Introduction
Hypertriglyceridemia (HTG) refers to serum
triglyceride (TG) levels at ≥150 mg/dl or ≥1.7 mmol/l, and its
prevalence increases with age (1).
Previous studies have reported that HTG is one of the risk factors
for numerous cardiovascular diseases (CVD) (2,3).
Glycerin and fatty acids (FAs) are common metabolites of TGs, and
serve a role in energy metabolism and maintaining physiological
functions in multiple organisms (4,5).
However, the effects and mechanisms of TGs or how their metabolites
alter the structure and function of vascular smooth muscle cells
(VSMCs) require further investigation.
The abnormal proliferation and migration of VSMCs is
one of the characteristics of atherosclerosis (6–8). Zhou
et al (9) revealed that high
TG serum levels could stimulate the proliferation of human fetal
VSMCs and affect the ultrastructure of cells, but results from Yin
et al (10) suggested that
high TG serum levels did not significantly promote the
proliferation of VSMCs. Furthermore, Mattern and Hardin (11) reported that oleic acid (OA) could
reduce the apoptosis of VSMCs induced by palmitic acid. Our
previous studies indicated that triolein (12) and medium-chain TGs (13) can promote (at low concentrations) or
inhibit (at high concentrations) the proliferation of rat aortic
VSMCs in a time- and concentration-dependent manner.
OA, a monounsaturated FA, representing ~80% of
plasma phospholipid monounsaturated FAs, is one of the primary FA
components of the Mediterranean diet and olive oil, is involved in
the regulation of cell proliferation (14,15).
Moreover, it is one of the metabolites derived from TGs (15). However, the effects and mechanisms
via which OAs regulate cell proliferation remain to be
elucidated.
Hepatocyte growth factor (HGF) is a cytokine with
multiple biological effects, such as promoting cell survival and
the regeneration of tissues, and suppressing and improving chronic
inflammation and fibrosis (16),
and serves a role in promoting or inhibiting cell proliferation
(17). However, to the best of our
knowledge, whether the HGF signaling pathway is involved in the
OA-induced proliferation of VSMCs is largely unknown, and this
issue was first discussed by Greene et al (18).
MAPK is a key signaling pathway that regulates cell
proliferation, apoptosis, inflammatory responses and
differentiation by activating a series of downstream regulatory
factors (19,20). The MAPK family includes the JNK and
p38 families (21,22). Furthermore, p38 MAPK signaling is
closely associated with atherosclerosis, as it has been
demonstrated to serve a role in VSMC (23) and H441 cell proliferation (24), HGF secretion (25) and connexin 43 expression (26).
Therefore, the main aim of the present study was to
elucidate the relationship between the effect of OA on VSMCs and
the HGF and p38 MAPK-signaling pathways.
Materials and methods
Reagents
High glucose (25 mmol/l) DMEM was purchased from
Gibco (Thermo Fisher Scientific, Inc.). OA was purchased from the
Cayman Chemical Company. Cell Counting Kit-8 (CCK-8) assays were
purchased from the Beyotime Institute of Biotechnology, while PI
was purchased from Sigma-Aldrich (Merck KGaA). Commercial HGF ELISA
kit (cat. no. EK1301) was purchased from Boster Biological
Technology. RevertAid First Strand cDNA Synthesis kits were
purchased from Hangzhou Woosen Biotechnology Co., Ltd., and SYBR
Green quantitative PCR (qPCR) mix for reverse transcription-qPCR
(RT-qPCR) was purchased from Takara Biotechnology Co., Ltd.
Transwell chambers were obtained from Corning, Inc. Rabbit
anti-human polyclonal antibody against HGF (cat. no. ab83760) was
purchased from Abcam, p38 (cat. no. AM8123) and phosphorylated p38
(p-p38; cat. no. AM1195) antibodies were obtained from Hunan
Auragene Biotechnology Co., Ltd., and monoclonal antibodies against
GAPDH (cat. no. 8884) were purchased from Cell Signaling
Technology, Inc. The PVDF membrane and ECL detection kits were
obtained from EMD Millipore. Goat anti-rabbit (cat. no. SA009) and
mouse (cat. no. SA001) IgG secondary antibodies conjugated to HRP,
the BCA protein assay kit, DMSO, SDS-PAGE loading buffer,
TRIzol® reagent and RIPA buffer were obtained from Hunan
Auragene Biotechnology Co., Ltd. Penicillin and streptomycin were
purchased from MP Biomedicals, LLC. The HGF inhibitor PHA665752
(27) and the p38 MAPK inhibitor
SB203580 were obtained from Selleck Chemicals.
Cell line and culture
A7r5 cells, a smooth muscle cell line derived from
the rat thoracic aorta, were obtained from The Cell Bank of Type
Culture Collection of the Chinese Academy of Sciences. Cells were
cultured in high glucose (25 mmol/l) DMEM supplemented with 10% FBS
(Hunan Auragene Biotechnology Co., Ltd.) (28), 100 U/ml penicillin and 100 mg/ml
streptomycin in a humidified atmosphere of 5% CO2 at
37°C.
Cell proliferation assays
A7r5 cells were seeded in a 96-well tissue culture
plate at a density 5×103 cells/well. DMSO (50 mmol/l)
was used as a solvent to dissolve OA. The final concentration of
DMSO in all experimental groups was not >1 mmol/l (<0.1%)
(29,30). Several studies have suggested that
treatment with 1–25 µM SB203580 (31,32)
and 0.1 µmol/l PHA665752 (30,33,34)
does not induce cell death and does not reduce the number of cells
cultured. Thus, these cells were then incubated at 37°C with
different concentrations of OA (0, 0.5, 5, 50, 200 or 800 µmol/l)
for 24 and 48 h, or 0 µmol/l OA, 50 µmol/l OA, 50 µmol/l OA+ 0.1
µmol/l PHA665752 or 50 µmol/l OA+ 2 µmol/l SB203580 for 24 h.
Concentrations of 0.1 and 2.0 µmol/l represented the
IC50 of PHA665752 and SB203580, respectively. Moreover,
0 µmol/l OA (containing 1 mmol/l DMSO) served as a control. After
treatment, the medium was removed from the wells, and 0.1 ml high
glucose DMEM containing 10% CCK-8 was added to each well for 3 h at
37°C according to the manufacturer's instructions. Cell viability
was determined by measuring the absorbance at 450 nm using averages
from quintuplicate wells, and normalized to a control well. The
absorbance at each time point was determined using a microplate
reader. All experiments were performed ≥3 times.
