Introduction
Evidence is mounting to indicate that chronic heart
failure (CHF) has become one of the primary causes of morbidity and
mortality in modern society (1,2).
Unfortunately, the specific mechanisms underlying CHF remain
unknown and the currently available treatments may only delay the
progression of disease. Cardiac hypertrophy is a critical
compensated stage that involves pathological remodeling of the
myocardium, ultimately resulting in CHF (3). Therefore, it is vital to be able to
prevent and treat cardiac hypertrophy to prevent the progression of
cardiac remodeling into CHF. In our previous study, it was shown
that the imbalance in the modification of histone H3K9ac, a process
mediated by histone acetylases (HATs), is involved in phenylephrine
(PE)-induced cardiomyocyte hypertrophy (4). Several studies have shown that cardiac
hypertrophy can be induced by the activation of multiple signaling
pathways (5–8). The extracellular signal-regulated
protein kinase (ERK) signaling pathway is considered to play a
particularly important role in regulating pathological cardiac
hypertrophy (9–11). Our previous study demonstrated that
the HAT inhibitor, anacardic acid (AA), could attenuate PE-induced
cardiac hypertrophy by regulating the modification of histone
H3K9ac acetylation (4); however,
the upstream signaling pathways were not determined.
To construct an animal model of myocardial
hypertrophy, three commonly accepted methods are partial ligation
of the thoracic aorta, arteriovenous fistula and subcutaneous
injection of PE (12–15). In the present study, PE was used to
induce cardiomyocyte hypertrophy in neonatal mice. Multiple studies
have confirmed that PE can activate ERK1/2 pathway (16–18).
Thus, the aim of the present study was to determine whether the
interactions between the ERK1/2 signaling pathway and
P300/CBP-associated factor (PCAF) served a key role in mediating
H3K9ac acetylation in PE-induced cardiomyocyte hypertrophy.
Materials and methods
Experimental mice
All experimental animals were provided by the
Experimental Animal Center of Chongqing Medical University. Clean
and healthy neonatal Kunming male and female mice were used (1–3
days-of-age, weighing 2.3–2.7 g). All animal experiments were
approved by the Animal Protection and Use Committee of Zunyi
Medical University (Zunyi, China), and complied with Directive
2010/63/EU of the European Parliament (19).
Cell culture and treatment
Under aseptic conditions, Kunming mice (1–3
days-of-age) were sacrificed by decapitation. The ventricular
tissue was immediately removed using ophthalmic scissors and cut
into 1–2 mm3 pieces. The tissue was then ground in 1 ml
0.05% collagenase type II (Worthington Biochemical Corporation) for
5 min at room temperature (repeated 8–10 times). Next, the
supernatant was centrifuged at 1,500 × g for 10 min at room
temperature, the supernatant was discarded and the cells
resuspended in DMEM/F12 supplemented with 20% FBS (HyClone;
Cytiva). After 1.5 h of culture in an incubator at 37°C with 5%
CO2, the adherent fibroblasts were discarded and the
cell suspension was transferred to a new culture flask. 5-BrdU
(Beijing Solarbio Science & Technology Co., Ltd.) was added to
a final concentration of 0.1 mmol/l to prevent the growth of
fibroblasts, and the cells were further cultured in an incubator.
According to previous studies (20–24),
the cells were treated with 100 µmol/l PE (MedChemExpress; cat. no.
HY-B0769), 50 µmol/l AA (Sigma-Aldrich; Merck KGaA; cat. no.
16611-84-0) or 10 µmol/l U0126 (Selleck Chemicals; cat. no. S1102).
Cardiomyocytes were treated with the aforementioned drugs
individually or in combination, dependent on the treatment
groups.
Cell viability assay
Cardiomyocytes were plated in a 96-well plate at a
density of 2×104 cells/well. Next, 10 µl Cell Counting
Kit-8 (CCK-8) solution (Beijing Solarbio Science & Technology
Co., Ltd.) was added and cells were cultured for 4 h at 37°C in the
dark. Finally, the absorbance was measured at 450 nm using a
Universal Microplate Spectrophotometer (Bio-Rad Laboratories,
Inc.).
Western blotting
Cardiomyocytes were used in western blotting at a
density of 3–4×106 cells/well. Nuclear protein was
harvested from mouse cardiomyocytes using a nuclear protein
extraction kit (Merck KGaA). Nucleic proteins were loaded on a 6 or
12% SDS-gel, resolved using SDS-PAGE and then transferred to a PVDF
membrane (Merck KGaA). Membranes were blocked at 4°C with 5% BSA
for 1 h and then incubated with a series of rabbit polyclonal
antibodies, which included anti-ANP (1:5,000; Abcam; cat. no.
ab189921), anti-H3K9ac (1:5,000; Abcam; cat. no. ab4441), anti-H3
(1:5,000; Abcam; cat. no. ab1791), anti-β-MHC (1:5,000; Abcam; cat.
