Introduction
As one of the severe microvascular complications of
type 2 diabetes mellitus, diabetic nephropathy (DN) is the leading
cause of end-stage renal disease (1). Although diagnosis and treatment
technologies have improved, the incidence of DN is increasing
worldwide (2). Therefore, there is
an urgent need to clarify the molecular pathogenesis of DN and to
identify therapeutic targets.
Autophagic dysfunction is considered to be involved
in DN (3). Autophagy is a cellular
recycling pathway that is essential for maintaining cellular
integrity (3). Under stress
conditions, human proximal tubular epithelial (HK-2) cells exploit
the autophagic machinery to clear damaged organelles and protein
aggregates. The degradation and recycling process of autophagy
contributes to maintaining normal cellular functions and delays the
progression of diabetic renal injury (4). Previous research has demonstrated that
during autophagy, lysosomes contribute to the regulation of the
degradation of damaged cellular components as an adaptive response,
playing an important role in the pathogenesis of DN (3,5). As a
result, the high glucose (HG)-induced impairment of autophagy in
renal tubular epithelial cells has emerged as an important process
responsible for the occurrence and development of diabetic renal
injury.
Multiple regulatory mechanisms modulate autophagy in
DN, including nutrient signaling pathways (6). Among these, Sirtuin (Sirt)1 functions
as an important regulator of autophagy (7). As a member of the Sirt family, Sirt1
has been reported to contribute to the clearance of damaged
organelles and the maintenance of energy homeostasis (8). Sirt3 belongs to the same family as
Sirt1, and is capable of regulating signaling pathways when stress
occurs, thereby inducing autophagy (9,10). In
a previous study, in a model of sepsis-induced acute kidney injury,
Sirt3 was demonstrated to promote autophagy, alleviating acute
kidney injury, tubular cell apoptosis and inflammatory responses in
the kidneys (11). Moreover,
through the activation of the process of autophagy, Sirt3
significantly attenuates the occurrence and development of
diabetes-related microvascular complications (12). Sirt3 has also been shown to inhibit
mitochondrial injury and cardiomyocyte apoptosis through the
activation of autophagy and mitophagy in HG-stimulated
cardiomyocytes (13). Mesenchymal
stem cells have been shown to attenuate diabetic lung fibrosis by
modulating Sirt3-mediated stress responses, such as the enhancement
of autophagy and inhibition of oxidative stress, inflammation,
apoptosis and endoplasmic reticulum stress (14).
With regards to the pathways regulating autophagy,
Notch homolog 1 (Notch-1)/hairy and enhancer of split-1 (Hes-1) has
been proven to be involved in cell proliferation, differentiation
and death (15). Moreover, this
pathway has been shown to regulate the equilibrium between
autophagosome formation and clearance by lysosomes in podocytes
(16). However, whether this
pathway plays a role in the autophagic process in renal tubular
epithelial cells is not yet fully understood, and the function of
Sirt3 in modulating autophagy in HG-stimulated proximal tubular
epithelial cell injury remains undetermined. Thus, the present
study aimed to evaluate the renoprotective effects of Sirt3 against
DN. Additionally, whether Sirt3 overexpression attenuates kidney
injury via modulating autophagy in HG-stimulated proximal tubular
epithelial cells was determined.
Materials and methods
Cell culture and treatment
HK-2 cells were purchased from the American Type
Culture Collection and cultured as previously described (17). All cells were cultured in DMEM
(Invitrogen; Thermo Fisher Scientific, Inc.) with 10% FBS (HyClone;
Cygtiva) at 37°C, 5% CO2 in a humidified incubator.
Cells (5×105 cells/well) were seeded in 6-well plates at
80% confluency and continued to be cultured in DMEM with 2% FBS.
The HK-2 cells were serum-starved for 24 h, followed by exposure to
DMEM with 5.5 mM glucose [normal glucose (NG)], 30 mM glucose (HG)
or NG + hyperosmotic medium [24.5 mmol/l mannitol (M)]. Following
washing three times with 1X PBS, the HK-2 cells were harvested for
the cell viability assay. This was followed by transfection with
overexpression plasmid and the inhibition of the Notch-1/Hes-1
pathway.
Cell viability assay
HK-2 cells were cultured in the designated medium
for an additional 6, 12, 24, 48 and 72 h. Cell viability was
determined using a Cell Counting Kit-8 (CCK-8; MedChemExpress)
assay. Briefly, the HK-2 cells were washed and plated at a density
of 1×103−104 cells per well in a 96-well
plate. Cells were incubated in fresh medium at 37°C for an
additional 48 h. Subsequently, CCK-8 solution (10 µl) was added to
each well of the culture medium at each time point, according to
the manufacturer's instructions. The plates were kept at 37°C with
5% CO2 in an incubated plate holder for 1 h. The
absorbance was determined using a microplate reader (BioTek
microplate reader; BioTek Instruments, Inc.) at 450 nm.
Transfection with overexpression
plasmid
HK-2 cells were seeded in a 6-well plate at a
density of 5×105 cells/well for 48 h. Plasmid
transfection was performed using FuGENE 6 (Promega Corporation)
according to the manufacturer's instructions. The plasmids used in
the present study were described previously and confirmed by DNA
sequencing (18). Cells were
transfected with 2 µg pCMV-SIRT3 or empty pCMV-vector (Integrated
Biotech Solutions) at 37°C. Following 48 h of transfection, the
HK-2 cells were stimulated with HG for 48 h at 37°C. The
transfection efficacy was determined by reverse
transcription-quantitative PCR (RT-qPCR) and western blot
analysis.
Inhibition of the Notch-1/Hes-1
pathway
To activate the Notch-1/Hes-1 pathway, Jagged1-FC
(final concentration, 0.5 µg/ml; cat. no. 1277-JG; R&D Systems,
Inc.) was added to the medium and the incubation was continued for
a further 2 h at room temperature.
