Introduction
Heart failure (HF), also known as congestive HF,
occurs when the cardiac muscles cannot pump enough blood and oxygen
to support other systems (1). HF
was considered a contributing cause of 1 in 8 deaths in the United
States in 2017 (2). HF is a
detrimental and progressive pathological condition, which seriously
affects the daily life of patients. Patients with HF present a
stage-related continuous decline in exercise tolerance that
disrupts the ability to function independently (3). In the severe or acute stages of HF,
the associated complications, including edema, dyspnea or
infection, can result in death (4).
At present, there is no effective treatment to cure HF or prevent
cardiomyocytes from constant injury. The existing medications for
cardiac dysfunction may only temporarily improve symptoms or slow
the rate of decline (5). Several
factors associated with HF, including narrowed coronary arteries or
hypertension, gradually leave the heart too weak to pulse
efficiently. The majority of patients with HF concurrently suffer
from hyperlipemia, which is considered the most common cause of
cardiovascular disease (6). It was
previously suggested that an important way to prevent HF may be to
target lipid metabolism (7). A
deeper understanding of the biological behaviors and molecular
basis of HF is required to develop therapeutic strategies.
The cell types within the heart include
cardiomyocytes, cardiac fibroblasts (CFs), vascular smooth muscle
cells and endothelial cells. CFs serve critical roles in supporting
normal cardiac function and in pathological remodeling of cardiac
tissue, including during HF (8).
CFs have various functions, including the synthesis and deposition
of inflammatory factors and cell-cell communications with other
cellular populations (9). In a
recent study, fibroblast activation was reported to contribute to
shaping the microenvironment of cardiac tissues during cardiac
dysfunction (10). Under such
conditions, CFs have been shown to exert proinflammatory activity,
which has been identified to correlate with disease escalation.
Notably, cardiomyocytes make up ~40% of the total cell population
in cardiac tissues, whereas CFs account for up to 60% intermingling
with cardiomyocytes (11). The
cross-talk between these two populations can be regulated by direct
cell-cell contact via microtubules and by indirect interactions via
a wide range of cytokines (11,12).
Therefore, targeting CFs may be considered a novel therapeutic
approach to treat HF.
Inflammasomes have been reported to be crucial for
their roles in cardiovascular disorders, neuroinflammation,
tumorigenesis and host defense against pathogenic invasion
(13–15). An inflammasome is assembled in
response to pathogeneses or tissue damage by the Nod-like receptor
protein or absent in melanoma 2-like receptors (16). Numerous proinflammatory cytokines,
such as IL1β, IL10 and IL18, and pyroptosis are induced by
inflammasome activation (17).
Inflammasomes regulate innate immunity, particularly by acting as
platforms for activation of caspase-1, caspase-8, caspase-11 and
IL1R-associated kinase. The role of the innate immune system has
been identified in the etiology of chronic inflammation in HF.
Previously, the best-studied inflammasome, the NLRP3 inflammasome,
was revealed to be activated in cardiomyocytes during HF and
produced a set of cytokines to promote inflammation (18). However, it remains poorly understood
as to whether CFs undergo inflammasome activation during cardiac
dysfunction. A recent study indicated that lipid metabolism was
closely associated with inflammasome functions (19). Hyperlipemia has consistently been
identified to be associated with severe outcomes, including the
development of HF, mortality and other cardiovascular events, and
low-density lipoprotein (LDL) is considered the most common risk
factor (6). However, how
hyperlipemia and LDL may influence cardiomyocyte function remains
unclear. The present study revealed that the LDL-induced NLRC3
inflammasome was extensively activated in the CFs of patients with
HF, which resulted in the release of excessive amounts of IL10 and
TNF-α, thus impairing the viability of cardiomyocytes and promoting
neonatal cardiomyocytes apoptosis. Inhibition of the LDL-induced
inflammasome proved beneficial and exerted protective effects on
cardiomyocytic function, thereby providing a novel target for
therapeutic intervention.
Materials and methods
Patients and samples
Serum samples were collected from 62 patients with
HF (mean age, 71.2 years; age range, 57–93 years; male:
Female=0.63:1) and 20 healthy controls (age range, 50–87 years;
male: Female=0.67:1) at the Department of Cardiology, Feicheng
Mining Center Hospital (Feicheng, China) between October 2015 and
June 2020. The diagnosis and classification of HF was conducted
according to the Framingham criteria (20). None of the patients had received
treatments prior to blood collection. The patient information,
including general characteristics, stages and prognosis, was
obtained from the medical records or outpatient follow-up records,
and is summarized in Table I. In
addition, three left ventricle (LV) samples were collected from
donors with HF following death and three LV samples were obtained
from donors dying of injurious accidents (age range, 37–61 years;
male: Female=2:1) between January 2015 and March 2019 at Feicheng
Mining Center Hospital, (Feicheng, China). The specimens were
immediately frozen in liquid nitrogen and stored until further
study. For IHC staining, tissues were embedded in paraffin. The
samples were numbered as ‘type (HF or control) + pathology no. +
region (LV) + section no.’, such as HF-15328-LV1. Written informed
consent for use of LV tissues was provided by the dying donors
themselves or their family members. The aforementioned procedures
were approved by the Research Ethics Committee of Feicheng Mining
Center Hospital. All patients provided written informed consent
according to the Declaration of Helsinki.
 | Table I.Characteristics of patients with
heart failure (62 cases). |
Table I.
Characteristics of patients with
heart failure (62 cases).
|
Characteristics | No. (%) | OR |
|---|
| Sex |
| 0.66 |
|
Male | 24 (38.71) |
|
|
Female | 38 (61.29) |
|
| Age, years |
| 1.39 |
|
<60 | 2 (3.23) |
|
|
60–80 | 37 (59.68) |
|
|
>80 | 23 (37.10) |
|
| Prognosis |
| 0.88 |
|
Alive | 49 (79.03) |
|
|
Dead | 13 (20.97) |
|
| Stage |
| 1.52 |
|
Mild | 28 (45.16) |
|
|
Severe | 34 (54.84) |
|
| IL10 (pg/ml) |
| 2.09 |
|
<3,500 | 25 (40.32) |
|
|
≥3,500 | 37 (59.68) |
|
| TNF-α (pg/ml) |
| 1.68 |
|
<3,000 | 23 (37.10) |
|
|
≥3,000 | 39 (62.90) |
|
Mice
The C57/BL6 wild type (WT) mice (n=6; male; age, 3
months; weight, 22–25 g) were acquired from Jackson Laboratory.
Mice were housed in the SPF animal room at 26°C with 50% humidity,
12-h light/dark cycles and free access to food/water. The WT mice
were treated with lipopolysaccharide (LPS; cat. no. QN0166; Beijing
Biolab Technology Co., Ltd.; 4 mg/kg) or PBS (control group) via
intraperitoneal injection once a day for 3 days. A total of 2 days
after LPS treatment, mice without any serious adverse reactions
were euthanized using CO2. Anesthesia was induced with
15% CO2 for 2–3 min and the chamber was then filled with
100% CO2 at a flow rate of 10–30% per min for ~10 min.
The euthanasia of neonatal pups was via decapitation. Bilateral
mydriasis and asystole were used to verify the death of animals. In
addition, Dahl salt-sensitive (Dahl/ss) HF mice (n=6; male; age,
3–4 months; weight, 22–25 g) were purchased from Beijing Vital
River Laboratory Animal Technology Co., Ltd.. These mice were
housed at 26°C with 50% humidity, 12-h light/dark cycles and free
access to 0.4% NaCl food. The Dahl/ss hypertensive model is a
well-established rodent model of salt-sensitive hypertension and
congestive HF. At 15 weeks of age, female Dahl/ss/obese mice
presented with LV diastolic dysfunction, as determined by marked LV
hypertrophy and fibrosis, associated with increased cardiac
oxidative stress and inflammation (21). The animal experiments were conducted
between November 2018 and September 2019. All animals were
maintained at the Laboratory Animal Facility, Feicheng Mining
Center Hospital. The animal studies were approved by the Animal
Care Committee of Feicheng Mining Center Hospital.
Cells
Cells were isolated as previously reported, with
some minor modifications (22). CFs
were derived from the adult WT or Dahl/ss mice. Briefly, heart
tissues were dissociated using the collagenase II enzyme (cat. no.
