Introduction
Intracerebral hemorrhage (ICH) is a stroke subtype
with a high mortality rate, which often results in neurological
impairments that can cause long-term disability (1). In the USA, the mortality rate is
35–52% within 30 days of ICH, with half of deaths occurring in the
first 2 days, and only 20% expectation of functional independence
at 6 months among an estimated 67,000 patients (2,3).
Non-traumatic ICH is due to the rupture of blood vessels in the
brain and subsequent tissue compression (4). The initial bleed causes hematoma mass
that results in physical disruption of brain tissue; the
pathophysiological processes of secondary injury following ICH are
characterized by ischemia, edema, apoptosis and necrosis
surrounding the hematoma (5,6).
Autophagy is a process by which cells recycle
cellular components, and degrade excess or defective organelles,
thereby participating in cytoplasmic component turnover and protein
quality control, which can prompt responses to nutrient depletion
and pathological stress (7,8). Autophagy is characterized by
degradation of macromolecular intracellular material by
double-membrane autophagic vesicles (known as autophagosomes) and
also serves a constitutive role in the elimination of mitochondria
(9). This specific regulated
mechanism of selective mitochondria degradation via autophagy is
defined as mitophagy (10).
Mitophagy is involved in mitochondrial maintenance, cellular
differentiation and cell survival in mammalian systems (11). An association between mitophagy and
mitochondrial function maintenance may underly the pathology of
certain diseases, aging, neurodegeneration and stroke (12). PTEN-induced putative kinase 1
(PINK1), Parkin, BCL/adenovirus E1B 19 kDa-interacting protein 3
(BNIP3), NIX/BNIP3L (BNIP3-like) and FUN14 domain containing 1 have
been identified as mitophagy receptors in mammalian cells (13). Previous studies (14,15) of
stroke have demonstrated that enhanced mitophagy serves a key role
in ameliorating secondary cell death and associated complications.
A study involving traumatic brain injury (TBI) showed that
enhancing mitophagy following TBI by melatonin treatment
ameliorates neuronal death and behavioral deficit by negatively
regulating inflammation activation and IL-1β secretion via
autophagy of damaged mitochondria through the mTOR pathway
(14). One study established that
activated mitophagy may inhibit inflammasome-mediated cell death
and decrease reactive oxygen species generation following induction
of subarachnoid hemorrhage (16).
Furthermore, mitophagy may contribute to the pro-survival apoptosis
pathway by decreasing cytochrome c release capacity
(17). However, the question of
whether mitophagy contributes to cell death in stroke remains
controversial.
Electroacupuncture (EA) is a modern technique based
on a combination of the meridian theory of Traditional Chinese
Medicine and transcutaneous electrical stimulation therapy
(18). Recommended as a
complementary treatment for stroke, EA has shown promise in stroke
rehabilitation (19). The benefits
of EA are attributed to its readily quantifiable stimulation
parameters of frequency, intensity and duration (20,21).
The technique has been applied to ICH models in a previous study
(22) and has been reported to
effectively decrease blood-brain barrier permeability and improve
neurological recovery (23).
Researchers have focused on the regulation of autophagy by EA
treatment in stroke, showing that EA may affect ultrastructure and
autophagy-associated factors (24,25).
For example, EA treatment was demonstrated to exert a
neuroprotective effect following ischemic stroke by inhibiting
autophagy via restricting autophagosome formation and mediating the
mTORC1-unc-51-like autophagy activating kinase 1 complex-Beclin1
pathway (26). Other research has
shown that EA (at GV20) pretreatment may exert a protective effect
on cerebral ischemic and reperfusion injury by upregulating
autophagy via enhanced expression of LC3 and downregulated
phosphorylated-mTOR (27). The
present study focused on acupoint GV20-GB7 (Baihui-Qubin). Our
previous study (28) investigated
the effectiveness of GV20-GB7 using an ICH rat model and found that
acupuncture at GV20-GB7 may suppress neuronal differentiation of
neural stem cells by inhibiting the Notch-Hes signaling pathway and
maintaining neural stem cell proliferation. Furthermore,
acupuncture at GV20-GB7 has been reported to improve the recovery
of neurological function by decreasing the inflammatory response
via inhibition of TNF-α/NFκB expression levels (29). The present study aimed to elucidate
the mechanism by which EA at GV20-GB7 regulates mitophagy in ICH
and whether it serves a neuroprotective role. It was hypothesized
that EA at GV20-GB7 could inhibit apoptotic cell death following
ICH by regulating the mitophagy process, thereby alleviating
symptoms of neurological impairment induced by ICH.
Materials and methods
Experimental animals
Male Sprague-Dawley rats (weight, 300–350 g) were
obtained from the Experimental Animal Center of Heilongjiang
University of Chinese Medicine [license no. SYXK (Hei) 2017061001].
Animals were housed in controlled conditions under a 12-h
light/dark cycle, at 22±2°C, 60–70% humidity and noise <60 dB,
with ad libitum access to food and water. All experiments
were approved by the Animal Care and Use Committee of Heilongjiang
University of Chinese Medicine and performed in accordance with the
National Institutes of Health Guide for the Care and Use of
Laboratory Animals (30).
ICH model and groups
Rats were anesthetized with pentobarbital [60 mg/kg;
intraperitoneal (i.p.) injection], then shaved at the scalp.
Aseptic techniques were used for surgical procedures, as described
in our previous study (29).
Briefly, a ~1-cm midline incision was made, exposing a
perpendicular intersection point of the coronal and sagittal
suture. A 0.1-mm hole was drilled in the skull (0.2 mm anterior and
3.5 mm laterally to the right of bregma). Subsequently, 50 µl
autologous whole blood from the tail artery was injected into the
basal ganglia via Hamilton syringe at 5 mm depth below the skull
surface and the needle was left in place for 5 min. The
sham-operated group received intracerebral needle insertion only,
without blood injection. The burr hole was sealed with dental zinc
phosphate cement and the skin was sutured. Following surgery, each
animal was placed in a clean cage with free access to food and
water. The success rate of the autologous blood injection
operation, which indicated ICH was successfully induced, was ~95%.
Rats in the control group of each time point did not undergo
surgery.
Since the morphology of mitophagy has been observed
in previous studies (31–34), the experiments were repeated at
least three times to confirm mitophagy as a phenomenon in ICH. A
total of 5 rats/group were used for transmission electron
microscopy (TEM), as previously described (35,36). A
total of 345 rats were used in the study. In order to determine the
time-dependent effect of ICH on mitophagy, 120 rats were randomly
and evenly divided into a control and ICH model group (n=60/group).
Tests were performed 6 and 24 h, and 3 and 7 days after ICH. In
order to evaluate the effect of EA on mitophagy following ICH, 225
rats were randomly assigned to five treatment groups, each of which
comprised four experimental subgroups (6 and 24 h, and 3 and 7 days
after ICH; Fig. S1). The treatment
groups were as follows: Sham-operated, ICH model, ICH +
3-methyladenine (3-MA), ICH + EA (EA group), ICH + 3-MA + EA (3-MA
+ EA group). The experimental process is as illustrated in Fig. S1.
Administration of 3-MA
3-MA is a PI3K inhibitor that inhibits autophagy at
the initiation stage and is used as an inhibitor of mitophagy
(32,37). In the present study, 3-MA (400
nm/µl; Selleck Chemicals) was dissolved in 0.9% saline by heating
to 60–70°C until completely dissolved and cooling to room
temperature immediately before treatment. Rats in the 3-MA and 3-MA
+ EA groups were anesthetized as aforementioned and placed on
stereotaxic apparatus, then 10 µl 3-MA was injected into the
lateral ventricles (1.5 mm anterior and 0.8 mm laterally to the
right of bregma) 15 min before whole blood-induced ICH surgery.
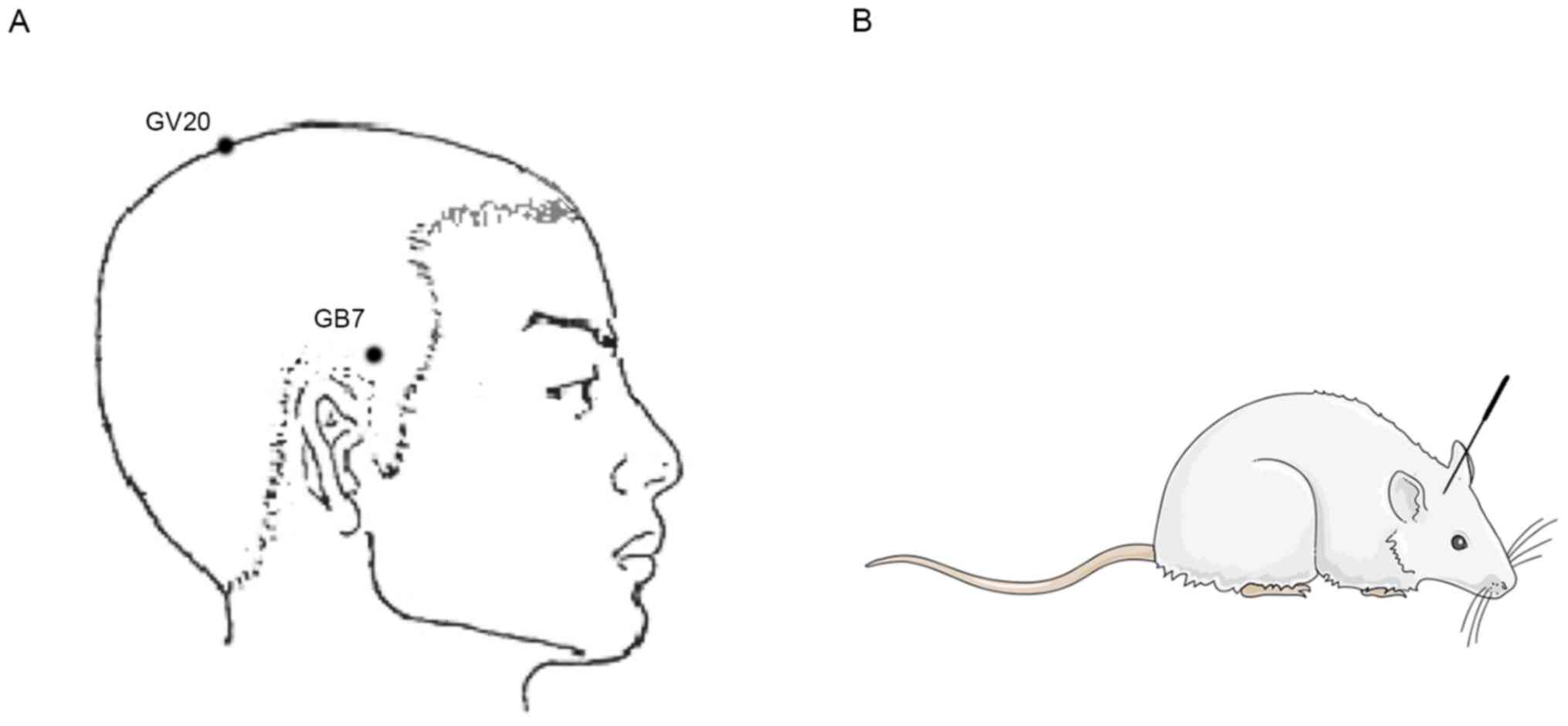
EA treatment
Rats were placed into a rat fixator (Beijing Ji
Nuotai Science and Technology Development Co., Ltd.). Treatment was
performed 2 h after the ICH model was established, and once every
24 h thereafter. Baihui (GV20) was located according to the world
acupuncture and moxibustion congress (38,39),
and acupoint Qubin (GB7) was determined using a trans-positional
method (40) (Fig. 1). Briefly, GB7 was located on the
connecting line of the orbital margin and porus acusticus externus.