no. Ab207926) anti-BNP (1:1,000; Abcam; cat. no. ab239510),
anti-β-actin (1:1,000; Abcam; cat. no. ab8226) and anti-PCAF
(1:2,000; Abcam; cat. no. ab176316), anti-ERK (1:2,000; Cell
Signaling Technology, Inc.; cat. no. 4695) or anti phospho-(p-)ERK
(1:2,000; Cell Signaling Technology, Inc.; cat. no. 4370); β-actin
and H3 were served as an internal controls. All antibodies were
diluted in Tris-buffered saline containing 5% skimmed milk, and
incubated with the membrane overnight at 4°C. HRP-labeled goat
anti-rabbit antibody (1:2,000; Santa Cruz Biotechnology, Inc.; cat.
no. sc2004) was used as the secondary antibody and incubated with
the membrane at 4°C for 2 h. The results were detected with
enhanced chemiluminescence reagents (Wanleibio Co., Ltd.). Finally,
membranes were scanned using a Bio-Rad image analyzer; and
densitometry analysis was performed using Quantity One version 4.4
(Bio-Rad Laboratories, Inc.).
RNA extraction and reverse
transcription-quantitative (RT-q)PCR
Cardiomyocytes were used for qPCR at a density of
2–3×106 cells/well. Total RNA was extracted from
myocardial cells using an RNA Extraction kit (BioTeke Corporation),
according to the manufacturer's protocol. Total RNA was then
reverse transcribed into single-stranded cDNA using an AMV Reverse
Transcription system, according to the manufacturer's protocol
(Takara Bio, Inc.). cDNA was amplified using a SYBR Green dye kit
and gene-specific primers (Takara Bio, Inc.). The primer sequences
of MEF2C and β-actin were: MEF2C forward,
5′-CCTTTTCCTTTTCTGGGGACTTGTT-3′ and reverse
5′-TGCCGCTGTGAGCCTCTATTTG-3′; and β-actin forward,
5′-CCTTTATCGGTATGGAGTCTGCG-3′ and reverse,
5′-CTGACATGACGTTGTTGGCA-3′. β-actin was used as a standardized
reference, and the 2−ΔΔCq method was used to determine
relative gene expression (25).
Immunofluorescence
Cardiomyocytes were seeded into 6-well plates
(1×105 cells/well) for 24 h and incubated with 50 µmol/l
AA for 1 h, then 10 µmol/l U0126 was added. After 48 h, 100 µmol/l
PE was added. Subsequently, the cells were fixed with 4%
paraformaldehyde for 15 min at room temperature (RT), treated with
0.3% Triton X-100 in PBS at RT for 20 min, and then incubated with
10% goat serum (Solarbio Science & Technology Co., Ltd.; cat.
no. SL038) at 37°C for 30 min. Subsequently, the cardiomyocytes
were incubated at 4°C overnight with primary antibodies against
α-actin (1:100; ProteinTech Group, Inc.; cat. no. 23660-1-AP),
H3K9ac (1:1,000 Abcam; cat. no. ab4441) and anti-PCAF (1:250;
Abcam; cat. no. ab176316). The following morning, the cells were
incubated at 37°C in the dark for 1 h with Alexa Fluor 594 goat
anti-mouse IgG secondary antibody (1:1,000; Thermo Fisher
Scientific, Inc.; cat. no. A-11005) and Alexa Fluor 488 goat
anti-rabbit IgG (1:200; Invitrogen; Thermo Fisher Scientific, Inc.;
cat. no. A-11008) secondary antibody. Finally, the cardiomyocytes
were counterstained with DAPI for 5 min at RT. All images were
obtained using a confocal microscope (magnification, ×40) with
standardized imaging parameters. Finally, the images were
quantified based on fluorescence using ImageJ 1.49 software
(National Institutes of Health).
Co-immunoprecipitation (Co-IP)
Co-IP was performed as described previously
(26), using anti-phospho-(p-)ERK,
anti-PCAF and anti-H3K9ac rabbit polyclonal antibodies with
Dynabeads protein G magnetic beads (Invitrogen; Thermo Fisher
Scientific, Inc.) for the immunoprecipitation and western blotting
of primary cardiomyocytes. First, the primary antibody was bound to
Protein G magnetic beads (according to the manufacturer's
protocol). Next, at pH 7.4, a magnet along with 1% Triton X-100,
0.5% NP-40, 20 mM HEPES, 50 mM NaCl and protease inhibitor (1:50;
Beijing Solarbio Science & Technology Co., Ltd.), were used to
immunoprecipitate the target antigen (p-ERK) in the buffer. The
sample was then washed three times with a lysis buffer. The
immobilized protein complex was eluted, denatured in 5X SDS sample
buffer at 95°C for 10 min, and then subjected to western blotting
analysis with anti-p-ERK (1:2,000; Cell Signaling Technology, Inc.;
cat. no. 4370), anti-PCAF and anti-H3K9ac antibodies. IgG served as
a negative control.
Chromatin immunoprecipitation
(ChIP)
ChIP was performed as described previously (26). Formaldehyde (1%) was added to the
homogenized cardiomyocytes to cross-link the DNA-protein complex,
and incubate at 37°C for 15 min. ChIP determination was then
performed using a specific kit (Merck KGaA). Next, the cross-linked
complex DNA was sheared with ultrasound and precipitated with
monoclonal antibodies (anti-MEF2C, 1:1,000; anti-H3K9ac, 1:2,000;
and anti-PCAF, 1:2,000) overnight at 4°C. DNA purification kits
(Merck KGaA) were then used to extract the final DNA, according to
the manufacturer's protocol. Normal mouse IgG (1:500;
Sigma-Aldrich, Merck KGaA) was used as a negative control.