17101015; 160 U/ml; Thermo Fisher Scientific, Inc.) at 37°C for 30
min. After washing three times, the cell suspension was passed
through a Corning® 70-µm cell strainer (Sigma-Aldrich;
Merck KGaA). Subsequently, the endothelial cells were removed using
magnetic beads according to a previously described protocol
(23). Cells were subsequently
cultured in a humidified incubator (Thermo Fisher Scientific, Inc.)
containing 5% CO2 at 37°C for 1 h. The adherent cells
could be identified as CFs based on the morphology via light
microscopy. Finally, the CFs were cultured with Dulbecco's modified
Eagle's medium (DMEM)-F12 (Gibco; Thermo Fisher Scientific, Inc.)
supplemented with 10% fetal calf serum (FCS; Gibco; Thermo Fisher
Scientific, Inc.), L-glutamine (cat. no. G7513; 20 mM;
Sigma-Aldrich; Merck KGaA) and penicillin/streptomycin (cat. no.
V900929; 100 U/ml; Sigma-Aldrich; Merck KGaA). Cells from passages
1–3 were used for subsequent experiments. When CFs were stimulated
by LPS (1 µg/ml) for 6 h at 37°C, SBC-115076 (cat. no. SML1712; 10
ng/ml; Sigma-Aldrich; Merck KGaA) was utilized to treat the cells
for 24 h at 37°C to inhibit LDL production.
Neonatal cardiomyocytes were dissociated as
previously described (24).
Briefly, hearts from 5 days postnatal (P5) mice (born from the WT
mice acquired from Jackson Laboratory) were collected, minced and
digested using collagenase II for 30 min at 37°C (160 U/ml). After
washing three times, cells were suspended in medium 199 (cat. no.
M4530; Sigma-Aldrich; Merck KGaA) with 10% FCS. The cardiomyocytes
were enriched using the pre-plating approach to remove
contaminating cells before being seeded in cell culture plates.
After 3 days, cells formed a confluent monolayer consisting of 95%
cardiomyocytes pulsing in synchrony, which were used in the
subsequent experiments. To assess the viability of cardiomyocytes,
the pulses of random cells were counted in 1 min under a light
microscope. Neonatal cardiomyocytes were cultured and treated with
IL10 at 37°C for 24 h (cat. no. I3019; 1 µg/ml; Sigma-Aldrich;
Merck KGaA) and TNF-α (cat. no. 654245; 1 µg/ml; Sigma-Aldrich;
Merck KGaA). The control group was treated with PBS. The
supernatant of LPS-stimulated CF cultures was harvested and mixed
with neonatal cardiomyocytes (1:1).
Immunohistochemistry (IHC)
Human or mouse tissues were dehydrated utilizing an
ethanol gradient (70, 80 and 95%; 5 min each) followed by
incubation with 100% ethanol three times for 5 min per incubation
at room temperature. The paraffin-embedded blocks were sliced into
5 µm sections and washed in a water bath at 40°C for 15 min. Prior
to dehydration and paraffin embedding, sections were fixed in 4%
paraformaldehyde (Image-iT™ Fixation/Permeabilization kit;
Invitrogen; Thermo Fisher Scientific, Inc.) at 4°C overnight. The
sections were heated in media containing 5% FBS (Gibco; Thermo
Fisher Scientific, Inc.) at 75°C for 30 min to reduce unspecific
background. Briefly, to investigate the expression of
fibroblast-activated protein (FAP), NLRC3 and LDL in the heart
tissues, the prepared sections were stained with antibodies against
FAP (cat. no. 66562; 1:200; Cell Signaling Technology, Inc.), NLRC3
(cat. no. ab77817; 1:100; Abcam) and LDL (cat. no. ab14519; 1:100;
Abcam) at 4°C overnight. The sections were then washed with PBS and
incubated with horseradish peroxidase-labeled goat anti-rabbit/or
anti-mouse IgG (cat. nos. A0208 and A0216; 1:100; Beyotime
Institute of Biotechnology) for 1 h at room temperature. Staining
was evaluated using a light microscope (magnification, ×400).
To assess the proliferation of cardiomyocytes, Ki-67
staining was performed. Cells were fixed with 4% paraformaldehyde
at room temperature for 30 min. Cells were incubated with the Ki-67
primary antibody (cat. no. 9449; 1:100; Cell Signaling Technology,
Inc.) at 4°C overnight, followed by incubation with a horseradish
peroxidase-labeled goat anti-rabbit/or anti-mouse IgG (cat. nos.
A0208 and A0216; 1:100; Beyotime Institute of Biotechnology) for 1
h at room temperature. Cells were counterstained with an anti-mouse
IgG (H+L) immunofluorescence antibody (cat. no. 4410; 1:200; Cell
Signaling Technology, Inc.) and DAPI. The scores of stained cells
(Ki-67+) were calculated by counting the average number
of positive cells in 500 nuclei in four random high-power
magnifications using a Nikon TE200 fluorescence microscope
(magnification, ×400).
Both human and mouse tissue were fixed using 10%
formalin at room temperature for 24–36 h. Tissues sections (8-µm
thick) were stained using hematoxylin and eosin at room temperature
for 3 min, respectively. Staining was observed using a light
microscope (magnification, ×400).
ELISA
The levels of IL1β, IL10, IL18 and TNF-α were
examined by utilizing ELISA kits (cat. nos. RAB0275 RAB0244,
RAB0267 and RAB1089; Sigma-Aldrich; Merck KGaA). Briefly, 100 µl
cell culture supernatant or sera was added into duplicate wells and
incubated at 37°C for 90 min. After washing three times, the
indicated primary antibodies (1:200) were added and incubated at
37°C for 60 min. Samples were washed a further three times, and 100
µl indicated secondary antibodies were added from the ELISA kits
and incubated at 37°C for 60 min. Following a color reaction with
TMB substrate for 15 min on ice, absorbance was detected at 450 nm
using a microplate reader (800TS; BioTek Corporation).
Western blot analysis
Tissues and cells were lysed in NP-40 splitting
buffer (GeneTex, Inc.; pH, 7.4) containing protease-inhibitor
cocktail (cat. no. 5871; Cell Signaling Technology, Inc.). Protein
concentrations were determined using a NanoDrop spectrophotometer
(Thermo Fisher Scientific. Inc.). Proteins (5 µg) were separated by
SDS-PAGE using 10% gels and were transferred onto PVDF membranes
(cat. no. ISEQ00010; EMD Millipore). Membranes were subsequently
incubated with antibodies against IL1β (cat. no. 12703; 1:500; Cell
Signaling Technology, Inc.), IL10 (cat. no. 12163; 1:500; Cell
Signaling Technology, Inc.), IL18 (cat. no. K002143p; 1:500;
Beijing Solarbio Science & Technology Co., Ltd.), TNF-α (cat.
no. ab183218; 1:500; Abcam), LDL (cat. no. ab14519; 1:1,000;
Abcam), NLRC3 (cat. no. ab77817; 1:1,000; Abcam) and GAPDH (cat.
no. ab8245; 1:3,000; Abcam) overnight at 4°C, and were then probed
with goat-anti-rabbit secondary antibodies (cat. no. A0545; 0.4
µg/ml; 1:2,500; Sigma-Aldrich; Merck KGaA) or rabbit-anti-mouse
secondary antibodies (cat. no. A9044; 0.4 µg/ml; 1:2,500;
Sigma-Aldrich; Merck KGaA) for 1 h at room temperature. The
immunoblotting results were visualized using TMB (cat. no. PR1210;
Beijing Solarbio Science & Technology Co., Ltd.).
RNA extraction and reverse
transcription-quantitative PCR (RT-qPCR)
RNA was isolated using TRIzol® reagent
(Invitrogen; Thermo Fisher Scientific. Inc.) according to the
manufacturer's protocol. cDNA was synthesized using iScript RT
system (Invitrogen; Thermo Fisher Scientific, Inc.) and 2 µg RNA
according to the manufacturer's protocol. mRNA expression levels
were determined using the iQ SYBR Green Supermix kit (Bio-Rad
Laboratories, Inc.) and the Bio-Rad iQ5 Multicolor Real-Time PCR
detection system (Bio-Rad Laboratories, Inc.), and were normalized
to the mRNA expression levels of GAPDH. qPCR was performed
based on the following conditions: Initial denaturation at 95°C for
5 min; followed by 35 cycles at 95°C for 30 sec and 60°C for 30
sec. The 2−∆∆Cq method (25) was performed to calculate the fold
change and all experiments were conducted at least three times.