Cell cycle analysis
A7r5 cells were seeded in a 6-well tissue culture
plate at a density 2.5×105 cells/well. These cells were
then incubated at 37°C with three different concentrations of OA
(0, 50 or 800 µmol/l) for 24 h; 0 µmol/l OA (containing 1 mmol/l
DMSO) served as a control. After treatment, cells were harvested
using trypsin. The cells were centrifuged at 300 × g for 5 min at
25°C, washed twice with cold PBS and fixed with 75% cool ethanol at
−20°C overnight (not <16 h). The fixed cells were then
centrifuged at 200 × g for 5 min at 25°C and washed twice with cold
PBS. Then, cells were stained with 50 µg/ml PI containing 10 µg/ml
RNase A for 30 min on ice. Finally, the distributions of cells in
different cell cycle phases were assessed using specific amounts of
cellular DNA (2×105 cells/tube) and flow cytometry
(CytoFLEX; Beckman Coulter, Inc.). In total, >10,000 cells were
counted per sample, and DNA histograms for cell cycle analysis were
analyzed using Cell Quest software version 2.0 (BD Biosciences).
The percentage of cells in the G0/G1 phase, S
phase and G2/M phase were analyzed, and all experiments
were repeated in triplicate.
Transwell assays
A7r5 cells in logarithmic growth phase were
harvested with trypsin, and then starved in serum-free medium.
Next, a cell suspension (2×104 cells) in 0.3 ml
high-glucose DMEM was seeded into the upper well of a Transwell
chamber (0.33 cm2 growth surface area; 8-pm pore size),
and these cells were then treated at 37°C with three concentrations
of OA (0, 50 or 800 µmol/l), or 0 µmol/l OA, 50 µmol/l OA, 50
µmol/l OA+ 0.1 µmol/l PHA665752 and 50 µmol/l OA+ 2 µmol/l SB203580
for 24 h. Moreover, 0 µmol/l OA (containing 1 mmol/l DMSO) served
as a control. In the lower chamber, 0.5 ml high-glucose DMEM with
10% FBS was added. After incubation for 24 h at 37°C in a
humidified incubator with 5% CO2, the cells on the upper
side of the upper chamber were removed using a cotton swab. The
lower side of the upper chamber was fixed with 3% methanol for 5
min at 25°C, and stained with 2% crystal violet for 10 min at 25°C.
The number of cells penetrating across the membrane was counted
using an inverted microscope at ×100 magnification in three random
visual fields, and all experiments were performed in
triplicate.
HGF level analysis using ELISA
After stimulation at 37°C with three different
concentrations of OA (0, 50 or 800 µmol/l) for 24 h, A7r5 cells
(1×106 cells) were collected and centrifuged at 1,000 ×
g for 10 min at 25°C to remove debris. The 0 µmol/l OA (containing
1 mmol/l DMSO) served as a control. HGF protein levels in these
cells were quantified using an ELISA kit (Abcam), according to the
manufacturer's instructions, and all samples were measured three
times. All experiments were repeated ≥3 times.
Western blotting
After stimulation at 37°C with three different
concentrations of OA (0, 50 or 800 µmol/l), or 0 µmol/l OA, 50
µmol/l OA, 50 µmol/l OA+ 0.1 µmol/l PHA665752 and 50 µmol/l OA+ 2
µmol/l SB203580 for 24 h, cells (1×106 cells) were
washed with PBS and then lysed in RIPA buffer for 30 min at 4°C.
The 0 µmol/l OA (containing 1 mmol/l DMSO) served as a control. The
total protein concentrations were measured using a BCA protein
assay kit. Equal amounts of protein (20 µg/well) were then mixed
with SDS-PAGE loading buffer and boiled for 10 min. These samples
were separated using 12% SDS-PAGE, and then transferred to a PVDF
membrane. Membranes were incubated with appropriate concentration
of primary antibody for HGF (1:100 dilution), p38 (1:1,000
dilution), p-p38 (1:1,000 dilution) or GAPDH (1:1,000 dilution)
overnight at 4°C. After washing three times with 1X TBS-0.1% Tween
20, these membranes were incubated for 1 h with goat anti-rabbit or
anti-mouse IgG secondary antibodies (1:15,000 dilution) conjugated
to HRP for evaluating the expression levels of HGF and GAPDH.
Proteins were visualized using an ECL detection kit, and the
relative band intensities were analyzed using Image Pro Plus 6.0
software (Media Cybernetics, Inc.). All results were verified using
≥3 independent experiments.
RNA isolation, RT and RT-qPCR
Total RNA was isolated from A7r5 cells
(1×106 cells), after stimulation at 37°C with varying
concentrations of OA (0 µmol/l OA, 50 µmol/l OA and 50 µmol/l OA+
0.1 µmol/l PHA665752) for 24 h, using TRIzol reagent, according to
the manufacturer's instructions. The 0 µmol/l OA (containing 1
mmol/l DMSO) served as a control. cDNAs were synthesized using 1 µg
total RNA with the RevertAid First Strand cDNA Synthesis kit,
following the manufacturer's instructions. RT-qPCR was performed
using SYBR Green qPCR mix, following the manufacturer's
instructions. The β-actin gene was measured as an internal
quantitative control. All reactions were carried out on an
ABI® 7300 Real-Time PCR system (Applied Biosystems;
Thermo Fisher Scientific, Inc.) in duplicate. The reaction
conditions were as follows: Initial denaturation at 95°C for 3 min;
followed by 40 cycles of denaturation at 95°C for 10 sec, annealing
and elongation at 60°C for 10 sec; and final extension at 72°C for
30 sec. Melt curves were analyzed for each sample. The average
value in each duplicate was used to calculate the relative amount
of HGF using the 2−ΔΔCq method (35). The primer sequences were as follows:
HGF forward, 5′-TCATTGGTAAAGGAGGCA-3′ and reverse,
5′-GTCACAGACTTCGTAGCG-3′; and β-actin forward,
5′-AGGCCCCTCTGAACCCTAAG-3′ and reverse,
5′-CCAGAGGCATACAGGGACAAC-3′. All experiments were conducted ≥3
times.
Statistical analysis
Data are presented as the mean ± SD. All statistical
analyses were performed using SPSS 20.0 software (IBM Corp.).
