Introduction
Heat stroke, which has a mortality rate of 10–15%
worldwide (1), arises from acute
and irreversible multiple organ dysfunction induced by severe heat
stress (HS) (2). Severe HS-induced
multiple organ dysfunction is thought to be associated with
excessive death of endothelial cells (e.g., via apoptosis,
autophagy or necrosis), which constitute the greatest surface area
of the human circulatory system (3–5).
Notably, HS can cause damage to cells in various tissues, including
endothelial cells. In response to the direct injurious effect of HS
and the increased levels of inflammatory mediators caused by HS,
endothelial cell apoptosis is an early event in heat stroke,
suggesting its critical role in the pathogenesis of this condition
(2,3,6).
Endothelial cell apoptosis is closely associated with microvascular
dysfunction, including impaired permeability, coagulation,
fibrinolysis, vascular tone and leukocyte recruitment (7). Microvascular dysfunction causes
interstitial leakage, impaired microcirculatory blood flow and
tissue hypoperfusion, and directly contributes to life-threatening
organ failure (8–10). A more detailed understanding of
HS-induced endothelial cell apoptosis may be helpful for future
therapy and in limiting HS-induced organ failure.
MicroRNA (miRNA/miR) is a type of endogenous small
noncoding RNA molecules that regulate gene expression by pairing
with a sequence-specific target or via other mechanisms; therefore,
it has been suggested that miRNAs can control diverse cellular and
signalling pathways (11). In
numerous pathological conditions, miRNA and miRNA-related
regulatory networks serve a crucial role in the prevention and
treatment of endothelial cell death (11–14).
Several miRNA molecules have been reported to be aberrantly
expressed in endothelial cells following HS, and their
dysregulation is thought to be linked to pathological phenotypes of
endothelial cells, such as proapoptotic, proinflammatory,
proadhesive and procoagulant phenotypes (12). However, despite extensive
investigation of these molecules in other diseases, exploration of
their roles and the underlying pathogenic mechanisms in HS-induced
endothelial cell death is limited.
In endothelial cells, the mitogen-activated protein
kinase (MAPK) pathway is reported to be one of the most frequently
activated pathways in several pathogenic conditions (13,14).
Accumulating evidence has indicated that the MAPK pathway is
involved in HS-induced organ injury and endothelial cell apoptosis;
therefore, this pathway is thought to be a valuable therapeutic
target in heat stroke (15).
Several miRNA molecules, such as miR-199b-3p, miR-30-3p and
miR-9-5p in endothelial cells, have been proposed to negatively
regulate the MAPK pathway and inhibit endothelial cell apoptosis
(16–18). However, whether MAPK can be
negatively regulated by relevant miRNAs in endothelial cells after
HS and its role remains poorly understood.
Our previous study demonstrated that miR-3613-3p was
suppressed in HS-treated human umbilical vein endothelial cells
(HUVECs) (19). In our preliminary
study, it was revealed that a decrease in miR-3613-3p might promote
cell apoptosis by negative post-transcriptional regulation of the
target gene MAPK kinase kinase 2 (MAP3K2) (19), a component of the MAPK pathway.
However, whether the miR-3613-3p/MAP3K2 axis, along with downstream
signalling molecules, contributes to HS-induced apoptosis remains
unclear.
The aim of the present study was to investigate
whether miR-3613-3p is a critical mediator in the progression of
the HS-induced apoptosis of HUVECs, and whether this may involve
changes in the activity of MAP3K2 and its downstream molecules.
Materials and methods
Cell culture and treatment
HUVECs were purchased from the American Type Culture
Collection and cultured in RPMI-1640 medium (Invitrogen; Thermo
Fisher Scientific, Inc.) supplemented with 5% FBS (Invitrogen;
Thermo Fisher Scientific, Inc.), 1% Eco Growth Supplement
(Promocell GmbH), 100 U/ml penicillin and 100 µg/ml streptomycin
(Invitrogen; Thermo Fisher Scientific, Inc.) in a humidified
atmosphere at 37°C and 5% CO2. All experiments were
carried out with the 3–6 generation HUVECs. For induction of HS,
HUVECs were heated in a humidified incubator at different
temperatures (39, 41, 43 or 45°C for 2 h followed by 37°C for 6 h)
or for different heat exposure times (43°C for 1, 2, 3 or 4 h
followed by 37°C for 6 h), then transferred to normal culture
conditions (5% CO2 at 37°C) for 6 h before analysis.
Cells cultured at 37±0.5°C for 2 h were used as control cells. The
use of non-immortal HUVECs has received ethical approval from the
Ethics Committee of Hefei Boe Hospital Co., Ltd. (approval no.
20190106).
Transient transfection
The miR-3613-3p mimic (cat. no. miR10017991-1-5,
https://www.ribobio.com/en/product_detail/?sku=miR10017991-1-5),
negative control (NC) miRNA (miR-NC; cat. no. miR1N0000001-1-5,
https://www.ribobio.com/en/product-search/?Category=all&species=all&keywords=micron
mimic NC), miR-36-13-3p inhibitor (cat. no. miR20017991-1-5,
http://www.ribobio.com/en/product_detail/?sku=miR20017991-1-5)
and anti-miR-NC (cat. no. miR2N0000001-1-5, https://www.ribobio.com/en/product-search/?category=all&species=all&keywords=micrOFF
inhibitor NC#22) were purchased from Guangzhou RiboBio Co., Ltd..
For construction of recombinant DNA or interference with gene
expression, the pcDNA3.1 plasmid (cat. no. E0648; Sigma-Aldrich;
Merck KGaA) was used as the vector. The pcDNA3.1-MAP3K2 plasmid was
constructed by Shanghai GeneChem Co., Ltd.. The empty pcDNA3.1
vector was used as the control. The MAP3K2 small interfering RNA
(siRNA; si-MAP3K2; cat. no. siB06111516204-1-5) and corresponding
NC (si-NC; cat. no. siN0000001-1-5) were purchased from Guangzhou
RiboBio Co., Ltd.. Cells were seeded into 6-well plates at a
density of ~1.5×105 cells/well. Following the
manufacturer's instructions, transient transfection was carried out
with Lipofectamine® 3000 reagent (Invitrogen; Thermo
Fisher Scientific, Inc.) at 37°C for 24 h. Different concentrations
of miR-3613-3p mimic or miR-3613-3p inhibitor were used
(miR-3613-3p mimic at 25, 50 and 100 nM; miR-3613-3p inhibitor at
25, 50 and 100 nM) to determine the effect of HS on apoptosis of
HUVECs. The appropriate concentrations of these factors were
selected and used in further experiments The concentrations of
other reagents were as follows: miR-NC, 50 nM; anti-miR-NC, 100 nM;
pcDNA3.1-MAP3K2, 100 nM; pcDNA3.1, 100 nM; si-MAP3K2, 50 nM; and
si-NC, 50 nM. After transfection, cells were treated with 43°C or
control heat treatments. Then, transfected cells were collected for
subsequent analysis.
RNA extraction and reverse
transcription-quantitative PCR (RT-qPCR)
Total RNA was isolated from HUVECs
(~5×106 cells) using TRIzol® (Thermo Fisher
Scientific, Inc.) and the miRNA easy mini kit (Qiagen, Inc.),
according to the manufacturer's protocols. A Nanodrop™
spectrophotometer (Thermo Fisher Scientific, Inc.) was used to
measure the quantity and quality of RNA, and 1% agarose gel
electrophoresis was used to detect RNA integrity. The expression
levels of miRNA were detected via RT-qPCR, and the primers were
synthesized and purchased from Shanghai Gene Pharmaceutical Co.,
Ltd. U6 was employed as the endogenous control for miRNA
expression. The primer sequences were as follows: miR-3613-3p
sense, 5′-CGTCCCTTCCCAACCCGAAAAAAA-3′ and antisense,
5′-CGCAGGGTCCGAGGTATTC-3′; and U6 sense, 5′-CTCGCTTCGGCAGCACA-3′
and antisense, 5′-AACGCTTCACGAATTTGCGT-3′. Briefly, specific
stem-loop primers and a TaqMan® MicroRNA Reverse
Transcription kit (Applied Biosystems; Thermo Fisher Scientific,
Inc.) were used to reverse transcribed sample total RNA (10 ng)
into cDNA. In a 15-µl reaction volume, mixtures were incubated for
30 min at 16°C, 30 min at 42°C and 5 min at 85°C, and held at 4°C.
Following the RT reaction, qPCR was carried out with an ABI 7300
Real-Time PCR system (Bio-Rad Laboratories, Inc.) and SYBR Green
(Invitrogen; Thermo Fisher Scientific, Inc.) according to the
manufacturer's protocol. The thermocycling conditions were 95°C for
10 min, followed by 40 cycles of 95°C for 15 sec and 60°C for 60
sec in a 20-µl reaction volume (20,21).
The relative level of each miRNA was determined by the
2−ΔΔCq method (22). All
results were normalized to U6 expression levels, which were
analyzed simultaneously. All experiments were performed at least in
triplicate.