A resistance detector was used to measure the electrical
characterization along the proposed line (resistance of acupoints
is lower compared with non-acupoints) (41). The posterior 2/3 point of the line
connecting the orbital margin and porus acusticus externus was
confirmed as the GB7. Sterilized acupuncture needles (0.25×15.00 mm
Hwato disposable acupuncture needle; Suzhou Medical Products
Factory Co., Ltd.) were inserted at Baihui (GV20) with the needle
tip penetrating ~8 mm ipsilateral (focal side; contralateral side
of the paralyzed limb) acupoint Qubin (GB7) (Fig. 1). The needle was connected to an EA
instrument (model G6805) and EA was performed with continuous-wave
at 4 Hz stimulation for 15 min/day at an intensity of 1 mA. Rats of
all groups were fixed to the fixator in the same way.
Assessment of neurobehavioral
deficits
Modified neurological severity score (mNSS) was used
to evaluate neurological deficits. mNSS tests were performed as
described by Chen et al (42) immediately following recovery from
anesthesia, and at 6 and 24 h and 3 and 7 days following EA
treatment. Briefly, the evaluation criteria comprises 18 points as
follows: Motor tests, including raising rats by the tail and floor
walk tests, 6 points; sensory tests, including placing and
proprioceptive tests, 2 points; beam balance test, 6 points
(normal=0; maximum=6); absent reflex and abnormal movement tests,
including pinna, corneal and startle reflexes, and seizures, 4
points. The test data were assessed by an experimenter who was
blinded to the experimental design and treatment procedure. The
total mNSS was classified as follows: 13–18, serious injury; 7–12,
moderate injury; 1–6, slight injury. Animals scoring 7–13 points
following the operation were considered to indicate a successful
ICH model and were selected for neurobehavioral deficit assessment
after treatment. These animals exhibited symptoms such as tipping
over to the paralyzed side, hind leg buckling, falling from the
balance beam and disappearance of corneal reflex.
TEM
Rats were euthanized with pentobarbital (120 mg/kg,
i.p.) after 6 and 24 h, 3 days and 7 days respectively, then
perfused with pre-cooled (4°C) physiological saline, followed by
PBS containing 4% paraformaldehyde. The brain was removed and cut
into 1 cm3 cubes, which were fixed with 2.5%
glutaraldehyde for 4 h, washed four times in 0.1 M PBS (pH 7.4)
overnight at 40°C, then postfixed in 1% osmium tetroxide for 2 h at
30°C and washed with PBS again. Following dehydration by graded
alcohol and dry acetone, samples were embedded in Epon/Araldite
mixture (Merck KGaA); polymerization was performed at 70°C for 2
days. Ultrathin sections (70 nm) were cut using a Leica Ultracut
microtome (Leica Microsystems, Inc.), and stained with 3% lead
citrate and uranyl acetate staining solution for 10 and 30 min
respectively, then drying at room temperature. The samples were
observed under a Philips 201 electron microscope (Philips Medical
Systems B.V.) and images were captured.
Western blot analysis
Following euthanasia with pentobarbital (120 mg/kg,
i.p.), rats were decapitated and the brain was removed. The focal
area was dissected and placed on ice. Subsequently, samples were
homogenized in lysis buffer (cat. no. M334-100ML; Amresco, LLC),
followed by centrifugation at 15,100 × g for 10 min at 4°C. The
protein concentration was determined using a BCA protein assay kit
(cat. no. WLA004; Wanleibio, Co., Ltd.). Equal amounts of denatured
protein (50 µg) were separated by SDS-PAGE on 10% gels, then
transferred onto PVDF membranes (EMD Millipore). The membranes were
blocked for 2 h with 5% non-fat milk in 0.1% TBS-Tween-20 (cat. no.
WLA025. Wanleibio Co., Ltd.) at room temperature and then incubated
overnight at 4°C with the following primary antibodies: BNIP3
(1:400; cat. no. bs-4239R; BIOSS), PINK1 (1:1,000; cat. no.
23274-1-AP; ProteinTech Group, Inc.), Parkin (1:400; cat. no.
WL02512; Wanleibio Co., Ltd.), translocase of outer mitochondrial
membrane 20 homolog (1:500; TOMM20; cat. no. WL03626; Wanleibio
Co., Ltd.), cytochrome c oxidase IV (1:400; COX IV; cat. no.
WL02203; Wanleibio Co., Ltd.), Bcl-2 (1:500; cat. no. 12789-1-AP;
ProteinTech Group, Inc.), BAX (1:400; cat. no. WL01637; Wanleibio
Co., Ltd.) cleaved caspase-3 (1:300; cat. no. 19677-1-AP;
ProteinTech Group, Inc.) and p53 (1:300; cat. no. 10442-1-AP;
ProteinTech Group, Inc.). The membranes were washed in TBST and
incubated with horseradish peroxidase-conjugated (HRP) secondary
goat anti-rabbit IgG (1:5,000; cat. no. 7074; Cell Signaling
Technology, Inc.) for 1 h at 37°C, then the PVDF membranes were
added to TBST bufferat room temperature, and rotated for 15 min.
Immunoblotting was detected by electrochemiluminescence (cat. no.
WLA003, Wanleibio Co., Ltd.), and densitometry was analyzed using a
Bio-image Analysis system (Bio-Rad Laboratories, Inc.). β-actin
(1:1,000; cat. no. WL01845; Wanleibio Co., Ltd.) was used as a
loading control. Results were calculated as the ratio of the
fluorescence intensity of target protein/β-actin.
Immunohistochemistry
Rats were euthanized with pentobarbital (120 mg/kg,
i.p.), perfused as aforementioned and the brain was removed.
Paraffin-embedded brain tissue was cut into 4-µm sections. A
two-step method of immunohistochemistry was performed according to
the instructions of kit (cat. no. PV-6002; ZSGB-BIO, Inc). Briefly,
sections were deparaffinized and rehydrated, then incubated in 3%
H2O2 for 10 min, followed by immersion in 10
mmol/l citrate buffer (pH 6.0) and heating to boil for 2 min then
cooling to room temperature. The sections were then incubated with
primary antibodies against p53 (1:300; cat. no. 10442-1-AP;
ProteinTech Group, Inc.) overnight at 4°C. After washing in PBS,
sections were incubated with anti-rabbit biotinylated secondary
antibody (1:500; cat. no. bs-0295G; BIOSS) at 37°C for 30 min, then
stained with 4% diaminobenzidine and 5% hematoxylin for 30 min at
room temperature. Sections were visualized using a Motic3000 light
microscope (Motic Incorporation, Ltd.) at ×400 magnification. Cells
were counted in five randomly selected fields of view in each
group. Nuclei were stained blue and p53-positive cells were
visualized as yellow-brown granules.
Immunofluorescence staining
After rats were euthanized with pentobarbital (120
mg/kg, i.p.) and perfused as aforementioned, brains were collected
and immersed in gradient alcohol, followed by immersion in
dimethylbenzene. Paraffin-embedded brain tissue was then cut into
5-µm sections. Following deparaffinization, sections were
permeabilized with gradient alcohol. The permeabilization solution
was then removed and samples were washed three times in PBS for 5
min each. Subsequently, the sections were blocked with 10% goat
serum for 15 min at room temperature (cat. no. SL083; Beijing
Solarbio Science & Technology Co., Ltd.) and slides were
incubated overnight with primary antibody anti-LC3II (1:100; cat.
no. bs-2912R; BIOSS) at 4°C. The sections were washed with PBS and
incubated with fluorescein isothiocyanate-labeled goat anti-rabbit
antibody (1:200; cat. no. A0516; Beyotime Institute of
Biotechnology) for 1 h at room temperature in the dark. In order to
detect the morphology of the nucleus, the nuclei were stained with
DAPI (0.1 µg/ml) for 15 min at room temperature and detected by
blue fluorescence. After rinsing with PBS three times (5 min each),
the sections were mounted on slides. Confocal images were captured
at ×400 magnification using a fluorescence microscope (DP73;
Olympus Corporation).
TUNEL
Rats were sacrificed with pentobarbital (120 mg/kg,
i.p.) and perfused as aforementioned, after which, the brains were
cut into sections (10 µm) and TUNEL assay was performed using a
TUNELAP kit (OriGene Technologies, Inc.) according to the
manufacturer's instructions. Briefly, the sections were washed with
PBS and reacted with TdT enzyme/buffer at 37°C for 1 h, followed by
enzyme-labeled anti-dUTP antibody reaction at room temperature for
30 min. Then, the sections were rinsed with 3,3-diaminobenzidine
and dehydrated with dimethylbenzene. After being air-dried, the
sections were observed and images were captured at ×400
magnification using a Motic3000 light microscope; six fields of
view were quantitatively assessed from each section (n=6) using
Image-pro Plus 6.0 Pathological Image Analysis system (Media
Cybernetic, Inc.). Brown staining of nuclei was considered to
indicate apoptotic cells.
Statistical analysis
Data are presented as the mean ± SEM (n=5). P-values
for western blotting, immunohistochemistry and TUNEL assay were
determined by one-way ANOVA followed by Tukey's post hoc test. Data
are presented as median (IQR). P-values for mNSS were determined by
Kruskal-Wallis test, followed by post hoc Dunn's multiple
comparisons test. GraphPad Prism 8.0 software (GraphPad Software,
Inc.) was used for all histograms and statistical analysis.
P<0.05 was considered to indicate a statistically significant
difference.
Results
Mitophagy is activated following
ICH
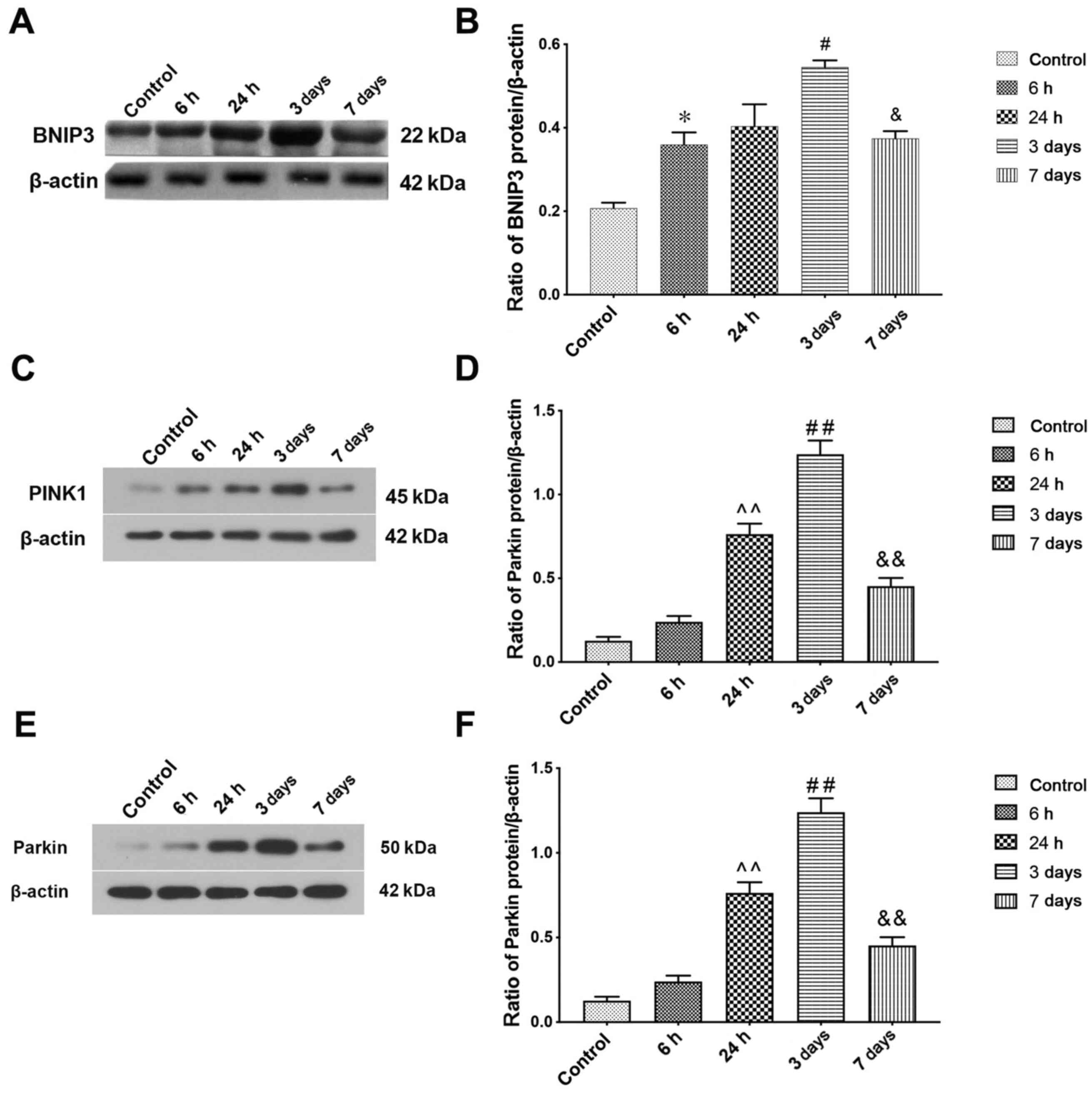
The induction of mitophagy following ICH was
evaluated by detecting the protein expression levels of BNIP3, a
protein that carries only one Bcl-2-homology-3 domain of the Bcl-2
family and mitochondrial autophagy receptor (43). Immunoblotting showed ICH caused a
time-dependent increase in BNIP3 (Fig.