Differences between two groups were compared using a two-tailed
Student's t-test, and the one-way ANOVA followed by Tukey's test
was performed to compare data between multiple groups. P<0.05
was considered to indicate a statistically significant
difference.
Results
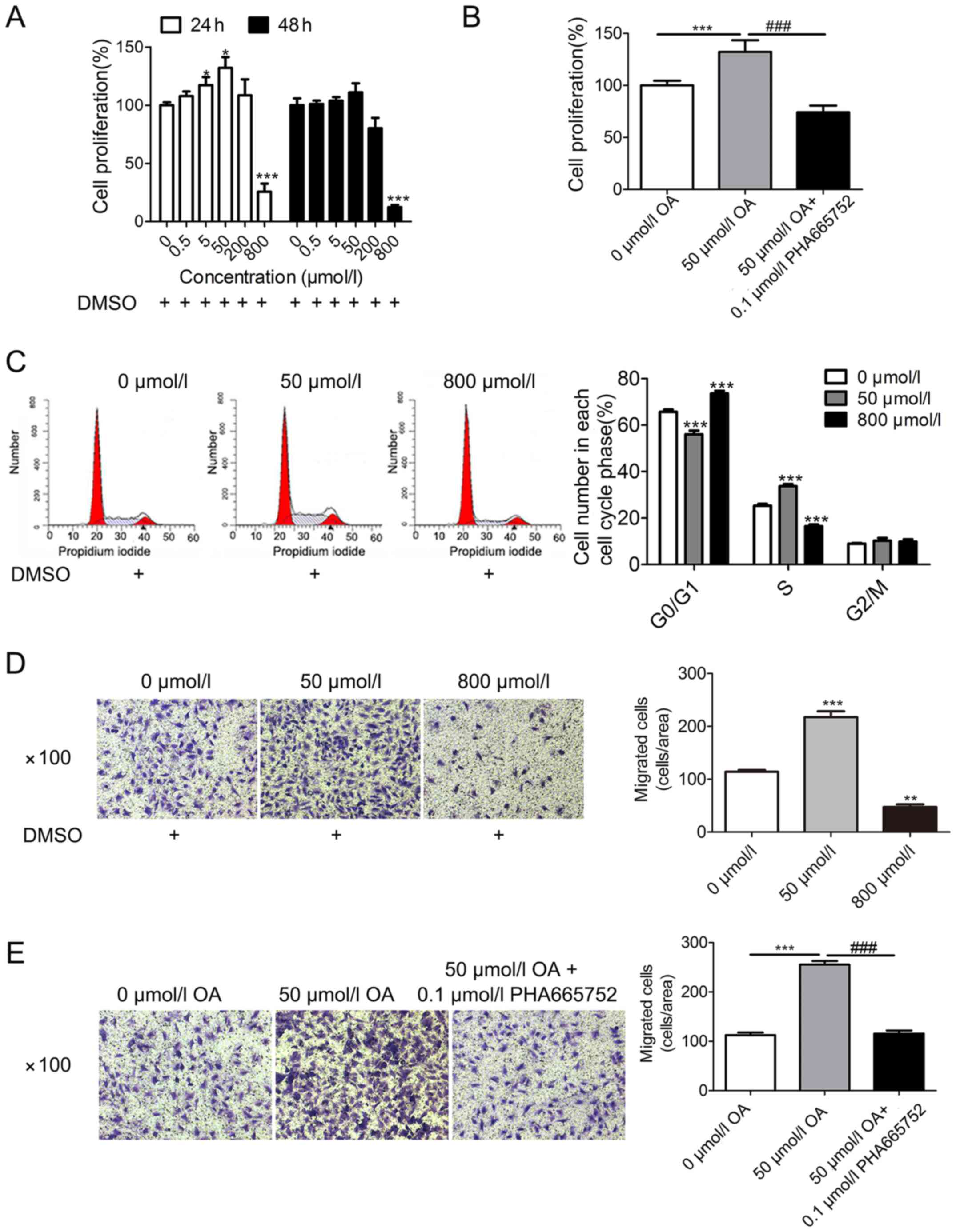
Effects of OA on A7r5 cell
proliferation and migration
First, A7r5 cells were treated with increasing
concentrations of OA (0, 0.5, 5, 50, 200 and 800 µmol/l). Then,
CCK-8 assays were conducted to evaluate the cell viability at two
time points (24 and 48 h post-treatment), based on findings from
previous studies (12,13). The current results demonstrated that
OA treatment promoted A7r5 cell proliferation at lower
concentrations (5 and 50 µmol/l), but inhibited A7r5 cell
proliferation at higher concentrations (800 µmol/l), in a time- and
concentration-dependent manner (Fig.
1A). The optimal parameters for promoting cell proliferation
were determined to be 24 h and a dose of 50 µmol/l OA. According to
the aforementioned results, three concentrations of OA (0, 50 and
800 µmol/l) and the treatment period of 24 h, were selected for
subsequent experiments. Treatment with 0 µmol/l OA (containing 1
mmol/l DMSO) served as a control group.
Next, A7r5 cells were incubated with three different
OA conditions (0 µmol/l OA, 50 µmol/l OA or 50 µmol/l + 0.1 µmol/l
PHA665752) for 24 h. The data indicated that the effect of OA (at
50 µmol/l) in promoting cell proliferation was mitigated by
PHA665752 (0.1 µmol/l) (Fig. 1B).
Subsequently, A7r5 cells were treated with three different
concentrations of OA (0, 50 and 800 µmol/l) for 24 h. Compared with
the 0 µmol/l OA group (G0/G1 phase, 65.72±1%;
S phase, 25.31±0.3%), the results of flow cytometry demonstrated
that low concentrations of OA (at 50 µmol/l) reduced the number of
cells in the G0/G1 phase (56.06±1.6%) and
increased the number of cells in the S phase (33.71±0.9%), while
high concentrations of OA (at 800 µmol/l) increased the number of
cells in the G0/G1 phase (73.64±1.1%) and
decreased the number of cells in the S phase (16.52±0.7%) (Fig. 1C).