Flow cytometric analysis of
apoptosis
Cell apoptosis was determined with an Annexin V-FITC
Apoptosis kit (Invitrogen; Thermo Fisher Scientific, Inc.)
according to the manufacturer's protocol. In total,
~1×106 cells were collected, washed with ice-cold PBS
prior to resuspension in binding buffer containing 5 µl Annexin
V-FITC for 10 min in the dark at room temperature. Subsequently,
the cells were pelleted by centrifugation at ~157 × g and 4°C for
10 min, the buffer was removed and the cells were resuspended in
reaction buffer containing 10 µl propidium iodide (PI). The
suspension was mixed and incubated in the dark at room temperature
for 15 min. Finally, cell apoptotic rates (early+late) were
determined using a flow cytometer (FACSCanto™ II; BD Biosciences).
FlowJo version 7.6.1 software (Tree Star, Inc.) was used to analyze
the data. The experiment was performed at least three times.
Protein extraction and western blot
analysis
Protein extraction, measurement of total protein
concentration and western blot analysis were performed as described
previously (19). The primary
antibodies used were as follows: Mouse anti-human MAP3K2 (cat. no.
SAB5300054), mouse anti-human p38 MAPK (cat. no. M8177), mouse
anti-human phosphorylated (p)-p38 MAPK (cat. no. MABS64), mouse
anti-human extracellular signal-regulated kinase (ERK1/2; cat. no.
M8159), mouse anti-human p-ERK1/2 (cat. no. M7802), mouse
anti-human c-Jun N-terminal kinase (JNK; cat. no. SAB4200176),
rabbit anti-human p-JNK (cat. no. ZRB1173), mouse anti-human
caspase-3 (cat. no. C5737) and mouse anti-human actin (cat. no.
A4700) (all from Sigma-Aldrich; Merck KGaA). All primary antibodies
were diluted 1:1,000. Goat anti-mouse horseradish
peroxidase-conjugated immunoglobulin G (cat. no. SAB3701029) and
goat anti-rabbit horseradish peroxidase-conjugated immunoglobulin G
(cat. no. AP156P) (both from Sigma-Aldrich; Merck KGaA) were
diluted 1:8,000 and used as secondary antibodies. Protein
expression levels were normalized to those of actin as the
endogenous control.
Luciferase reporter assay
The luciferase vector reporter plasmids pGL3-MAP3K2
with the wild-type 3′-untranslated region (3′-UTR) (MAP3K2-WT) or
mutant 3′-UTR (MAP3K2-MUT) were purchased from Guangzhou RiboBio
Co., Ltd. miR-NC or anti-miR-NC were used as the NCs. HUVECs
(~1×105 cells/well) were seeded in 24-well plates and
cultured for 24 h prior to transfection. Cells were co-transfected
with luciferase vectors (MAP3K2-WT or MAP3K2-MUT reporter plasmid)
and either miR-3613-3p mimic or miR-NC at room temperature for 24
h. Simultaneously, cells were co-transfected with luciferase
vectors (MAP3K2-WT or MAP3K2-MUT reporter plasmid) and either
miR-3613-3p inhibitor or anti-miR-NC at room temperature for 24 h.
Lipofectamine® 3000 reagent was used according to the
manufacturer's protocol. The dual-luciferase activity was measured
at 48 h after transfection. The dual-Luciferase reporter system
(Promega Corporation) was used to quantify Firefly and
Renilla luciferase activities according to manufacturer's
protocol. All experiments were performed in triplicate; n=3–6 for
each experiment. The final concentrations were as follows:
MAP3K2-WT, 100 nM; MAP3K2-MUT, 100 nM; miR-3613-3p mimic, 50 nM;
miR-3613-3p inhibitor, 100 nM; miR-NC, 50 nM; and anti-miR-NC, 100
nM. All experiments were performed in triplicate; n=3–6 for each
experiment.
Treatments with selective pathway
inhibitors
SB203580 (an inhibitor of p38 MAPK), PD98059 (an
inhibitor of ERK) and SP600125 (an inhibitor of JNK) were from
Sigma-Aldrich (Merck KGaA). All inhibitors were diluted in DMSO
(stock concentrations 10 and 20 mM) and kept at −20°C. During the
experiments, they were used at a final concentration of 10 µM. They
were respectively added in culture medium 24 h prior to HS exposure
at 37°C.
Caspase-3 activity assay
A caspase-3 activity fluorescent assay kit (Enzo
Life Sciences, Inc.) was used according to the manufacturer's
protocol. Cells were seeded at a density of 1×105
cells/well in 96-well plates. After 24 h, cells were exposed to HS
or control heat. Cells were then lysed in caspase-3 sample lysis
buffer, and total cellular protein was extracted and quantified
using a BCA Protein assay kit (Thermo Fisher Scientific, Inc.)
according to the manufacturer's protocol. Prior to the caspase-3
assay, samples containing equal amounts of total protein were
incubated with the DEVD substrate conjugate provided in the kit at
37°C for 2 h. The samples were then evaluated by measurement at an
excitation wavelength of 400 nm and emission wavelength of 505 nm
in an automatic microplate reader (SpectraMax M5; Molecular
Devices, LLC).
DNA fragmentation assay
A Cellular DNA Fragmentation ELISA kit (Roche
Applied Science; cat. no. 11585045001) was used according to the
manufacturer's instructions. This assay is based on the
quantitative detection of 5-bromo-2′-deoxyuridine (BrdU)-labeled
DNA fragments. Cells were seeded at a density of 1×105
cells/well in 96-well plates. After 24 h of growth, cells were
exposed to different treatments. Subsequently, 10 µM BrdU was added
to each well, and the cells were cultured with BrdU for 24 h. The
complexes were centrifuged at ~250 × g for 10 min at 4°C, the
supernatant carefully removed. After DNA labelling, the cells were
lysed in 200 µl incubation buffer and soluble DNA fragments were
quantified using the Cellular DNA Fragmentation ELISA kit,
according to the manufacturer's instructions. Absorbance values
were detected spectrophotometrically at 450 nm using an ELx808
ELISA reader (BioTek Instruments). All experiments were performed
in triplicate.
Statistical analysis
Data are presented as the mean ± standard error of
the mean from at least three independent experiments performed in
duplicate. Statistical analysis was performed using SPSS version
20.0 (IBM Corp.). Differences between two groups were compared
using unpaired Student's t-test. Comparisons among multiple groups
were performed with one-way analysis of variance followed by
Tukey's test. P<0.05 was considered to indicate a statistically
significant difference.
Results
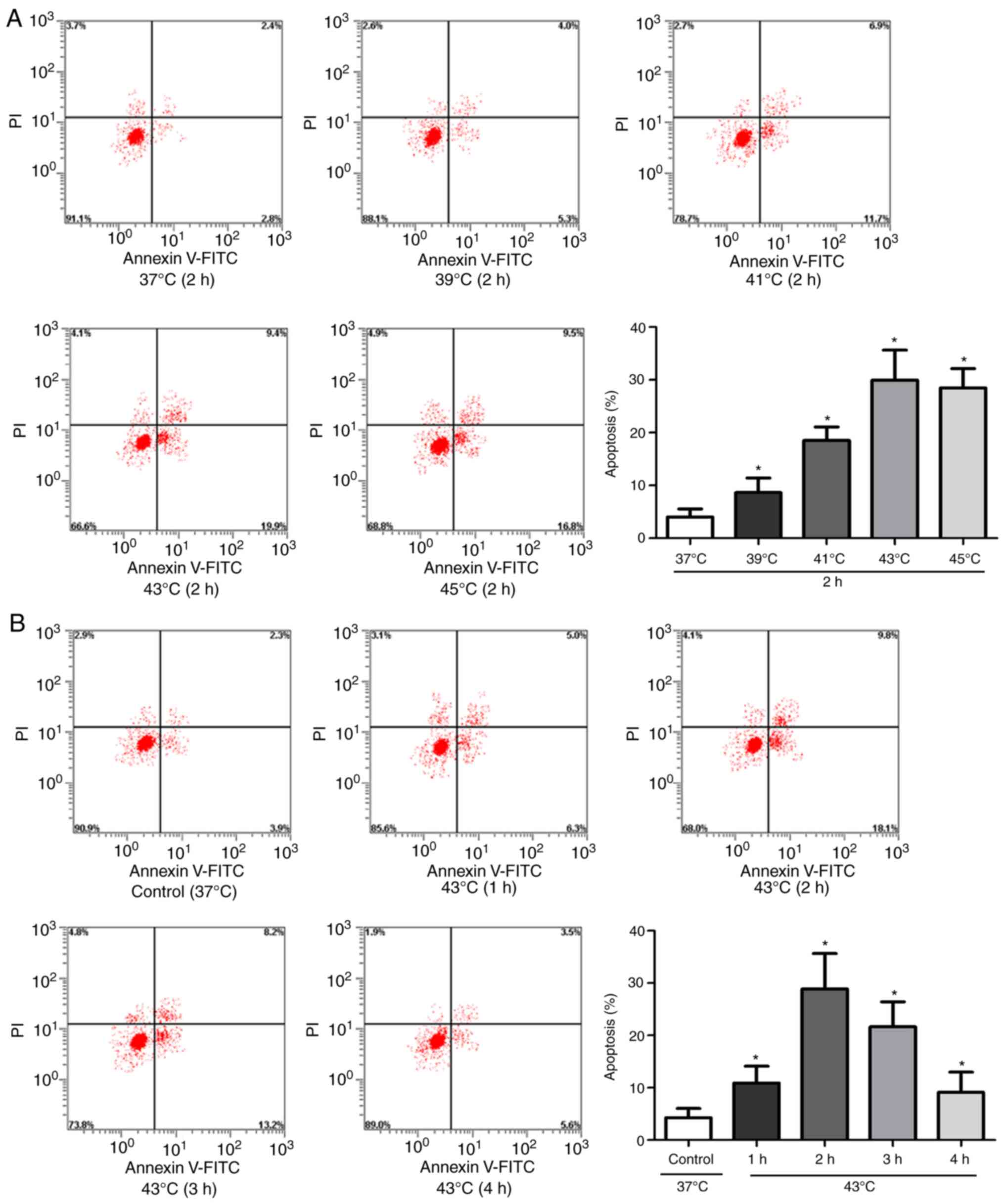
Apoptosis is induced by HS in
HUVECs
To evaluate the apoptosis of HUVECs following HS,
Annexin V-fluorescein isothiocyanate/PI staining and flow cytometry
were performed. Cells were incubated at different temperatures (39,
41, 43, or 45°C for 2 h followed by 37°C for 6 h) or for different
heat exposure times (43°C for 1, 2, 3 or 4 h followed by 37°C for 6
h). The apoptotic rate was increased with increasing temperature or
heat exposure time, peaking at 43°C or 2 h in the respective
experiments (Fig. 1A and B). These
results suggested that HUVEC apoptosis was enhanced by HS.