Western blot analysis
Western blot analysis was performed as previously
described (19). The HK-2 cells
were washed with 1X PBS three times and lysed with RIPA lysis
buffer (Beyotime Institute of Biotechnology). Total protein
extracts were collected and the concentration was quantified using
a BCA assay (Pierce; Thermo Fisher Scientific, Inc.). Total protein
(30 µg) was loaded onto 10% SDS-PAGE (Beijing Solarbio Science
& Technology Co., Ltd.) followed by transfer onto PVDF
membranes (Bio-Rad Laboratories, Inc.). The membranes were blocked
with TBS containing 0.1% Tween-20 (TBST) containing 5% non-fat milk
for 1 h at room temperature, followed by primary antibody
incubation overnight at 4°C. The antibodies were as follows:
Anti-Beclin-1 (cat. no. ab210498; 1:1,000; Abcam), anti-LC-3II
(cat. no. ab48394; 1:1,000; Abcam), anti-p62 (cat. no. ab56416;
1:1,000; Abcam), anti-Sirt3 (cat. no. ab217319; 1:1,000; Abcam),
anti-Notch-1 (cat. no. ab52627; 1:1,000; Abcam), anti-Hes-1 (cat.
no. ab108937; 1:1,000; Abcam) and anti-β-actin (cat. no. ab8227;
1:1,000; Abcam). Following washing with TBST three times for 15
min, the membranes were then incubated with HRP-conjugated
secondary antibodies (cat. no. 7074 and 7076; 1:1,000; Cell
Signaling Technology, Inc.) for 2 h at room temperature. Proteins
were visualized using the ECL method (Thermo Fisher Scientific,
Inc.). Semi-quantification of the western blot bands was performed
using ImageJ software (version 1.8.0; National Institutes of
Health).
RT-qPCR analysis
Total cellular mRNA of Beclin-1, LC-3II, p62, Sirt3,
Notch-1 and Hes-1 was isolated using TRIzol® reagent
(Invitrogen; Thermo Fisher Scientific, Inc.), according to the
manufacturer's instructions. RT-qPCR was performed as previously
described (15). Total RNA (2 µg)
extracted from the cultured cells was used as a template for cDNA
synthesis. The primer sequences were as follows: Beclin-1 forward,
5′-AATGACTTTTTTCCTTAGGGGG-3′ and reverse,
5′-GTGGCTTTTGTGGATTTTTTCT-3′; LC-3II forward,
5′-AGTGCCTGTGTTGTTACGGA-3′ and reverse, 5′-GCAGAAGGGAGTGTGTCTGA-3′;
p62 forward, 5′-AGTCGGAGCGGGTTCTCTAT-3′ and reverse,
5′-GTGACACACATTCCAGCGAT-3′; Notch-1 forward,
5′-CAGACAGGCAGGTGGGGTCGTGGTA-3′ and reverse,
5′-GCGACAACGCCTACCTCTG-3′; Hes-1 forward,
5′-CAACACGACACCGGATAAAC-3′ and reverse, 5′-TTCAGCTGGCTCAGACTTTC-3′;
and β-actin forward, 5′-TGACGTGGACATCCGCAAAG-3′ and reverse,
5′-CTGGAAGGTGGACAGCGAGG-3′. The conditions for amplification were
as follows: Initial denaturation at 95°C for 5 min, followed by 40
cycles each at 95°C for 10 sec, followed by 60°C for 30 sec, and
72°C for 45 sec. β-actin mRNA served as an endogenous control. The
relative quantities of products were determined using the
2−ΔΔCq method (20). The
experiments were performed in triplicate for each group.
Cell immunofluorescence
Following seeding on coverslips in 6-well plates
(2×105 cells/well), the cells were fixed with 4%
paraformaldehyde for 10 min at 4°C. Subsequently, the cells were
blocked with 10% goat serum (Beijing Solarbio Science &
Technology Co., Ltd.) for 15 min at room temperature, and subjected
to immunofluorescence staining with anti-Sirt3 polyclonal antibody
(1:500; cat. no. ab40963; Abcam) as the primary antibody. Following
incubation overnight with anti-Sirt3 antibody at 4°C, the HK-2
cells were then incubated with Cy3-labeled IgG (cat. no. TA130020;
1:100; OriGene Technologies, Inc.) for 1 h at room temperature. The
following day, the nuclei were stained with DAPI for 10 min at room
temperature. Immunofluorescence was observed and captured using a
fluorescence microscope (Olympus Corporation).
Statistical analysis
All experiments were carried out in triplicate. The
analyses were performed using SPSS software (version 23.0; IBM
Corp.). Data are expressed as the mean ± SD. For multiple
comparisons between groups, a one-way ANOVA followed by Tukey's
post hoc test was performed. P<0.05 was considered to indicate a
statistically significant difference.
Results
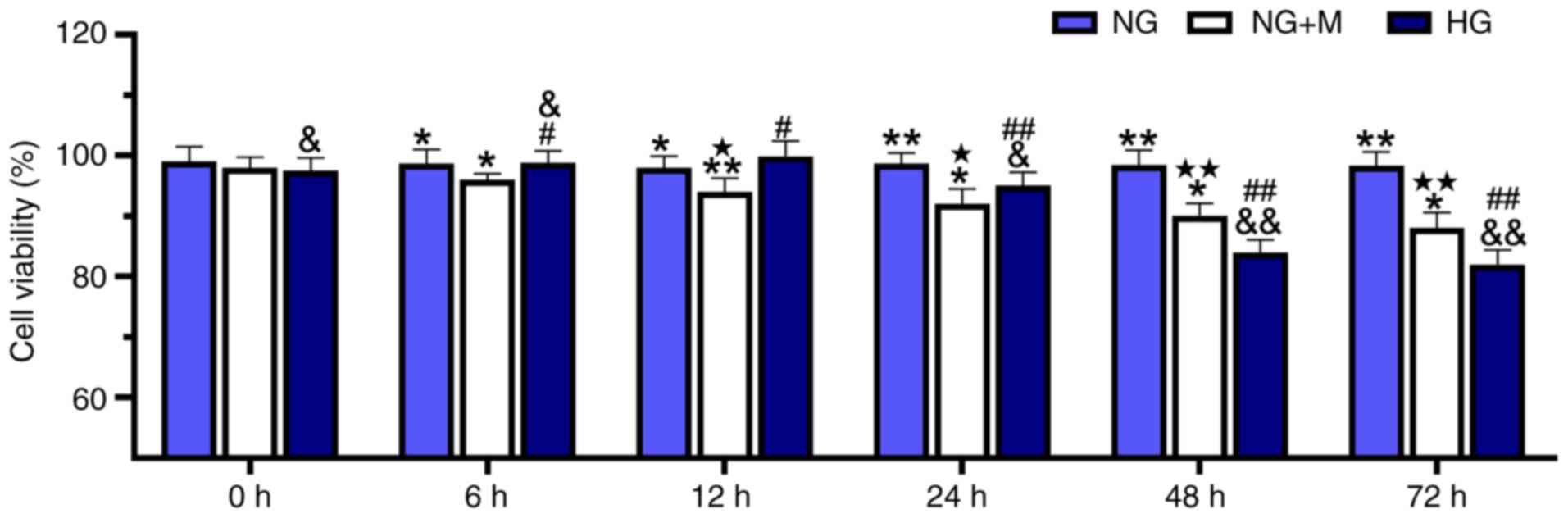
HG reduces the viability of HK-2
cells
HK-2 cell cultivation was performed in NG, HG or a
hyperosmotic environment, and cell viability was then determined
using a CCK-8 assay (Fig. 1). When
the HK-2 cells were incubated in HG culture medium for 6 and 12 h,
it was observed that there was a slight increase in cell viability
compared with cells cultivated in NG. However, treatment with NG +
M decreased cell viability compared with the NG group over the
culturing time. Of note, HG markedly decreased cell viability
compared with the cells treated with NG and NG + M with the
duration of culture.