The following primer sequences were used:
IL10, forward 5′-AGAAGTACCTGGACTCGCCA-3′, reverse
3′-CTGGAAGTCCCACTTCATCTGT-5′; TNFA, forward
5′-TCTCCCAGGAGCCGACTG-3′, reverse3′-ATCCCAAAAGCGACCCAGTG-5′;
NLRP1, forward 5′-GCCTTGGTGAAACCAGGAGA-3′, reverse
3′-GCTGCTCTCGATACTGGTCC-5′; NLRP3, forward
5′-CCTGAGCAGCCTCATCAGAA-3′, reverse 3′-GCAAGTGCTGCAGTTTCTCC-5′;
NLRP4, forward 5′-TGCTGGCCTTCTAACCAACA-3′, reverse
3′-CACACCCACATCTCCGATTTC-5′; NLRP5, forward
5′-GGCTGGCTGTGGTTTTCTTG-3′, reverse 3′-AGGTGTCTGCTCCTCGAGAT-5′;
NLRP8, forward 5′-GAGAGGCTGTCGCAGAGTAA-3′, reverse
3′-TTGGTTTTTGCGGAGCATGG-5′; NLRP9, forward
5′-CGGATTTTGGCTTGTTGTGGT-3′, reverse 3′-ACAAGATAAACAGCGGCCCA-5′;
NLRP10, forward 5′-TTTGTCGGAGACGGAGAAGC-3′, reverse
3′-TAACTGTCCATGCGCTGGTT-5′; NLRP12, forward
5′-GCCAAAGTGCACCCAGAATG-3′, reverse 3′-GTCAGCCTCCCTTCCTTCAT-5′;
NLRP14, forward 5′-ACCAACACAGACGTGGATTGT-3′, reverse
3′-CAACTGGCATCCTCACAGGT-5′; NOD2, forward
5′-GTACGAGATGCAGGAGGAGC-3′, reverse 3′-GCCAATGTCACCCACAGAGT-5′;
NLRC1, forward 5′-GTCTGGACGAGTTCCGTTGT-3′, reverse
3′-TGTTCCCGTTAAGCTGGTCC-5′; NLRC3, forward
5′-TCTCCGGCACCTGGATCTTA-3′, reverse 3′-GGGCCAAGGATTTAGTGGCT-5′; and
GAPDH, forward 5′-GGGTGATGCAGGTGCTACTT-3′ and reverse
3′-GGCAGGTTTCTCAAGACGGA-5′.
Flow cytometry (FCM)
FCM was performed using the Annexin V-FITC/PI
Apoptosis Detection kit (cat. no. C1062S; Beyotime Institute of
Biotechnology). Briefly, cells were diluted to
1×105/ml/per well, dissociated in 1X binding buffer on
ice for 30 min, and were subsequently stained with Annexin V-FITC
(10 µmol/µl) and propidium iodide (PI; 10 µmol/µl) in the dark at
room temperature for 20 min. The cells were then analyzed using FCM
(FACSCanto II; BD Biosciences) and FlowJo software (version 10.0;
BD Biosciences). The apoptotic rate was calculated as the
percentage of Annexin V-FITC+/PI+ cells in
total cells.
Lentivirus production
Lentiviruses were prepared using LDL short hairpin
RNA (shRNA;
5′-CCGGAGTCGCCATTCTCCCTTAATACTCGAGTATTAAGGGAGAATGGCGACTTTTTTG-3′),
NLRC3
(5′-CCGGACCACGGTCTGCACCATATTACTCGAGTAATATGGTGCAGACCGTGGTTTTTTG-3′)
or scrambled shRNA
(5′-CCTAAGGTTAAGTCGCCCTCGCTCGAGCGAGGGCGACTTAACCTTAGG,CAGGTCGAAGCGGTCGCTAAAAAGCTGGCTCTATTAAACAGGTCG-3′)
(all Sigma-Aldrich; Merck KGaA), 0.72 pmol pMD2.G, 1.3 pmol psPAX2
(Addgene, Inc.) and 1.64 pmol Col plasmids (Addgene, Inc.)
according to the standard protocol. Briefly, Lenti-X 293T packaging
cells (China National Infrastructure of Cell Line Resource) were
seeded at 3.8×106/6-well plate in DMEM and incubated at
37°C in an atmosphere containing 5% CO2 for 20 h. The
medium was then gently aspirated and 10 ml fresh DMEM containing 25
µM chloroquine diphosphate was added to the cells for 5 h at 37°C.
Subsequently, diluted polyethylenimine (5%) was added dropwise to
the cells while gently flicking the diluted DNA tube. 293T cells
were incubated for 15–20 min at room temperature. The transfection
mix was transferred to the Lenti-X 293T packaging cells with
continuous incubation for 18 h 37°C. Viruses were harvested at 48,
72, and 96 h post-transfection. The viral supernatant was
centrifuged at 500 × g for 5 min at 4°C to pellet any packaging
cells that were collected during harvesting. The supernatant was
filtered through a 0.45 µm PES filter. Murine CFs
(5×106/ml) were infected with the prepared lentivirus at
37°C for ~5 h, cultured with complete DMEM, harvested after 72 h,
then subjected to RT-qPCR and subsequent experiments.
MTT assay
Neonatal cardiomyocytes (1,000 cells/well) were
seeded into 96-well plates (Thermo Fisher Scientific, Inc.) and
incubated for 24 h at 37°C in an atmosphere containing 5%
CO2. Cell viability was evaluated by performing an MTT
assay. Briefly, 0.5 mg Thiazolyl Blue Tetrazolium Bromide (cat. no.
M2128; Sigma-Aldrich; Merck KGaA) was added to each well for 4 h at
4°C. After removing the supernatant, 100 µl dimethyl sulfoxide
(cat. no. 1912433; Johnson & Johnson Services, Inc.) was added
to each well and incubated for 10 min at the room temperature. The
purple mixture in each well was then measured at 490 nm using a
microplate reader (800TS; BioTek Corporation). The inhibition rate
(%) was analyzed using the following formula: [(Control
OD490-Experimental OD490)/Control OD490] ×100.
Statistical analysis
Data are presented as the mean ± standard deviation,
and were analyzed by Student's t-test using SPSS v.19.0 (IBM Corp.)
and GraphPad Prism v.8.0 (GraphPad Software, Inc.). The unpaired
t-test was used for comparison between two groups, whereas
comparison of mean values between multiple groups was evaluated by
one-way ANOVA followed by Student-Newman-Keuls post hoc test. The
functional experiments were performed at least three times. The
correlation between NLRC3 mRNA expression and IL10 or
TNFA mRNA expression was analyzed by Pearson's correlation.
Logistic regression was performed to analyze the clinical risk
factors. P<0.05 was considered to indicate a statistically
significant difference.
Results
CFs are activated during HF resulting
in microenvironmental inflammation
An inflammatory microenvironment has been
characterized as an important feature of HF (26). To observe the inflammation in
patients with HF, a total of 62 patients with HF and 20 healthy
controls were included in the current cohort: 28 (45.16%) patients
suffered from mild HF and 34 (54.84%) were in the severe stage
(Table I). Sera were harvested from
these patients and healthy controls to examine the levels of
well-known inflammatory factors, including IL1β, IL10, IL18 and
TNF-α. As shown in Fig. 1A, all of
these cytokines were elevated in patients with mild and severe HF,
and were progressively increased in the severe stage. Furthermore,
IL10 and TNF-α were most significantly increased in the severe
stage (Fig. 1A; Table I). As evidenced by logistic
regression analysis, the expression levels of IL10 and TNF-α, as
well as age and HF stage, affected the prognosis, which indicated
that IL10 and TNF-α could be considered valuable prognostic
indicators for patients with HF (Table
I).