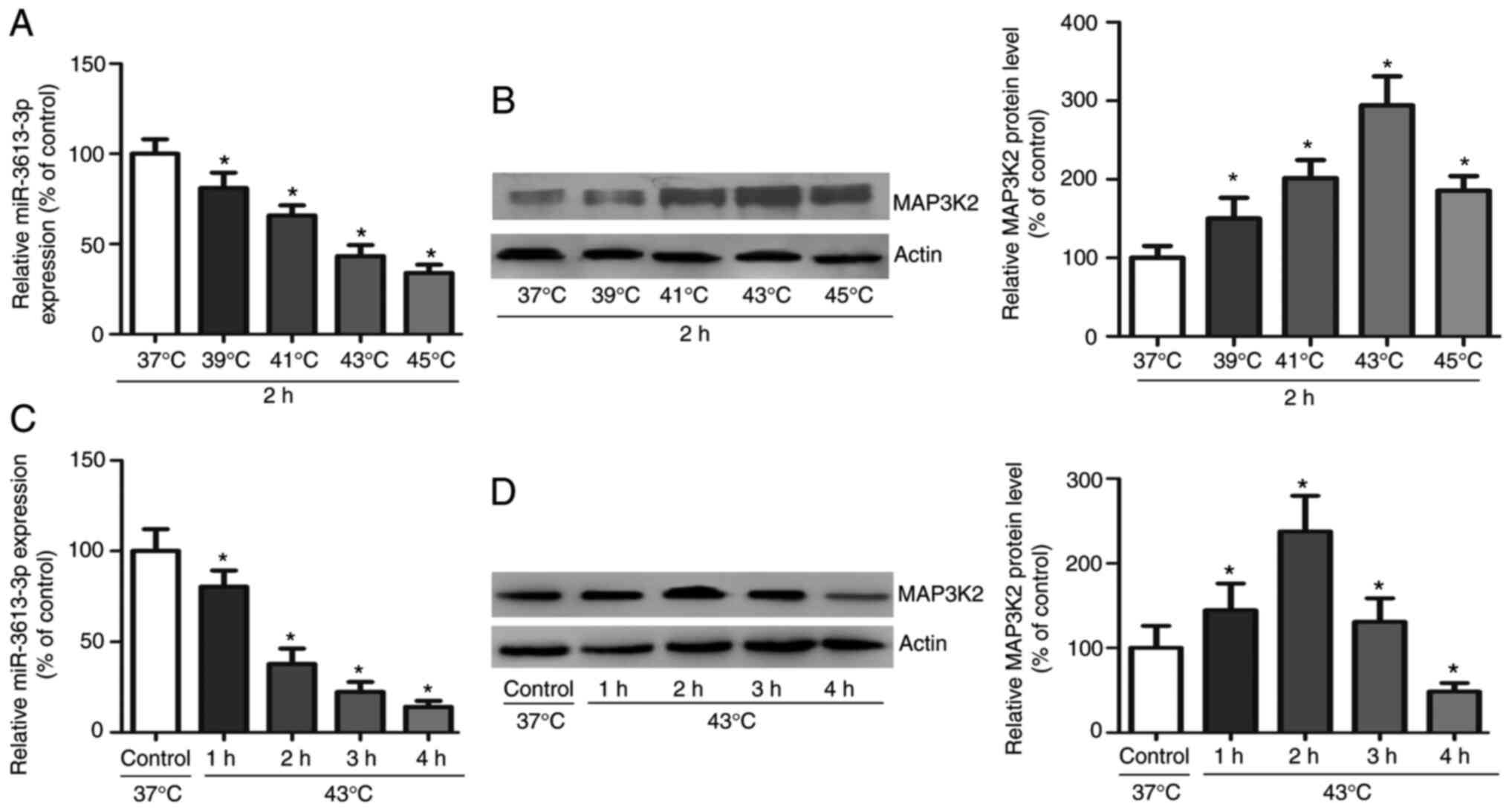
HS induces miR-3613-3p downregulation
and MAP3K2 upregulation in HUVECs
To evaluate the expression levels of miR-3613-3p and
MAP3K2 in HUVECs during HS exposure, RT-qPCR and western blot
analysis were performed. In HUVECs treated with different
temperatures or for different heat exposure durations, the
expression levels of miR-3613-3p were decreased, whereas those of
MAP3K2 were increased. As shown in Fig.
2A and B, the expression levels of miR-3613-3p were
significantly decreased and those of MAP3K2 were significantly
increased at all time points. Furthermore, as shown in Fig. 2C and D, the expression levels of
miR-3613-3p were consistently decreased at all durations, and those
of MAP3K2 were significantly increased after HUVECs were exposed to
HS for 2 h. These results indicated that HS induced opposing
changes in the expression levels of miR-3613-3p and MAP3K2 in
HUVECs; however, whether miR-3613-3p directly or indirectly targets
MAP3K2 remains unknown.
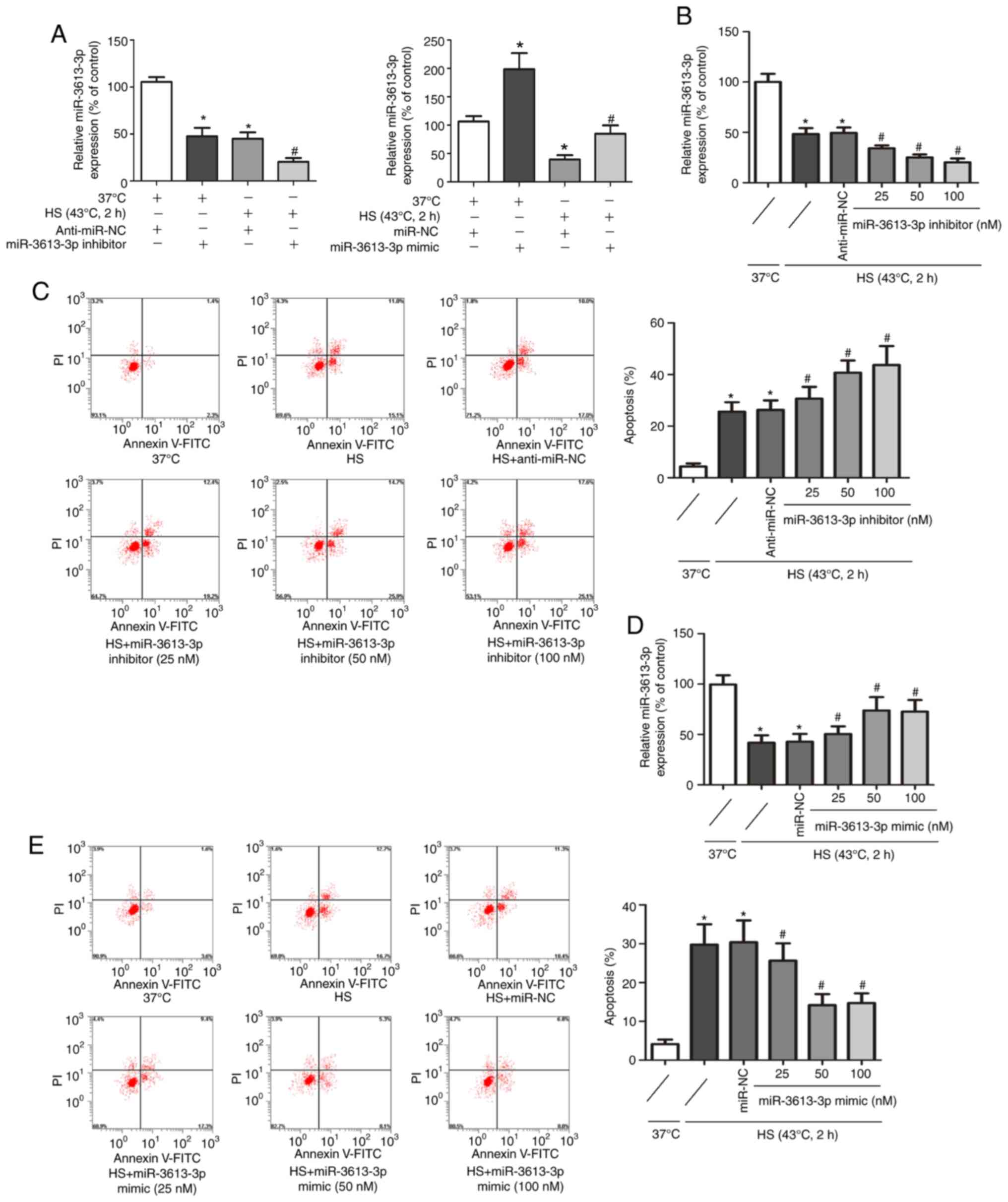
miR-3613-3p regulates the HS-induced
apoptosis of HUVECs
To further understand the relationship between
miR-3613-3p and the apoptosis of HS-exposed HUVECs, HUVECs were
transfected with a miR-3613-3p inhibitor or miR-3613-3p mimic.