Transwell assay results indicated that low
concentrations of OA (at 50 µmol/l) promoted cell migration, but
high concentrations of OA (at 800 µmol/l) suppressed cell migration
(Fig. 1D). Additionally, when A7r5
cells were incubated with 0 µmol/l OA, 50 µmol/l OA or 50 µmol/l OA
+0.1 µmol/l PHA665752 for 24 h, the results suggested that the
effect of OA (at 50 µmol/l) in promoting cell migration was
mitigated by treatment with PHA665752 (0.1 µmol/l) (Fig. 1E).
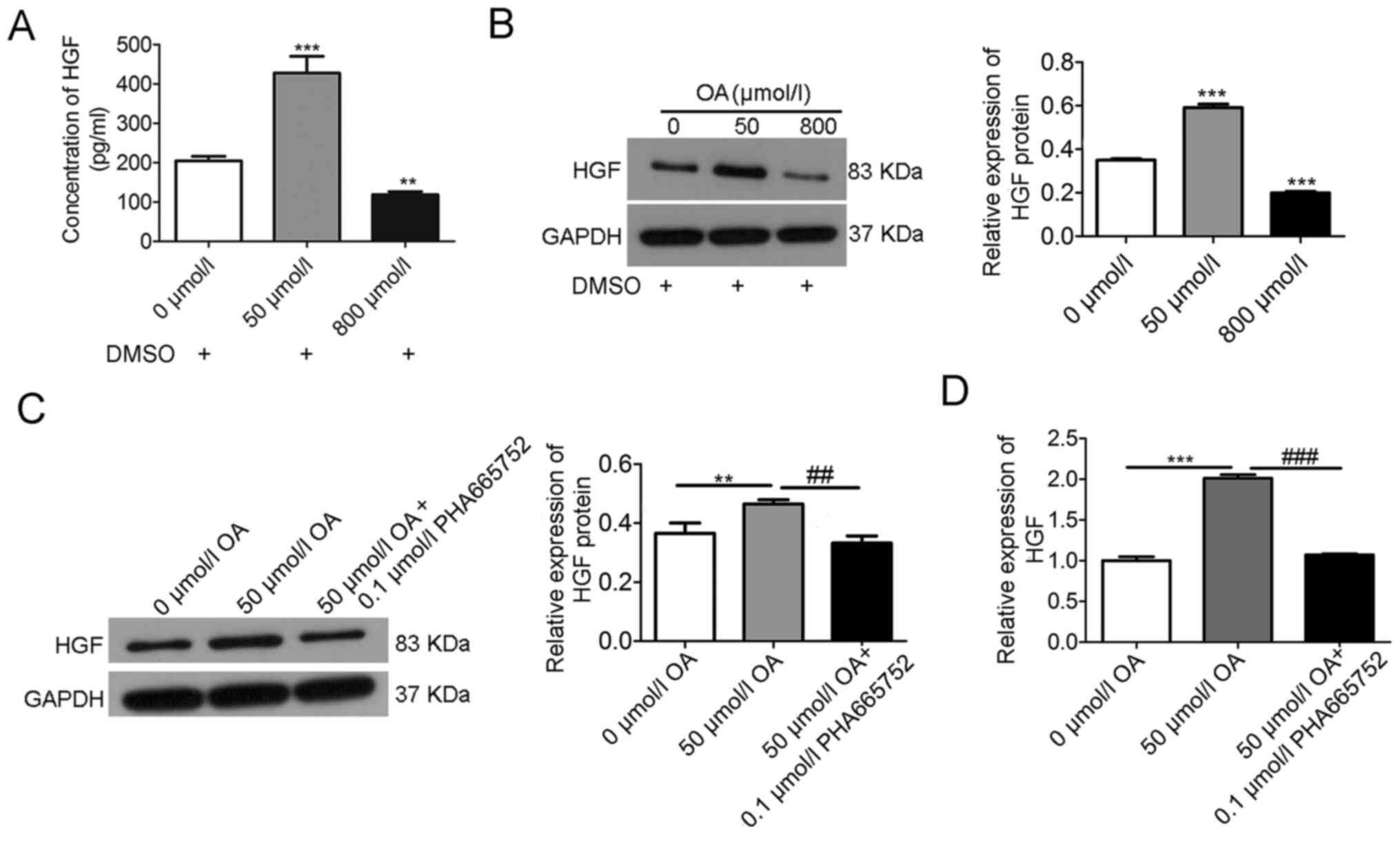
Effects of OA on HGF expression in
A7r5 cells
OA treatment increased HGF expression at low
concentrations (50 µmol/l OA) but downregulated it at high
concentrations (800 µmol/l OA), as determined by ELISA (Fig. 2A) and western blotting (Fig. 2B). Moreover, the increased
expression of HGF at low concentrations of OA (50 µmol/l) was
mitigated by 0.1 µmol/l PHA665752, as observed via western blotting
(Fig. 2C) and RT-qPCR (Fig. 2D).