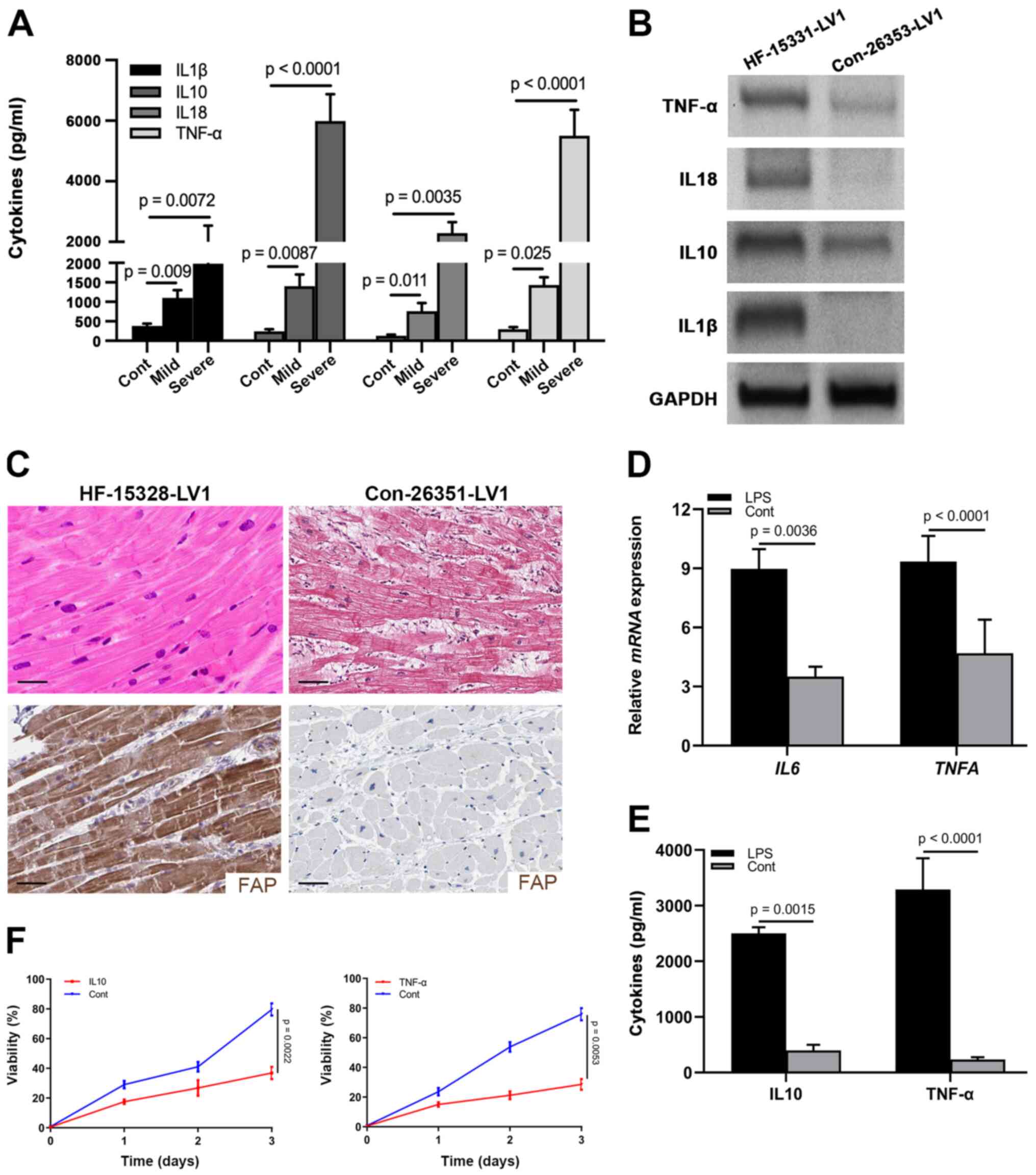 | Figure 1.CFs are activated in the heart
tissues of patients with HF. (A) Levels of IL1β, IL10, IL18 and
TNF-α in the sera from healthy controls (n=20), and patients with
mild (n=28) and severe (n=34) HF, as determined by ELISA. (B)
Protein expression levels of IL1β, IL10, IL18 and TNF-α in
myocardial samples from a control individual (Con-26353-LV1) and
patient with HF (HF-15331-LV1), as determined by western blotting.
GAPDH was used as the internal control. (C) Hematoxylin and eosin
staining and immunohistochemical staining of FAP in myocardial
samples from a control individual (Con-26351-LV1) and patient with
HF (HF-15328-LV1) (scale bar, 10 µm). (D) mRNA expression levels of
IL10 and TNFA in cardiac tissues from LPS-treated
mice. (E) Levels of IL10 and TNF-α in the supernatant of CFs
treated with LPS. (F) MTT assay showing the viability of neonatal
cardiomyocytes treated with IL10 and TNF-α. CFs, cardiac
fibroblasts; HF, heart failure; FAP, fibroblast-activated
protein. |
Similarly, western blot analysis detected higher
expression levels of IL1β, IL10, IL18 and TNF-α in heart tissues
from donors with HF (Fig. 1B).
Previously, activated fibroblasts have been reported to represent a
common pathological characteristic of cardiac fibrosis and heart
dysfunction (27,28). Accordingly, to detect the activation
of CFs in the cardiac tissues of HF hearts, the expression of FAP
was detected by IHC in human donor samples. A large number of CFs
(FAP+) were present across the cardiac tissues of HF
donors compared with in the normal controls, which were positively
associated with fibrosis in the hematoxylin and eosin staining
(Fig. 1C).
To confirm the presence of inflammation in cardiac
tissues, LPS was used to treat mice for 3 days and the heart
tissues were harvested to examine the mRNA expression levels of
IL10 and TNFA. As shown in Fig. 1D, the mRNA expression levels of
IL10 and TNFA were upregulated in the heart tissues
of LPS-stimulated mice compared with those in the control group,
indicating that LPS could induce inflammation. To further identify
the role of CFs in the production of IL10 and TNF-α, CFs were
cultured with LPS stimulation and the supernatant was collected to
examine the concentration of IL10 and TNF-α. As expected, IL10 and
TNF-α were significantly elevated in the LPS treatment group
compared with those in the control group (Fig. 1E), which indicated that activated
fibroblasts could provide specific inflammatory factors. With the
goal of identifying the effects of IL10 and TNF-α on the viability
of neonatal cardiomyocytes, cells purified from P5 mice were
cultured and treated with IL10 and TNF-α. Subsequently, the results
of an MTT assay revealed that cell viability was significantly
suppressed by these two cytokines (Fig.
1F). Taken together, these data suggested that IL10 and TNF-α
derived from the activated CFs may attenuate the viability of
cardiomyocytes.
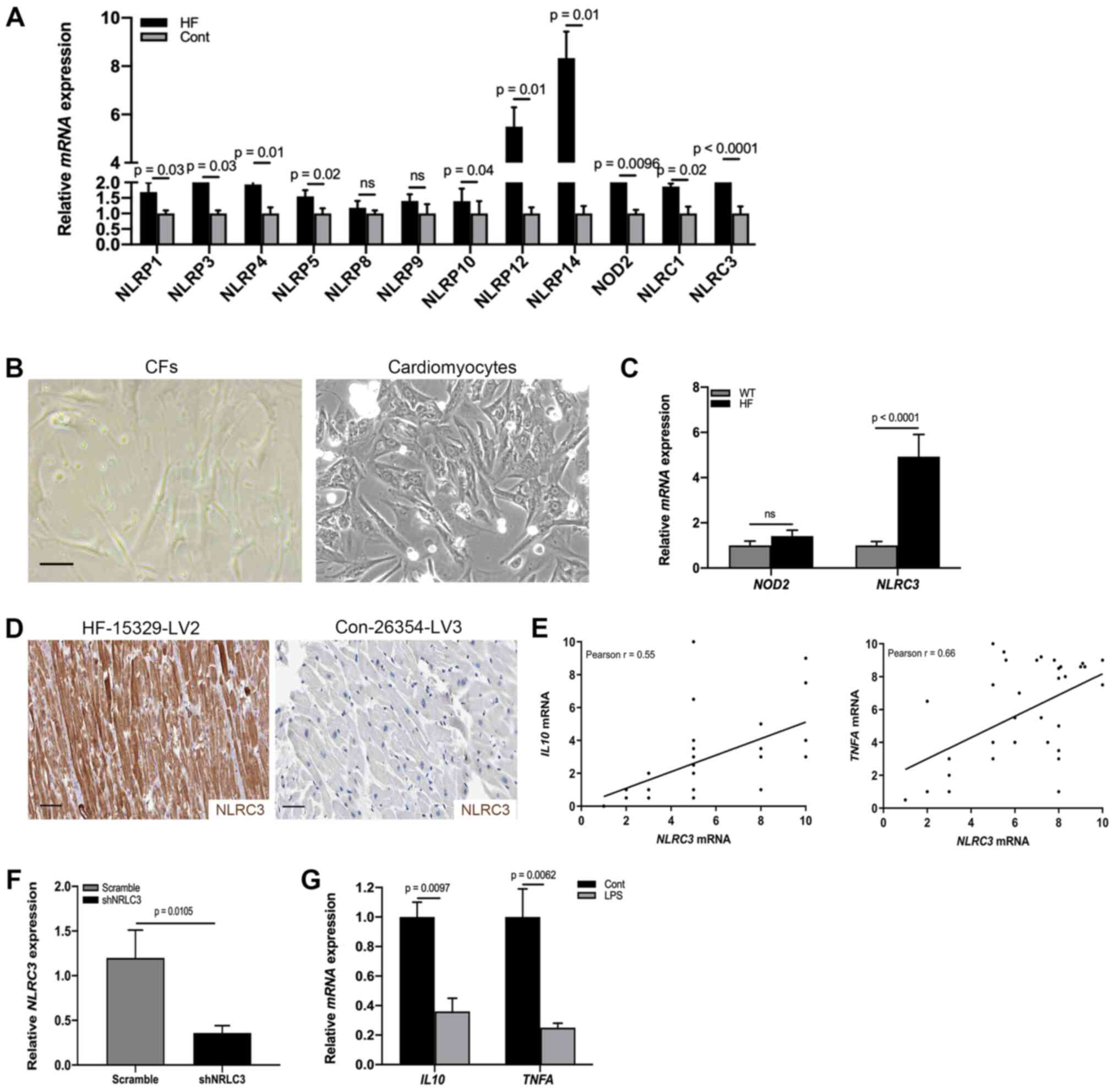
NLRC3 inflammasome is activated in CFs
of HF
Having identified fibroblast activation in the heart
tissues of patients with HF, the present study aimed to detect the
molecular mechanism underlying the proinflammatory phenotype of
CFs. Previous studies reported that inflammasome activation
contributed to the release of a wide variety of cytokines,
including IL1β, IL18 and TNF-α in the absence of cell death
(16,29). To detect specific activation of the
inflammasome during cardiac dysfunction, RT-qPCR was used to
analyze the heart tissues of patients with HF and healthy control
donors; the results indicated that the mRNA expression levels of
NOD2 and NLRC3 were significantly increased compared
with those in the control group (Fig.