Long-term culture of HK-2 cells in HG
inhibits the expression of Sirt3 and autophagy
After the cells were subjected to the different
treatments (NG, NG + M and HG), the expression level of Sirt3 was
detected (Fig. 2A). The expression
of Sirt3 exhibited an unremarkable change in the HK-2 cells
cultured in NG + M compared with the NG group. Compared with the NG
group, the protein expression level of Sirt3 was significantly
decreased in the HK-2 cells following stimulation with HG for 48 h
(Fig. 2B). A similar trend was
observed in the cell immunofluorescence assay (Fig. 2J). Short-term HG culture (6 and 12
h) led to a significant elevation in Sirt3 protein expression.
However, the expression level of Sirt3 tended to decrease with the
increasing incubation time. No further changes in Sirt3 protein
expression were observed at 48 h. Sirt3 mRNA expression exhibited a
similar trend with that of its protein expression determined by
western blot analysis (Fig.
2C).
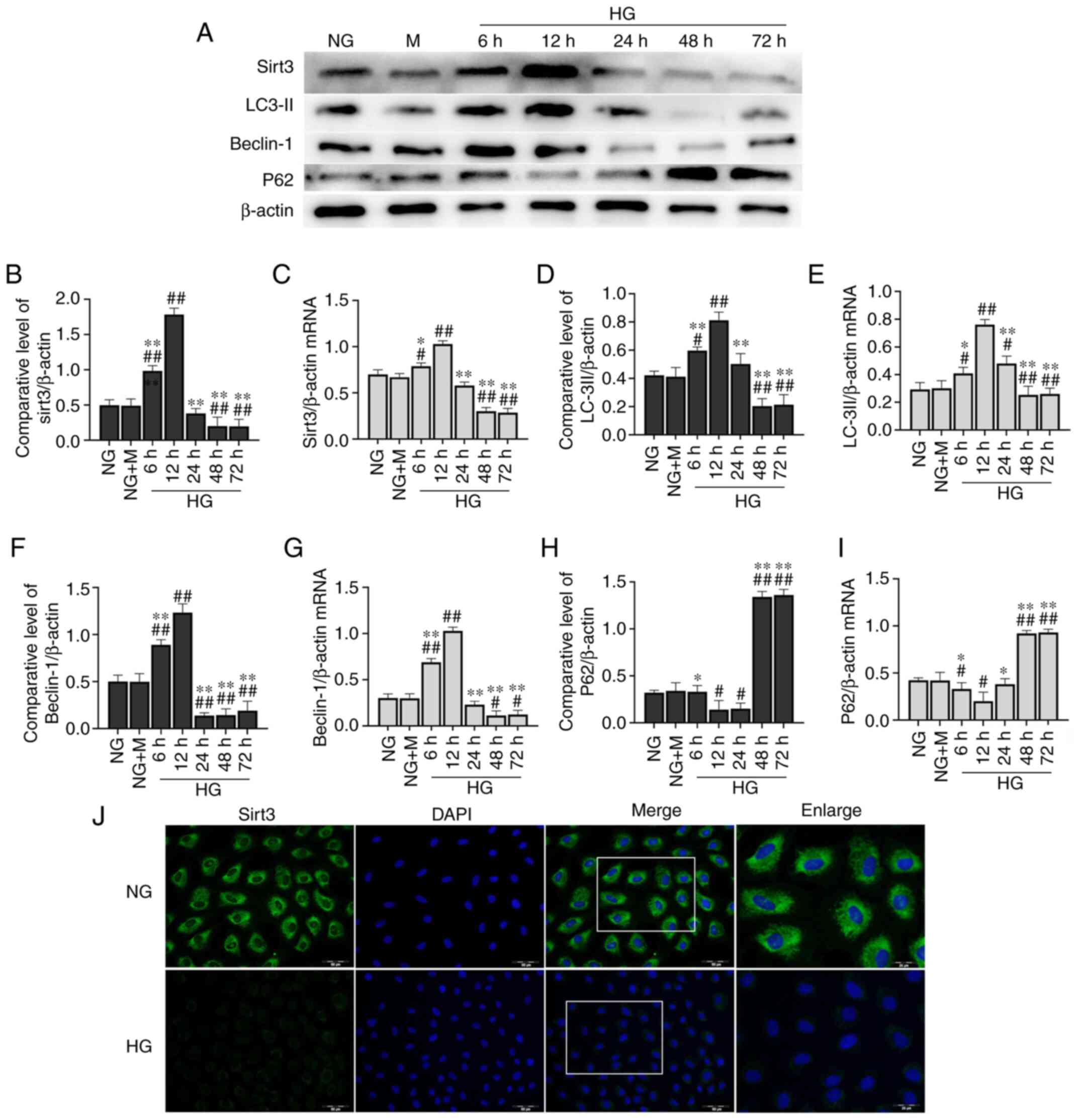 | Figure 2.Long-term culture of HK-2 cells in HG
inhibits expression of Sirt3 and autophagy. (A) Representative
western blots of proteins examined. Expression levels of (B) Sirt3,
(D) LC-3II, (F) Beclin-1 and (H) p62 were determined by western
blotting. β-actin was used as an internal control. mRNA expression
levels of (C) Sirt3, (E) LC-3II, (G) Beclin-1 and (I) p62 were
determined by reverse transcription-quantitative PCR. (J) Effects
of HG on the expression of Sirt3 were determined by
immunofluorescence staining [scale bar, 50 µm; the enlarged image
is a larger scale image of the area in the merged image represented
by the white box (scale bar, 25 µm)]. Sirt3 (green) was defined by
staining cells with anti-Sirt3 antibody. DAPI staining (blue) was
used to determine the position of the nucleus.