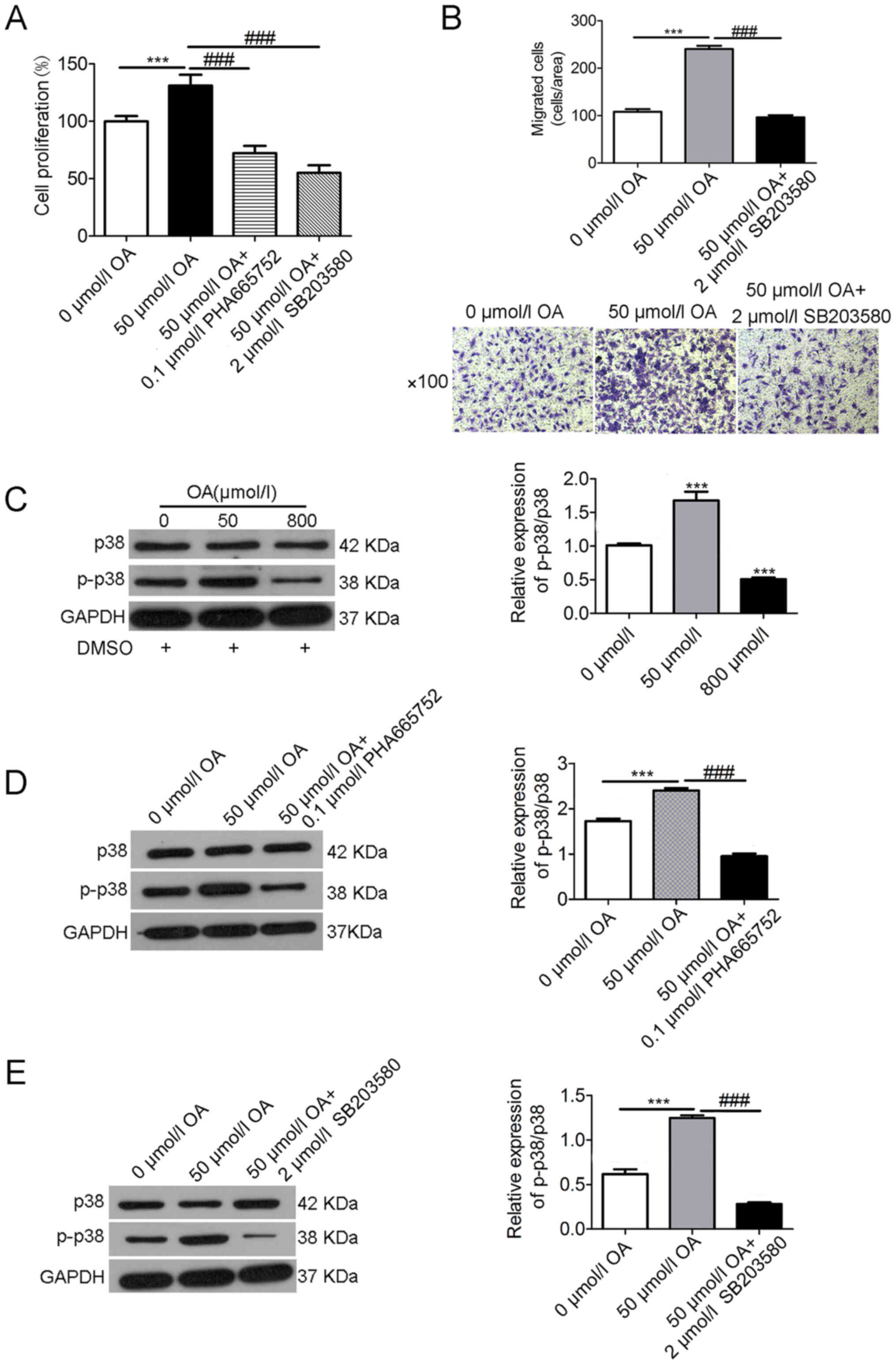
Effects of HGF inhibition and p38 MAPK
inhibition on OA-induced A7r5 cell proliferation and migration
To investigate the mechanism of OA-induced A7r5 cell
proliferation and migration via HGF signaling, the p38 inhibitor
SB203580 was used, which inhibits PKB phosphorylation. The results
demonstrated that the A7r5 cell proliferation induced by OA (50
µmol/l) was mitigated by PHA665752 (0.1 µmol/l) or SB203580 (2
µmol/l) (Fig. 3A). Moreover, A7r5
cell migration was mitigated by SB203580 (2 µmol/l) (Fig. 3B). The effects of OA on p38 and
p-p38 expression levels were then determined via western blotting
(Fig. 3C), and the data revealed
that OA treatment increased p-p38 expression at low concentrations
(50 µmol/l OA) but downregulated it at high concentrations (800
µmol/l OA). Moreover, the increased expression of p-p38 at low
concentrations of OA (50 µmol/l) was mitigated by PHA665752 (0.1
µmol/l) (Fig. 3D) or SB203580 (2
µmol/l) (Fig. 3E). By contrast,
there was no notable difference in the expression of p38.
In addition, via preliminary experiments, it was
observed that 1 mmol/l DMSO, 2 µmol/l SB203580 or 0.1 µmol/l
PHA665752 did not affect the proliferation and migration of A7r5
cells (data not shown), which was consistent with previous studies
(29–34).
Discussion
The present study demonstrated that OA could promote
A7r5 cell proliferation at a low concentration and over a short
time length, but OA inhibited A7r5 cell proliferation at a high
concentration and over longer times, as demonstrated using CCK-8
assays, cell cycle analysis and Transwell assays. Interestingly,
the A7r5 cell proliferation and migration induced by OA (50 µmol/l)
were mitigated by PHA665752 (0.1 µmol/l), a selective inhibitor of
HGF. Using ELISA and western blotting, it was observed that HGF
expression was increased in A7r5 cells at low doses of OA (50
µmol/l) but was decreased using high doses of OA (800 µmol/l).
However, the increased expression of HGF induced by OA (50 µmol/l)
was also mitigated by the HGF inhibitor PHA665752 (0.1 µmol/l).
HGF, an angiogenic factor, is associated with the
risk of coronary heart disease, stroke and atherosclerosis
(36,37). Circulating HGF has been proposed as
a potential clinical biomarker for CVD (38). Previous studies have shown that high
concentrations of HGF were significantly associated with the
progression of atherosclerosis (39). HGF and its receptor c-MET serve an
important role in endothelial injury and repair, angiogenesis, cell
migration, cell survival and anti-inflammatory responses in
multiple cell types (16,40). The present study used the HGF
inhibitor PHA665752 (27,41,42) to
examine whether there was an association between the effect of OA
and the HGF signaling pathway. The current results suggested that
the effect of OA on proliferation and migration in A7r5 cells was
mitigated by PHA665752. Moreover, the mRNA and protein expression
levels of HGF induced by OA were suppressed by PHA665752 in A7r5
cells. These results indicated that OA promoted cell proliferation
and migration via HGF signal transduction, which is helpful for
further understanding the effects and mechanisms driving OA's
effect on CVD (38).
p38 MAPK signaling has been reported to serve a role
in VSMC proliferation (23) and HGF
secretion from human astrocytoma cells (25). The present study used the p38 MAPK
inhibitor SB203580 to evaluate whether there was an association
between the effects of OA and p38 MAPK signaling, and p38 MAPK
served as the positive control for several of the assays. The
current data demonstrated that A7r5 cell proliferation and
migration induced by OA (50 µmol/l) were suppressed by SB203580 (2
µmol/l), and the increased expression level of p-p38 at low
concentrations of OA (50 µmol/l) was also mitigated by PHA665752
(0.1 µmol/l) or SB203580 (2 µmol/l). However, there was no notable
difference in the expression level of p38. The present findings
suggested that the effect of OA on cell proliferation and migration
was mitigated by SB203580 in A7r5 cells, and the expression of
p-p38 protein induced by OA was also suppressed by SB203580 in A7r5
cells. Furthermore, the results indicated that OA promoted cell
proliferation and migration by regulating p38 phosphorylation, and
that p38 MAPK signal transduction may serve an important role in
the VSMC proliferation and migration induced by OA. Collectively,
these data suggested that the OA-induced stimulation of the
proliferation and migration of VSMCs may be associated with
increased expression levels of HGF and p-p38. Therefore, the
results of other studies (19,23)
and the present study support the hypothesis that p38 MAPK, as a
downstream mediator, regulates the activity of HGF induced by OA in
A7r5 cells.