Compared with the NC groups, cells successfully transfected with
the miR-3613-3p inhibitor or miR-3613-3p mimic exhibited reduced or
increased miR-3613-3p expression levels, respectively (Fig. 3A). HS significantly reduced the
expressionlevel of miR-3613-3p in HUVECs. The use of miR-3613-3p
inhibitors further aggravated the reduction of miR-3613-3p in
HUVECs exposed to HS. Moreover, the effect of miR-3613-3p inhibitor
was dose-dependent, with the inhibition reaching its peak at 100 nM
(Fig. 3B). The miR-3613-3p
inhibitor, which led to a marked decrease in miR-3613-3p
expression, resulted in upregulation of HS-induced apoptosis. The
effects of the miR-3613-3p inhibitor were dose-dependent and peaked
at 100 nM (Fig. 3C). Transfection
of miR-3613-3p mimic could partially reverse the inhibition effect
of HS on miR-3613-3p in HUVECs. The effect was dose-dependent,
peaking at 50 nM (Fig. 3D). In
addition, transfection with the miR-3613-3p mimic reduced
HS-induced apoptosis of HUVECs in a dose-dependent manner. This
effect gradually increased with increasing miR-3613-3p mimic
concentration and reached a peak at 50 nM (Fig. 3E). These results indicated a
regulatory effect of miR-3613-3p on the HS-induced apoptosis of
HUVECs. The similar effects of the miR-3613-3p mimic at 50 and 100
nM were considered to be related to the degree of saturation of
intermediate binding sites between the miR-3613-3p mimic and mRNA.
In subsequent experiments, the optimal concentration of 100 nM
miR-3613-3p inhibitor or 50 nM miR-3613-3p mimic was used.
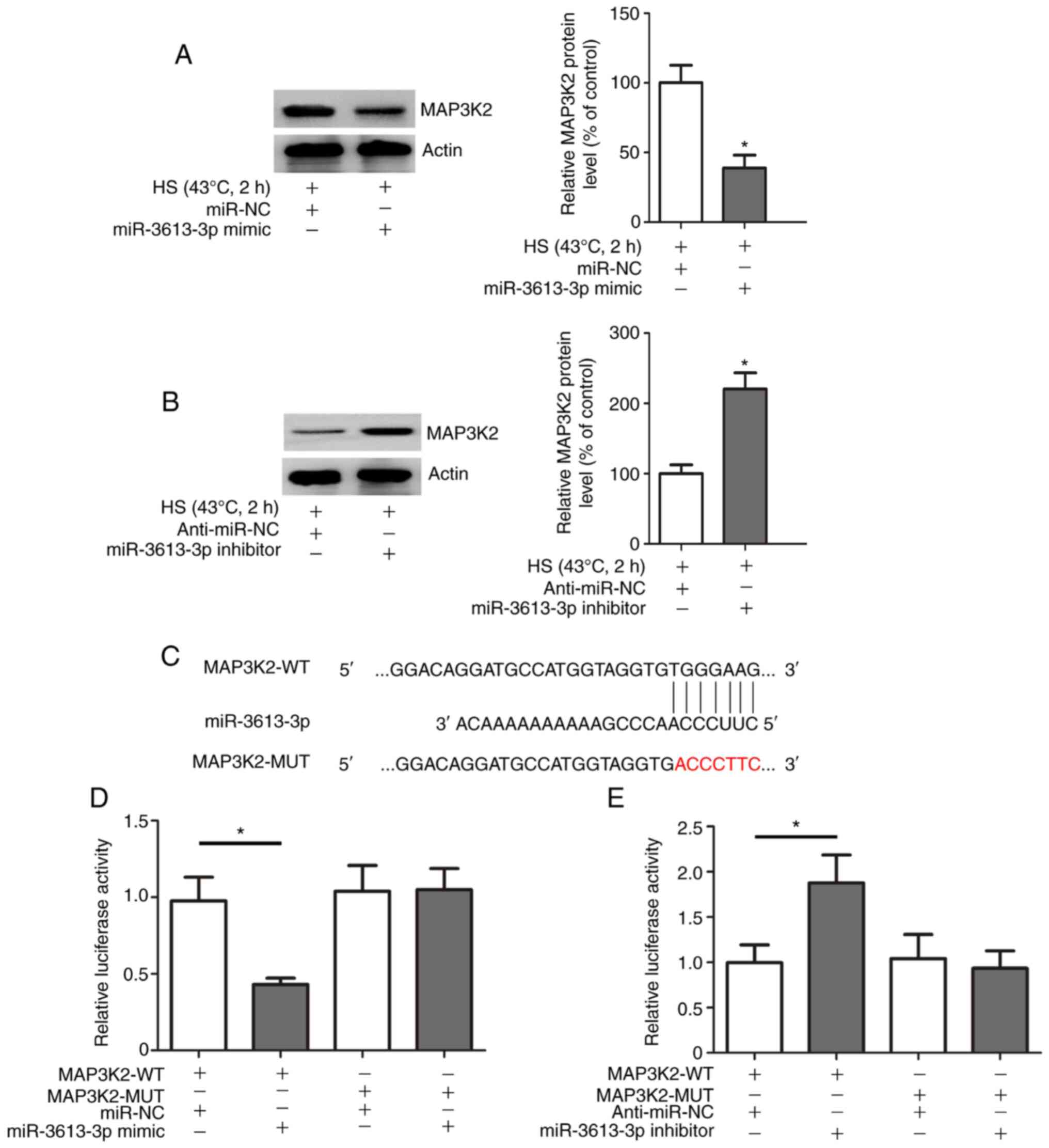
MAP3K2 is a target of miR-3613-3p in
HUVECs
Previous bioinformatics analysis indicated that
MAP3K2 may be a target gene of miR- 3613-3p (12). The present study investigated the
relationship between miR-3613-3p and MAP3K2 in the context of HS.
Overexpression of miR-3613-3p in HUVECs induced using the
miR-3613-3p mimic inhibited HS-induced MAP3K2 expression (Fig. 4A), whereas suppression of
miR-3613-3p induced using the miR-3613-3p inhibitor enhanced
HS-induced MAP3K2 expression in HUVECs (Fig. 4B). To further confirm whether the
predicted target sites of miR-3613-3p were located within the
3′-UTR of MAP3K2 mRNA, dual luciferase reporter assays were
performed with either the MAP3K2-WT or MAP3K2-MUT plasmid in HUVECs
at 37°C. A schematic representation of the putative miR-3613-3p
binding site in the MAP3K2 mRNA 3′-UTR is shown in Fig. 4C. Reduced luciferase activity was
detected in HUVECs transfected with MAP3K2-WT and the miR-3613-3p
mimic compared with that in HUVECs co-transfected with MAP3K2-WT
and miR-NC (Fig. 4D). Moreover, the
luciferase assay data indicated that luciferase activity was
significantly increased in HUVECs co-transfected with MAP3K2-WT and
the miR-3613-3p inhibitor compared with that in HUVECs
co-transfected with MAP3K2-WT and anti-miR-NC (Fig. 4E). Conversely, no change in the
luciferase activity of the MAP3K2-MUT vector was detected after
co-transfection with the miR-3613-3p mimic, the miR-3613-3p
inhibitor or the corresponding NC (Fig.
4D and E). Collectively, these data indicated that MAP3K2 was a
target of miR-3613-3p.