#P<0.05 and ##P<0.01 vs. NG; *P<0.05
and **P<0.01 vs. HG at 12 h. NG, normal glucose (5.5 mM
glucose); M, mannitol (5.5 mM glucose + 24.5 mmol/l mannitol); HG,
high glucose (30 mM glucose); Sirt3, sirtuin3. |
In order to evaluate the effects of HG on autophagic
activation in HK-2 cells, western blot analysis and RT-qPCR were
performed to determine the mRNA and protein expression levels of
Beclin-1, LC-3II and p62 (Fig. 2A and
D-I). HG increased the protein expression levels of Beclin-1
and LC-3II at 6 and 12 h compared with the NG group. However, at
later time points, HG significantly decreased Beclin-1 and LC-3II
protein expression levels. These levels exhibited a downward trend
from 24 h, with the most significant decrease observed at 48 h
(Fig. 2D and F). With regards to
p62 expression, the opposite trend was observed (Fig. 2H). Similar results were obtained by
RT-qPCR (Fig. 2E, G and I).
Long-term culture of HK-2 cells in HG
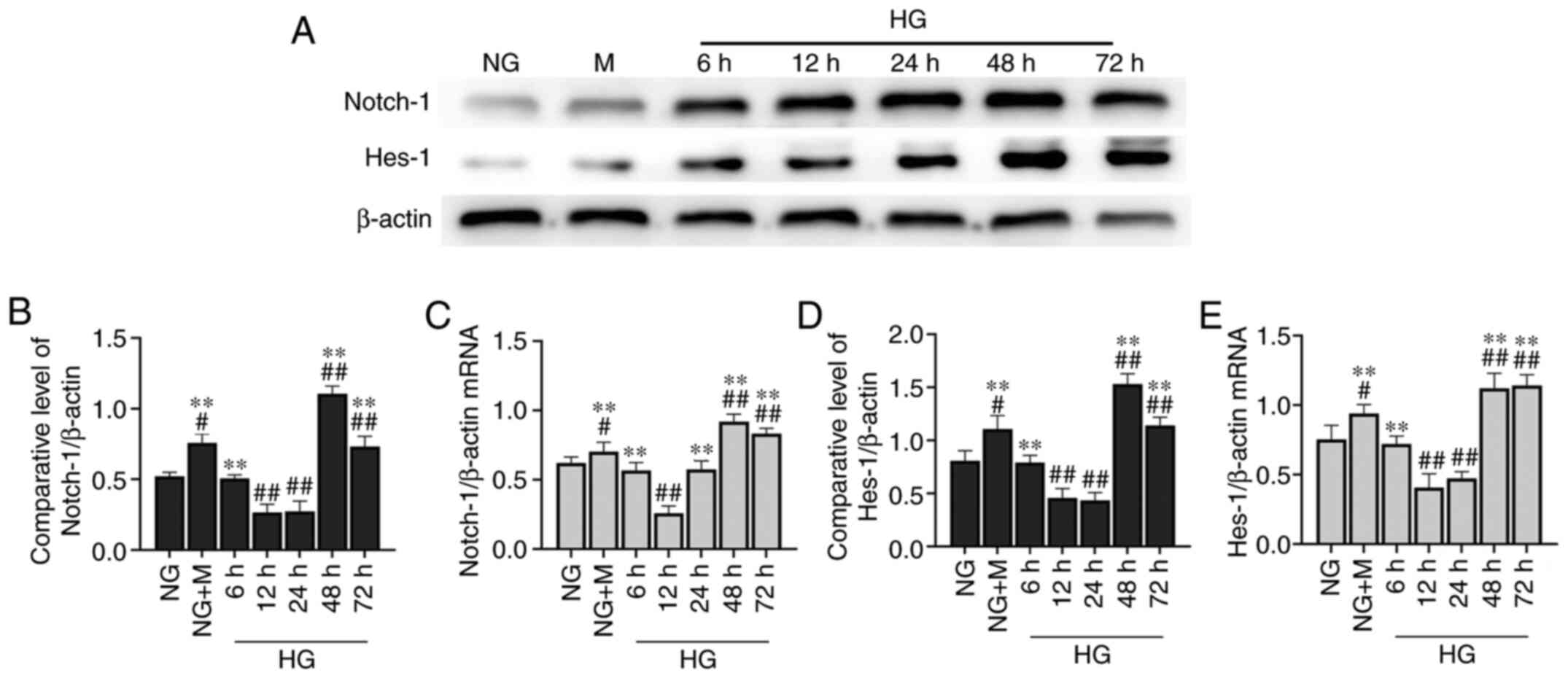
activates Notch-1/Hes-1 signaling
The expression levels of key proteins in the
Notch-1/Hes-1 pathway were moderately expressed in the HK-2 cells
(Fig. 3A). At 48 h, there was
significant activation of the Notch-1/Hes-1 pathway in the HK-2
cells cultured in NG + M compared with the NG group. Following a
phase of inhibition, the Notch-1/Hes-1 pathway was significantly
activated in the cells stimulated with HG for 48 h (Fig. 3B and D). Similar to the results
obtained by western blot analysis, HG upregulated the expression of
Notch-1/Hes-1 at the transcriptional level at 48 h (Fig. 3C and E).