2A). Semi-quantitative analysis of protein band density
indicated that BNIP3 protein expression levels increased at 6 h
after ICH compared with the sham group and peaked at 3 days. The
expression levels on day 7 after ICH induction were still higher
compared with the control group (Fig.
2B). Mitophagy protein markers PINK1 and Parkin were also
detected by western blotting to determine mitophagy activity
following ICH; the results were consistent with those of BNIP3
(Fig. 2C-F).
Autophagy following ICH was identified by
immunofluorescent techniques. LC3-II-positive cells were notably
increased at all time points in the ICH group compared with the
control group, indicating autophagy was activated (Fig. 3). The results showed that the
lighteness of LC3-II positive neurons (Fig. 3) at day 3 following ICH was
significantly higher than that in other groups. The observed
LC3-II-positive neurons were notably decreased at 7 days following
ICH. (Fig. 3A).
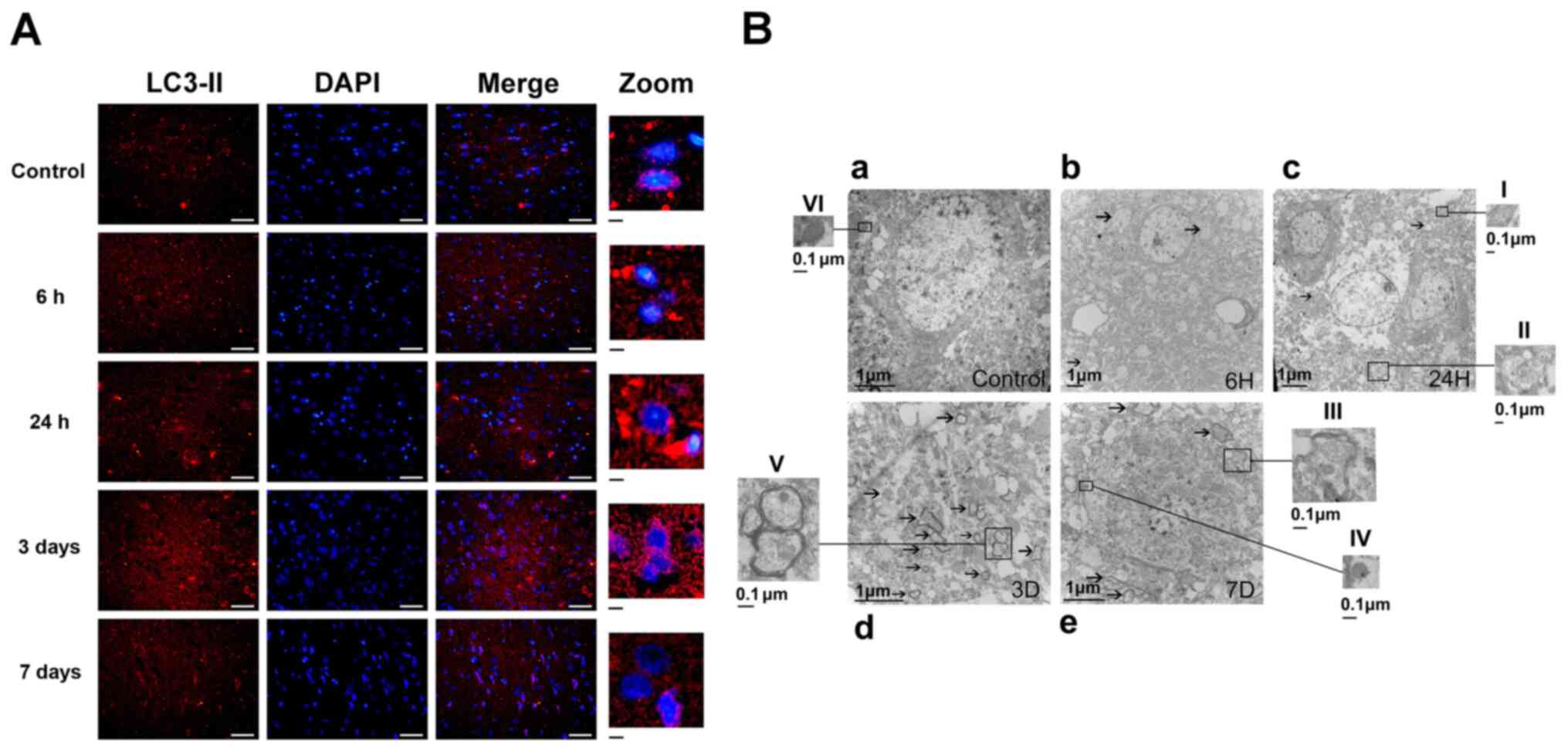 | Figure 3.Autophagy is activated following ICH.
(A) Representative immunofluorescence images of LC3-II (red)
surrounding the basal ganglia at different time points following
ICH. More LC3-II puncta were observed on day 3 compared with other
time points. Scale bar, 50 µm. Magnified scale bar, 5 µm. (B)
Transmission electron microscopy analysis of ultrastructural
changes at different time points following ICH. (a) Healthy
mitochondria with clear crista structure were observed in the sham
operation group. Magnification, ×10,000. (b-e) Morphological
changes in the basal ganglia of model groups at different time
points. Black arrows indicate double-membrane autophagic vacuoles,
including partially degraded mitochondria or other autophagic
content. Magnification, ×4,000, ×6,000, ×12,000 and ×12,000,
respectively. I, Large mitochondrion with disordered cristae; II,
autolysosome containing residues of mitochondria and lytic
organelles; III and V, autophagosomes containing mitochondria,
demonstrating ongoing mitophagy; IV, lysosome; VI, healthy
mitochondrion with normal size and clear crista structure. |
The ultrastructural phenotypes of autophagosomes,
autolysosomes and mitochondria were investigated by TEM (Fig. 3B). A large number of healthy
mitochondria with clear crista structures were observed in the
Control group (Fig. 3B-a).
Double-membrane autophagosomes containing mitochondria, which are a
typical morphological characteristic of mitophagy, were observed
following ICH (Fig. 3Bb-e). Greater
numbers of these autophagosomes and autolysosomes were observed at
day 3 following ICH, indicating increased mitophagy (Fig. 3B-d). Damaged and deformed
mitochondria with vague cristae and lost matrix granules were also
observed following ICH. These results indicated that mitophagy was
activated following ICH; this effect was most notable at 3 days
after ICH (Fig. 3B-b-e).
EA treatment upregulates mitophagy
following ICH
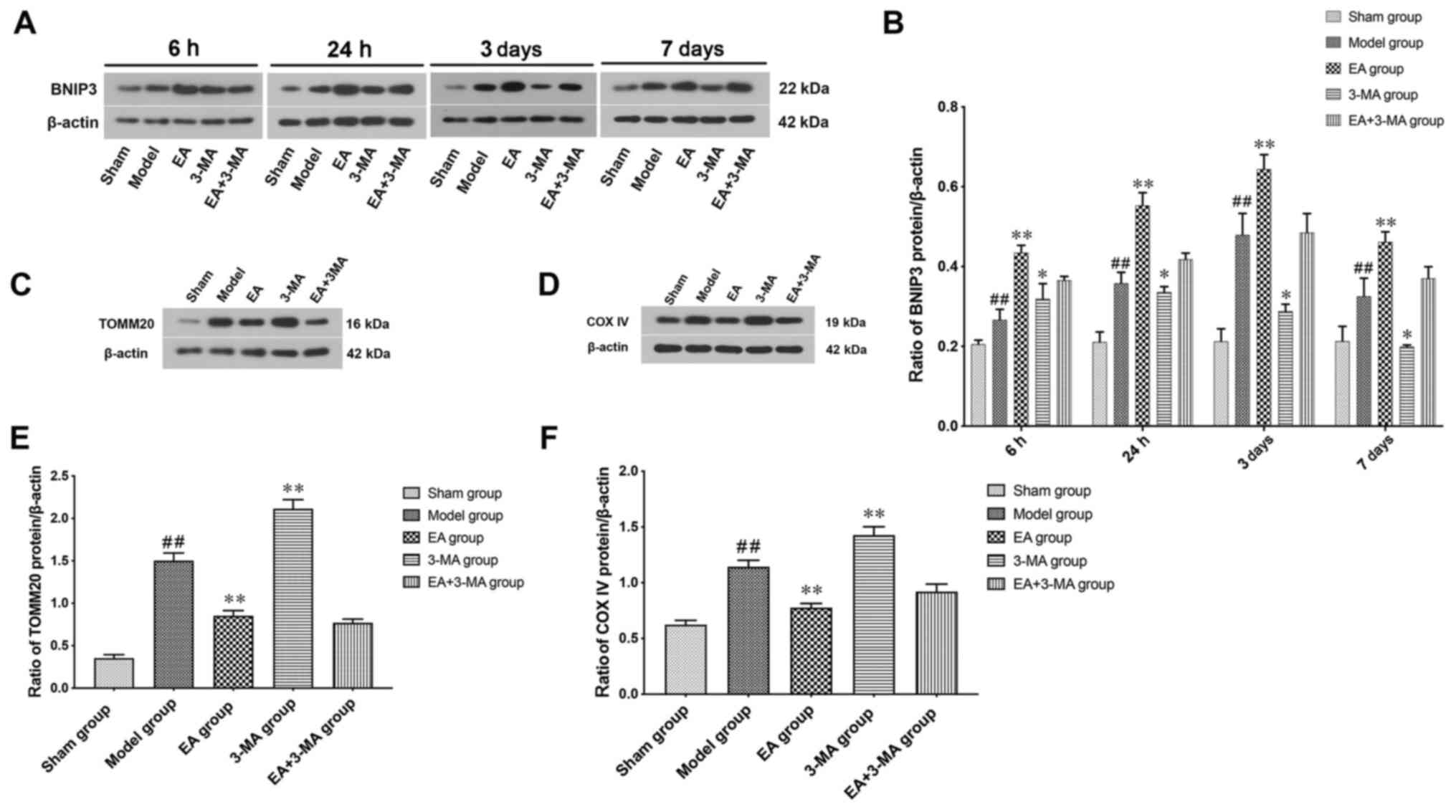
The effect of EA on mitophagy following ICH was
assessed by measuring BNIP3 expression levels (Fig. 4A and B). The expression levels of
BNIP3 were increased following ICH and significantly increased in
the EA group at 7 days compared with the model group. Compared with
the model group, the 3-MA group exhibited lower expression levels
of BNIP3 in the focal area at 7 days. Notably, the effects of EA
(GV20-GB7) were blocked by 3-MA.