2A). By contrast, the expression levels of NLRP1, NLRP3, NLRP4,
NLRP5, NLRP8, NLRP9, NLRP12, NLRP14 and NLRC1 were not
significantly altered. NOD2 and the NLRP3 inflammasome have been
reported to be activated in mesenchymal stromal cells in mice with
cardiomyopathy (30). However, it
is poorly understood whether CFs undergo NLRC3 inflammasome
activation to promote cardiac dysfunction. To address this,
fibroblasts were collected from HF model heart tissues (Fig. 2B) and the mRNA expression levels of
NOD2 and NLRC3 were examined by RT-qPCR. As shown in
Fig. 2C, significant differences
existed in NLRC3 mRNA expression. Similar to the results of
RT-qPCR analysis, a proportion (31.2%) of CFs expressed NLRC3 in
the cardiac tissues of HF mouse models (Fig. 2D). In order to ascertain the
association between NLRC3 inflammasome activation and the
proinflammatory phenotype, a correlation analysis was performed
using RT-qPCR data; a positive correlation was detected between
NLRC3 and IL10/TNFA mRNA expression (Fig. 2E). To explore the role of NLRC3 in
IL production, CFs were infected with a lentiviral plasmid
containing a NLRC3-specific shRNA, in order to reduce the
expression of NLRC3 (Fig. 2F).
After NLRC3-deficient cells were treated with LPS for 72 h, the
mRNA expression levels IL10 and TNFA were revealed to
be downregulated, demonstrating inactivation of CFs lacking NLRC3
(Fig. 2G). Collectively, these data
suggested that NLRC3 inflammasome activation may contribute to the
proinflammatory features of CFs.
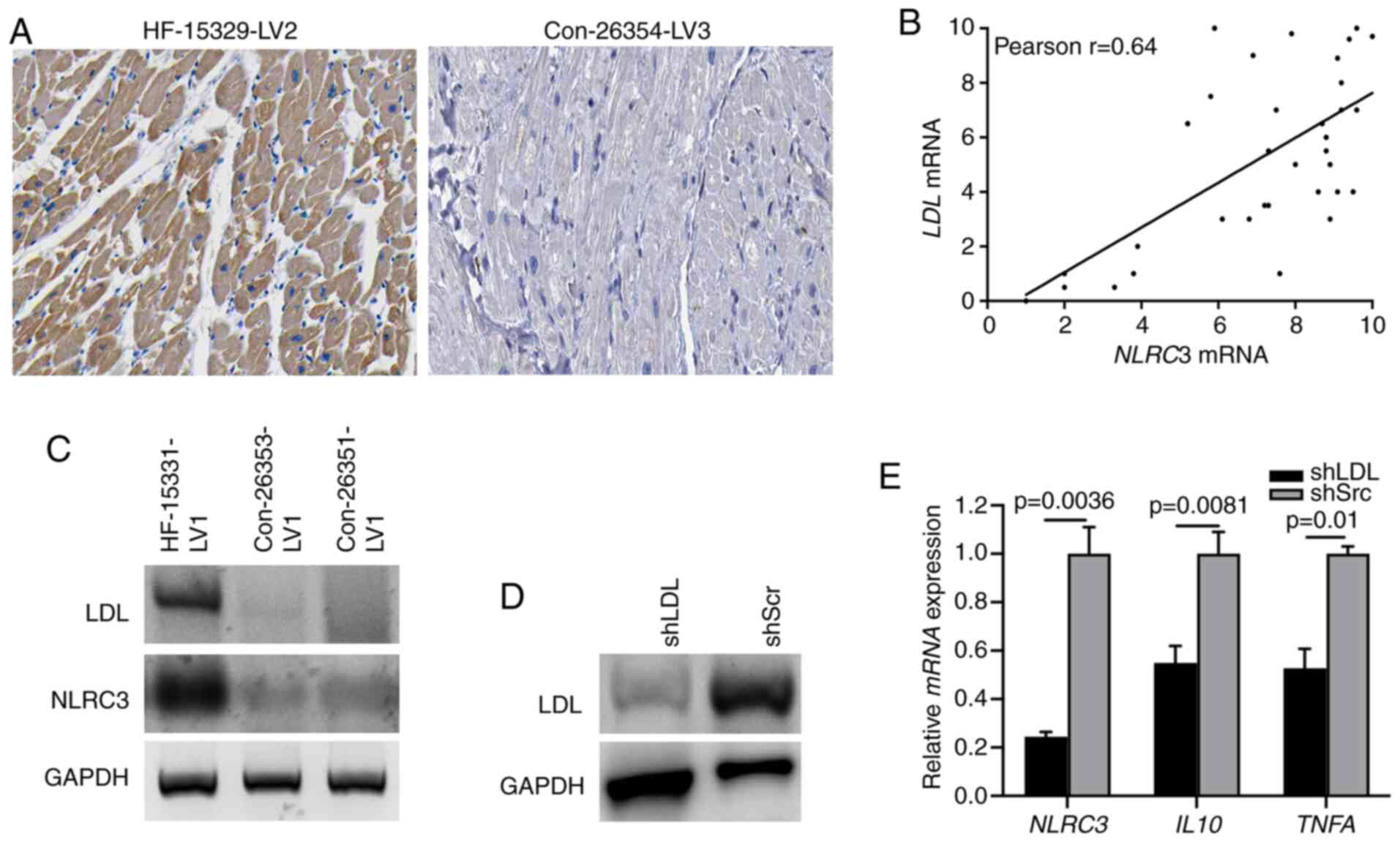
LDL is required for NLRC3 inflammasome
activation in CFs
Previously, lipid metabolism has been reported to be
involved in the progression of chronic inflammation (31). LDL, as a modulator for lipid
metabolism, serves a critical role in activating broadly diverse
pathways associated with inflammation and fibrosis (32). A recent study demonstrated that the
level of LDL was highly increased in activated fibroblasts
(33). Therefore, the present study
aimed to determine whether the expression of LDL could induce NLRC3
inflammasome activation. Diffuse LDL immunostaining was observed in
the cardiac tissues of patients with HF, supporting the evidence
linking LDL with cardiac dysfunction (Fig. 3A). Moreover, a strong correlation
between LDL and NLRC3 expression was determined based
on the RT-qPCR (Fig. 3B), which was
further indicated via immunoblotting of human HF heart tissues
(Fig. 3C). To further detect the
role of LDL in NLRC3 inflammasome activation, CFs were infected
with LDL shRNA or scrambled shRNA to knockdown LDL expression
(Fig. 3D). When treating
LDL-deficient cells with LPS, the results of RT-qPCR revealed that
the mRNA expression levels of NLRC3, IL10 and TNFA
were significantly suppressed compared with those in the control
group (also treated with LPS) (Fig.