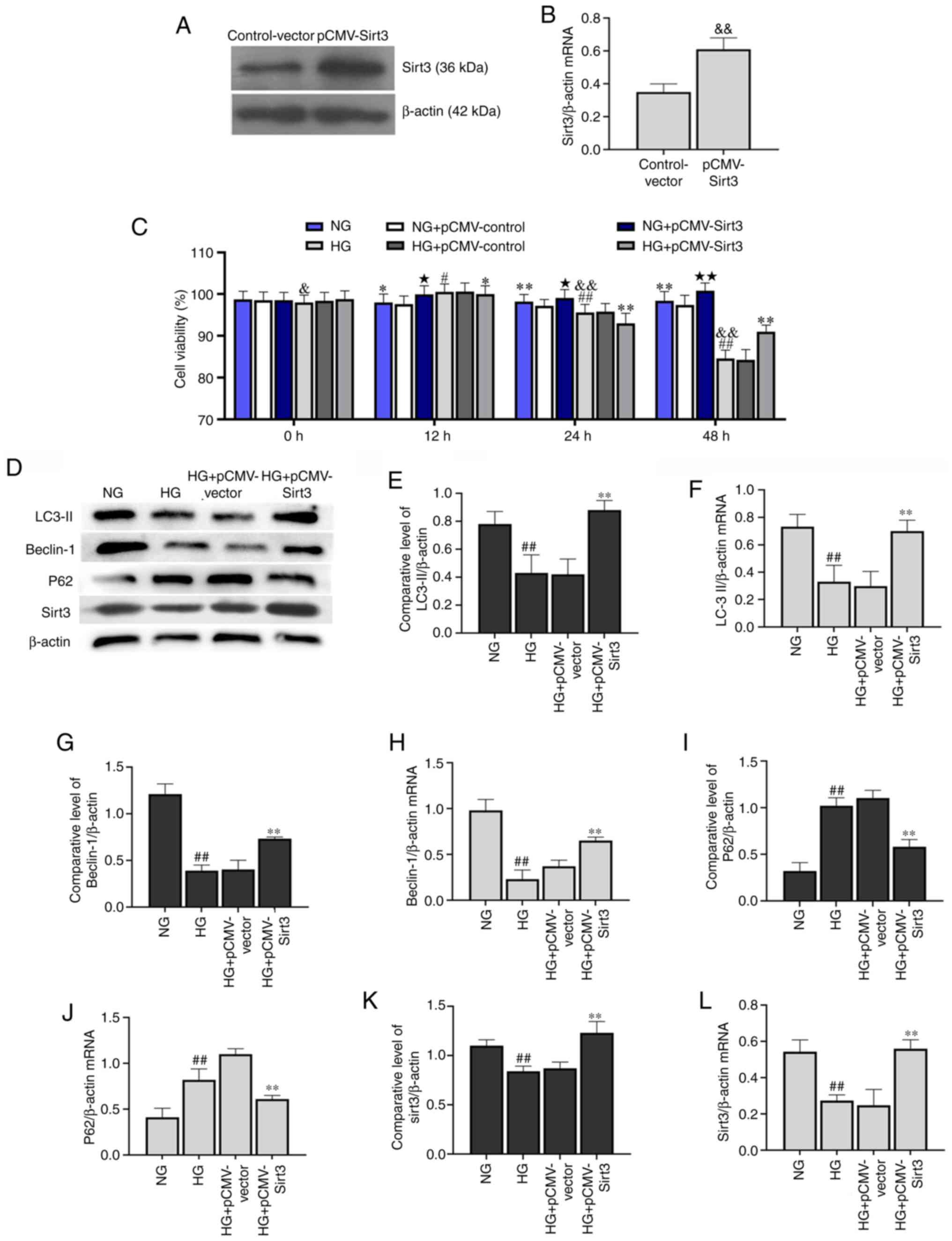
Sirt3 activates autophagy in
HG-stimulated HK-2 cells
As shown in Fig.
4D-J, HG significantly decreased Beclin-1 and LC-3II
expression, whereas it significantly increased p62 expression
compared with the HK-2 cells in the NG group. Following
transfection with pCMV-Sirt3, Sirt3 was significantly upregulated
compared with the control-vector group (Fig. 4A and B), and Sirt3 was effectively
activated when treated with HG + pCMV-Sirt3 compared with the HG +
pCMV-vector group (Fig. 4D).
Compared with NG and HG group, the overexpression of Sirt3 by
transfection with pCMV-Sirt3 significantly enhanced cell viability
at 48 h in the NG + pCMV-vector and HG + pCMV-vector groups,
respectively (Fig. 4C). In
addition, the expression of Beclin-1 and LC-3II was significantly
elevated at the transcriptional and translational levels in the HG
+ pCMV-Sirt3 group compared with the HG group. Moreover, pCMV-Sirt3
significantly decreased p62 expression compared with the HG group
(Fig. 4E-J). These results
demonstrated that Sirt3 reversed the inhibition of autophagy
induced by HG. Moreover, the results presented in Fig. 4K and L also demonstrated that HG
inhibited the expression of Sirt3 at the transcriptional level
compared with the NG group.
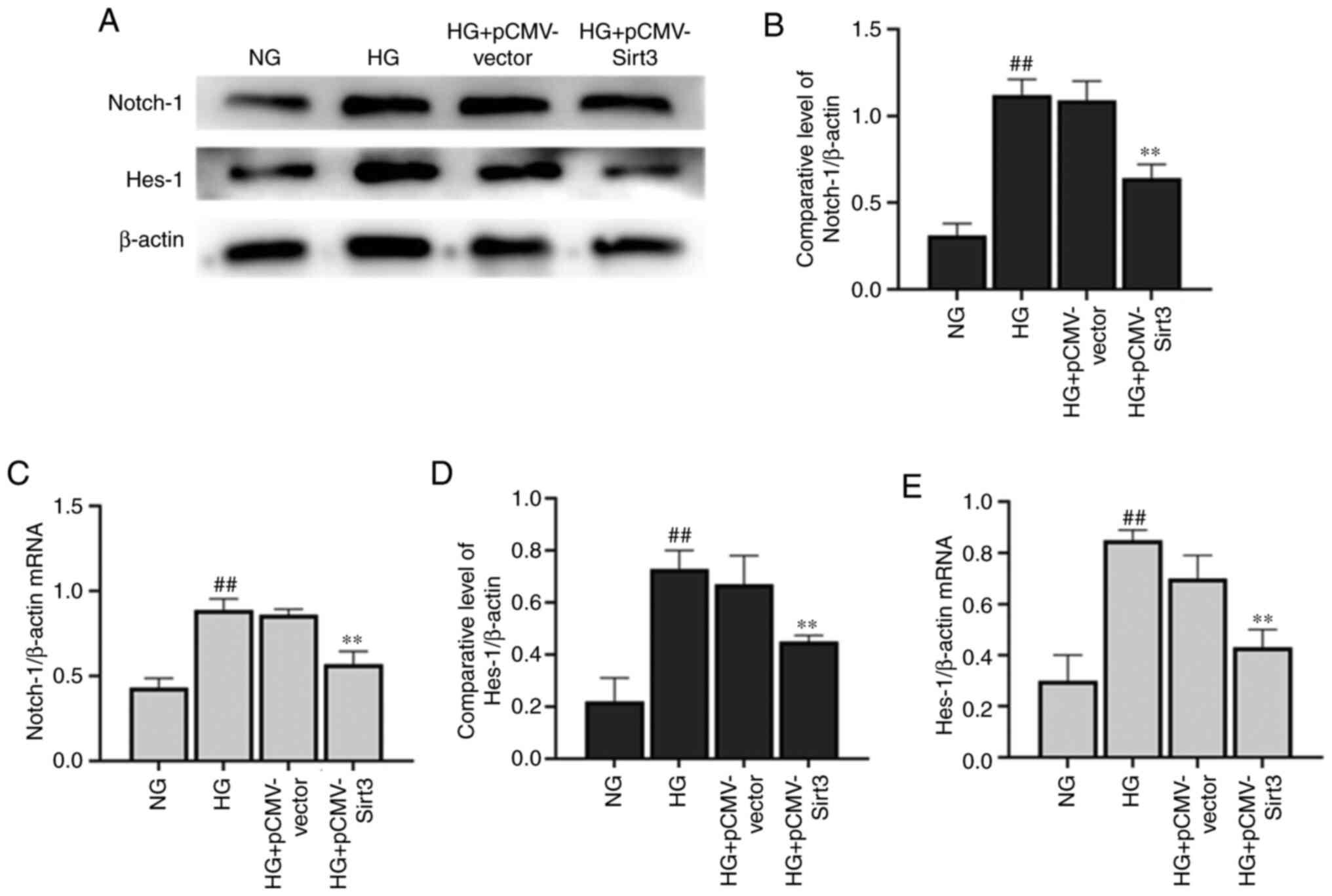
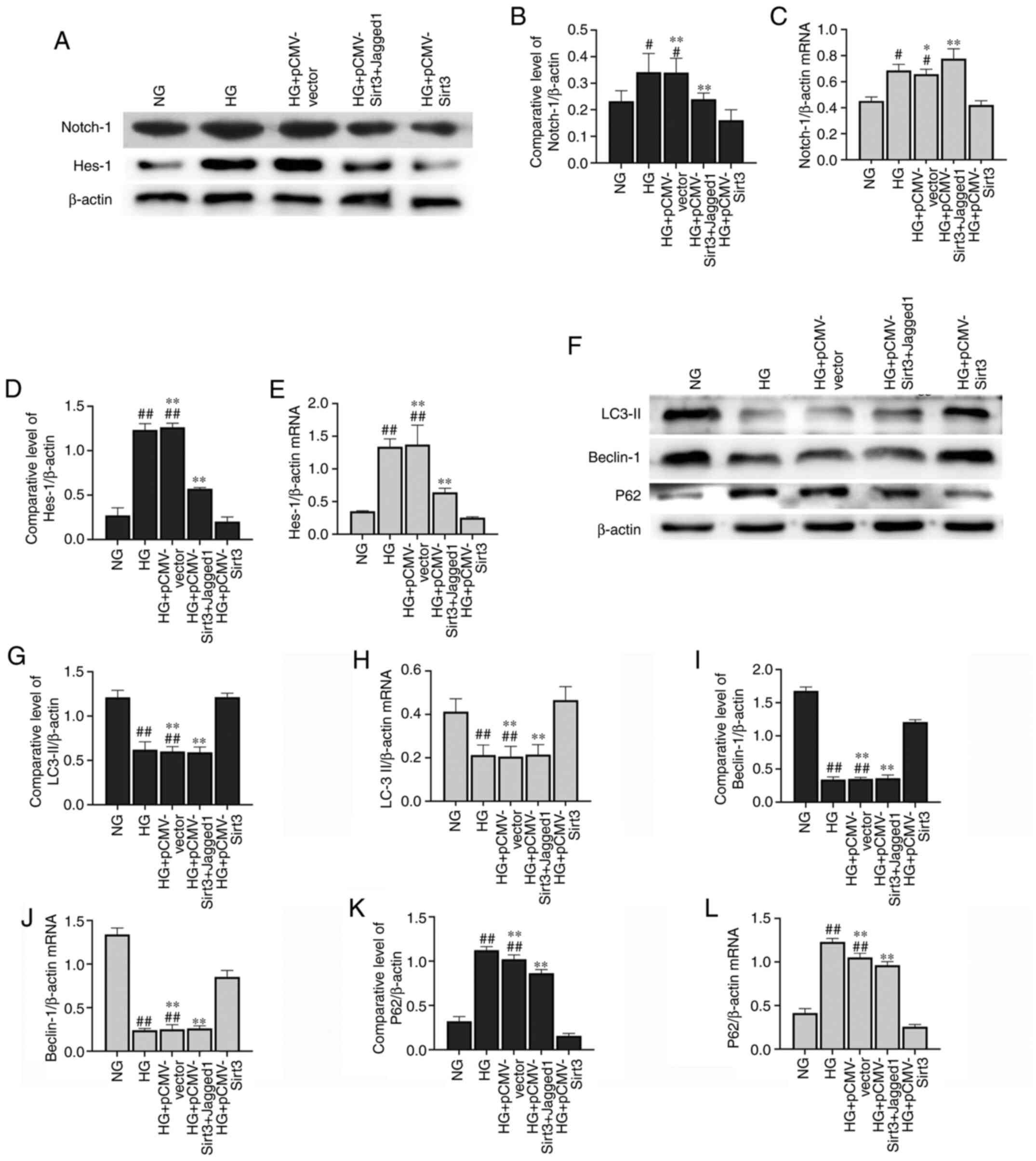
Sirt3 inhibits the Notch-1/Hes-1
pathway in HG-stimulated HK-2 cells