Statistical analysis
All experiments were repeated six time with six
independent samples. All data are expressed as the mean ± standard
deviation. All statistical analyses were performed using SPSS
version 18.0 (SPSS Inc.). Comparisons among multiple groups were
analyzed using one-way ANOVA followed by Tukey's post-hoc test.
P<0.05 was considered to indicate a statistically significant
difference.
Results
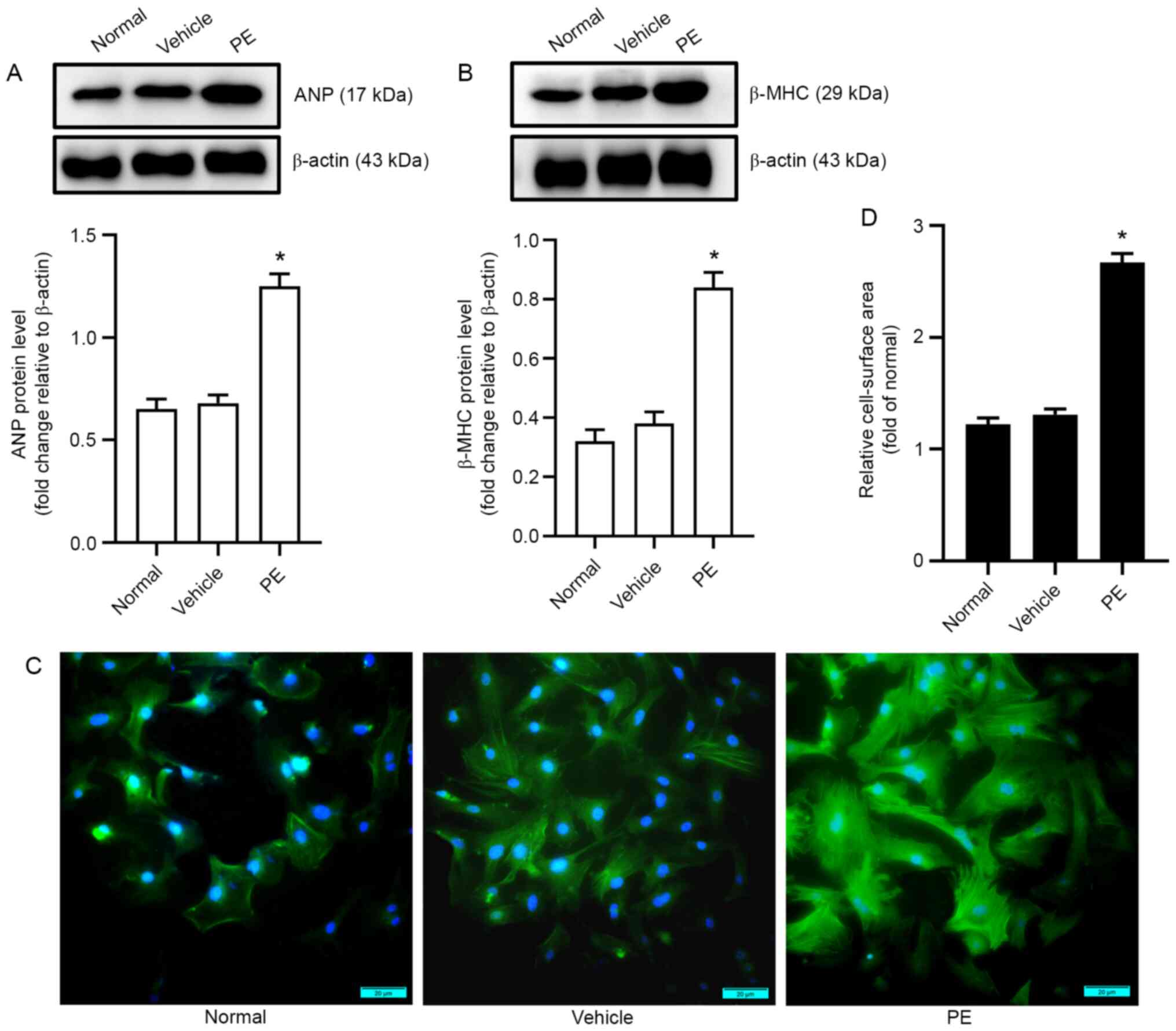
PE-induced cardiomyocyte hypertrophy
in neonatal mice
In order to establish a model of PE-induced
cardiomyocyte hypertrophy, first, the optimal PE exposure
concentration (100 µmol/l) was determined based on a previous study
(20). The treatment time of PE was
48 h in cultured myocardial cells, and the effects were determined
using western blotting. Data arising from the western blotting
experiments showed that the levels of biomarkers for myocardial
hypertrophy (ANP and β-MHC) in the PE group were significantly
higher than those in the vehicle group (Fig. 1A and B). Additionally, the
cell-surface area was also assayed using immunofluorescence
analysis. The results showed that myocardial cells in the PE group
were significantly larger than those in the vehicle group (Fig. 1C and D). These data showed that a
mouse model of PE-induced cardiomyocyte hypertrophy was
successfully established.