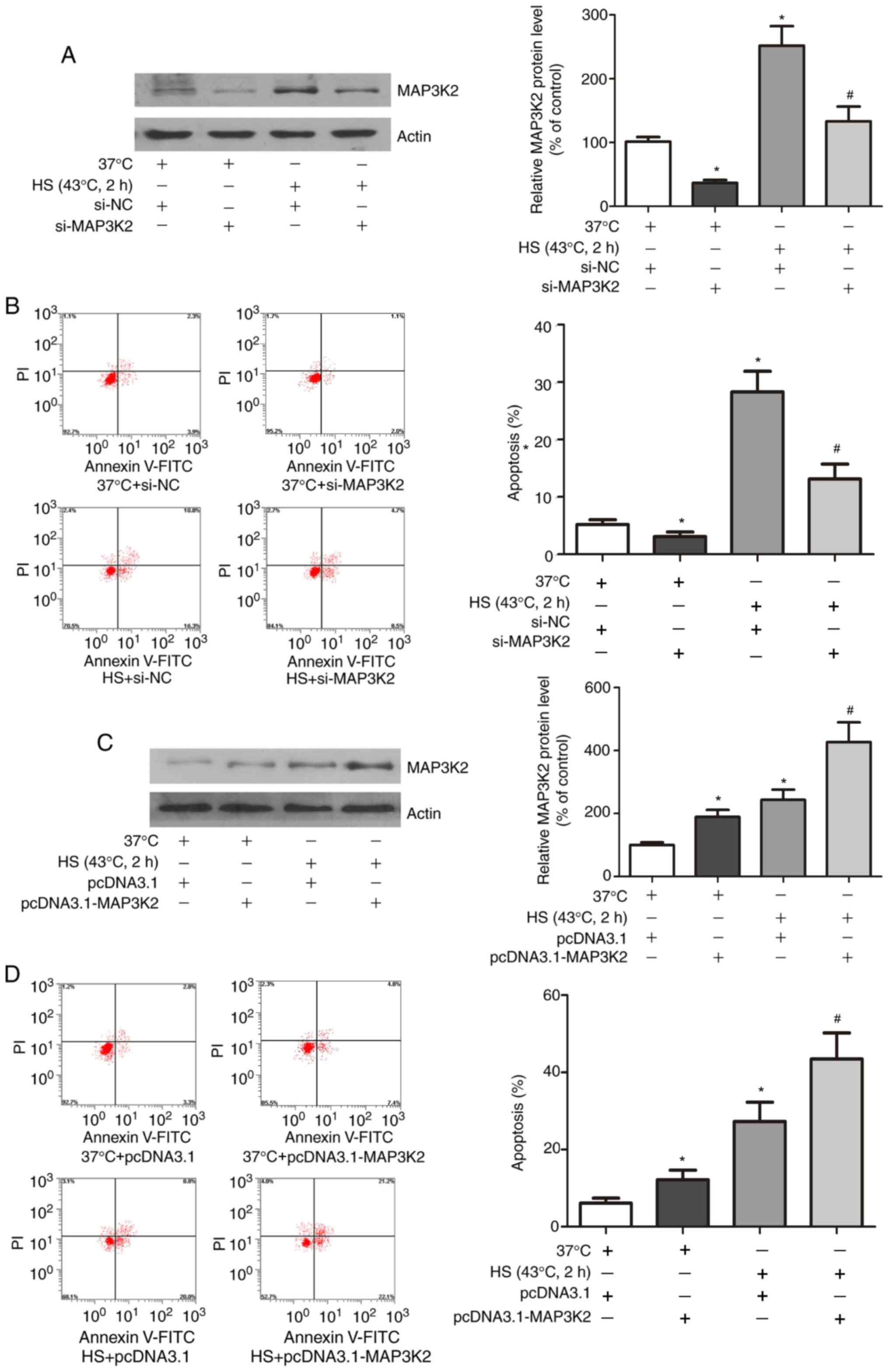
MAP3K2 mediates HS-induced HUVEC
apoptosis
The present study also aimed to determine whether
MAP3K2 was involved in the HS-induced apoptosis of HUVECs. As shown
in Fig. 5A and C, HUVECs were
successfully transfected with si-MAP3K2 or pcDNA3.1-MAP3K2;
compared with in the control groups, the transfected cells
exhibited decreased or increased protein expression levels of
MAP3K2, respectively. Notably, si-MAP3K2 treatment protected HUVECs
from HS-induced apoptosis (Fig.
5B), whereas pcDNA3.1-MAP3K2 treatment resulted in increased
HS-induced apoptosis of HUVECs (Fig.
5D).
miR-3613-3p acts through MAP3K2 to
influence HUVEC apoptosis
To determine whether MAP3K2 was involved in the
pathogenic role of miR-3613-3p in HS-induced HUVEC apoptosis, the
present study investigated the relationship among miR-3613-3p,
MAP3K2 and apoptosis in HS-exposed HUVECs. MAP3K2 expression and
apoptosis in HUVECs following HS were significantly inhibited by
the miR-3613-3p mimic. Transfection with pcDNA3.1-MAP3K2 partially
reversed the miR-3613-3p mimic-induced inhibition of MAP3K2
expression and apoptosis (Fig. 6A and
B), whereas transfection with si-MAP3K2 promoted the
miR-3613-3p mimic-induced inhibition of MAP3K2 expression and
apoptosis (Fig. 6C and D).
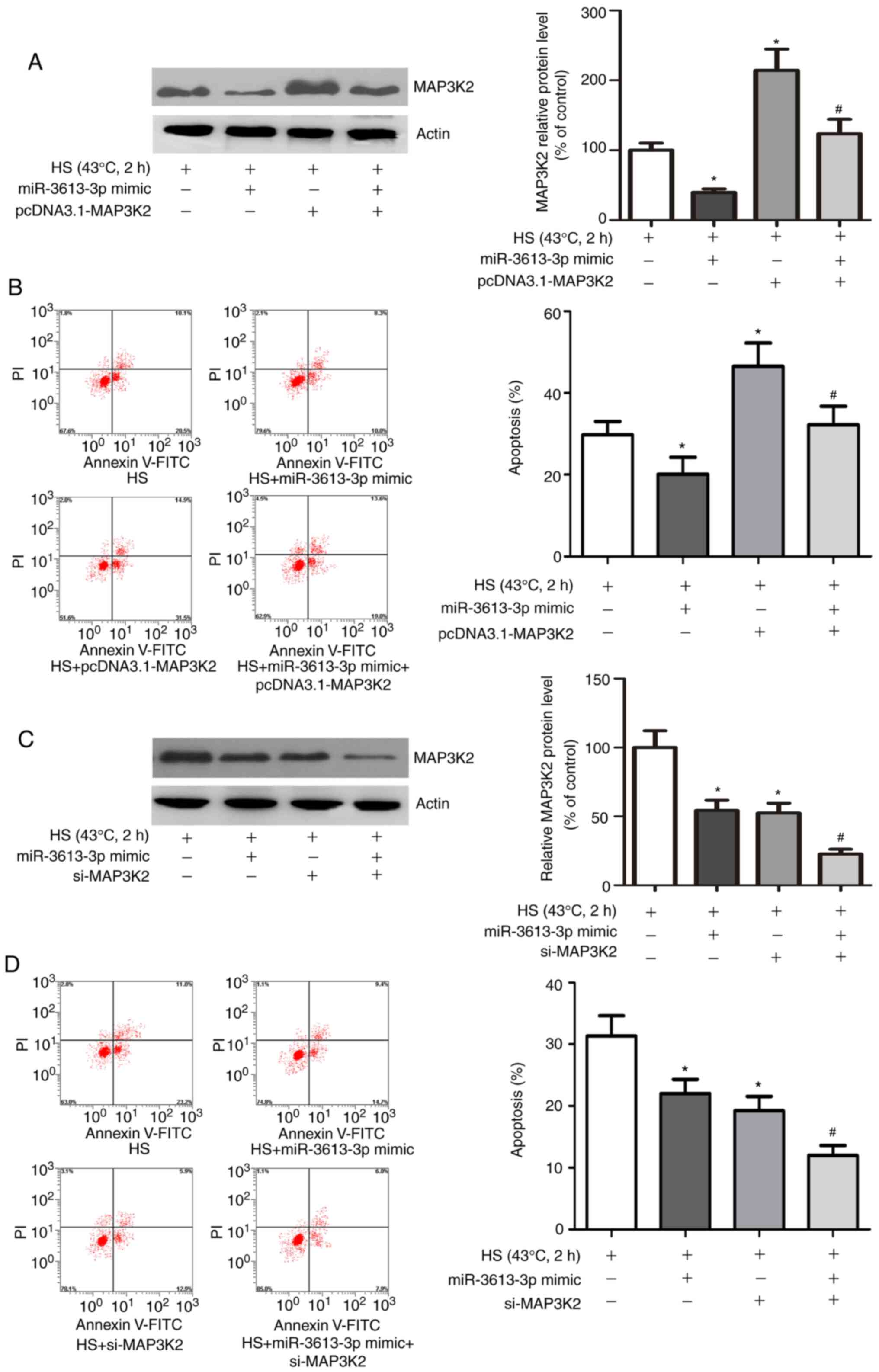 | Figure 6.miR-3613-3p acts through MAP3K2 to
influence HUVEC apoptosis. (A) Expression of MAP3K2 in HUVECs after
HS exposure, miR-3613-3p overexpression and/or MAP3K2
overexpression. (B) Apoptosis of HUVECs after HS exposure,
miR-3613-3p overexpression and/or MAP3K2 overexpression (C)
Expression of MAP3K2 in HUVECs after HS exposure, miR-3613-3p
overexpression and/or silencing MAP3K2. (D) Apoptosis of HUVECs
after HS exposure, miR-3613-3p overexpression and/or silencing
MAP3K2. *P<0.05 vs. HS group; #P<0.05 vs. HS +
miR-3613-3p mimic. FITC, fluorescein isothiocyanate; HUVECs, human
umbilical vein endothelial cells; HS, heat stress; MAP3K2,
mitogen-activated protein kinase kinase kinase 3; miR, microRNA;
NC, negative control; PI, propidium iodide; si, small
interfering. |
MAP3K2 affects the HS-induced
apoptosis of HUVECs by increasing p38 phosphorylation and caspase-3
activity
To elucidate the molecular mechanisms by which
MAP3K2 mediates HUVEC apoptosis, the underlying signalling pathways
were assessed. As shown in Fig. 7A,
HS had no effect on the expression levels of p38, JNK and ERK, but
significantly upregulated their phosphorylation. Moreover, HS
resulted in cleavage of caspase-3 in HUVECs. To further investigate
the effects of different MAPKs on the HS-induced apoptosis of
HUVECs, HUVECs were exposed to HS and treated with a range of
specific inhibitors, including SB203580, PD98059 and SP600125. As
shown in Fig. 7B, inhibition of p38
MAPK, but not ERK or JNK, diminished the production of cleaved
caspase-3 and DNA fragments in HS-treated HUVECs. In addition, the
relationship between MAP3K2 and p38 was determined. Western blot
analysis revealed that HS increased the phosphorylation of p38 and
activation of caspase-3; notably, knockdown of MAP3K2 expression
significantly suppressed the HS-induced increases in p38
phosphorylation and caspase-3 activation, whereas overexpression of
MAP3K2 markedly promoted these effects (Fig. 7C). Collectively, these findings
indicated that MAP3K2 may accelerate HUVEC apoptosis induced by HS
via regulation of the p38/caspase-3 signalling pathway.