The aforementioned results support the following two
principal findings. First, low (physiological) concentrations of OA
induce VSMC proliferation and migration, which is consistent with
previous studies (43,44). Second, the proliferation and
migration of VSMCs induced by OA appear to be mediated by HGF and
the p38 MAPK pathway. To the best of our knowledge, the current
study demonstrated for the first time that OA stimulated the
proliferation and migration of VSMCs via the HGF and p38 MAPK
pathways in vitro.
Several signaling pathways, including the PI3K/Akt
(28), reactive oxygen species
(ROS) (45), AMP-activated protein
kinase (AMPK)/endothelial nitric oxide synthase/FAS (46) and AMPK pathways (47), have been reported to serve a role in
the proliferation and migration of VSMCs. The present findings
suggest that HGF and p38 MAPK represent a new signaling pathway
that participates in the proliferation and migration of VSMCs
stimulated by OA.
The present study also suggested that the effect of
OA on the proliferation and migration of A7r5 cells could be
regulated by the HGF and p38 MAPK signaling pathways, and the
proliferation and migration of VSMCs induced by OA were associated
with increased expression levels of HGF and p-p38.
Several studies have revealed that OA treatment can
induce apoptosis in neuronal cells (48), carcinoma cells (49) and VSMCs (50,51),
and is respectively associated with dephosphorylation of Bad,
increasing ROS production, or caspase 3 activity, autophagy and
LOX-1 upregulation. These results may explain the toxicity or
inhibition of treatment with OA at high concentrations and over
longer time frames on the proliferation and migration of A7r5
cells, which was observed in the current experiments. There are
five limitations to the present study. First, the mechanisms
involved in the up- or downregulation of the expression levels of
HGF and p-p38 in A7r5 cells induced by OA need to be studied in the
future. Second, the effects on cell cycle markers using small
interfering RNAs or short hairpin RNAs, and other related signaling
molecules, need to be evaluated further. Third, the present data
indicated that OA had an effect on both the proliferation and
migration of A7r5 cells, but the results of migration experiments
may have been affected by differentiated proliferation (52). Fourth, the expression levels of
contractile and synthetic markers in VSMCs should be evaluated.
Fifth, there was the absence of PHA665752 or SB203580 alone control
groups in this study.
In conclusion, the effect of OA on A7r5 cell
proliferation and migration was both time- and
concentration-dependent, and low concentrations and short time
frames promoted cell proliferation and migration, while high
concentrations and long time frames inhibited cell proliferation
and migration. Treatment with OA at 24 h could promote A7r5 cell
proliferation and migration, but the effect of OA in promoting cell
proliferation and migration disappeared at 48 h, which was
consistent with, but not precisely replicated, in our prior
experiments (12,13). These findings suggested that the
promoting or inhibiting effects of glyceryl trioleate and medium
chain TG on the proliferation of A7r5 cells (low concentrations and
short time periods promoting cell proliferation; high
concentrations and long time periods inhibiting cell proliferation)
were not associated with cell apoptosis. Our previous findings
(12,13) are indirectly supported by a study
from Belal et al (53),
which revealed that OA can promote the proliferation of bovine
satellite cells without apoptosis or necrosis. Oxidative stress and
ROS production may explain the current experiment results obtained
from treatment with OA for 48 h or at high concentrations (49), and long treatment durations or high
concentrations of OA treatment may activate the MAPK pathway by
oxidative stress to inhibit cell proliferation (19,20).
Collectively, it was indicated OA, HGF and p38 MAPK may be
potential therapeutic targets for CVDs (such as atherosclerosis)
(37). The present results provide
novel insights into the biological effects of OA, HGF and p38 MAPK,
and new evidence for the effect and mechanism of OA regulation on
the proliferation and migration of VSMCs.
Acknowledgements
Not applicable.
Funding
This work was partly supported by grant (grant no.
jsdxrcyjkyxm201304) from the Jishou University and a grant (grant
no. CX2018B711) from the Education Bureau of Hunan Province,
China.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
MY conceived and designed the study. JL performed
the experiments and acquired the data. MY and JL confirm the
authenticity of all the raw data. MY, JL and TC analyzed the data.
MY, JL and TC drafted of the manuscript. MY and TC contributed to
the critical revision of the manuscript for important intellectual
content. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
CCK-8
|
Cell Counting Kit-8
|
|
CVD
|
cardiovascular diseases
|
|
FAs
|
fatty acids
|
|
HGF
|
hepatocyte growth factor
|
|
HTG
|
hypertriglyceridemia
|
|
OA
|
oleic acid
|
|
p-p38
|
phosphorylated p38
|
|
RT-qPCR
|
reverse transcription-quantitative
PCR
|
|
ROS
|
reactive oxygen species
|
|
TG
|
triglyceride
|
|
VSMCs
|
vascular smooth muscle cells
|
References
|
1
|
Zhao WH, Zhang J, You Y, Man QQ, Li H,
Wang CR, Zhai Y, Li Y, Jin SG and Yang XG: Epidemiologic
characteristics of dyslipidemia in people aged 18 years and over in
China. Zhonghua Yu Fang Yi Xue Za Zhi. 39:306–310. 2005.(In
Chinese). PubMed/NCBI
|
|
2
|
Després JP and Lemieux I: Abdominal
obesity and metabolic syndrome. Nature. 444:881–887. 2006.
View Article : Google Scholar
|
|
3
|
Xie C, Wang ZC, Liu XF and Yang MS: The
common biological basis for common complex diseases: Evidence from
lipoprotein lipase gene. Eur J Hum Genet. 18:3–7. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hardy S, Langelier Y and Prentki M: Oleate
activates phosphatidylinositol 3-kinase and promotes proliferation
and reduces apoptosis of MDA-MB-231 breast cancer cells, whereas
palmitate has opposite effects. Cancer Res. 60:6353–6358.