p-ERK1/2 interacts with PCAF and
altered H3K9ac acetylation in hypertrophic cardiomyocytes induced
by PE
Studies have confirmed that the p-ERK1/2 signaling
pathway plays an important role in pathological myocardial cell
hypertrophy (27,28). Thus, the effects of the ERK1/2
signaling pathway on the manner by which AA attenuates PCAF
mediated-H3K9ac hyperacetylation in PE-induced hypertrophic
cardiomyocytes was determined. First, according to the previous
literature (21–24), the optimum concentration of the
histone acetylase inhibitor AA was determined (50 µmol/l), as well
as for the ERK inhibitor U0126 (10 µmol/l), in accordance with
H3K9ac levels, and the viability of myocardial cells using western
blotting and CCK-8 examination, respectively (Fig. 2A and B). Additionally, the
inhibitory activity of AA decreased as the concentration of AA was
increased. As the concentration of the inhibitor increases, its
inhibitory effect will gradually increase, but when the
concentration of the inhibitor is higher than a certain level, its
inhibitory effect will be weakened (29,30).
After hypertrophic cardiomyocytes had been treated with AA and/or
U0126, the expression of total-(T-)ERK1/2 and p-ERK1/2 was
determined by western blotting, and the results showed that the
expression levels of p-ERK1/2 in the PE group were significantly
higher than those in the control group. Additionally, it was also
found that the HATs inhibitor AA, and the ERK inhibitor U0126 could
ameliorate the increase in p-ERK1/2 levels induced by PE in primary
cultured myocardial cells; however, the expression of T-ERK1/2
remained unchanged (Fig. 2C). In
addition, our previous study found that an imbalance in the
modification of histone H3K9ac, as mediated by PCAF, was involved
in the pathological hypertrophy of myocardial cells (31). Next, whether the ERK1/2 signaling
pathway interacted with PCAF in order to regulate the modification
of histone H3K9ac acetylation and further enhance pathological
cardiac hypertrophy was determined. Co-IP experiments were not used
as an analytical method, but instead performed to verify the