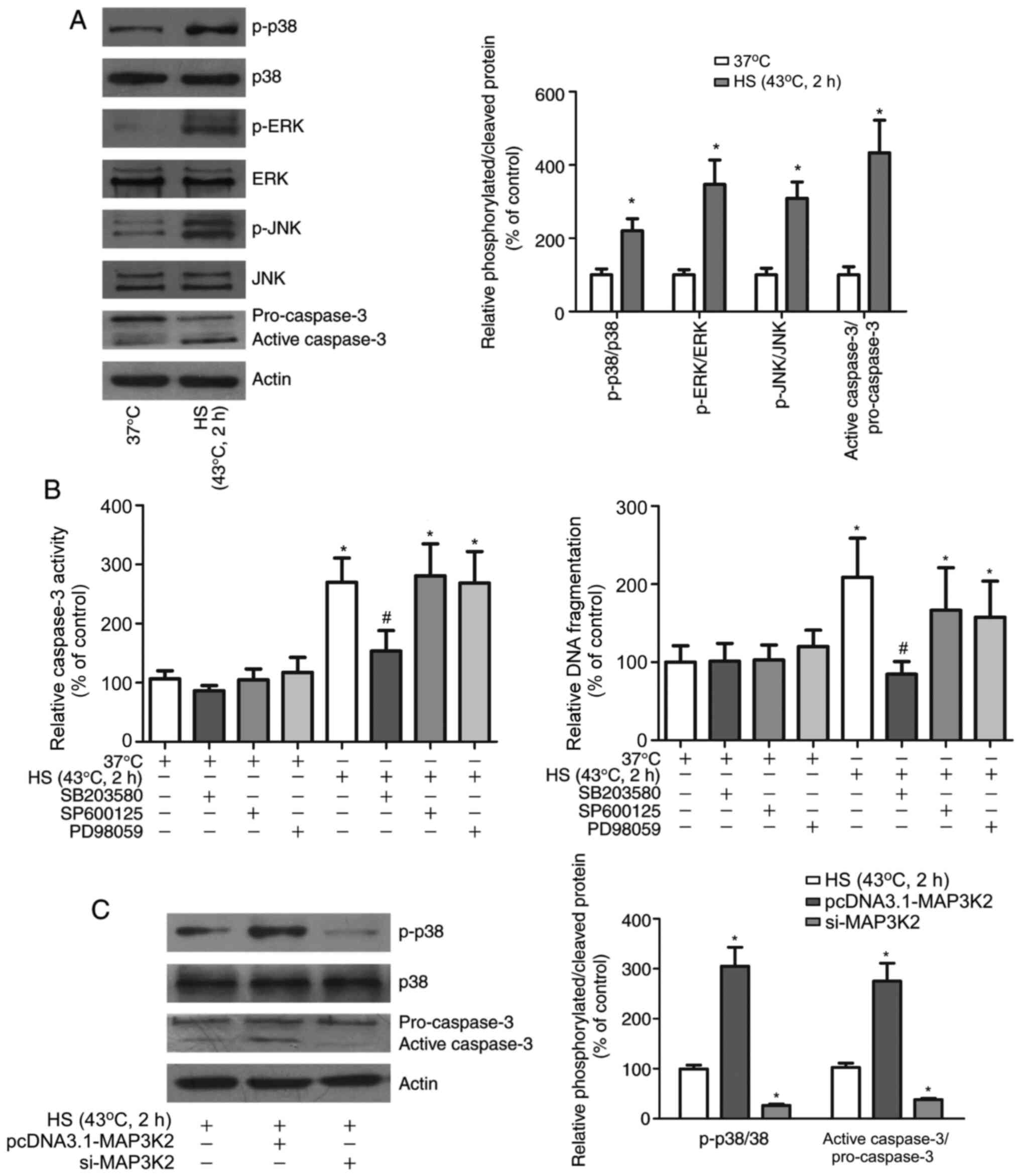 | Figure 7.MAP3K2 affects the HS-induced
apoptosis of HUVECs by upregulating p38 phosphorylation and
caspase-3 activity. (A) Expression and phosphorylation of p38, ERK
and JNK, and the level of cleaved caspase-3 in HUVECs after HS
exposure. (B) Caspase-3 activity and DNA fragmentation in HUVECs
after HS exposure and treatment with SB203580, SP600125 or PD98059.
*P<0.05 vs. 37°C group; #P<0.05 vs. HS group. (C)
Expression and phosphorylation of p38 and the level of cleaved
caspase-3 in HUVECs after HS exposure and MAP3K2 overexpression or
knockdown. *P<0.05 vs. HS group. ERK, extracellular
signal-regulated kinase; HUVECs, human umbilical vein endothelial
cells; HS, heat stress; JNK, c-Jun N-terminal kinase; MAP3K2,
mitogen-activated protein kinase kinase kinase 3; p-,
phosphorylated; si, small interfering. |
Discussion
Accumulating evidence has indicated that miRNA and
miRNA-mediated regulatory networks serve a vital role in the onset
and development of endothelial cell-related diseases and may
represent therapeutic targets (23–25).
Our previous miRNA expression profiling data in HUVECs indicated
that miR-3613-3p may be a HS-related miRNA (12). Furthermore, using an in vivo
approach, our previous study revealed a regulatory relationship
between miR-3613-3p and MAP3K2 that mediated the apoptosis of
HS-treated HUVECs (19).
The present study confirmed that miR-3613-3p could
target MAP3K2 to suppress its protein expression and participate in
HS-mediated HUVEC apoptosis. This conclusion was confirmed by the
following experiments: i) A negative association between
miR-3613-3p and MAP3K2 expression was observed in HUVECs upon
exposure to HS. ii) Inhibition of miR-3613-3p promoted MAP3K2
expression, whereas upregulation of miR-3613-3p suppressed MAP3K2
expression. iii) miR-3613-3p negatively regulated MAP3K2 expression
in HUVECs via binding to a site in the 3′-UTR of MAP3K2 mRNA.
Notably, luciferase activity induced by the MAP3K2-WT plasmid was
specifically responsive to miR-3613-3p, unlike the MAP3K2-MUT
plasmid. iv) In a HS model, inhibition of MAP3K2 decreased
HS-induced apoptosis, whereas HS-induced apoptosis was increased
when MAP3K2 expression was enhanced. v) Restoration of MAP3K2
expression partially restored the suppression of apoptosis induced
by miR-3613-3p overexpression, whereas inhibition of MAP3K2
enhanced miR-3613-3p-induced suppression of apoptosis. Thus, the
present results indicated that MAP3K2 was a target of miR-3613-3p
in terms of molecular mechanism and function.
There are three types of MAPK in the evolutionarily
conserved MAPK cascades: MAPK kinase kinases (MAP3Ks), MAPK kinases
(MAP2Ks) and MAPKs. By sequentially activating each other, MAPKs
transduce signals from the activated receptors to the nucleus
(26,27). MAP3K2 is an upstream kinase in the
MAPK signalling pathway and has been reported to participate in
numerous important cellular processes, such as apoptosis,
proliferation and differentiation (28–30).