3E). These findings indicated that LDL may confer
proinflammatory signatures to CFs through the NLRC3
inflammasome.
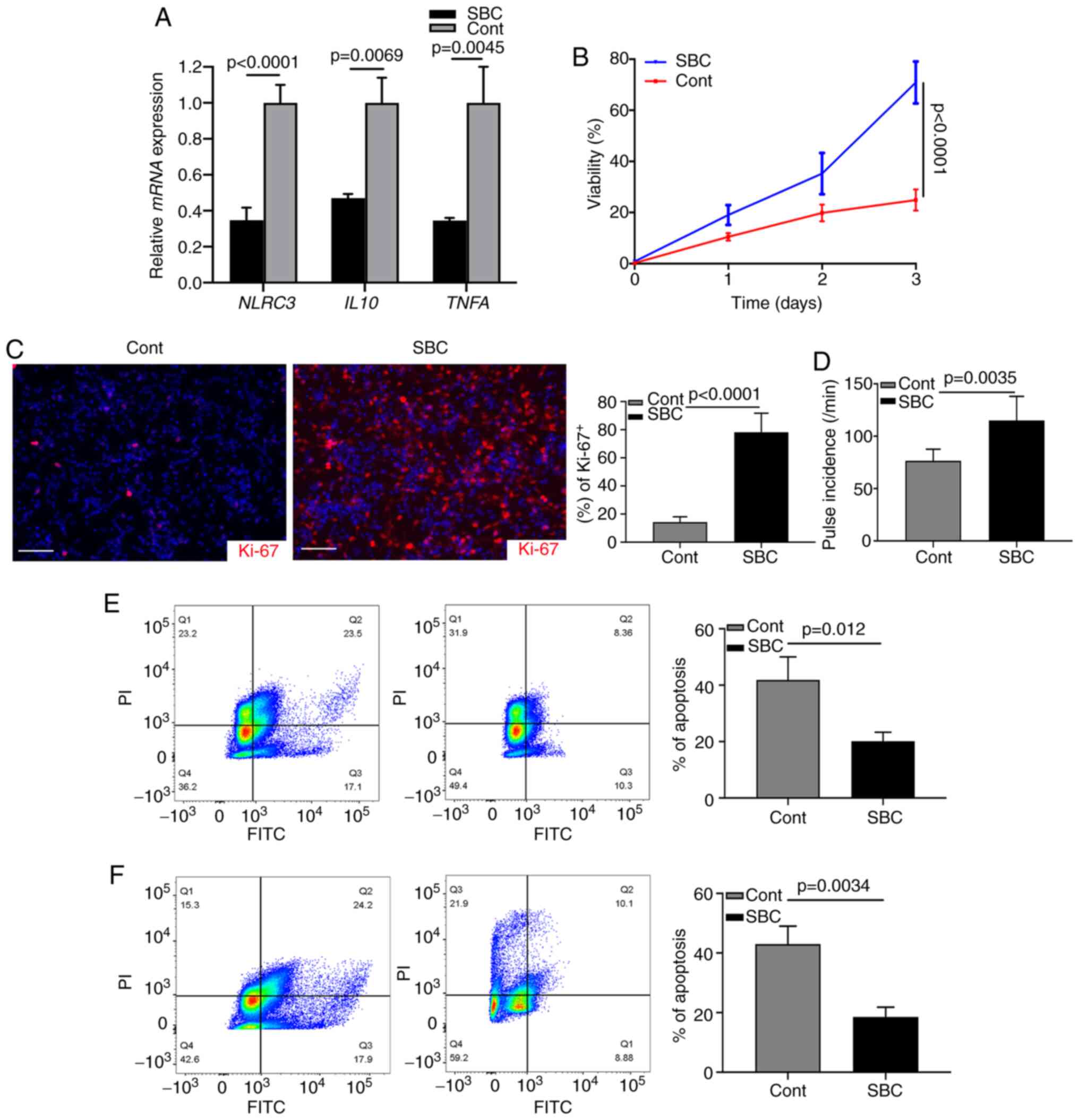
Targeting LDL improves cardiomyocyte
function
The aforementioned data indicated that activated CFs
may contribute to the progression of cardiac chronic inflammation.
The present study investigated whether cardiomyocyte injury could
be inhibited by targeting the LDL-induced NLRC3 inflammasome. A
proprotein convertase subtilisin/kexin type 9 (PCSK9) inhibitor
(SBC-115076) was used to inhibit the levels of LDL in CF cultures
(34). Following LPS stimulation,
the mRNA expression levels of NLRC3, IL10 and TNFA
were significantly suppressed by inhibition of LDL (Fig. 4A). Subsequently, the supernatant of
LPS-stimulated CF cultures was harvested and mixed with neonatal
cardiomyocytes (1:1); the results of a subsequent MTT assay
revealed that cell viability was reduced by 70% in the untreated
group (Fig. 4B). Ki-67 index was
examined by immunostaining and revealed that the expression levels
of Ki-67 were decreased in the control group without treatment
(Fig. 4C). Most cardiomyocytes were
actively proliferating (Ki-67+) after SBC-115076
treatment (Fig. 4C). When counting
the pulse of cardiomyocytes, it was similarly revealed that
SBC-115076 treatment significantly improved the viability of cells
(Fig. 4D). These data suggested
that inhibiting chronic inflammation via targeting LDL may provide
beneficial effects on cardiomyocytic function. In addition, when
neonatal cardiomyocytes were cultured with the supernatant of
LPS-stimulated CFs or with fibroblasts from the hearts of a mouse
model of HF, FCM analysis revealed that SBC-115076 treatment
markedly inhibited the apoptosis of cardiomyocytes compared with
that in the control group (Fig. 4E and
F). These data demonstrated that activated CFs may construct an
inflammasome-associated cytokine microenvironment promoting cardiac
dysfunction.
Discussion
HF progresses over time as the cardiac pulse loses
power; this results in a decline in exercise tolerance and the
dysfunction of other systems, which seriously affects daily life.
In the United States, ~6.0 million people suffer from HF, and both
children and adults can be diagnosed with this disorder; however,
the symptoms and clinical management differ. Notably, cardiac
dysfunction is not a normal part of aging nor is it a disease of
old age. HF has emerged as the seventh leading cause of death in
the United States and ~300,000 Americans <60 years old suffer
from mild HF (35). Consistent with
these previous findings, the present study reported that age and
risk stages affected the prognosis of patients with HF. However, in
China, there remains a shortage of systematic databases to
comprehensively analyze the features of Chinese patient with HF.
The development of a corresponding system may help to improve the
diagnosis and treatment of HF.
A well-known risk factor of HF is cardiovascular
events. The majority of patients with HF also suffer from
cardiovascular pathophysiological conditions, such as hypertension,
coronary heart disease and hyperlipemia. For patients with chronic
HF, the cutoff for total cholesterol is between 190 and 200 mg/dl
for the diagnosis of chronic HF (36). Previous retrospective trials have
reported that statins may have beneficial effects on a subset of
patients with HF, particularly with regards to mortality and
disease worsening (37,38). LDL or cholesterol can build up in
the walls of arteries and increase the risk of heart disorders.
Cholesterol levels should be measured at least once every 2 years
for all individuals >20 years old (39). After measuring the 10-year risk,
doctors will recommend a percentage by which the patient should try
to lower LDL levels via improving diet, exercise and medication if
necessary (40). In addition to
statins, a PCSK9 inhibitor has recently been reported to
significantly decrease the levels of LDL (34). In terms of microenvironmental
effects, the present study ruled out the molecular basis regarding
the correlation between lipid metabolism and HF, and highlighted
the contribution of LDL-induced inflammasome activation to
cardiomyocytic dysfunction. The proinflammatory phenotype could be
partly attributed to this mechanism. Similarly, Stoekenbroek et
al (34) identified the
TREM2-ApoE pathway as the main mediator of the inflammatory
phenotype in microglia in neurodegenerative conditions and
indicated that targeting lipid metabolism could restore homeostatic
fibrosis components.
HF is currently at the forefront of biomedical
research. Scientists have discovered numerous aspects of HF and
other cardiovascular diseases, and research advances have provided
information on how cardiac pathogenesis affects heart function.
Numerous approaches are currently under investigation worldwide,
with the aim of better understanding the molecular basis of HF
(41,42). Potentially, cholesterol or LDL may
lead to chronic inflammation, which is a serious cause of HF
progression. When high levels of cholesterol occur in the blood,
excessive LDL can seep into the myocardium or arterial inner walls,
triggering an inflammatory response and enhancing the activation of
numerous cellular components (43).