formation of a complex between the ERK1/2 signaling pathway and
PCAF mediated-H3K9ac acetylation, and it was successfully
demonstrated that there was an interaction in the primary cultured
myocardial cells. Collectively, these data indicated that the
ERK1/2 signaling pathway may interact with PCAF mediated-H3K9ac
acetylation (Fig. 2D).
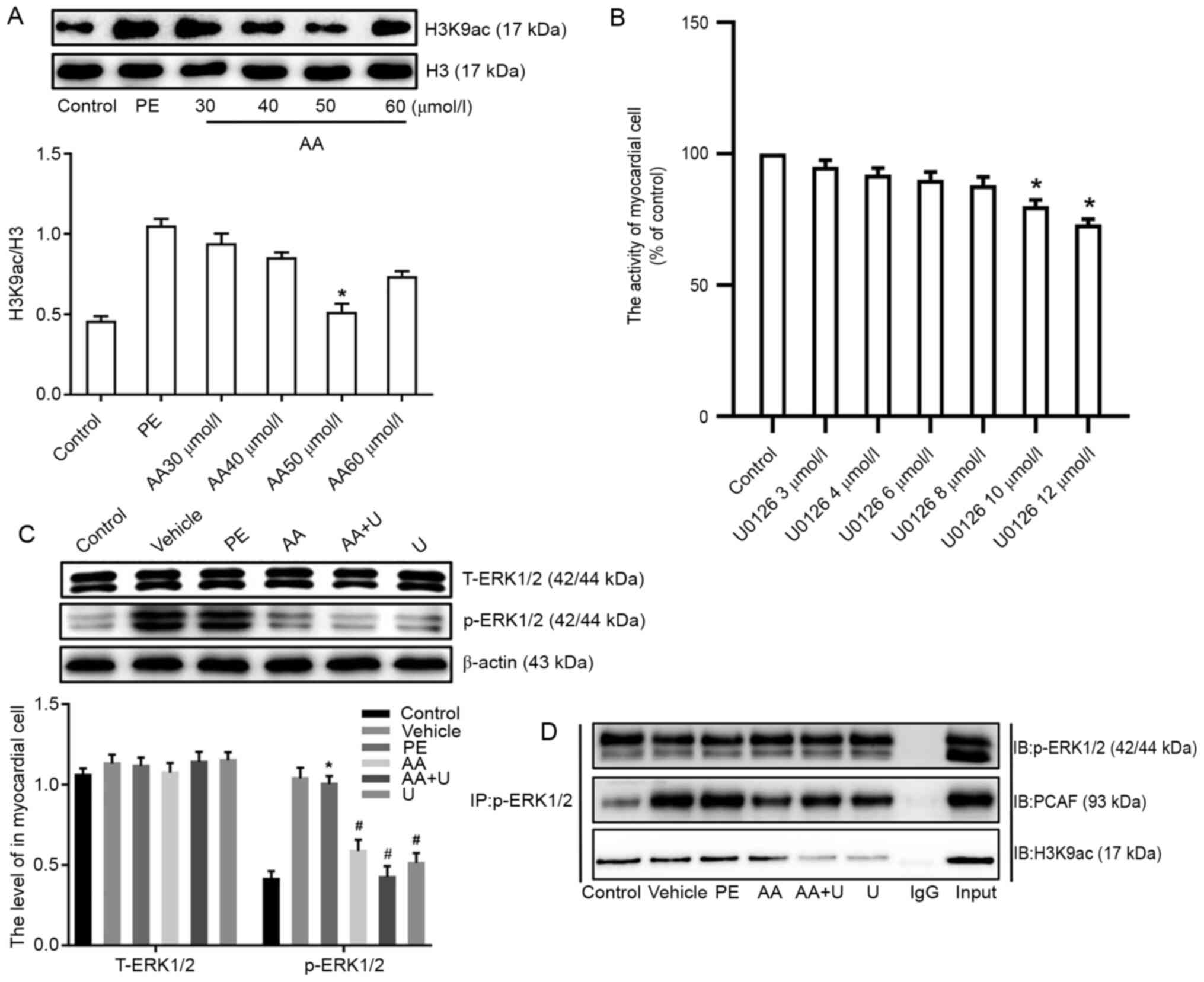 | Figure 2.p-ERK1/2 interacted with PCAF and
modified H3K9ac acetylation in hypertrophic cardiomyocytes induced
by PE. (A) Different concentrations of AA (30, 40, 50 and 60
µmol/l) were used to identify the optimal concentration of AA; 50
µmol/l was selected for subsequent experiments, based on the levels
of histone H3K9ac. (B) Effects of different concentrations of the
ERK inhibitor U0126 (2, 4, 6, 8 and 10 µmol/l) on cell viability in
neonatal mouse cardiomyocytes. (C) Expression of T-ERK1/2 and
p-ERK1/2 in myocardial cells from neonatal mice. (D)
Co-immunoprecipitation in cell lysates of mouse myocardial cells
exposed to six different experimental conditions with
anti-p-ERK1/2-protein G magnetic beads and IB with anti-PCAF,
anti-H3K9ac or anti-p-ERK1/2 antibodies to evaluate protein
expression. Input, positive control; IgG, negative control. n=6.
*P<0.05 vs. control group; #P<0.05 vs. PE group.
P-, phospho-; PCAF, P300/CBP-associated factor; PE, phenylephrine;
H3K9ac, histone 3 acetylation K9; AA, anacardic acid; T-, total-;
IB, immunoblotting; ERK, extracellular signal-regulated protein
kinase; PE, 100 µmol/l phenylephrine for 48 h; Vehicle, 100 µmol/l
phenylephrine + equal volume of DMSO for 48 h; AA, 50 µmol/l AA for
30 min + 100 µmol/l phenylephrine for 48 h; AA + U, 50 µmol/l AA +
10 µmol/l U0126 for 30 min + 100 µmol/l phenylephrine for 48 h; U,
10 µmol/l U0126 for 30 min + 100 µmol/l phenylephrine for 48 h;
Control, no drug. |
Expression of PCAF in primary cultured
myocardial cells
Our previous study showed that PCAF plays a critical
role in pathological cardiac hypertrophy in a manner that is
dependent on the modification of histone acetylation, and it was