The relative number of mitochondria at day 3
following ICH was assessed by levels of mitochondrial protein
markers TOMM20 and COX IV (Fig. 4C and
D). Expression levels of both TOMM20 and COX IV increased
following ICH; this was alleviated by EA treatment. Treatment with
3-MA increased the expression levels of TOMM20 and COX IV compared
with the Model group (Fig. 4E and
F), indicating inhibition of mitophagy. These results suggested
that EA treatment may enhance autophagy-associated mitochondrial
elimination following ICH.
TEM was performed to investigate the effect of EA on
stimulating focal mitophagy. Since mitophagy was most activated on
day 3 after ICH, this time point was selected for the TEM
experiments. There was no notable observation of autophagic
vacuoles in the sham group but autophagosomes and autolysosomes
containing mitochondria were observed in the model group. Swollen
and dilated mitochondria and rough endoplasmic reticulum were also
observed, compared with the sham group (Fig. 5A and B). Larger numbers of
autophagosomes with mitochondria and autolysosomes with partially
degraded mitochondria, which are characteristic of mitophagy, were
observed in the electron micrographs from the EA group, but mild
neuron injury was still visible (Fig.
5C). Morphological changes were found in the 3-MA group,
including decreased numbers of autophagosomes, shrunken nuclei and
disrupted cell membranes, which indicated early-stage apoptosis
(Fig. 5D). In addition, treatment
with 3-MA neutralized the effect of EA on mitophagy enhancement
(Fig. 5E). These results indicated
that inhibiting mitophagy may lead to neuron apoptosis; enhancing
mitophagy via EA decreased accumulation of injured mitochondria,
which may delay neuron death. Collectively, these results
demonstrated that mitophagy was augmented by EA (GV20-GB7).
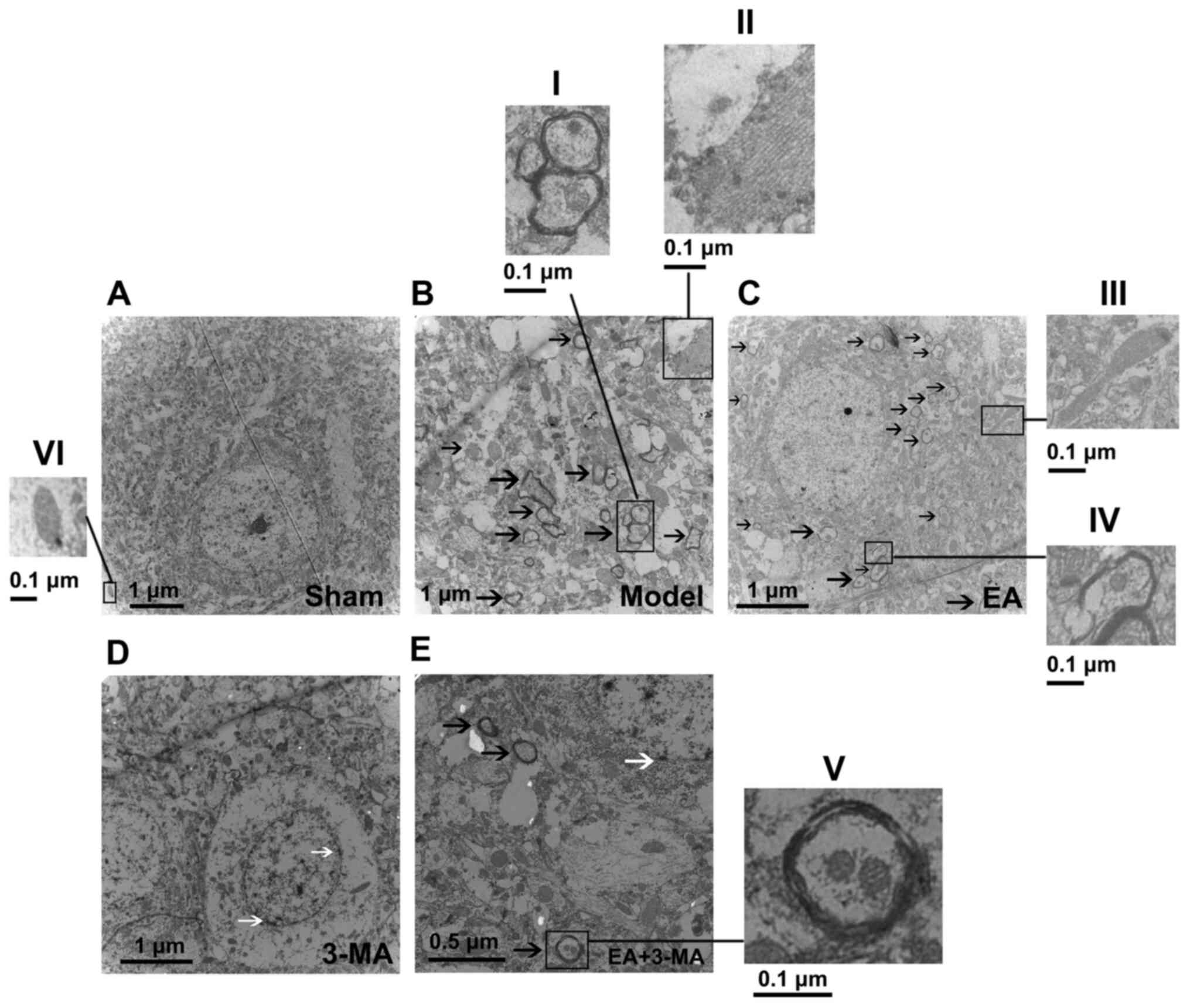 | Figure 5.Transmission electron microscopy
images of focal bleeding areas of rats in each group. Images were
captured at (A) ×5,000, (B) ×12,000, (C and D) ×7,000 and (E)
×15,000 magnification. Black arrows indicate double-membrane
autophagic vacuoles, which include partially degraded mitochondria
or other autophagic content. White arrows indicate disrupted cell
membranes. I and V, Typical mitophagic mitochondrion encapsulated
by double-membrane autophagosomes; II, rough endoplasmic reticulum;
III, deformed mitochondrion; IV, autolysosome; VI, normal
mitochondrion with tight, orderly cristae. |
EA decreases mitochondrial apoptosis
following ICH
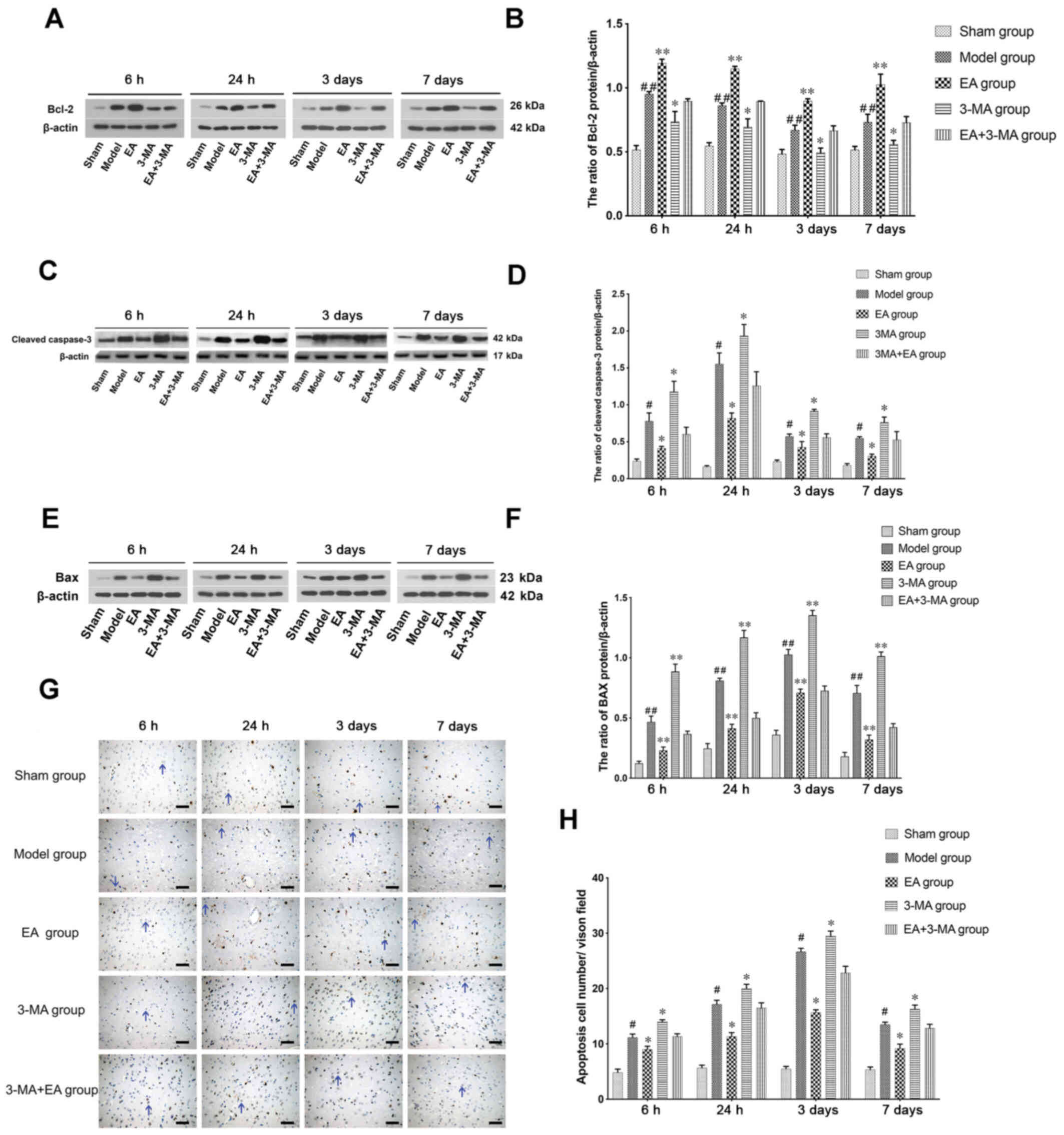
The anti-apoptotic protein Bcl-2 is activated in
mitochondrial apoptosis (44). In
the present study, Bcl-2 was upregulated at 6 h following ICH in
the model group compared with the sham group (Fig. 6A and B) and declined after 24 h to
reach a minimum level 3 days after ICH. EA treatment significantly
increased Bcl-2 expression relative to the model group.
Cleaved caspase-3, an executioner protease of
apoptosis, was also detected in the present study (Fig. 6C and D). Cleaved caspase-3
expression was increased following ICH, whereas EA (GV20-GB7)
treatment decreased cleaved caspase-3 expression levels at 6 and 24
h.
Bax was also detected by western blot analysis
(Fig. 6E and F). The expression of
Bax was upregulated after ICH was initiated and enhanced by EA
treatment compared to the Model group. Additional 3-MA treatment
reversed the effects of EA; in addition, 3-MA decreased Bcl-2
expression levels, and increased those of Bax and cleaved caspase-3
compared with the model group. Notably, cleaved caspase-3 peaked at
24 h after ICH, which may be due to the apoptotic function of
cleaved caspase-3 in the early stages after stroke (45,46).
TUNEL assay demonstrated that the number of apoptotic cells
increased in ICH rats. Apoptosis was reduced by EA treatment,
whereas increased by administration of 3-MA, of which in accordance
with the western blotting results (Fig.
6G and H).