The expression of Notch-1 (Fig. 5A-C) and Hes-1 (Fig. 5A, D and E) was significantly
elevated in the HK-2 cells following stimulation with HG for 48 h
compared with the NG group. Nevertheless, the HG-induced activation
of the Notch-1/Hes-1 pathway was attenuated when the cells were
transfected with pCMV-Sirt3, which suggested that Sirt3 inhibited
the Notch-1/Hes-1 pathway in the HG-stimulated HK-2 cells.
Sirt3 upregulates autophagy in HK-2
cells via the inhibition of Notch-1/Hes-1 signaling
As illustrated in Fig.
6A-E, HG could induce activation of the Notch-1/Hes-1 signaling
pathway, which was inhibited by Sirt3. Notch-1/Hes-1 pathway
activation via the Notch-1/Hes-1 pathway activator, Jagged-1,
significantly upregulated Notch-1/Hes-1 protein expression compared
with the HG + pCMV-Sirt3 group. As shown in Fig. 6F, a marked decrease in Beclin-1 and
LC-3II expression was observed in the HK-2 cells following
stimulation with HG for 48 h (Fig.
6G-J). The accumulation of p62 was also detected in the
HG-stimulated cells (Fig. 6K and
L). However, the autophagic process was activated when the
cells were treated with HG + pCMV-Sirt3. A significant decrease in
Beclin-1 and LC-3II, and a pronounced rise in P62 was detected in
the HG + pCMV-Sirt3 + Jagged-1 group compared with the HG +
pCMV-Sirt3 group (Fig. 6G-L). These
results indicated that Sirt3 positively regulated autophagy via the
inhibition of the Notch-1/Hes-1 pathway.