In addition, the deposited cholesterol may rupture and cause
cardiovascular events and strokes via blood clots, during which
chronic inflammation may have a role. Inflammation has been
identified as the link between numerous disorders and conditions,
particularly those influencing the cardiovascular and nervous
systems (44). In addition to HF or
other heart diseases, diabetes, dementia and cancer have been
associated with inappropriate and chronic inflammation (45). For example, microglia- or
astrocyte-derived inflammatory factors have been reported to
contribute to inhibiting the proliferation of neural stem cells in
the hippocampus of patients with Alzheimer's disease (46). Lipid-triggered neuroinflammation has
also been reported in the majority of malignant brain tumors, where
it may promote progression (47).
Both cancer cells and non-malignant stromal cells can exhibit a
proinflammatory phenotype with the goal of accelerating tumor
growth and resisting chemoradiotherapy (48). Although studies regarding
cardio-inflammation have suggested that a number of patients
develop hyperlipemia as they age, those with HF tend to develop far
more and in a predictable pattern, beginning in the areas important
for exercise tolerance. However, the exact role of chronic
inflammation in cardiomyocyte dysfunction remains to be completely
elucidated, although it has been hypothesized that it may damage
pulse function and promote the processes of cardiac fibrosis.
Fibroblasts represent an intrinsic and dynamic cell
population in the cardiac microenvironment, which produce cytokines
and extracellular matrix components (28). Fibroblast activation exerts both
detrimental and beneficial effects on various disorders (42). Appropriate cardiomyocyte-fibroblast
interactions are essential for cardiac function. In addition, via
their effects on stromal components, fibroblasts are involved in
cancer proliferation and migration (49). Notably, characterization of the
features of CFs may provide comprehensive knowledge of treatment of
cardiovascular diseases. Activated CFs are considered a common
pathological characteristic of HF. Previous studies have suggested
that proinflammation of CFs in HF cardiac tissues may be increased
because fibroblast-mediated inflammatory associated pathways are
upregulated (10,28). Recently, Sen et al (41) described a novel CF subtype
associated with HF by analyzing single-cell transcripts, which
suggested further investigation into CF subgroups using the
advanced single-cell technology. The results of the present study
demonstrated that CFs were activated in the cardiac tissues of
patients with HF. The specifically activated CFs presented a
proinflammatory phenotype secreting NLRC3 inflammasome-induced IL10
and TNF-α, in order to promote the progression of cardiac
dysfunction. These findings highlight the essential roles of CFs in
microenvironmental inflammation and may offer more opportunities
for future therapeutic strategies.
In the current study, to investigate the mechanism
underlying the proinflammatory phenotype of CFs, activation of the
NLRC3 inflammasome was detected by analyzing mRNA and protein
expression. To further detect the molecular basis of inflammasome
activation, CFs obtained from adult mice were manipulated by shRNA
technology to determine the critical role of LDL in triggering
inflammasome-mediated cytokine production. An inflammatory
microenvironment is considered one of the characteristics of HF
progression (26). The present
findings indicated the important effects of LDL on inducing
inflammasome activation during HF progression, indicating the
importance of managing chronic inflammatory reactions in other
cardiovascular pathogenic conditions. However, it is still under
debate whether inflammasome activation can benefit or impair
cardiac functions. Cardiomyocytes function well if they are
supported by other cell populations in the microenvironment,
including CFs, endothelial cells and immune cells (50). Nonetheless, the molecular basis
underlying the communication between cells and cell activation are
not well defined. Recently, numerous studies have provided insights
into the involvement of newly discovered non-pathological
functional signalosome complexes in microenvironmental
inflammation, such as inflammasomes, necrosomes and apoptosomes
(29,51); in particular, the key roles served
by inflammasomes, including NLRC4. In conclusion, the present study
demonstrated the crucial role of NLRC3 inflammasome activation in
CFs and revealed that CFs-induced inflammation promoted HF
progression.
Acknowledgements
The authors would like to thank Dr Hai Wang
(Feicheng Mining Center Hospital) for providing valuable
suggestions about data processing.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
WZ and PW contributed to the conception and design
of this study. WZ and PW confirm the authenticity of all the raw
data. PW was responsible for the details of experimental
performance and performed the experiments. PW, WBZ and WZ performed
the histological diagnosis and IHC staining evaluation. JZ and ZF
collected and analyzed the clinical data. YS interpreted the
pathological data. PW and WZ analyzed the clinical data. PW
completed the manuscript, figures and tables. All authors read and
approved the final manuscript.
Ethics approval and consent to
participate
The studies involving human tissues were approved by
the Research Ethics Committee of Feicheng Mining Center Hospital.
All patients provided written informed consent according to the
Declaration of Helsinki. Written informed consent for use of LV
tissues was provided by the dying donors themselves or their family
members. The animal studies were approved by the Animal Care
Committee of Feicheng Mining Center Hospital.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
King KC and Goldstein S: Congestive heart
failure and pulmonary edema. StatPearls. StatPearls Publishing;
Treasure Island, FL: 2021
|
|
2
|
Ingram DD, Malec DJ, Makuc DM,
Kruszon-Moran D, Gindi RM, Albert M, Beresovsky V, Hamilton BE,
Holmes J, Schiller J and Sengupta M: National center for health
statistics guidelines for analysis of trends. Vital Health Stat.
2:1–71. 2018.PubMed/NCBI
|
|
3
|
Malik A, Brito D and Chhabra L: Congestive
heart failure. StatPearls. StatPearls Publishing; Treasure Island,
FL: 2021
|
|
4
|
Skrzypek A, Mostowik M, Szeliga M,
Wilczyńska-Golonka M, Dębicka-Dąbrowska D and Nessler J: Chronic
heart failure in the elderly: Still a current medical problem.
Folia Med Cracov. 58:47–56. 2018.PubMed/NCBI
|
|
5
|
Côté E: Feline congestive heart failure:
Current diagnosis and management. Vet Clin North Am Small Anim
Pract. 47:1055–1064. 2017. View Article : Google Scholar
|
|
6
|
Bozkurt B, Aguilar D, Deswal A, Dunbar SB,
Francis GS, Horwich T, Jessup M, Kosiborod M, Pritchett AM,
Ramasubbu K, et al: Contributory risk and management of
comorbidities of hypertension, obesity, diabetes mellitus,
hyperlipidemia, and metabolic syndrome in chronic heart failure: A
scientific statement from the American heart association.
Circulation. 134:e535–e578. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Bedi KC Jr, Snyder NW, Brandimarto J, Aziz
M, Mesaros C, Worth AJ, Wang LL, Javaheri A, Blair IA, Margulies KB
and Rame JE: Evidence for intramyocardial disruption of lipid
metabolism and increased myocardial ketone utilization in advanced
human heart failure. Circulation. 133:706–716. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Piccoli MT, Gupta SK, Viereck J,
Foinquinos A, Samolovac S, Kramer FL, Garg A, Remke J, Zimmer K,
Batkai S and Thum T: Inhibition of the cardiac fibroblast-enriched
lncRNA Meg3 prevents cardiac fibrosis and diastolic dysfunction.
Circ Res. 121:575–583. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Souders CA, Bowers SL and Baudino TA:
Cardiac fibroblast: The renaissance cell. Circ Res. 105:1164–1176.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Frangogiannis NG: Cardiac fibrosis: Cell
biological mechanisms, molecular pathways and therapeutic
opportunities. Mol Aspects Med. 65:70–99. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhang P, Su J and Mende U: Cross talk
between cardiac myocytes and fibroblasts: From multiscale
investigative approaches to mechanisms and functional consequences.
Am J Physiol Heart Circ Physiol. 303:H1385–H1396. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Batista-Almeida D, Martins-Marques T,
Ribeiro-Rodrigues T and Girao H: The role of proteostasis in the
regulation of cardiac intercellular communication. Adv Exp Med
Biol. 1233:279–302. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Karki R, Man SM and Kanneganti TD:
Inflammasomes and cancer. Cancer Immunol Res. 5:94–99. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zhou W, Chen C, Chen Z, Liu L, Jiang J, Wu
Z, Zhao M and Chen Y: NLRP3: A novel mediator in cardiovascular
disease. J Immunol Res. 2018:57021032018. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
van de Veerdonk FL, Joosten LA and Netea
MG: The interplay between inflammasome activation and antifungal
host defense. Immunol Rev. 265:172–180. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Man SM and Kanneganti TD: Regulation of
inflammasome activation. Immunol Rev. 265:6–21. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Palomo J, Dietrich D, Martin P, Palmer G
and Gabay C: The interleukin (IL)-1 cytokine family-Balance between
agonists and antagonists in inflammatory diseases. Cytokine.