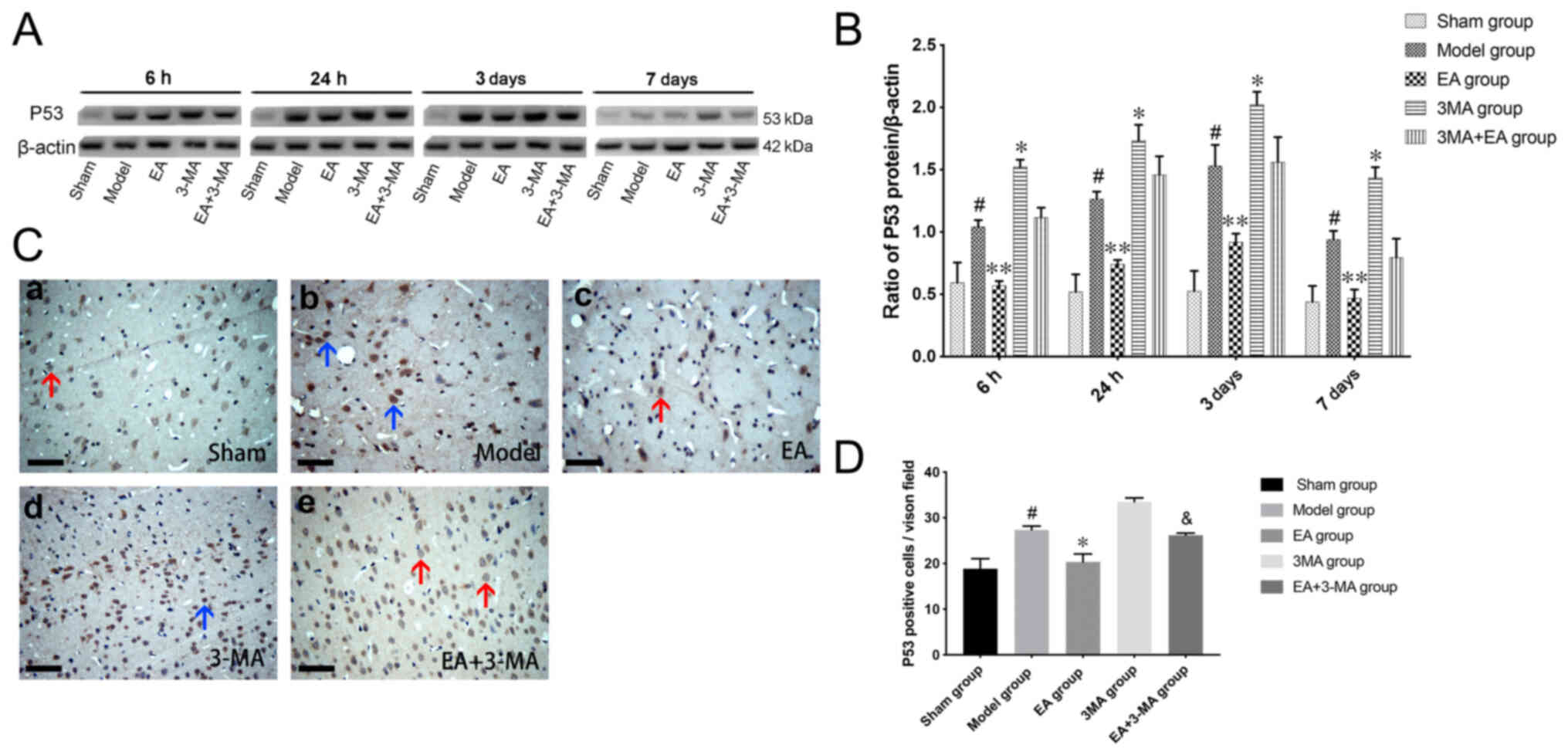
EA suppresses expression of p53
Tumor suppressor protein p53 regulates apoptosis by
interacting with both anti- and pro-apoptotic Bcl-2 family proteins
(47). Activation of the p53 system
has also been found to induce autophagy (48). In the present study, the expression
levels of p53 were evaluated by western blot analysis (Fig. 7A and B); the expression levels of
p53 in the focal area were significantly elevated at 6 h after ICH,
peaked at 3 days and remained high at 7 days. EA treatment
significantly prevented the elevation of p53 levels compared with
the model group at each time point, whereas 3-MA promoted p53
expression and counteracted the effect of EA treatment.
Immunohistochemical analysis was performed to confirm the
distribution of p53 (Fig. 7C and
D); numbers of p53-positive cells were consistent with the
results of western blot analysis. Positive signals of p53 were
strongly deposited in the cytoplasm of neuronal cells on day 3
after ICH.
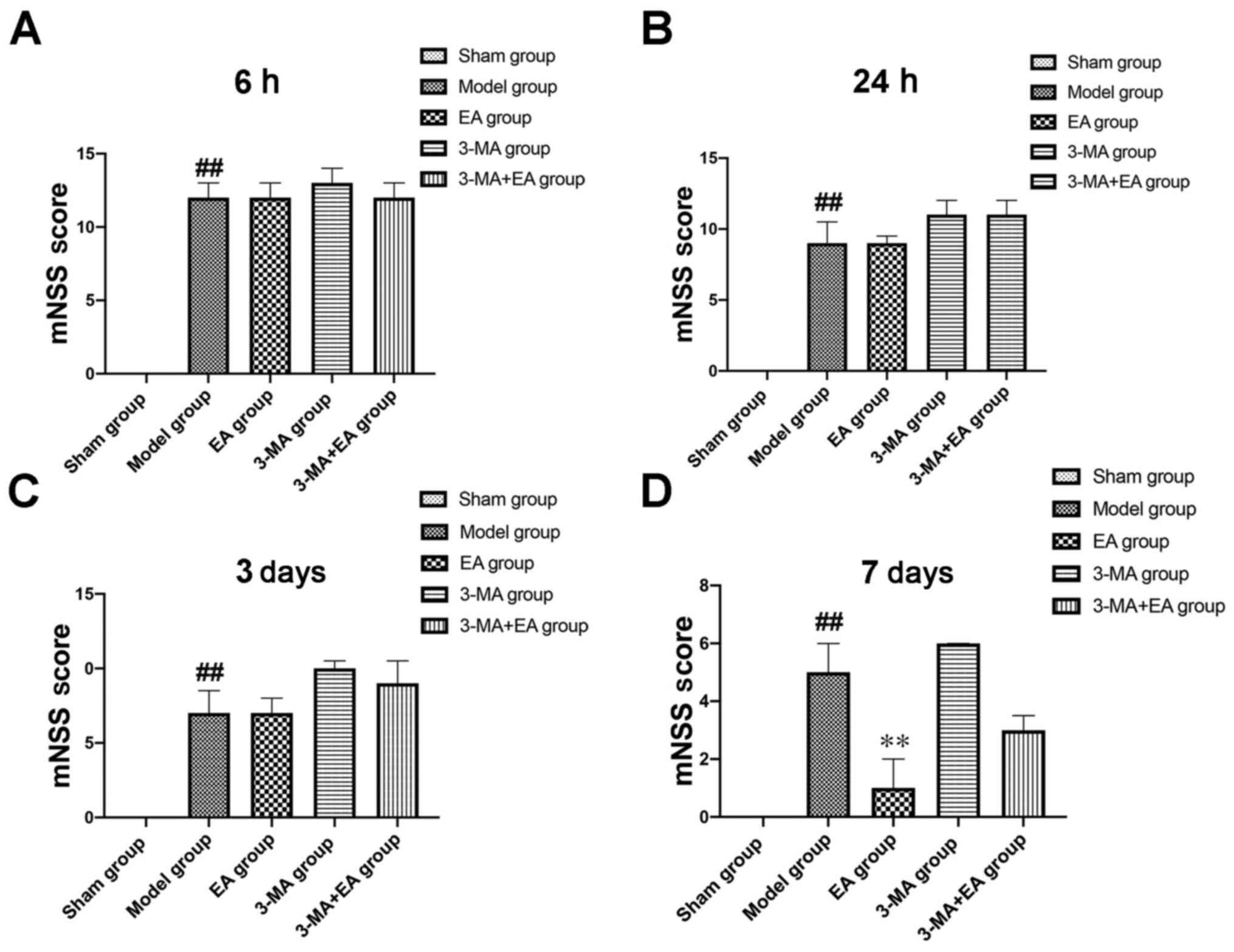
Neurobehavioral deficits following
ICH
Neurobehavioral deficits were evaluated to
investigate the effect of EA on recovery and mitophagy following
ICH. Neurobehavioral deficits were assessed by mNSS tests at 6 and
24 h and 3 and 7 days after ICH in the presence or absence of EA
treatment. Rats in each group exhibited neurological deficits at 6
h following induction of ICH, which improved over time, as
indicated by decreased mNSS test scores (Fig. 8). However, mNSS in the EA group
showed no significant difference compared with the model group
until day 7 after EA treatment (Fig.
8). Alleviation of neurological deficits was counteracted by
3-MA.
Discussion
Mitochondrial dysfunction is associated with the
pathogenesis of stroke (49). The
autophagic degradation of dysfunctional mitochondria is
commensurate with mitophagy, which is a mechanism of mitochondrial
quality control and is key for maintaining intracellular
homeostasis (50). Previously,
mitophagy has attracted attention as a novel therapeutic target in
stroke (51,52). It has been extensively researched in
ischemic stroke (53–55), but there are few studies in the
context of ICH.
Scalp acupuncture (SA) is a specialized acupuncture
technique used to treat patients with ischemic stroke (56). Baihui (GV20) to Qubin (GB7) is one
of the most effective stimulation lines or areas (57). Although SA treatment for acute ICH
has been disputed (58), clinical
trials have proved its efficacy and safety (59). A randomized controlled study
demonstrated that SA, combined with conventional treatment,
increased the hematoma absorption ratio and alleviated neurological
impairment compared with conventional treatment alone (60). A clinical study by Sun et al
(61) of 500 cases of patients with
cerebrovascular hemiplegia treated via acupuncture on Baihui-Qubin
achieved a satisfactory therapeutic effect. A previous study
involving Baihui-Qubin (GV20-GB7) treatment suggest inhibited
Notch-Hes signaling pathway transduction in rat basal ganglia after
ICH, thereby inhibiting neuronal differentiation and maintaining
neural stem cell proliferation (28). The present study focused on
investigating the underlying mechanism of the neuroprotective
effect of GV20-GB7 EA on mitophagy following ICH.
The PI3K inhibitor 3-MA is an autophagy inhibitor
that works in the early stage to suppress autophagy (62). 3-MA has been administered
intracerebroventricularly in a number of studies of brain disease
(63,64); it has also been administered i.p. in
a number of studies on pulmonary and hepatic disease (65,66),
but few studies have assessed its impact on cerebral disease
(67,68). In addition, the standard dosage of
i.p. and intraventricular injection was different in previous
studies (69); intraperitoneally
injected 3-MA was 150–300 µg/kg, whereas intraventricularly
injected 3-MA was 3–10 µl/rat, which was notably lower. Further
studies are required to determine the effect of this. Evidence has
indicated the role of 3-MA as an inhibitor of mitophagy (70,71);
to the best of our knowledge, high mortality rate caused by
intraventricular 3-MA treatment has not previously been reported.
Accordingly, cella lateralis injection of 3-MA was selected in the
present study.
In the present study, evidence of autophagy was
detected in focal tissue by observing the expression of LC3-II;
mitophagy was reflected by the expression levels of mitophagy
protein markers, such as PINK1, Parkin and BNIP3, which indicated
that mitophagy increased gradually following ICH and peaked at day
3. In order to investigate whether mitophagy was triggered
following ICH, morphological changes of neurons were observed by
TEM. Autophagic vacuoles appeared shortly following ICH; lysosomes
and degraded mitochondria were also detected, indicating that
mitochondrial autophagy was activated.