2000.PubMed/NCBI
|
|
5
|
Sargsyan E, Artemenko K, Manukyan L,
Bergquist J and Bergsten P: Oleate protects beta-cells from the
toxic effect of palmitate by activating pro-survival pathways of
the ER stress response. Biochim Biophys Acta. 1861:1151–1160. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ross R: The pathogenesis of
atherosclerosis - an updata. N Engl J Med. 314:488–500. 1986.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Basatemur GL, Jørgensen HF, Clarke MCH,
Bennett MR and Mallat Z: Vascular smooth muscle cells in
atherosclerosis. Nat Rev Cardiol. 16:727–744. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Bennett MR, Sinha S and Owens GK: Vascular
smooth muscle cells in atherosclerosis. Circ Res. 118:692–702.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zhou XX, Zhou XH, Yang HM and Su PQ: High
triglyceride serum promotes the proliferation of vascular smooth
muscle cells. Chin J Cardiol. 28:3162000.(In Chinese).
doi:10.3760/j:issn:0253-3758.2000.04.024.
|
|
10
|
Yin Z, Gao HK, Li LS, Luan RH and Wang HC:
Effects of atorvastatin on hyperlipemic serum induced proliferation
of vascular smooth muscle cells in rats. Chin Hear J. 20:180–183.
2008.(In Chinese).
|
|
11
|
Mattern HM and Hardin CD: Vascular
metabolic dysfunction and lipotoxicity. Physiol Res. 56:149–158.
2007.PubMed/NCBI
|
|
12
|
Wang YQ and Yang MS: Effects of glyceryl
trioleate on the proliferation of rat aortic smooth muscle cells. J
Chongqing Med Univ. 39:1384–1390. 2014.(In Chinese).
|
|
13
|
Yang MS and Wang YQ: Bidirectional effects
of medium chain triglyceride on the proliferation of vascular
smooth muscle cells. Chin J Arterioscler. 24:551–556. 2016.(In
Chinese). doi: 10.13406/jcnki.cyxb.001052. PubMed/NCBI
|
|
14
|
Liotti A, Cosimato V, Mirra P, Calì G,
Conza D, Secondo A, Luongo G, Terracciano D, Formisano P, Beguinot
F, et al: Oleic acid promotes prostate cancer malignant phenotype
via the G protein-coupled receptor FFA1/GPR40. J Cell Physiol.
233:7367–7378. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Yang C, Lim W, Bazer FW and Song G: Oleic
acid stimulation of motility of human extravillous trophoblast
cells is mediated by stearoyl-CoA desaturase-1 activity. Mol Hum
Reprod. 23:755–770. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Nakamura T, Sakai K, Nakamura T and
Matsumoto K: Hepatocyte growth factor twenty years on: Much more
than a growth factor. J Gastroenterol Hepatol. 26 (Suppl
1):S188–S202. 2011. View Article : Google Scholar
|
|
17
|
Forte G, Minieri M, Cossa P, Antenucci D,
Sala M, Gnocchi V, Fiaccavento R, Carotenuto F, De Vito P, Baldini
PM, et al: Hepatocyte growth factor effects on mesenchymal stem
cells: Proliferation, migration, and differentiation. Stem Cells.
24:23–33. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Greene EL, Lu G, Zhang D and Egan BM:
Signaling events mediating the additive effects of oleic acid and
angiotensin II on vascular smooth muscle cell migration.
Hypertension. 37:308–312. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wan Q, Liu Z and Yang Y: Puerarin inhibits
vascular smooth muscle cells proliferation induced by fine
particulate matter via suppressing of the p38 MAPK signaling
pathway. BMC Complement Altern Med. 18:1462018. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kyriakis JM and Avruch J: Mammalian MAPK
signal transduction pathways activated by stress and inflammation:
A 10-year update. Physiol Rev. 92:689–737. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Cuenda A and Rousseau S: p38 MAP-kinases
pathway regulation, function and role in human diseases. Biochim
Biophys Acta. 1773:1358–1375. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Karin M and Gallagher E: From JNK to pay
dirt: Jun kinases, their biochemistry, physiology and clinical
importance. IUBMB Life. 57:283–295. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Jacob T, Ascher E, Alapat D, Olevskaia Y
and Hingorani A: Activation of p38MAPK signaling cascade in a VSMC
injury model: Role of p38MAPK inhibitors in limiting VSMC
proliferation. Eur J Vasc Endovasc Surg. 29:470–478. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Awasthi V and King RJ: PKC, p42/p44 MAPK,
and p38 MAPK are required for HGF-induced proliferation of H441
cells. Am J Physiol Lung Cell Mol Physiol. 279:L942–L949. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Chattopadhyay N, Tfelt-Hansen J and Brown
EM: PKC, p42/44 MAPK and p38 MAPK regulate hepatocyte growth factor
secretion from human astrocytoma cells. Brain Res Mol Brain Res.
102:73–82. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Yao J, Ke J, Zhou Z, Tan G, Yin Y, Liu M,
Chen J and Wu W: Combination of HGF and IGF-1 promotes connexin 43
expression and improves ventricular arrhythmia after myocardial
infarction through activating the MAPK/ERK and MAPK/p38 signaling
pathways in a rat model. Cardiovasc Diagn Ther. 9:346–354. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Christensen JG, Schreck R, Burrows J,
Kuruganti P, Chan E, Le P, Chen J, Wang X, Ruslim L, Blake R, et
al: A selective small molecule inhibitor of c-Met kinase inhibits
c-Met-dependent phenotypes in vitro and exhibits cytoreductive
antitumor activity in vivo. Cancer Res. 63:7345–7355.
2003.PubMed/NCBI
|
|
28
|
Yun MR, Lee JY, Park HS, Heo HJ, Park JY,
Bae SS, Hong KW, Sung SM and Kim CD: Oleic acid enhances vascular
smooth muscle cell proliferation via phosphatidylinositol
3-kinase/Akt signaling pathway. Pharmacol Res. 54:97–102. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Qi W, Ding D and Salvi RJ: Cytotoxic
effects of dimethyl sulphoxide (DMSO) on cochlear organotypic
cultures. Hear Res. 236:52–60. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Xiang Q, Zhen Z, Deng DY, Wang J and Chen
Y, Li J, Zhang Y, Wang F, Chen N, Chen H and Chen Y: Tivantinib
induces G2/M arrest and apoptosis by disrupting tubulin
polymerization in hepatocellular carcinoma. J Exp Clin Cancer Res.