76:25–37. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Qiu Z, He Y, Ming H, Lei S, Leng Y and Xia
ZY: Lipopolysaccharide (LPS) aggravates high glucose- and
hypoxia/reoxygenation-induced injury through activating
ROS-Dependent NLRP3 inflammasome-mediated pyroptosis in H9C2
cardiomyocytes. J Diabetes Res. 2019:81518362019. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhang YZ, Sui XL, Xu YP, Gu FJ, Zhang AS
and Chen JH: NLRP3 inflammasome and lipid metabolism analysis based
on UPLC-Q-TOF-MS in gouty nephropathy. Int J Mol Med. 44:172–184.
2019.PubMed/NCBI
|
|
20
|
King M, Kingery J and Casey B: Diagnosis
and evaluation of heart failure. Am Fam Physician. 85:1161–1168.
2012.PubMed/NCBI
|
|
21
|
Murase T, Hattori T, Ohtake M, Abe M,
Amakusa Y, Takatsu M, Murohara T and Nagata K: Cardiac remodeling
and diastolic dysfunction in DahlS.Z-Lepr(fa)/Lepr(fa) rats: A new
animal model of metabolic syndrome. Hypertens Res. 35:186–193.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Strutz F, Okada H, Lo CW, Danoff T, Carone
RL, Tomaszewski JE and Neilson EG: Identification and
characterization of a fibroblast marker: FSP1. J Cell Biol.
130:393–405. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Rui T, Feng Q, Lei M, Peng T, Zhang J, Xu
M, Abel ED, Xenocostas A and Kvietys PR: Erythropoietin prevents
the acute myocardial inflammatory response induced by
ischemia/reperfusion via induction of AP-1. Cardiovasc Res.
65:719–727. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Yao Y, Xu X, Zhang G, Zhang Y, Qian W and
Rui T: Role of HMGB1 in doxorubicin-induced myocardial apoptosis
and its regulation pathway. Basic Res Cardiol. 107:2672012.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Strassheim D, Dempsey EC, Gerasimovskaya
E, Stenmark K and Karoor V: Role of inflammatory cell subtypes in
heart failure. J Immunol Res. 2019:21640172019. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Chen G, Bracamonte-Baran W, Diny NL, Hou
X, Talor MV, Fu K, Liu Y, Davogustto G, Vasquez H, Taegtmeyer H, et
al: Sca-1+ cardiac fibroblasts promote development of
heart failure. Eur J Immunol. 48:1522–1538. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Travers JG, Kamal FA, Robbins J, Yutzey KE
and Blaxall BC: Cardiac fibrosis: The fibroblast awakens. Circ Res.
118:1021–1040. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Malik A and Kanneganti TD: Inflammasome
activation and assembly at a glance. J Cell Sci. 130:3955–3963.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Miteva K, Pappritz K, Sosnowski M,
El-Shafeey M, Müller I, Dong F, Savvatis K, Ringe J, Tschöpe C and
Van Linthout S: Mesenchymal stromal cells inhibit NLRP3
inflammasome activation in a model of Coxsackievirus B3-induced
inflammatory cardiomyopathy. Sci Rep. 8:28202018. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Bennett M and Gilroy DW: Lipid mediators
in inflammation. Microbiol Spectr. 4(6)2016.
|
|
32
|
Storey BC, Staplin N, Haynes R, Reith C,
Emberson J, Herrington WG, Wheeler DC, Walker R, Fellström B,
Wanner C, et al: Lowering LDL cholesterol reduces cardiovascular
risk independently of presence of inflammation. Kidney Int.
93:1000–1007. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kattoor AJ, Kanuri SH and Mehta JL: Role
of Ox-LDL and LOX-1 in atherogenesis. Curr Med Chem. 26:1693–1700.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Stoekenbroek RM, Lambert G, Cariou B and
Hovingh GK: Inhibiting PCSK9-biology beyond LDL control. Nat Rev
Endocrinol. 15:52–62. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Rivera AL, Estañol B, Fossion R,
Toledo-Roy JC, Callejas-Rojas JA, Gien-López JA, Delgado-García GR
and Frank A: Loss of breathing modulation of heart rate variability
in patients with recent and long standing diabetes mellitus type
II. PLoS One. 11:e01659042016. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Afsarmanesh N, Horwich TB and Fonarow GC:
Total cholesterol levels and mortality risk in nonischemic systolic
heart failure. Am Heart J. 152:1077–1083. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Foody JM, Shah R, Galusha D, Masoudi FA,
Havranek EP and Krumholz HM: Statins and mortality among elderly
patients hospitalized with heart failure. Circulation.
113:1086–1092. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Lee MMY, Sattar N, McMurray JJV and
Packard CJ: Statins in the prevention and treatment of heart
failure: A review of the evidence. Curr Atheroscler Rep. 21:412019.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Bahiru E, de Cates AN, Farr MR, Jarvis MC,
Palla M, Rees K, Ebrahim S and Huffman MD: Fixed-dose combination
therapy for the prevention of atherosclerotic cardiovascular
diseases. Cochrane Database Syst Rev. 3:Cd0098682017.PubMed/NCBI
|
|
40
|
Vodonos A, Ostapenko I, Toledano R, Henkin
Y, Zahger D, Wolak T, Sherf M and Novack V: Statin adherence and
LDL cholesterol levels. Should we assess adherence prior to statin
upgrade? Eur J Intern Med. 26:268–272. 2015.PubMed/NCBI
|
|
41
|
Sen S, Petraco R, Mayet J and Davies J:
Wave intensity analysis in the human coronary circulation in health
and disease. Curr Cardiol Rev. 10:17–23. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Tanai E and Frantz S: Pathophysiology of
heart failure. Compr Physiol. 6:187–214. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Radulović B, Potočnjak I, Dokoza Terešak
S, Trbušić M, Vrkić N, Huršidić Radulović A, Starčević N, Milošević
M, Degoricija V and Frank S: Cholesterol and chloride in acute
heart failure. Acta Clin Croat. 58:195–201. 2019.
|
|
44
|
Zhang J, Zu Y, Dhanasekara CS, Li J, Wu D,
Fan Z and Wang S: Detection and treatment of atherosclerosis using
nanoparticles. Wiley Interdiscip Rev Nanomed Nanobiotechnol.
9:10.1002/wnan.1412. 2017. View Article : Google Scholar
|
|
45
|
Ferrucci L and Fabbri E: Inflammageing:
Chronic inflammation in ageing, cardiovascular disease, and
frailty. Nat Rev Cardiol. 15:505–522. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Sochocka M, Donskow-Łysoniewska K, Diniz
BS, Kurpas D, Brzozowska E and Leszek J: The gut microbiome
alterations and inflammation-driven pathogenesis of alzheimer's
disease-a critical review. Mol Neurobiol. 56:1841–1851. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Mount CW, Majzner RG, Sundaresh S, Arnold
EP, Kadapakkam M, Haile S, Labanieh L, Hulleman E, Woo PJ, Rietberg
SP, et al: Potent antitumor efficacy of anti-GD2 CAR T cells in
H3-K27M+ diffuse midline gliomas. Nat Med. 24:572–579.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Sowers JL, Johnson KM, Conrad C, Patterson
JT and Sowers LC: The role of inflammation in brain cancer. Adv Exp
Med Biol. 816:75–105. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Wei L, Ye H, Li G, Lu Y, Zhou Q, Zheng S,
Lin Q, Liu Y, Li Z and Chen R: Cancer-associated fibroblasts
promote progression and gemcitabine resistance via the SDF-1/SATB-1
pathway in pancreatic cancer. Cell Death Dis. 9:10652018.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Kanbay M, Yerlikaya A, Sag AA, Ortiz A,
Kuwabara M, Covic A, Wiecek A, Stenvinkel P and Afsar B: A journey
from microenvironment to macroenvironment: The role of
metaflammation and epigenetic changes in cardiorenal disease. Clin
Kidney J. 12:861–870. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Parry TL, Melehani JH, Ranek MJ and Willis
MS: Functional amyloid signaling via the inflammasome, necrosome,
and signalosome: New therapeutic targets in heart failure. Front
Cardiovasc Med. 2:252015. View Article : Google Scholar : PubMed/NCBI
|