To explore the biological function of MAP3K2 in the HS-induced
apoptosis of HUVECs, the present study assessed the downstream
genes of MAP3K2. HS stimulation increased ERK, JNK and p38 MAPK
phosphorylation in HUVECs when apoptosis was notably increased,
which was accompanied by increased cleavage of caspase-3. Moreover,
inhibition of p38, but not JNK or ERK, markedly suppressed
HS-induced production of cleaved caspase-3 (a key executor of
apoptosis) and DNA fragmentation. In addition, in the present
study, the HS-induced increases in p38 phosphorylation and
caspase-3 activation in HUVECs were partially reversed by
suppression of MAP3K2. Moreover, upregulation of MAP3K2 enhanced
the HS-induced phosphorylation of p38 and activation of caspase-3
in HUVECs. The p38 MAPK pathway has been reported to enhance
caspase-3 activity by promoting heat shock protein (Hsp)27–78
phosphorylation and MAPK-activated protein kinase 2 (MK2)
expression in neural stem cells (31). In nerve cells, targeting other
oncogenes downstream of p38, such as Bcl-2-like protein 11, has
been reported to increase caspase-3 activity (32). These findings collectively suggest
that p38 may act as the downstream target of MAP3K2 and could
promote apoptosis via caspase-3 in HUVECs upon exposure to HS.
p38 MAPK is first activated by typical MEKK or
mixed-lineage kinases, then activates mitogen-activated protein
kinase kinase 3 (MKK3) and MKK6, which are highly selective for p38
MAPK (33,34). In general, MAP3K2 phosphorylation
was previously reported to preferentially regulate the JNK and ERK5
pathways by phosphorylating and activating MKK5 as well as MAP
kinase 7 (35–38). However, the functional experiments
in the present study revealed that HS-induced activation of the
MAP3K2 pathway in a p38-dependent manner and resulted in HUVEC
apoptosis. Consistent with the present findings, some previous
studies have reported that MAP3K2 is associated with the activation
of p38 (28,39). For example, sublytic C5b-9 promoted
the apoptosis of glomerular mesangial cells by activating the
MAP3K2/p38 MAPK axis (28). These
findings indicated that the pathways engaged by MAP3K2 are highly
context dependent, and vary by both cell type and stimulus.
Liu et al (40) observed that significant
phosphorylation of p38 occurred in HUVECs after 15 min at 43°C. Li
et al (41) investigated the
time course of p38 MAPK phosphorylation in HUVECs stimulated by HS;
p38 phosphorylation was detected as early as 1 h after heat shock
and continued to the 9-h time point, which was also consistent with
the time period during which caspase-9 and caspase-3 activation,
Bcl-2 ubiquitination and apoptosis occurred. Similar to the
alterations observed in HUVECs, p38 MAPK was shown to be rapidly
activated after heat shock in glial cells; its phosphorylation
peaked at 3 h and was maintained to the 12-h time point in glial
cells, consistent with the time course of HS-induced apoptosis
(42). Additionally,
phosphorylation of p38 has been shown to be elevated at 6 h after
HS in intestinal epithelial cells (43), and HS-induced activation of p38 was
measurable in cardiomyocytes 1 h after HS exposure (44). In this study, phosphorylation of p38
was at a high level 2 h after HS, coinciding with the decrease in
miR-3613-3p and increase in MAP3K2 expression. p38 MAPK is a
downstream molecule of multiple signalling pathways, and is
eventually activated through signalling pathways composed of
extracellular signalling molecules, specific receptors on the
surface of the membrane and intracellular signalling molecules
(45–47). Notably, the initiation time and
duration of p38 MAPK activation are irregular and may vary with
different cell types or environmental stresses.
In addition to the present findings, previous
studies have revealed that HS can activate different MAPK
signalling pathways in a variety of cell types. p-MAPKs, which are
downstream genes of MAPKs, and some of their downstream molecules,
were the focus of these studies. Li et al (31) indicated that p38/MK2/Hsp27-78
signalling was activated during HS-induced apoptosis and autophagy
progression in neural stem cell. In addition, Liu et al
(44) reported that HS promoted the
activation of p38, leading to cardiomyocyte apoptosis and cardiac
dysfunction. Huang et al (15) indicated that the ERK pathways
participated in the HS-induced receptor-interacting protein
1/3-dependent necroptosis of pulmonary vascular endothelial cells,
which exacerbated lung injury. A previous study also indicated that
activation of p38/c-Jun served a proapoptotic role in HS-induced
intestinal epithelial cell apoptosis, which eventually led to
reduced epithelial barrier integrity and increased intestinal
permeability, whereas ERK conferred resistance to apoptosis
(43). In summary, these data
suggested that both the activation of MAPKs and the biological
outcome of MAPK activation are dependent on the intensity/duration
of HS and are cell type- and tissue-specific. However, the
relationship between HS and MAPK signalling is complex, and whether
a single MAPK pathway drives functions in various physiological
processes, or whether the same physiological process involves
multiple MAPK pathways requires further research.
Some limitations of the current study must be
mentioned: i) The present study used HUVECs to reveal the mechanism
underlying HS-induced apoptosis. All experiments were in cell
models, which may have major differences from in vivo models
and cannot perfectly imitate the clinical course of HS. Future
in vivo studies are required to determine the effect of
endothelial cell apoptosis on heat-related organ dysfunction. ii)
Endothelial cells in different organs may exhibit different
responses to HS due to differences in organ function and the local
internal environment, although the organ specificity of endothelial
cells was not considered in the present study. iii) miRNA and
signalling pathways are connected by complex interactions and
regulatory mechanisms that form positive and/or negative regulatory
networks. The effects of other miRNA molecules and signalling
pathways were not considered. iv) Although SB203580 has been widely
used in previous studies to inhibit protein, siRNA can inhibit
protein production as they directly inhibit mRNA. In future
studies, siRNA targeting p38 will be used to obtain improved
results.
In conclusion, the present study revealed that HS
induced HUVEC apoptosis via regulation of the
miR-3613-3p/MAP3K2/p38/caspase-3 axis. The present findings also
provided insight into the role of miR-3613-3p during heat stroke,
suggesting the modulation of miR-3613-3p as a potential therapeutic
strategy for heat stroke.
Acknowledgements
The authors would like to thank Dr Qiping Lu
(Department of General Surgery, Central Theater General Hospital of
the People's Liberation Army of China, Wuhan, China) for her advice
and Dr Guoguo Zhu (Department of Emergency, Central Theater General
Hospital of the People's Liberation Army of China, Wuhan, China)
for her help in preparing the manuscript.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analysed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
JL participated in the design of the study, carried
out experiments and data analysis, performed drafting and editing
of the manuscript, and was a major contributor to the writing of
the manuscript. SX and SL participated in the experiments, data
analysis and statistical analysis. BC conceived and designed the
experiments, edited and revised the manuscript, and approved the
final version of the manuscript. All authors read and approved the
final manuscript. JL and BC confirm the authenticity of all the raw
data.
Ethics approval and consent to
participate
Ethical approval was obtained for the use of
non-immortal HUVECs from the Ethics Committee of Hefei Boe Hospital
Co., Ltd. (approval no. 20190106).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
miR-3613-3p
|
microRNA-3613-3p
|
|
HS
|
heat stress
|
|
MAP3K2
|
mitogen-activated protein kinase
kinase kinase 2
|
|
HUVEC
|
human umbilical vein endothelial
cell
|
|
3′-UTR
|
3′-untranslated region
|
|
MAPK
|
mitogen-activated protein kinase
|
References
|
1
|
Jin H, Chen Y, Ding C, Lin Y, Chen Y,
Jiang D and Su L: Microcirculatory disorders and protective role of
Xuebijing in severe heat stroke. Sci Rep. 8:45532018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Bouchama A and Knochel JP: Heat stroke. N
Engl J Med. 346:1978–1988. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Roberts GT, Ghebeh H, Chishti MA,
Al-Mohanna F, El-Sayed R, Al-Mohanna F and Bouchama A:
Microvascular injury, thrombosis, inflammation, and apoptosis in
the pathogenesis of heatstroke: A study in baboon model.
Arterioscler Thromb Vasc Biol. 28:1130–1136. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Lam NN, Hung TD and Hung DK: Acute
respiratory distress syndrome among severe burn patients in a
developing country: Application result of the berlin definition.
Ann Burns Fire Disasters. 31:9–12. 2018.PubMed/NCBI
|
|
5
|
Haines RJ, Wang CY, Yang CGY, Eitnier RA,
Wang F and Wu MH: Targeting palmitoyl acyltransferase ZDHHC21
improves gut epithelial barrier dysfunction resulting from
burn-induced systemic inflammation. Am J Physiol Gastrointest Liver
Physiol. 313:G549–G557. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Bouchama A, Hammami MM, Haq A, Jackson J
and al-Sedairy S: Evidence for endothelial cell activation/injury
in heatstroke. Crit Care Med. 24:1173–1178. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Oshima K, Han X, Ouyang Y, El Masri R,
Yang Y, Haeger SM, McMurtry SA, Lane TC, Davizon-Castillo P, Zhang
F, et al: Loss of endothelial sulfatase-1 after experimental sepsis
attenuates subsequent pulmonary inflammatory responses. Am J
Physiol Lung Cell Mol Physiol. 317:L667–L677. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Colbert JF and Schmidt EP: Endothelial and
microcirculatory function and dysfunction in sepsis. Clin Chest
Med. 37:263–275. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Bombeli T, Mueller M and Haeberli A:
Anticoagulant properties of the vascular endothelium. Thromb
Haemost. 77:408–423. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Donadello K, Piagnerelli M, Reggiori G,
Gottin L, Scolletta S, Occhipinti G, Zouaoui Boudjeltia K and
Vincent JL: Reduced red blood cell deformability over time is
associated with a poor outcome in septic patients. Microvasc Res.
101:8–14. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Bartel DP: MicroRNAs: Genomics,
biogenesis, mechanism, and function. Cell. 116:281–297. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Liu J, Zhu G, Xu S, Liu S, Lu Q and Tang
Z: Analysis of miRNA expression profiling in human umbilical vein
endothelial cells affected by heat stress. Int J Mol Med.