confirmed that p-ERK1/2 could interact with PCAF mediated-H3K9ac
acetylation (31). Thus, in the
present study, the expression of PCAF was assessed by
immunofluorescence and western blotting, and a notable increase in
PCAF expression in hypertrophic cardiomyocytes was observed when
induced by PE. In contrast, exposure to AA reversed the
upregulation of PCAF in primary myocardial cells, as did the ERK
inhibitor, U0126 (Fig. 3).
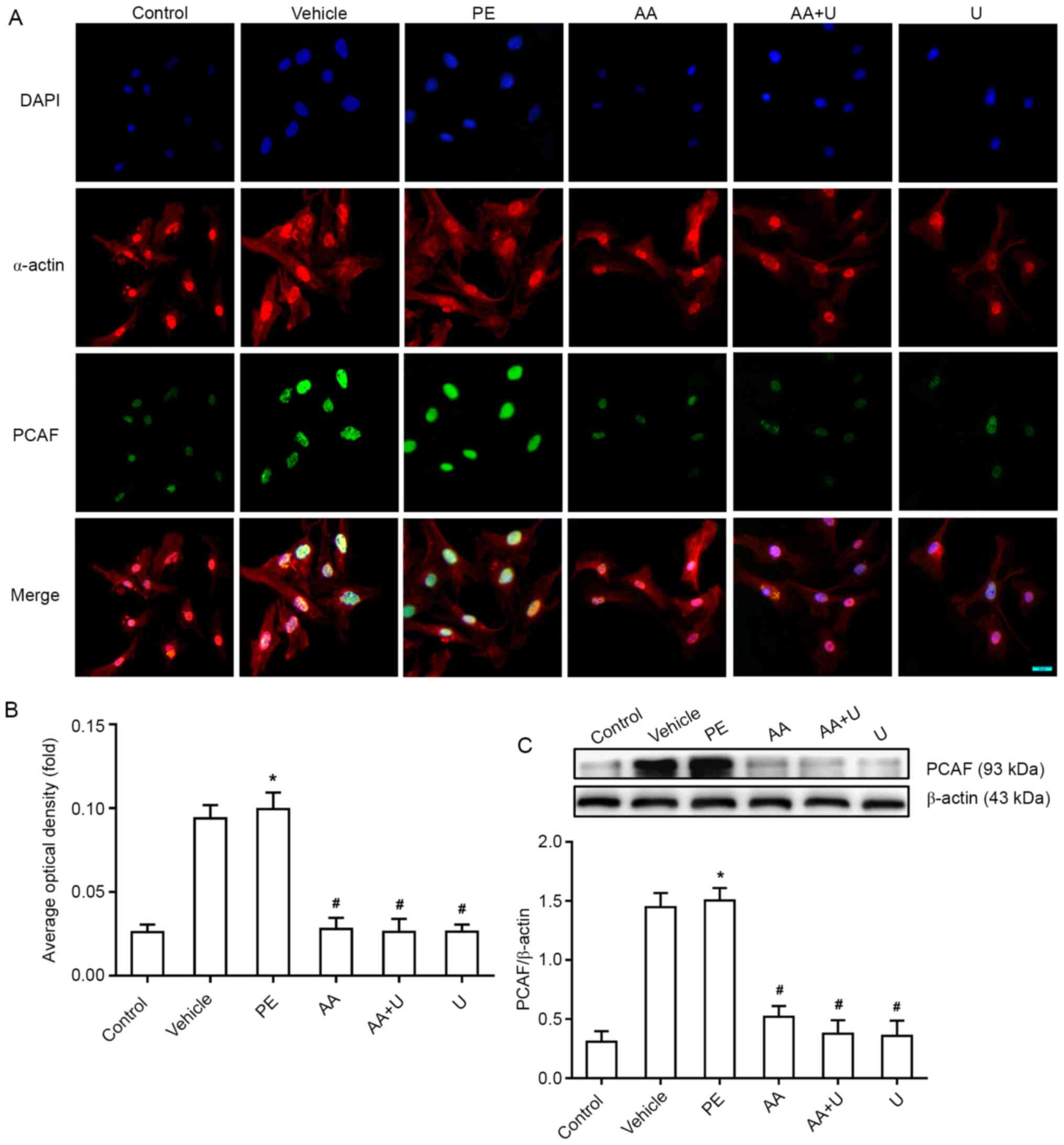 | Figure 3.Effects of AA and U0126 on the
expression of PCAF in PE-induced cardiomyocyte hypertrophy. (A)
PCAF (green fluorescence) and α-actin (red fluorescence), in
combination with DAPI staining (blue fluorescence), in
cardiomyocytes exposed to six different conditions. Scale bar, 20
µm. (B) Mean optical density of PCAF immunofluorescence in the six
groups. (C) Western blotting of PCAF expression, showed PCAF levels
were significantly higher in the hypertrophic cardiomyocytes
induced by PE, whereas AA and/or U0126 prevented this effect. n=6.
*P<0.05 vs. control group; #P<0.05 vs. PE group.
PCAF, P300/CBP-associated factor; PE, phenylephrine; AA, anacardic
acid; PE, 100 µmol/l phenylephrine for 48 h; Vehicle, 100 µmol/l
phenylephrine + equal volume of DMSO for 48 h; AA, 50 µmol/l AA for
30 min + 100 µmol/l phenylephrine for 48 h; AA + U, 50 µmol/l AA +
10 µmol/l U0126 for 30 min + 100 µmol/l phenylephrine for 48 h; U,
10 µmol/l U0126 for 30 min + 100 µmol/l phenylephrine for 48 h;
Control, no drug. |
Levels of histone H3K9ac acetylation
in primary cultured myocardial cells
Our previous study demonstrated that an imbalance in
the modification of histone H3K9ac acetylation can result in
pathological cardiac hypertrophy (32). In the present study,
immunofluorescence and western blotting data showed that the levels
of H3K9ac acetylation in the PE group were significantly higher
than that in the control group. Additionally, it was shown that the
HATs inhibitor AA, or the ERK inhibitor U0126, could downregulate
the hyperacetylation of H3K9ac induced by PE in primary myocardial
cells (Fig. 4).