The efficient autophagic degradation of mitochondria
has been reported to require the participation of BNIP3, thus
indicating that mitophagy is specifically activated by BNIP3
(72). BNIP3 is a pro-apoptotic
member of the Bcl-2 family with an atypical BH3 domain, which can
also function as a receptor for targeting autophagosomes to
mitochondria (73,74). Due to its links with both mitophagy
and apoptosis (75), BNIP3 was
selected in the present study as a target to investigate changes in
mitophagy following ICH. EA treatment promoted mitophagy by
upregulating BNIP3; the therapeutic effect was mitigated by 3-MA.
Inhibition of expression of BNIP3 or mitophagy by 3-MA may be due
to the blockage of the LC3-interacting region of BNIP3, which
facilitates a direct interaction between BNIP3 and LC3 to promote
clearance of dysfunctional mitochondria (72). The present study demonstrated that
inhibition of mitophagy by 3-MA may enhance mitochondrial
accumulation, as evidenced by decreased levels of the mitophagy
marker BNIP3, and increased levels of mitochondrial markers TOMM20
and COX IV in the ICH + 3-MA group after 3 days. The results also
suggested that the effect of EA treatment may be achieved by
enhancing autophagic clearance of damaged mitochondria, which was
consistent with a study by Zhang et al (51). The protective role of autophagy
during the reperfusion phase following ischemic stroke may be
attributable to mitophagy-associated mitochondrial clearance and
inhibition of downstream apoptosis, reflected by decreased COX IV
and TOMM20 and increased LC3-II/GAPDH following oxygen-glucose
deprivation/reperfusion, 3-MA treatment also increases infarct
volume (51). These results
indicated that autophagic mitochondrial recycling may serve a
protective role following ICH.
Evidence has indicated that mitophagy and apoptosis
are interconnected (76,77). Bcl-2 family proteins and p53 serve
dual roles in regulating autophagy and apoptosis (78), yet p53 and Bcl-2 alteration
mechanisms are found to overlap. As a pro-apoptotic protein with
homology to Bcl-2 in the BH3 domain, BNIP3 is considered to be a
regulator of mitophagy (79).
Previous studies demonstrated that p53 may function primarily as a
transrepressor of BNIP3 to protect against hypoxia-induced
apoptotic cell death both in vitro and in vivo,
suggesting that BNIP3 is associated with dysfunction of
mitochondria and neuronal death induced by stroke (80,81).
Taken in conjunction with these earlier reports, the present
results supported an ICH model in which BNIP3 and p53 modify their
activity in a negative feedback loop; EA (GV20-GB7) enhanced
mitophagy by upregulating BNIP3, while decreased p53 contributed to
a neuroprotective effect.
In order to investigate whether increased mitophagy
by EA (GV20-GB7) could serve a neuroprotective role against ICH
stress, the effects of increased mitophagy on apoptosis were
evaluated. The results showed that, in parallel with the
alterations in BNIP3 levels and ultrastructural changes of
mitophagy, EA (GV20-GB7) may also inhibit apoptosis by upregulating
Bcl-2, as well as decreasing the expression levels of cleaved
caspase-3 and Bax. Previous research has revealed that the caspase
and Bcl-2 families jointly participate in the intrinsic apoptosis
pathway mediated by mitochondria (50,82).
In addition, mitochondrial dysfunction may cause the rupture of the
outer mitochondrial membrane and cytochrome c release, which
could activate caspase-3 release (83). The potential mechanism may be due to
BNIP3 acting as a dual regulator of Bcl-2 family proteins and
suppressing apoptosis by inhibiting pro-apoptotic protein Bcl-2 and
activating Bax protein, as well as harboring an LC3-interacting
region that can initiate mitophagy (43,72).
Combined with the present results, it was suggested that mitophagy
enhanced by EA (GV20-GB7) may have an effect on degradation of
apoptotic cell death after ICH. However, the mechanism underlying
the effects of EA (GV20-GB7) on mitophagy and apoptosis remains
unclear. In addition, in test of cleaved caspase-3 of western blot
of this study, the EA and EA + 3-MA group only differed at 24 h.
Since 3-MA primarily functions to inhibit mitophagy, the
neutralizing effect of 3-MA on EA-induced downregulation of cleaved
caspase-3 may differ from that on other factors, such as BNIP3.
This may be because the apoptotic function of cleaved caspase-3 is
only active in the early stages following stroke. Expression levels
of cleaved caspase-3 were low on day 3 and 7. During the stages of
low expression by the day 3 and 7, either EA or 3-MA treatment may
not have an obvious effect on the expression levels of cleaved
caspase-3. Therefore, 3-MA only significantly reversed the effect
of EA treatment at 24 h.
The tumor suppressor and transcription factor p53
triggers apoptosis and modulates autophagy by acting at the
mitochondrial level (84,85). During the neuronal apoptosis
process, p53 has been reported to act as a key factor in early
apoptotic signaling in neurons acting upstream of mitochondrial
damage and may cause mitochondrial membrane disruption, which can
lead to caspase activation (86).
As a master regulator of autophagy, the inhibition of autophagy by
p53 has been shown to be associated with its nuclear-to-cytosolic
redistribution (87).
Cytoplasmic p53 may translocate to the mitochondria
and induce mitochondrial membrane permeabilization by activating
pro-apoptotic Bcl-2 family members to inhibit anti-apoptotic Bcl-2
family proteins (88,89). In the present study, EA(GV20-GB7)
treatment increased the expression of anti-apoptosis Bcl-2 protein
and downregulated the expression of cleaved caspase-3 and Bax;
these results coincided with a significant decrease of p53. From
these results, it was concluded that ICH-induced p53 expression in
rats was decreased by EA (GV20-GB7), which inhibited apoptosis. In
addition, immunohistochemical results showed that p53 was primarily
distributed in the cytoplasm, which illustrated that the
mitophagy-augmenting and apoptosis-restraining effects of EA
treatment may be associated with interactions between mitophagy and
apoptotic factors, and downregulated cytoplasmic expression levels
of p53 protein. Moreover, 3-MA inhibited mitophagy and abolished
the effects of EA, further indicating that in this study, the
protective mechanism of EA may involve mitophagy.
In the present study, anti-apoptotic Bcl-2 protein
was decreased in response to ICH, meanwhile, the pro-apoptotic
protein BNIP3 was increased; however increased BNIP3 also improved
mitophagy, which inhibited apoptotic cell death. As p53 is a
regulator of both autophagy and apoptosis, it may be suggested that
p53 induced apoptosis by interacting with Bcl-2 family proteins,
simultaneously inhibiting anti-apoptotic proteins, such as Bcl-2,
and activating pro-apoptotic proteins, such as Bax and Bak. BNIP3
may be a putative transcriptional target and downstream effector of
p53 in autophagy (90); BNIP3 is
also an autophagy (specifically mitophagy) inducer which has
similar effects to p53 when acting as a pro-apoptotic protein.
Therefore, EA-alleviated p53 may improve mitophagy and inhibit
apoptosis by increasing the expression levels of BNIP3 and
decreasing those of Bcl-2.
Neurobehavioral deficits were evaluated by mNSS
tests. The behavioral effect of EA was assessed by multiple
modalities of testing, including function, sensory (including
placing and proprioceptive), beam balance, reflex absence and
abnormal movement tests. In the present study, neurobehavioral
deficits were not significantly alleviated until day 7 after EA
treatment, which is consistent with our previous research (29,36),
further suggesting that the benefit of EA increases with treatment
duration. Consistent with our previous research (29), each sub-test was observed
independently, excluding vibrissae proprioception and symmetry of
limb, the assessment failed to demonstrate significantly improved
performance by GV20-GB7 EA treatment until 7 days after ICH. Longer
observation time and a larger sample size should be used in future
studies to improve the validity of the test. EA at GV20-GB7 may
improve neurological function following ICH by clearance of
mitochondria via mitophagy.
BNIP3 has a C-terminal transmembrane domain, which
is essential for its homodimerization and pro-apoptotic function.
Following homodimerization, BNIP3 is associated with mitochondrial
targeting where it functions in inner membrane depolarization and
permeabilization of the mitochondrial outer membrane to induce
mitochondrial dysfunction, and both non-apoptotic and apoptotic
cell death (91). A study of mouse
hepatocytes indicated that hypoxia may increase formation of the
BNIP3 homodimer but decrease the amount of the monomeric form of
BNIP3 (92). Another study also
suggested that the BNIP3 homodimer may function as an autophagy
receptor to facilitate the removal of organelles, such as
mitochondria and endoplasmic reticulum (93). This feature is suggested to serve a
wide role in the pathogenesis of various diseases, including
stroke. However, to the best of our knowledge, the mechanism
underlying activation of dimerized BNIP3, its translocation to the
mitochondria during mitophagy, and its potential regulation by
acupuncture or other medical intervention have not yet been studied
in an ICH model.
There are certain limitations to the present study.
All samples used were brain homogenates; thus, the point of
mitophagy activation and regulation by p53 cannot be precisely
located. Also, the underlying mechanism of EA-enhanced mitophagy
was not described here and merits further investigation.
Furthermore, it is difficult to conclude whether the effectiveness
of EA on mitophagy following ICH was achieved by regulating
mitophagy induced by ischemia during ICH secondary injury or by
improving mitochondrial metabolism.
Regulation of mitophagy to maintain the integrity
and homeostasis of mitochondria is a promising therapeutic target
in the future of ICH treatment but further studies are required to
reveal the potential mechanism underlying mitophagy following ICH.
For example, the exact mechanism by which PINK1 phosphorylates
itself and its substrates has not yet been fully elucidated. Parkin
recruitment and activation during mitophagy involves PINK1-mediated
phosphorylation of Parkin and E3-ubiquitin (94). GTPase mitofusin 2 functions as a
mitochondrial receptor for Parkin, which can mediate Parkin
recruitment to mitochondria and can further promote Parkin-mediated
ubiquitination by p-PINK1 (95).
Considering the importance of healthy mitochondria for recovery and
survival following ICH (96),
mitophagy is an important research area for the development of
novel treatments and to reveal how EA may improve neurobehavioral
deficits.
In conclusion, the present study identified the role
of mitophagy in an ICH model. The neuroprotective effect of EA on
Baihui-Qubin (GV20-GB7), which occurs via enhanced mitophagy, may
be induced by inhibiting apoptosis. The protective effect of p53
inhibition by EA on GV20-GB7 may occur via regulation of p53
interactions with Bcl-2 family proteins, which may promote the
expression of BNIP3 and the activation of anti-apoptotic Bcl-2.
Furthermore, 3-MA pretreatment inhibited mitophagy, aggravated
apoptotic cell death and reversed improvement on neurological
deficits induced by EA (GV20-GB7) treatment. These findings
contribute to knowledge concerning mitophagy induction in ICH and
suggest that regulating the balance between mitophagy and apoptosis
may be one mechanism by which EA (GV20-GB7) improves recovery in
ICH.
Supplementary Material
Supporting Data
Acknowledgements
The authors would like to thank to Mr. Haorui Wang
of Royal College of Art for providing technical assistance and
useful discussion.
Funding
The present study was supported by the National
Natural Science Foundation of China (grant no. 81473764,81273824),
the Key Program of Natural Science Foundation of Heilongjiang
Province of China (grant no. ZD 201204), the Doctoral Fund of
Ministry of Education of China (grant no. 20102327110003) and the
Science Foundation for Young Scholars of Zhongshan Hospital, Fudan
University (grant no. 2020ZSQN65).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
RG performed the experiments and statistical
analysis, collected and analyzed data and wrote the manuscript. ZL
performed the experiments, proofread the manuscript and constructed
figures. XL, PL and SD collected samples and data, and performed
the experiments, including rat ICH surgery. WZ contributed to the
conception and design of the study and obtained funding. XY, XD,
XL, HL, QC and WT participated in conceptualization and design of
the study and critically revised the manuscript for important
intellectual content. RG and WZ confirm the authenticity of all the
raw data. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
The present study was approved by the Ethics
Committee of the Heilongjiang University of Chinese Medicine
(approved no. 2017061001).
Patient consent for publication
Not applicable.
Competing interests
The authors declare they have no competing
interests.
