regulation. PLoS Biol.
10:e10013202012. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Zhou B, Lu X, Hao Y and Yang P: Real-time
monitoring of the regulatory volume decrease of cancer cells: A
model for the evaluation of cell migration. Anal Chem.
91:8078–8084. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Algharabil J, Kintner DB, Wang Q, Begum G,
Clark PA, Yang SS, Lin SH, Kahle KT, Kuo JS and Sun D: Inhibition
of Na(+)-K(+)-2Cl(−) cotransporter isoform 1 accelerates
temozolomide-mediated apoptosis in glioblastoma cancer cells. Cell
Physiol Biochem. 30:33–48. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Ernest NJ and Sontheimer H: Extracellular
glutamine is a critical modulator for regulatory volume increase in
human glioma cells. Brain Res. 1144:231–238. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Haas BR and Sontheimer H: Inhibition of
the sodium-potassium-chloride cotransporter isoform-1 reduces
glioma invasion. Cancer Res. 70:5597–5606. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Hamann S, Herrera-Perez JJ, Zeuthen T and
Alvarez-Leefmans FJ: Cotransport of water by the
Na+-K+−2Cl(−) cotransporter NKCC1 in
mammalian epithelial cells. J Physiol. 588:4089–4101. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Mach C and Dollfus S: Scale for assessing
negative symptoms in schizophrenia: A systematic review. Encephale.
42:165–171. 2016.(In French). View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Tandon R, Gaebel W, Barch DM, Bustillo J,
Gur RE, Heckers S, Malaspina D, Owen MJ, Schultz S, Tsuang M, et
al: Definition and description of schizophrenia in the DSM-5.
Schizophr Res. 150:3–10. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Guessoum SB, Le Strat Y, Dubertret C and
Mallet J: A transnosographic approach of negative symptoms
pathophysiology in schizophrenia and depressive disorders. Prog
Neuropsychopharmacol Biol Psychiatry. 99:1098622020. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Gonzalez-Burgos G and Lewis DA: GABA
neurons and the mechanisms of network oscillations: implications
for understanding cortical dysfunction in schizophrenia. Schizophr
Bull. 34:944–961. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Lewis DA, Hashimoto T and Volk DW:
Cortical inhibitory neurons and schizophrenia. Nat Rev Neurosci.
6:312–324. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Arion D and Lewis DA: Altered expression
of regulators of the cortical chloride transporters NKCC1 and KCC2
in schizophrenia. Arch Gen Psychiatry. 68:21–31. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Blanquie O, Liebmann L, Hubner CA, Luhmann
HJ and Sinning A: NKCC1-mediated GABAergic signaling promotes
postnatal cell death in neocortical cajal-retzius cells. Cereb
Cortex. 27:1644–1659. 2017.PubMed/NCBI
|
|
99
|
Lewis DA and Sweet RA: Schizophrenia from
a neural circuitry perspective: Advancing toward rational
pharmacological therapies. J Clin Invest. 119:706–716. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
100
|
de Los Heros P, Kahle KT, Rinehart J,
Bobadilla NA, Vázquez N, San Cristobal P, Mount DB, Lifton RP,
Hebert SC and Gamba G: WNK3 bypasses the tonicity requirement for
K-Cl cotransporter activation via a phosphatase-dependent pathway.
Proc Natl Acad Sci USA. 103:1976–1981. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
de Los Heros P, Alessi DR, Gourlay R,
Campbell DG, Deak M, Macartney TJ, Kahle KT and Zhang J: The
WNK-regulated SPAK/OSR1 kinases directly phosphorylate and inhibit
the K+-Cl− co-transporters. Biochem J.
458:559–573. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Vorontsova I, Donaldson PJ, Kong Z,
Wickremesinghe C, Lam L and Lim JC: The modulation of the
phosphorylation status of NKCC1 in organ cultured bovine lenses:
Implications for the regulation of fiber cell and overall lens
volume. Exp Eye Res. 165:164–174. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Alessi DR, Zhang J, Khanna A, Hochdorfer
T, Shang Y and Kahle KT: The WNK-SPAK/OSR1 pathway: Master
regulator of cation-chloride cotransporters. Sci Signal. 7:re32014.
View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Conway LC, Cardarelli RA, Moore YE, Jones
K, McWilliams LJ, Baker DJ, Burnham MP, Bürli RW, Wang Q, Brandon
NJ, et al: N-Ethylmaleimide increases KCC2 cotransporter activity
by modulating transporter phosphorylation. J Biol Chem.
292:21253–21263. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Glover M, Zuber AM and O'Shaughnessy KM:
Renal and brain isoforms of WNK3 have opposite effects on NCCT
expression. J Am Soc Nephrol. 20:1314–1322. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Lu DC, Hannemann A, Wadud R, Rees DC,
Brewin JN, Low PS and Gibson JS: The role of WNK in modulation of
KCl cotransport activity in red cells from normal individuals and
patients with sickle cell anaemia. Pflugers Arch. 471:1539–1549.
2019. View Article : Google Scholar : PubMed/NCBI
|