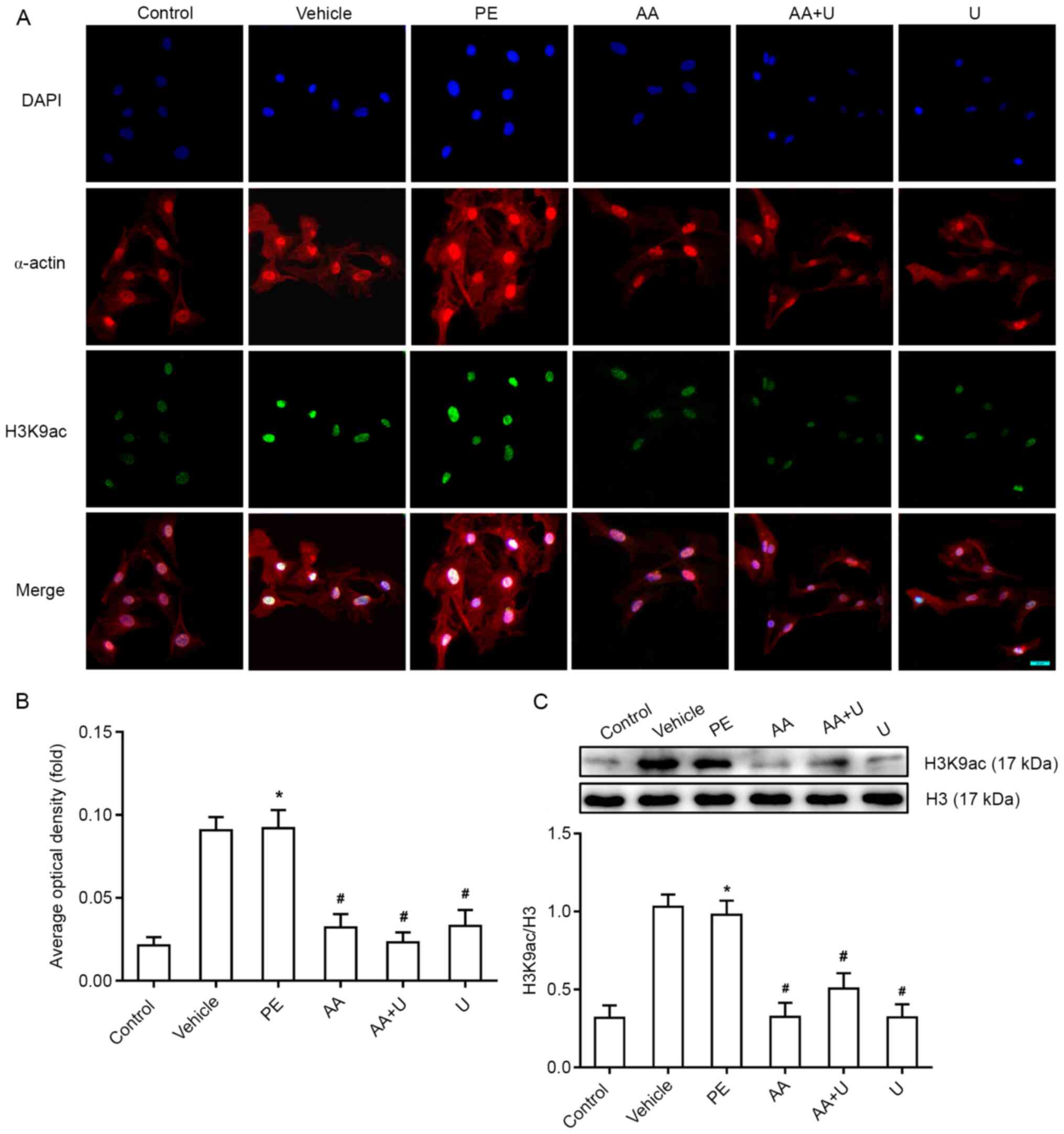 | Figure 4.Acetylation levels of histone H3K9ac
in mouse myocardial cells. (A) H3K9ac (green fluorescence) and
α-actin (red fluorescence), combined with DAPI staining (blue
fluorescence), in cardiomyocytes exposed to six different
conditions. Scale bar, 20 µm. (B) Mean optical density of H3K9ac
immunofluorescence in the six groups. (C) Western blotting data
showed that the levels of H3K9ac were significantly higher in the
hypertrophic cardiomyocytes induced by PE, whereas AA and/or U0126
prevented this effect. n=6. *P<0.05 vs. control group;
#P<0.05 vs. PE group. PCAF, P300/CBP-associated
factor; PE, phenylephrine; AA, anacardic acid; PE, 100 µmol/l
phenylephrine for 48 h; Vehicle, 100 µmol/l phenylephrine + equal
volume of DMSO for 48 h; AA, 50 µmol/l AA for 30 min + 100 µmol/l
phenylephrine for 48 h; AA + U, 50 µmol/l AA + 10 µmol/l U0126 for
30 min + 100 µmol/l phenylephrine for 48 h; U, 10 µmol/l U0126 for
30 min + 100 µmol/l phenylephrine for 48 h; Control, no drug;
H3K9ac, histone 3 acetylation K9. |
Overexpression of MEF2C is mediated by
p-ERK1/2-related H3K9ac hyperacetylation in myocardial cells
treated with PE
MEF2C is a nuclear transcription factor in
cardiac cells that is involved in pathological cardiac hypertrophy
and several other cardiovascular diseases (33,34).
Therefore, the binding of PCAF and histone H3K9ac acetylation in
the promoter region of MEF2C was assessed using ChIP-qPCR.
The data showed that the levels of PCAF promoter binding of
MEF2C in the PE group was higher than that in the control
group. Additionally, it was also found that the HATs inhibitor AA,
and the ERK inhibitor U0126 downregulated the binding of PCAF to
the promoter region of MEF2C. Meanwhile, the levels of
histone H3K9ac acetylation in the promoter region of MEF2C
was increased in the PE group. It was also evident that the HATs
inhibitor AA, and the ERK inhibitor U0126, could attenuate the
levels of histone H3K9ac acetylation in the promoter region of
MEF2C (Fig. 5A and B). The
mRNA expression levels of the MEF2C gene was determined
using RT-qPCR, and the results showed there was a notable increase
in gene expression in myocardial cells treated with PE. Exposure to
AA or U0126 reduced the overexpression of MEF2C mRNA in
primary cultured myocardial cells treated with PE (Fig. 5C).