References
|
1
|
Caplan LR: Intracerebral haemorrhage.
Lancet. 339:656–658. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Broderick J, Connolly S, Feldmann E,
Hanley D, Kase C, Krieger D, Mayberg M, Morgenstern L, Ogilvy CS,
Vespa P, et al: Guidelines for the management of spontaneous
intracerebral hemorrhage in adults: 2007 update: A guideline from
the American Heart Association/American Stroke Association Stroke
Council, High Blood Pressure Research Council, and the Quality of
Care and Outcomes in Research Interdisciplinary Working Group.
Stroke. 38:2001–2023. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Counsell C, Boonyakarnkul S, Dennis M,
Sandercock P, Bamford J, Burn J and Warlow C: Primary intracerebral
haemorrhage in the oxfordshire community stroke Project.
Cerebrovascular Dis. 5:26–34. 1995. View Article : Google Scholar
|
|
4
|
Keep RF, Hua Y and Xi G: Intracerebral
haemorrhage: Mechanisms of injury and therapeutic targets. Lancet
Neurol. 11:720–731. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Nehls DG, Mendelow AD, Graham DI, Sinar EJ
and Teasdale GM: Experimental intracerebral hemorrhage: Progression
of hemodynamic changes after production of a spontaneous mass
lesion. Neurosurgery. 23:439–444. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Belur PK, Chang JJ, He S, Emanuel BA and
Mack WJ: Emerging experimental therapies for intracerebral
hemorrhage: Targeting mechanisms of secondary brain injury.
Neurosurg Focus. 34:E92013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Levine B and Kroemer G: Autophagy in the
pathogenesis of disease. Cell. 132:27–42. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Niu M, Dai X, Zou W, Yu X, Teng W, Chen Q,
Sun X, Yu W, Ma H and Liu P: Autophagy, endoplasmic reticulum
stress and the unfolded protein response in intracerebral
hemorrhage. Transl Neurosci. 8:37–48. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Klionsky DJ and Emr SD: Autophagy as a
regulated pathway of cellular degradation. Science. 290:1717–1721.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wang K and Klionsky DJ: Mitochondria
removal by autophagy. Autophagy. 7:297–300. 2014. View Article : Google Scholar
|
|
11
|
Goldman SJ, Taylor R, Zhang Y and Jin S:
Autophagy and the degradation of mitochondria. Mitochondrion.
10:309–315. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Graef M and Nunnari J: A role for
mitochondria in autophagy regulation. Autophagy. 7:1245–1246. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wei H, Liu L and Chen Q: Selective removal
of mitochondria via mitophagy: Distinct pathways for different
mitochondrial stresses. Biochim Biophys Acta. 1853:2784–2790. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lin C, Chao H, Li Z, Xu X, Liu Y, Hou L,
Liu N and Ji J: Melatonin attenuates traumatic brain injury-induced
inflammation: A possible role for mitophagy. J Pineal Res.
61:177–186. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Yuan Y, Zheng Y, Zhang X, Chen Y, Wu X, Wu
J, Shen Z, Jiang L, Wang L, Yang W, et al: BNIP3L/NIX-mediated
mitophagy protects against ischemic brain injury independent of
PARK2. Autophagy. 13:1754–1766. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Li J, Lu J, Mi Y, Shi Z, Chen C, Riley J
and Zhou C: Voltage-dependent anion channels (VDACs) promote
mitophagy to protect neuron from death in an early brain injury
following a subarachnoid hemorrhage in rats. Brain Res. 1573:74–83.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Yang Y, Xing D, Zhou F and Chen Q:
Mitochondrial autophagy protects against heat shock-induced
apoptosis through reducing cytosolic cytochrome c release and
downstream caspase-3 activation. Biochem Biophys Res Commun.
395:190–195. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Colbert AP, Spaulding K, Larsen A, Ahn AC
and Cutro JA: Electrodermal activity at acupoints: Literature
review and recommendations for reporting clinical trials. J
Acupunct and Meridian Stud. 4:5–13. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhang S, Wu B, Liu M, Li N, Zeng X, Liu H,
Yang Q, Han Z, Rao P and Wang D; all Investigators, : Acupuncture
efficacy on ischemic stroke recovery: Multicenter randomized
controlled trial in China. Stroke. 46:1301–1306. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Langevin HM, Schnyer R, MacPherson H,
Davis R, Harris RE, Napadow V, Wayne PM, Milley RJ, Lao L,
Stener-Victorin E, et al: Manual and electrical needle stimulation
in acupuncture research: Pitfalls and challenges of heterogeneity.
J Altern Complement Med. 21:113–128. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zhou F, Guo J, Cheng J, Wu G and Xia Y:
Electroacupuncture increased cerebral blood flow and reduced
ischemic brain injury: Dependence on stimulation intensity and
frequency. J Appl Physiol (1985). 111:1877–1887. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhu Y, Deng L, Tang H, Gao X, Wang Y, Guo
K, Kong J and Yang C: Electroacupuncture improves neurobehavioral
function and brain injury in rat model of intracerebral hemorrhage.
Brain Res Bull. 131:123–132. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Li HQ, Li Y, Chen ZX, Zhang XG, Zheng XW,
Yang WT, Chen S and Zheng GQ: Electroacupuncture exerts
neuroprotection through caveolin-1 mediated molecular pathway in
intracerebral hemorrhage of rats. Neural Plast. 2016:73082612016.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ting Z, Jianbin Z and Luqi H: Protective
effect of electroacupuncture on neurons autophagy in perfusion
period of cerebral ischemia. Neurosci Lett. 661:41–45. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wu Z, Zou Z, Zou R, Zhou X and Cui S:
Electroacupuncture pretreatment induces tolerance against cerebral
ischemia/reperfusion injury through inhibition of the autophagy
pathway. Mol Med Rep. 11:4438–4446. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Liu W, Shang G, Yang S, Huang J, Xue X,
Lin Y, Zheng Y, Wang X, Wang L, Lin R, et al: Electroacupuncture
protects against ischemic stroke by reducing autophagosome
formation and inhibiting autophagy through the mTORC1-ULK1
complex-Beclin1 pathway. Int J Mol Med. 37:309–318. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Wu ZQ, Cui SY, Zhu L and Zou ZQ: Study on
the mechanism of mTOR-Mediated autophagy during electroacupuncture
pretreatment against cerebral ischemic injury. Evid Based
Complement Alternat Med. 2016:91215972016. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zou W, Chen QX, Sun XW, Chi QB, Kuang HY,
Yu XP and Dai XH: Acupuncture inhibits Notch1 and Hes1 protein
expression in the basal ganglia of rats with cerebral hemorrhage.
Neural Regen Res. 10:457–462. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Liu H, Sun X, Zou W, Leng M, Zhang B, Kang
X, He T and Wang H: Scalp acupuncture attenuates neurological
deficits in a rat model of hemorrhagic stroke. Complement Ther Med.
32:85–90. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Health N: Guide for the care and use of
laboratory animals. NIH contract No. No1-RR-2-2135. 11–28.
1985.
|
|
31
|
Guan R, Zou W, Dai X, Yu X, Liu H, Chen Q
and Teng W: Mitophagy, a potential therapeutic target for stroke. J
Biomed Sci. 25:872018. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Li Q, Zhang T, Wang J, Zhang Z, Zhai Y,
Yang GY and Sun X: Rapamycin attenuates mitochondrial dysfunction
via activation of mitophagy in experimental ischemic stroke.
Biochem Biophys Res Commun. 444:182–188. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Di Y, He YL, Zhao T, Huang X, Wu KW, Liu
SH, Zhao YQ, Fan M, Wu LY and Zhu LL: Methylene blue reduces acute
cerebral ischemic injury via the induction of mitophagy. Mol Med.
21:420–429. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Jing CH, Wang L, Liu PP, Wu C, Ruan D and
Chen G: Autophagy activation is associated with neuroprotection
against apoptosis via a mitochondrial pathway in a rat model of
subarachnoid hemorrhage. Neuroscience. 213:144–153. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Liu XY, Dai XH, Zou W, Yu XP, Teng W, Wang
Y, Yu WW, Ma HH, Chen QX, Liu P, et al: Acupuncture through Baihui
(DU20) to Qubin (GB7) mitigates neurological impairment after
intracerebral hemorrhage. Neural Regen Res. 13:1425–1432. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Zhang B, Dai XH, Yu XP, Zou W, Teng W, Sun
XW, Yu WW, Liu H, Wang H, Sun MJ and Li M: Baihui
(DU20)-penetrating-Qubin (GB7) acupuncture inhibits apoptosis in
the perihemorrhagic penumbra. Neural Regen Res. 13:1602–1608. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Wang C, Hu Q and Shen HM: Pharmacological
inhibitors of autophagy as novel cancer therapeutic agents.
Pharmacol Res. 105:164–175. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Hua XB: On animal acupoints. J Tradit Chi
Med. 7:301–304. 1987.PubMed/NCBI
|
|
39
|
Jittiwat J: Laser Acupuncture at GV20
improves brain damage and oxidative stress in animal model of focal
ischemic stroke. J Acupunct Meridian Stud. 10:324–330. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Yin CS, Jeong HS, Park HJ, Baik Y, Yoon
MH, Choi CB and Koh HG: A proposed transpositional acupoint system
in a mouse and rat model. Res Vet Sci. 84:159–165. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Han HJ, Park SJ, Soh KS, Myoung HS, Lee
KJ, Ogay V and Lee YH: Electrical characterization of proposed
transpositional acupoints on the urinary bladder meridian in a rat
model. Evid Based Complement Alternat Med. 2011:2954752011.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Chen J, Sanberg PR, Li Y, Wang L, Lu M,
Willing AE, Sanchez-Ramos J and Chopp M: Intravenous administration
of human umbilical cord blood reduces behavioral deficits after
stroke in rats. Stroke. 32:2682–2688. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Choe SC, Hamacher-Brady A and Brady NR:
Autophagy capacity and sub-mitochondrial heterogeneity shape
Bnip3-induced mitophagy regulation of apoptosis. Cell Commun
Signal. 13:372015. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Gross A, McDonnell JM and Korsmeyer SJ:
BCL-2 family members and the mitochondria in apoptosis. Genes Dev.
13:1899–1911. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Chen B, Wang G, Li W, Liu W, Lin R, Tao J,
Jiang M, Chen L and Wang Y: Memantine attenuates cell apoptosis by
suppressing the calpain-caspase-3 pathway in an experimental model
of ischemic stroke. Exp Cell Res. 351:163–172. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Wagner DC, Riegelsberger UM, Michalk S,
Hartig W, Kranz A and Boltze J: Cleaved caspase-3 expression after
experimental stroke exhibits different phenotypes and is
predominantly non-apoptotic. Brain Res. 1381:237–242. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Yan J, Yun H, Yang Y, Jing B, Feng C and
Song-bin F: Upregulation of BNIP3 promotes apoptosis of lung cancer
cells that were induced by p53. Biochem Biophys Res Commun.
346:501–507. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Tasdemir E, Chiara Maiuri M, Morselli E,
Criollo A, D'Amelio M, Djavaheri-Mergny M, Cecconi F, Tavernarakis
N and Kroemer G: A dual role of p53 in the control of autophagy.
Autophagy. 4:810–814. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Sims NR and Muyderman H: Mitochondria,
oxidative metabolism and cell death in stroke. Biochim Biophys
Acta. 1802:80–91. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Kubli DA and Gustafsson AB: Mitochondria
and mitophagy: The yin and yang of cell death control. Circ Res.