34:1182015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Sreekanth GP, Chuncharunee A,
Sirimontaporn A, Panaampon J, Noisakran S, Yenchitsomanus PT and
Limjindaporn T: SB203580 modulates p38 MAPK signaling and dengue
virus-induced liver injury by reducing MAPKAPK2, HSP27, and ATF2
phosphorylation. PLoS One. 11:e01494862016. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
He T, Liu S, Chen S, Ye J, Wu X, Bian Z
and Chen X: The p38 MAPK inhibitor SB203580 abrogates tumor
necrosis factor-induced proliferative expansion of mouse
CD4+Foxp3+ regulatory T cells. Front Immunol.
9:15562018. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Liu T, Li Q, Sun Q, Zhang Y, Yang H, Wang
R, Chen L and Wang W: MET inhibitor PHA-665752 suppresses the
hepatocyte growth factor-induced cell proliferation and
radioresistance in nasopharyngeal carcinoma cells. Biochem Biophys
Res Commun. 449:49–54. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Xiang QF, Zhan MX, Li Y, Liang H, Hu C,
Huang YM, Xiao J, He X, Xin YJ, Chen MS and Lu LG: Activation of
MET promotes resistance to sorafenib in hepatocellular carcinoma
cells via the AKT/ERK1/2-EGR1 pathway. Artif Cells Nanomed
Biotechnol. 47:83–89. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Bielinski SJ, Berardi C, Decker PA, Larson
NB, Bell EJ, Pankow JS, Sale MM, Tang W, Hanson NQ, Wassel CL, et
al: Hepatocyte growth factor demonstrates racial heterogeneity as a
biomarker for coronary heart disease. Heart. 103:1185–1193. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Bell EJ, Larson NB, Decker PA, Pankow JS,
Tsai MY, Hanson NQ, Wassel CL, Longstreth WT Jr and Bielinski SJ:
Hepatocyte growth factor is positively associated with risk of
stroke: The MESA (multi-ethnic study of atherosclerosis). Stroke.
47:2689–2694. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Gallo S, Sala V, Gatti S and Crepaldi T:
Cellular and molecular mechanisms of HGF/Met in the cardiovascular
system. Clin Sci (Lond). 129:1173–1193. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Bell EJ, Decker PA, Tsai MY, Pankow JS,
Hanson NQ, Wassel CL, Larson NB, Cohoon KP, Budoff MJ, Polak JF, et
al: Hepatocyte growth factor is associated with progression of
atherosclerosis: The multi-ethnic study of atherosclerosis (MESA).
Atherosclerosis. 272:162–167. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Nakamura T and Mizuno S: The discovery of
hepatocyte growth factor (HGF) and its significance for cell
biology, life sciences and clinical medicine. Proc Jpn Acad Ser B
Phys Biol Sci. 86:588–610. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Smolen GA, Sordella R, Muir B, Mohapatra
G, Barmettler A, Archibald H, Kim WJ, Okimoto RA, Bell DW, Sgroi
DC, et al: Amplification of MET may identify a subset of cancers
with extreme sensitivity to the selective tyrosine kinase inhibitor
PHA-665752. Proc Natl Acad Sci USA. 103:2316–2321. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Mukohara T, Civiello G, Davis IJ, Taffaro
ML, Christensen J, Fisher DE, Johnson BE and Jänne PA: Inhibition
of the met receptor in mesothelioma. Clin Cancer Res. 11:8122–8130.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Lu G, Meier KE, Jaffa AA, Rosenzweig SA
and Egan BM: Oleic acid and angiotensin II induce a synergistic
mitogenic response in vascular smooth muscle cells. Hypertension.
31:978–985. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Lu G, Greene EL, Nagai T and Egan BM:
Reactive oxygen species are critical in the oleic acid-mediated
mitogenic signaling pathway in vascular smooth muscle cells.
Hypertension. 32:1003–1010. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Ahn HJ, Park J, Song JS, Ju MK, Kim MS, Ha
H, Song KH and Kim YS: Mycophenolic acid inhibits oleic
acid-induced vascular smooth muscle cell activation by inhibiting
cellular reactive oxygen species. Transplantation. 84:634–638.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Ou TT, Lin MC, Wu CH, Lin WL and Wang CJ:
Gallic acid attenuates oleic acid-induced proliferation of vascular
smooth muscle cell through regulation of AMPK-eNOS-FAS signaling.
Curr Med Chem. 20:3944–3953. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Lin MC, Ou TT, Chang CH, Chan KC and Wang
CJ: Protocatechuic acid inhibits oleic acid-induced vascular smooth
muscle cell proliferation through activation of AMP-activated
protein kinase and cell cycle arrest in G0/G1 phase. J Agric Food
Chem. 63:235–241. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Zhu Y, Schwarz S, Ahlemeyer B, Grzeschik
S, Klumpp S and Krieglstein J: Oleic acid causes apoptosis and
dephosphorylates Bad. Neurochem Int. 46:127–135. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Carrillo C, Cavia Mdel M and Alonso-Torre
SR: Antitumor effect of oleic acid; mechanisms of action: A review.
Nutr Hosp. 27:1860–1865. 2012.PubMed/NCBI
|
|
50
|
Cheng CI, Lee YH, Chen PH, Lin YC, Chou MH
and Kao YH: Free fatty acids induce autophagy and LOX-1
upregulation in cultured aortic vascular smooth muscle cells. J
Cell Biochem. 118:1249–1261. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Artwohl M, Lindenmair A, Roden M,
Waldhäusl WK, Freudenthaler A, Klosner G, Ilhan A, Luger A and
Baumgartner-Parzer SM: Fatty acids induce apoptosis in human smooth
muscle cells depending on chain length, saturation, and duration of
exposure. Atherosclerosis. 202:351–362. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Zhang C, Wang W, Lin J, Xiao J and Tian Y:
lncRNA CCAT1 promotes bladder cancer cell proliferation, migration
and invasion. Int Braz J Urol. 45:549–559. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Belal SA, Sivakumar AS, Kang DR, Cho S,
Choe HS and Shim KS: Modulatory effect of linoleic and oleic acid
on cell proliferation and lipid metabolism gene expressions in
primary bovine satellite cells. Anim Cells Syst (Seoul).
22:324–333. 2018. View Article : Google Scholar : PubMed/NCBI
|