40:1719–1730. 2017.PubMed/NCBI
|
|
13
|
Corre I, Paris F and Huot J: The p38
pathway, a major pleiotropic cascade that transduces stress and
metastatic signals in endothelial cells. Oncotarget. 8:55684–55714.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Li M, van Esch BCAM, Wagenaar GTM, Garssen
J, Folkerts G and Henricks PAJ: Pro- and anti-inflammatory effects
of short chain fatty acids on immune and endothelial cells. Eur J
Pharmacol. 831:52–59. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Huang W, Xie W, Gong J, Wang W, Cai S,
Huang Q, Chen Z and Liu Y: Heat stress induces RIP1/RIP3-dependent
necroptosis through the MAPK, NF-κB, and c-Jun signaling pathways
in pulmonary vascular endothelial cells. Biochem Biophys Res
Commun. 528:206–212. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yong YX, Yang H, Lian J, Xu XW, Han K, Hu
MY, Wang HC and Zhou LM: Up-regulated microRNA-199b-3p represses
the apoptosis of cerebral microvascular endothelial cells in
ischemic stroke through down-regulation of MAPK/ERK/EGR1 axis. Cell
Cycle. 18:1868–1881. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhou Z, Chen Y, Zhang D, Wu S, Liu T, Cai
G and Qin S: MicroRNA-30-3p suppresses inflammatory factor-induced
endothelial cell injury by targeting TCF21. Mediators Inflamm.
2019:13421902019. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Yi J and Gao ZF: MicroRNA-9-5p promotes
angiogenesis but inhibits apoptosis and inflammation of high
glucose-induced injury in human umbilical vascular endothelial
cells by targeting CXCR4. Int J Biol Macromol. 130:1–9. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Liu J, Han X, Zhu G, Liu S, Lu Q and Tang
Z: Analysis of potential functional significance of
microRNA-3613-3p in human umbilical vein endothelial cells affected
by heat stress. Mol Med Rep. 20:1846–1856. 2019.PubMed/NCBI
|
|
20
|
Tang S, Allagadda V, Chibli H, Nadeau JL
and Mayer GD: Comparison of cytotoxicity and expression of metal
regulatory genes in zebrafish (Danio rerio) liver cells
exposed to cadmium sulfate, zinc sulfate and quantum dots.
Metallomics. 5:1411–1422. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Tang S, Cai Q, Chibli H, Allagadda V,
Nadeau JL and Mayer GD: Cadmium sulfate and CdTe-quantum dots alter
DNA repair in zebrafish (Danio rerio) liver cells. Toxicol
Appl Pharmacol. 272:443–452. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Li X, Xue X, Sun Y, Chen L, Zhao T, Yang
W, Chen Y and Zhang Z: MicroRNA-326-5p enhances therapeutic
potential of endothelial progenitor cells for myocardial
infarction. Stem Cell Res Ther. 10:3232019. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Xing X, Li Z, Yang X, Li M, Liu C, Pang Y,
Zhang L, Li X, Liu G and Xiao Y: Adipose-derived mesenchymal stem
cells-derived exosome-mediated microRNA-342-5p protects endothelial
cells against atherosclerosis. Aging (Albany NY). 12:3880–3898.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Deng X, Chu X, Wang P, Ma X, Wei C, Sun C,
Yang J and Li Y: MicroRNA-29a-3p reduces TNFα-induced endothelial
dysfunction by targeting tumor necrosis factor receptor 1. Mol Ther
Nucleic Acids. 18:903–915. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zehorai E and Seger R: Beta-like importins
mediate the nuclear translocation of mitogen-activated protein
kinases. Mol Cell Biol. 34:259–270. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Lee Y, Kim YJ, Kim MH and Kwak JM: MAPK
cascades in guard cell signal transduction. Front Plant Sci.
7:802016. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zhu G, Qiu W, Li Y, Zhao C, He F, Zhou M,
Wang L, Zhao D, Lu Y, Zhang J, et al: Sublytic C5b-9 induces
glomerular mesangial cell apoptosis through the cascade pathway of
MEKK2-p38 MAPK-IRF-1-TRADD-caspase 8 in rat Thy-1 nephritis. J
Immunol. 198:1104–1118. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Chen Z, Zhu R and Zheng J, Chen C, Huang
C, Ma J, Xu C, Zhai W and Zheng J: Cryptotanshinone inhibits
proliferation yet induces apoptosis by suppressing STAT3 signals in
renal cell carcinoma. Oncotarget. 8:50023–50033. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Chang X, Liu F, Wang X, Lin A, Zhao H and
Su B: The kinases MEKK2 and MEKK3 regulate transforming growth
factor-β-mediated helper T cell differentiation. Immunity.
34:201–212. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Li H, Liu Y, Wen M, Zhao F, Zhao Z, Liu Y,
Lin X and Wang L: Hydroxysafflor yellow A (HSYA) alleviates
apoptosis and autophagy of neural stem cells induced by heat stress
via p38 MAPK/MK2/Hsp27-78 signaling pathway. Biomed Pharmacother.
114:1088152019. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Zhuang Y, Xu H, Richard SA, Cao J, Li H,
Shen H, Yu Z, Zhang J, Wang Z, Li X and Chen G: Inhibition of EPAC2
attenuates intracerebral hemorrhage-induced secondary brain injury
via the p38/BIM/caspase-3 pathway. J Mol Neurosci. 67:353–363.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Barata AG and Dick TP: A role for
peroxiredoxins in H2O2- and MEKK-dependent
activation of the p38 signaling pathway. Redox Biol. 28:1013402020.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Deschodt-Lanckman M and Strosberg AD: In
vitro degradation of the C-terminal octapeptide of cholecystokinin
by ‘enkephalinase A’. FEBS Lett. 152:109–113. 1983. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Dermott JM, Ha JH, Lee CH and Dhanasekaran
N: Differential regulation of Jun N-terminal kinase and p38MAP
kinase by Galpha12. Oncogene. 23:226–232. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Raviv Z, Kalie E and Seger R: MEK5 and
ERK5 are localized in the nuclei of resting as well as stimulated
cells, while MEKK2 translocates from the cytosol to the nucleus
upon stimulation. J Cell Sci. 117:1773–1784. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Shen CT, Qiu ZL, Song HJ, Wei WJ and Luo
QY: miRNA-106a directly targeting RARB associates with the
expression of Na(+)/I(−) symporter in thyroid cancer by regulating
MAPK signaling pathway. J Exp Clin Cancer Res. 35:1012016.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Tsioumpekou M, Papadopoulos N, Burovic F,
Heldin CH and Lennartsson J: Platelet-derived growth factor
(PDGF)-induced activation of Erk5 MAP-kinase is dependent on Mekk2,
Mek1/2, PKC and PI3-kinase, and affects BMP signaling. Cell Signal.
28:1422–1431. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Shi X, Liu TT, Yu XN, Balakrishnan A, Zhu
HR, Guo HY, Zhang GC, Bilegsaikhan E, Sun JL, Song GQ, et al:
microRNA-93-5p promotes hepatocellular carcinoma progression via a
microRNA-93-5p/MAP3K2/c-Jun positive feedback circuit. Oncogene.
39:5768–5781. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Liu Y, Zhou G, Wang Z, Guo X, Xu Q, Huang
Q and Su L: NF-κB signaling is essential for resistance to heat
stress-induced early stage apoptosis in human umbilical vein
endothelial cells. Sci Rep. 5:135472015. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Li L, Tan H, Yang H, Li F, He X, Gu Z,
Zhao M and Su L: Reactive oxygen species mediate heat
stress-induced apoptosis via ERK dephosphorylation and Bcl-2
ubiquitination in human umbilical vein endothelial cells.
Oncotarget. 8:12902–12916. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Li H, Liu Y, Gu Z, Li L, Liu Y, Wang L and
Su L: p38 MAPK-MK2 pathway regulates the heat-stress-induced
accumulation of reactive oxygen species that mediates apoptotic
cell death in glial cells. Oncol Lett. 15:775–782. 2018.PubMed/NCBI
|
|
43
|
Liu Y, Wang Z, Xie W, Gu Z, Xu Q and Su L:
Oxidative stress regulates mitogen-activated protein kinases and
c-Jun activation involved in heat stress and
lipopolysaccharide-induced intestinal epithelial cell apoptosis.
Mol Med Rep. 16:2579–2587. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Liu ZF, Ji JJ, Zheng D, Su L and Peng T:
Calpain-2 protects against heat stress-induced cardiomyocyte
apoptosis and heart dysfunction by blocking p38 mitogen-activated
protein kinase activation. J Cell Physiol. 234:10761–10770. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Pu Y, Liu Z, Tian H and Bao Y: The
immunomodulatory effect of Poria cocos polysaccharides is mediated
by the Ca2+/PKC/p38/NF-κB signaling pathway in
macrophages. Int Immunopharmacol. 72:252–257. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Wang W, Weng J, Yu L, Huang Q, Jiang Y and
Guo X: Role of TLR4-p38 MAPK-Hsp27 signal pathway in LPS-induced
pulmonary epithelial hyperpermeability. BMC Pulm Med. 18:1782018.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Zhang X, Chen Q, Song H, Jiang W, Xie S,
Huang J and Kang G: MicroRNA-375 prevents TGF-β-dependent
transdifferentiation of lung fibroblasts via the MAP2K6/P38
pathway. Mol Med Rep. 22:1803–1810. 2020. View Article : Google Scholar : PubMed/NCBI
|