111:1208–1221. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Zhang X, Yan H, Yuan Y, Gao J, Shen Z,
Cheng Y, Shen Y, Wang RR, Wang X, Hu WW, et al: Cerebral
ischemia-reperfusion-induced autophagy protects against neuronal
injury by mitochondrial clearance. Autophagy. 9:1321–1333. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Shen Z, Zheng Y, Wu J, Chen Y, Wu X, Zhou
Y, Yuan Y, Lu S, Jiang L, Qin Z, et al: PARK2-dependent mitophagy
induced by acidic postconditioning protects against focal cerebral
ischemia and extends the reperfusion window. Autophagy. 13:473–485.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Baek SH, Noh AR, Kim KA, Akram M, Shin YJ,
Kim ES, Yu SW, Majid A and Bae ON: Modulation of mitochondrial
function and autophagy mediates carnosine neuroprotection against
ischemic brain damage. Stroke. 45:2438–2443. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Shi RY, Zhu SH, Li V, Gibson SB, Xu XS and
Kong JM: BNIP3 interacting with LC3 triggers excessive mitophagy in
delayed neuronal death in stroke. CNS Neurosci Ther. 20:1045–1055.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Wu M, Lu G, Lao YZ, Zhang H, Zheng D,
Zheng ZQ, Yi J, Xiang Q, Wang LM, Tan HS, et al:
Garciesculenxanthone B induces PINK1-Parkin-mediated mitophagy and
prevents ischemia-reperfusion brain injury in mice. Acta Pharmacol
Sin. 42:199–208. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Yin ZL, Meng ZX, Ge S, Zhang MJ and Huang
LH: Clinical observation of dynamic scalp acupuncture combined with
task-oriented mirror therapy for upper limbs function impairment in
patients with hemiplegia after ischemic stroke. Zhongguo Zhen Jiu.
40:918–922. 2020.(In Chinese). PubMed/NCBI
|
|
57
|
Wang WW, Xie CL, Lu L and Zheng GQ: A
systematic review and meta-analysis of Baihui (GV20)-based scalp
acupuncture in experimental ischemic stroke. Sci Rep. 4:39812014.
View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Zheng GQ, Zhao ZM, Wang Y, Gu Y, Li Y,
Chen XM, Fu SP and Shen J: Meta-analysis of scalp acupuncture for
acute hypertensive intracerebral hemorrhage. J Altern Complement
Med. 17:293–299. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Lang Y, Cui FY, Li KS, Tan ZJ and Zou YH:
Imaging observation of scalp acupuncture on brain gray matter
injury in stroke patients with cerebral infarction. Zhongguo Zhong
Xi Yi Jie He Za Zhi. 36:294–299. 2016.(In Chinese). PubMed/NCBI
|
|
60
|
Wang HQ, Bao CL, Jiao ZH and Dong GR:
Efficacy and safety of penetration acupuncture on head for acute
intracerebral hemorrhage: A randomized controlled study. Medicine
(Baltimore). 95:e55622016. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Sun ST, Li SR, Zhu YZ, Chen SL, Wan GZ,
Sun YZ, Hou GW and Yu ZH: Clinical study on 500 cases of
cerebro-vascular hemiplegia treated by acupuncture through baihui
to qubin. J Tradit Chin Med. 5:167–170. 1985.PubMed/NCBI
|
|
62
|
Wu Y, Wang X, Guo H, Zhang B, Zhang XB,
Shi ZJ and Yu L: Synthesis and screening of 3-MA derivatives for
autophagy inhibitors. Autophagy. 9:595–603. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Shao A, Wang Z, Wu H, Dong X, Li Y, Tu S,
Tang J, Zhao M, Zhang J and Hong Y: Enhancement of autophagy by
histone deacetylase inhibitor trichostatin a ameliorates neuronal
apoptosis after subarachnoid hemorrhage in rats. Mol Neurobiol.
53:18–27. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Xia DY, Li W, Qian HR, Yao S, Liu JG and
Qi XK: Ischemia preconditioning is neuroprotective in a rat
cerebral ischemic injury model through autophagy activation and
apoptosis inhibition. Braz J Med Biol Res. 46:580–588. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Ma H, Chen H, Dong A, Wang Y, Bian Y and
Xie K: Hydrogen-rich saline attenuates hyperalgesia and reduces
cytokines in rats with post-herpetic neuralgia via activating
autophagy. Xi Bao Yu Fen Zi Mian Yi Xue Za Zhi. 33:155–158.
2017.(In Chinese). PubMed/NCBI
|
|
66
|
Tang Y, Cai QH, Wang YJ, Fan SH, Zhang ZF,
Xiao MQ, Zhu JY, Wu DM, Lu J and Zheng YL: Protective effect of
autophagy on endoplasmic reticulum stress induced apoptosis of
alveolar epithelial cells in rat models of COPD. Biosci Rep.
37:BSR201708032017. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Wu Q, Gao C, Wang H, Zhang X, Li Q, Gu Z,
Shi X, Cui Y, Wang T, Chen X, et al: Mdivi-1 alleviates blood-brain
barrier disruption and cell death in experimental traumatic brain
injury by mitigating autophagy dysfunction and mitophagy
activation. Int J Biochem Cell Biol. 94:44–55. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Guo Z, Cao G, Yang H, Zhou H, Li L, Cao Z,
Yu B and Kou J: A combination of four active compounds alleviates
cerebral ischemia-reperfusion injury in correlation with inhibition
of autophagy and modulation of AMPK/mTOR and JNK pathways. J
Neurosci Res. 92:1295–1306. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Chen X, Wang L, Deng Y, Li X, Li G, Zhou
J, Cheng D, Yang Y, Yang Q, Chen G and Wang G: Inhibition of
autophagy prolongs recipient survival through promoting CD8(+) T
cell apoptosis in a rat liver transplantation model. Front Immunol.
10:13562019. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Cao S, Shrestha S, Li J, Yu X, Chen J, Yan
F, Ying G, Gu C, Wang L and Chen G: Melatonin-mediated mitophagy
protects against early brain injury after subarachnoid hemorrhage
through inhibition of NLRP3 inflammasome activation. Sci Rep.
7:24172017. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Zuo W, Zhang S, Xia CY, Guo XF, He WB and
Chen NH: Mitochondria autophagy is induced after hypoxic/ischemic
stress in a Drp1 dependent manner: The role of inhibition of Drp1
in ischemic brain damage. Neuropharmacology. 86:103–115. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Hanna RA, Quinsay MN, Orogo AM, Giang K,
Rikka S and Gustafsson AB: Microtubule-associated protein 1 light
chain 3 (LC3) interacts with Bnip3 protein to selectively remove
endoplasmic reticulum and mitochondria via autophagy. J Biol Chem.
287:19094–19104. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Zhang J and Ney PA: Role of BNIP3 and NIX
in cell death, autophagy, and mitophagy. Cell Death Differ.
16:939–946. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Tan H, Wu Z, Wang H, Bai B, Li Y, Wang X,
Zhai B, Beach TG and Peng J: Refined phosphopeptide enrichment by
phosphate additive and the analysis of human brain phosphoproteome.
Protemomics. 15:500–507. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Liu L, Sakakibara K, Chen Q and Okamoto K:
Receptor-mediated mitophagy in yeast and mammalian systems. Cell
Res. 24:787–795. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Kissová I, Plamondon L-T, Brisson L,
Priault M, Renouf V, Schaeffer J, Camougrand N and Manon S:
Evaluation of the roles of apoptosis, autophagy, and mitophagy in
the loss of plating efficiency induced by Bax expression in yeast.
J Biol Chem. 281:36187–36197. 2006. View Article : Google Scholar
|
|
77
|
Li XX, Tsoi B, Li YF, Kurihara H and He
RR: Cardiolipin and its different properties in mitophagy and
apoptosis. J Histochem Cytochem. 63:301–311. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Levine B, Sinha SC and Kroemer G: Bcl-2
family members: Dual regulators of apoptosis and autophagy.
Autophagy. 4:600–606. 2008. View Article : Google Scholar
|
|
79
|
Ney PA: Mitochondrial autophagy: Origins,
significance, and role of BNIP3 and NIX. Biochim Biophys Acta.
1853:2775–2783. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Feng X, Liu X, Zhang W and Xiao W: p53
directly suppresses BNIP3 expression to protect against
hypoxia-induced cell death. EMBO J. 30:3397–3415. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Hoshino A, Matoba S, Iwai-Kanai E,
Nakamura H, Kimata M, Nakaoka M, Katamura M, Okawa Y, Ariyoshi M,
Mita Y, et al: p53-TIGAR axis attenuates mitophagy to exacerbate
cardiac damage after ischemia. J Mol Cell Cardiol. 52:175–184.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Marino G, Niso-Santano M, Baehrecke EH and
Kroemer G: Self-consumption: The interplay of autophagy and
apoptosis. Nat Rev Mol Cell Biol. 15:81–94. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Jevtić G, Nikolić T, Mirčić A, Stojković
T, Velimirović M, Trajković V, Marković I, Trbovich AM, Radonjić NV
and Petronijević ND: Mitochondrial impairment, apoptosis and
autophagy in a rat brain as immediate and long-term effects of
perinatal phencyclidine treatment-influence of restraint stress.
Prog Neuropsychopharmacol Biol Psychiatry. 66:87–96. 2016.
View Article : Google Scholar
|
|
84
|
Chipuk JE, Kuwana T, Bouchier-Hayes L,
Droin NM, Newmeyer DD, Schuler M and Green DR: Direct activation of
Bax by p53 mediates mitochondrial membrane permeabilization and
apoptosis. Science. 303:1010–1014. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Livesey KM, Kang R, Vernon P, Buchser W,
Loughran P, Watkins SC, Zhang L, Manfredi JJ, Zeh HJ III, Li L, et
al: p53/HMGB1 complexes regulate autophagy and apoptosis. Cancer
Res. 72:1996–2005. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Culmsee C and Mattson MP: p53 in neuronal
apoptosis. Biochem Biophys Res Commun. 331:761–777. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Morselli E, Tasdemir E, Maiuri MC,
Galluzzi L, Kepp O, Criollo A, Vicencio JM, Soussi T and Kroemer G:
Mutant p53 protein localized in the cytoplasm inhibits autophagy.
Cell Cycle. 7:3056–3061. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Chipuk JE and Green DR: p53's believe it
or not: Lessons on transcription-independent death. J Clin Immunol.
23:355–361. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Moll UM and Zaika A: Nuclear and
mitochondrial apoptotic pathways of p53. FEBS Lett. 493:65–69.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Wang EY, Gang H, Aviv Y, Dhingra R,
Margulets V and Kirshenbaum LA: p53 mediates autophagy and cell
death by a mechanism contingent on Bnip3. Hypertension. 62:70–77.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Hendgen-Cotta UB, Esfeld S, Rudi K,
Miinalainen I, Klare JP and Rassaf T: Cytosolic BNIP3 dimer
interacts with mitochondrial BAX forming heterodimers in the
mitochondrial outer membrane under basal conditions. Int J Mol Sci.
18:6872017. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Namas RA, Metukuri MR, Dhupar R, Velosa C,
Jefferson BS, Myer E, Constantine GM, Billiar TR, Vodovotz Y and
Zamora R: Hypoxia-induced overexpression of BNIP3 is not dependent
on hypoxia-inducible factor 1α in mouse hepatocytes. Shock.
36:196–202. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Hanna Akram R: Bnip3 interacts with LC3 to
induce selective removal of endoplasmic reticulum and mitochondria
via autophagy. UC San Diego Electronic Theses & Dissertations.
12–33. 2011.https://escholarship.org/content/qt1wq0k372/qt1wq0k372.pdfJuly
1–2019
|
|
94
|
Durcan TM and Fon EA: The three ‘P's of
mitophagy: PARKIN, PINK1, and post-translational modifications.
Genes Dev. 29:989–999. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Chen Y and Dorn GW: PINK1-phosphorylated
mitofusin 2 is a parkin receptor for culling damaged mitochondria.
Science. 340:471–475. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Kim-Han J, Kopp S, Dugan L and Diringer M:
Perihematomal mitochondrial dysfunction after intracerebral
Hemorrhage. Stroke. 37:2457–2462. 2006. View Article : Google Scholar : PubMed/NCBI
|