Discussion
Autophagy is a basic process of physiological
metabolism that functions to remove impaired organelles and protein
aggregates, resulting in the recycling and remobilization of
nutrients (21). The dysfunction of
the process of autophagy has been reported to be an important
pathogenic mechanism and it plays a role in the occurrence and
development of several diseases (22). Under HG stress conditions, the
impairment of autophagy predisposes nephrocytes to accumulate
unfolded proteins and damaged organelles (23). The inhibition of autophagy induced
by HG is considered to be one of the key pathogenetic factors for
DN (24). Studies have suggested
that the alleviation of autophagic obstruction is involved in the
repair of the damage caused to renal tubular epithelial cells by
hyperglycemia (25,26). Although there is evidence that
focused on the autophagic process in DN, the molecular mechanisms
responsible for the regulation of autophagy remain unclear.
In the present study, it was found that cell
viability increased during the early phase of HG stimulation. The
viability of the HK-2 cells was decreased by extending the
incubation time. In parallel, autophagy was suppressed in the HK-2
cells. The inhibitory effect was most evident at 48 h and later
time points. A number of theoretical hypotheses have been proposed
to explain the molecular pathways regulating autophagy in the
diabetes-induced damage to renal tubular epithelial cells (24). Among these, Sirt1, which has been
implicated as a nicotinamide adenine dinucleotide-dependent
deacetylase, has been reported to be a positive regulator of
autophagy (27). Sirt1 serves as a
potential renoprotective factor. The low expression of Sirt1 has
been shown to lead to diabetic renal injury via the negative
regulation of autophagy (24).
Sirt1 and Sirt3 belong to the same family and can both mediate the
regulation of autophagy. In the present study, HG decreased Sirt3
expression in the HK-2 cells. To date, at least to the best of our
knowledge, there is no in-depth study available on the effects of
HG on Sirt3. The mechanisms through which Sirt3 functions in
response to environmental signals remain to be determined.
Previously, in animal experiments, Sirt3 was shown to ameliorate
autophagy dysfunction in acute tubular cell injury, resulting in
the upregulation of LC-3II and Beclin-1 expression (11). Additionally, a previous in
vitro study revealed that Sirt3 attenuated the inhibition of
autophagy induced by high levels of oxalic acid in renal tubular
epithelial cells; this led to the suppression of cell apoptosis and
necrosis, as well as in the protection of renal function (28). In the present study, Sirt3 activated
autophagy in the HG-stimulated HK-2 cells, as evidenced by the
upregulation of LC-3II and Beclin-1, and the downregulation of p62
expression, either at the transcriptional or post-transcriptional
levels in the HK-2 cells. These results are in accordance with
those of a similar previous study (11). However, unlike the present study,
the previous study focused on renal tubular cell damage caused by
sepsis rather than HG-induced renal tubular cell damage. Due to the
different pathogenic mechanisms, whether the two diseases have
anything in common in regulatory pathways warrants further
investigation.
With regards to the exact mechanisms through which
Sirt3 promoted autophagy, it has previously been reported that
Sirt3 regulates the autophagic process by activating different
downstream signaling pathways. Previously, in a cell model of
rotenone-induced Parkinson's disease, Sirt3 was found to protect
against neurodegenerative disease through the regulation of
autophagy via activating the liver kinase B1/adenosine
monophosphate-activated protein kinase (AMPK)/mTOR pathway
(29). Additionally, studies have
demonstrated that Sirt3 protects against cellar damage via
AMPK-mediated autophagy (11,30).
The present study demonstrated that Sirt3 promoted the autophagy of
HK-2 cells via the inhibition of Notch-1/Hes-1 signaling.
It is well-known that the Notch pathway is an
evolutionarily conserved intercellular signaling pathway, and is
involved in the course of renal development by regulating the
differentiation and maturation of podocytes (31,32). A
previous study demonstrated that autophagy in podocytes was
markedly diminished when Notch signaling was upregulated (33). However, studies on the association
between the Notch pathway and autophagy in renal tubular epithelial
cells are limited. In the present study, it was found that the
Notch-1/Hes-1 pathway was activated in HG-stimulated HK-2cells. The
promoting effects of Sirt3 on autophagy were attenuated following
the activation of the Notch-1/Hes-1 pathway. This provides further
evidence that Sirt3 activated autophagy in HK-2 cells via the
inhibition of Notch-1/Hes-1 pathway.
The findings presented herein are preliminary and
thus, further in-depth investigations are warranted. In future
experiments, the formation of autophagosomes should be traced using
the autophagic flux indicator RFP-GFP-LC3 under fluorescence
microscopy. Additionally, the formation of autophagosomes in
different stages of integration should be confirmed by electron
microscopy. Moreover, detecting changes in LC3-II and Beclin-1
levels alone does not appear sufficient for the evaluation of
autophagic activity. The induction of autophagy and the
accumulation of autophagosomes are not entirely representative of
the activation of the autophagic pathway. For an improved
evaluation of autophagic activity, more effective methods (e.g.
electron microscopy and LC3-GFP imaging) should be used for
detection of the degradation process.
In conclusion, the present study demonstrated that
the overexpression of Sirt3 induced an increase in the levels of
autophagy regulators in HK-2 cells stimulated with HG. Sirt3
activated autophagy at least partly, via the inhibition of the
Notch-1/Hes-1 pathway. Thus, Sirt3 may be a viable target in the
treatment of DN via the inhibition of Notch-1/Hes-1 signaling.
Acknowledgements
Not applicable.
Funding
This study was supported by the Natural Science Fund
of Inner Mongolia Autonomous Region (grant no. 2019MS08064).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
YW, JC and YL conceived and designed the present
study. YW and ZW were responsible for data analysis and performed
the experiments. YW, JC and YL wrote the manuscript. YL reviewed
and edited the manuscript. YW and YL confirm the authenticity of
all the raw data. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Warren AM, Knudsen ST and Cooper ME:
Diabetic nephropathy: An insight into molecular mechanisms and
emerging therapies. Expert Opin Ther Targets. 23:579–591. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Gilbertson DT, Liu J, Xue JL, Louis TA,
Solid CA, Ebben JP and Collins AJ: Projecting the number of
patients with end-stage renal disease in the United States to the
year 2015. J Am Soc Nephrol. 16:3736–3741. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Tang C, Livingston MJ, Liu Z and Dong Z:
Autophagy in kidney homeostasis and disease. Nat Rev Nephrol.
16:489–508. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Small DM, Bennett NC, Coombes J, Johnson
DW and Gobe GC: Mitochondrial homeostasis is impeded by degradation
and autophagy in oxidative stress-induced renal cell injury.
Revista Española De Reumatismo Y Enfermedades Osteoarticulares.
11:67–73. 2013.
|
|
5
|
Kitada M, Ogura Y, Monno I and Koya D:
Regulating autophagy as a therapeutic target for diabetic
nephropathy. Curr Diab Rep. 17:532017. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zhu SY, Yao RQ, Li YX, Zhao PY, Ren C, Du
XH and Yao YM: Lysosomal quality control of cell fate: A novel
therapeutic target for human diseases. Cell Death Dis. 11:8172020.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Guo J, Zheng HJ, Zhang W, Lou W, Xia C,
Han XT, Huang WJ, Zhang F, Wang Y and Liu WJ: Accelerated kidney
aging in diabetes mellitus. Oxid Med Cell Longev. 2020:12340592020.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lee IH, Cao L, Mostoslavsky R, Lombard DB,
Liu J, Bruns NE, Tsokos M, Alt FW and Finkel T: A role for the
NAD-dependent deacetylase Sirt1 in the regulation of autophagy.
Proc Natl Acad Sci USA. 105:3374–3379. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Li R, Xin T, Li D, Wang C, Zhu H and Zhou
H: Therapeutic effect of Sirtuin 3 on ameliorating nonalcoholic
fatty liver disease: The role of the ERK-CREB pathway and
Bnip3-mediated mitophagy. Redox Biol. 18:229–243. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zhang T, Liu J, Shen S, Tong Q, Ma X and
Lin L: SIRT3 promotes lipophagy and chaperon-mediated autophagy to
protect hepatocytes against lipotoxicity. Cell Death Differ.
27:329–344. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhao W, Zhang L, Chen R, Lu H, Sui M, Zhu
Y and Zeng L: SIRT3 protects against acute kidney injury via
AMPK/mTOR-regulated autophagy. Front Physiol. 9:15262018.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kitada M, Kume S, Takeda-Watanabe A,
Kanasaki K and Koya D: Sirtuins and renal diseases: Relationship
with aging and diabetic nephropathy. Clin Sci (Lond). 124:153–164.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Yu W, Gao B, Li N, Wang J, Qiu C, Zhang G,
Liu M, Zhang R, Li C, Ji G and Zhang Y: Sirt3 deficiency
exacerbates diabetic cardiac dysfunction: Role of
Foxo3A-Parkin-mediated mitophagy. Biochim Biophys Acta Mol Basis
Dis. 1863:1973–1983. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Chen Y, Zhang F, Wang D, Li L, Si H, Wang
C, Liu J, Chen Y, Cheng J and Lu Y: Mesenchymal stem cells
attenuate diabetic lung fibrosis via adjusting Sirt3-mediated
stress responses in rats. Oxid Med Cell Longev.
2020:80761052020.PubMed/NCBI
|
|
15
|
Artavanis-Tsakonas S, Rand MD and Lake RJ:
Notch signaling: Cell fate control and signal integration in
development. Science. 284:770–776. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yuri S, Nishikawa M and Yanagawa N, Jo OD
and Yanagawa N: Maintenance of mouse nephron progenitor cells in
aggregates with Gamma-secretase inhibitor. PLoS One.
10:e01292422015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wang Y, Li Y, Yang Z, Wang Z, Chang J,
Zhang T, Chi Y, Han N and Zhao K: Pyridoxamine treatment of HK-2
human proximal tubular epithelial cells reduces oxidative stress
and the inhibition of autophagy induced by high glucose levels. Med
Sci Monit. 25:1480–1488. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Jiao X, Li Y, Zhang T, Liu M and Chi Y:
Role of Sirtuin3 in high glucose-induced apoptosis in renal tubular
epithelial cells. Biochem Biophys Res Commun. 480:387–393. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wang Z, Li Y, Wang Y, Zhao K, Chi Y and
Wang B: Pyrroloquinoline quinine protects HK-2cells against high
glucose-induced oxidative stress and apoptosis through Sirt3 and
PI3K/Akt/FoxO3a signaling pathway. Biochem Biophys Res Commun.
508:398–404. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Mizushima N and Levine B: Autophagy in
human diseases. N Engl J Med. 383:1564–1576. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Jiang P and Mizushima N: Autophagy and
human diseases. Cell Res. 24:69–79. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kim KH and Lee MS: Autophagy-a key player
in cellular and body metabolism. Nat Rev Endocrinol. 10:322–337.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ding Y and Choi ME: Autophagy in diabetic
nephropathy. J Endocrinol. 224:R15–R30. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Liu WJ, Huang WF, Ye L, Chen RH, Yang C,
Wu HL, Pan QJ and Liu HF: The activity and role of autophagy in the
pathogenesis of diabetic nephropathy. Eur Rev Med Pharmacol Sci.
22:3182–3189. 2018.PubMed/NCBI
|
|
26
|
Lin F: Autophagy in renal tubular injury
and repair. Acta Physiol (Oxf). 220:229–237. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Ng F and Tang BL: Sirtuins' modulation of
autophagy. J Cell Physiol. 228:2262–2270. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Peng Y, Yang C, Shi X, Li L, Dong H, Liu
C, Fang Z, Wang Z, Ming S, Liu M, et al: Sirt3 suppresses calcium
oxalate-induced renal tubular epithelial cell injury via
modification of FoxO3a-mediated autophagy. Cell Death Dis.
10:342019. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zhang M, Deng YN, Zhang JY, Liu J, Li YB,
Su H and Qu QM: SIRT3 protects Rotenone-induced injury in SH-SY5Y
cells by promoting autophagy through the LKB1-AMPK-mTOR Pathway.
Aging Dis. 9:273–286. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Wang Y, Zhang X, Wang P, Shen Y, Yuan K,
Li M, Liang W and Que H: Sirt3 overexpression alleviates
hyperglycemia-induced vascular inflammation through regulating
redox balance, cell survival, and AMPK-mediated mitochondrial
homeostasis. J Recept Signal Transduct Res. 39:341–349. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Surendran K, Boyle S, Barak H, Kim M,
Stomberski C, McCright B and Kopan R: The contribution of Notch1 to
nephron segmentation in the developing kidney is revealed in a
sensitized Notch2 background and can be augmented by reducing Mint
dosage. Dev Biol. 337:386–395. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Vooijs M, Ong CT, Hadland B, Huppert S,
Liu Z, Korving J, van den Born M, Stappenbeck T, Wu Y, Clevers H
and Kopan R: Mapping the consequence of Notch1 proteolysis in vivo
with NIP-CRE. Development. 134:535–544. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zheng D, Tao M, Liang X, Li Y, Jin J and
He Q: p66Shc regulates podocyte autophagy in high glucose
environment through the Notch-PTEN-PI3K/Akt/mTOR pathway. Histol
Histopathol. 35:405–415. 2020.PubMed/NCBI
|