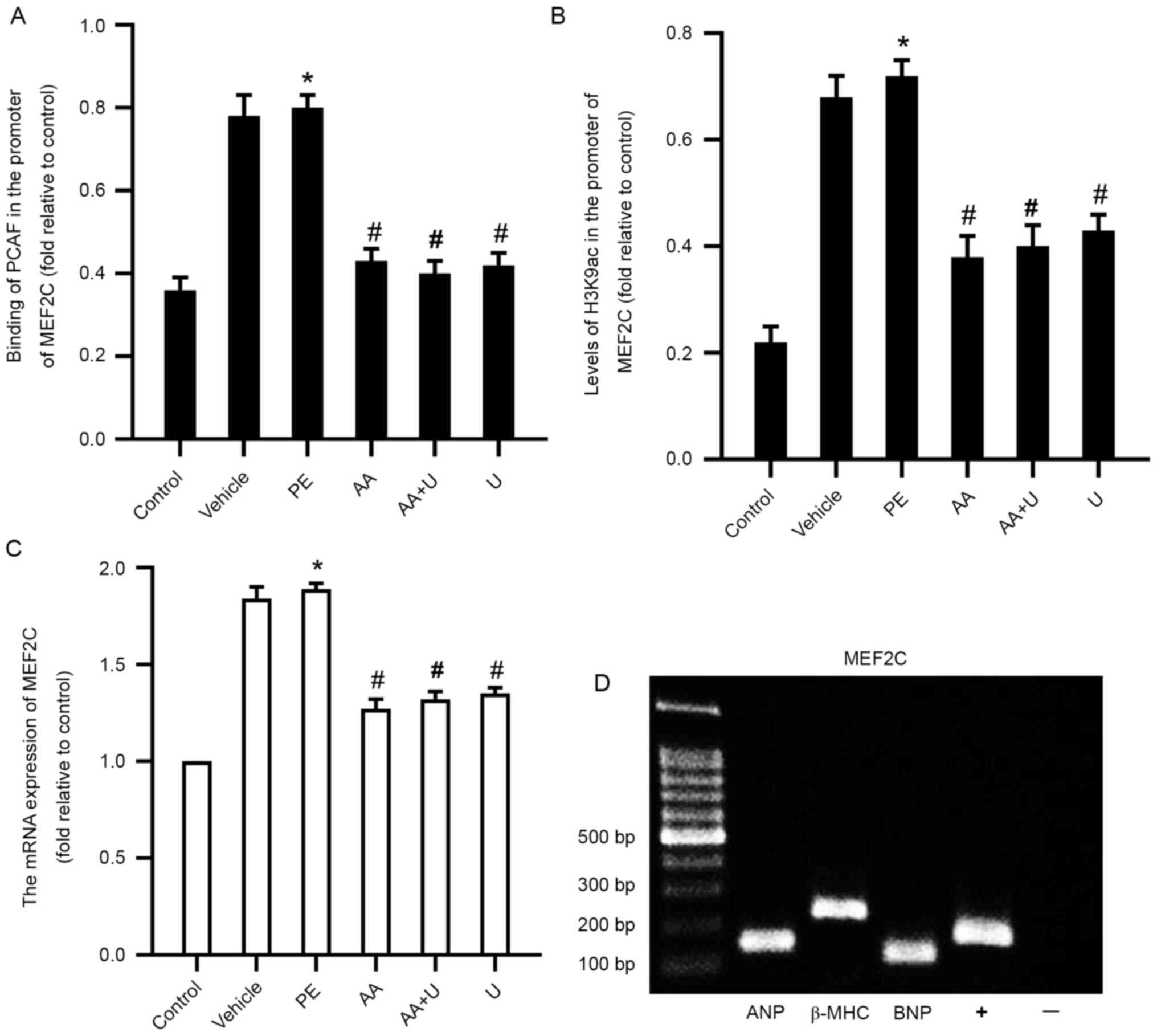 | Figure 5.PCAF binding, acetylation levels of
H3K9ac in the MEF2C promoter and the expression of
MEF2C in myocardial cells. (A) Binding of PCAF to the
promoter in MEF2C was assessed using ChIP-qPCR. (B)
Acetylation levels of histone H3K9ac in the promoter of
MEF2C were assessed using ChIP-qPCR. (C) mRNA expression of
MEF2C in cardiomyocytes exposed to six different conditions.
(D) ChIP-qPCR data showed that the cardiac nuclear transcription
factor MEF2C could bind to the promoter of biomarker genes
for cardiac hypertrophy (ANP, BNP and β-MHC). n=6.
*P<0.05 vs. control group; #P<0.05 vs. PE group.
PCAF, P300/CBP-associated factor; PE, phenylephrine; AA, anacardic
acid; PE, 100 µmol/l phenylephrine for 48 h; Vehicle, 100 µmol/l
phenylephrine + equal volume of DMSO for 48 h; AA, 50 µmol/l AA for
30 min + 100 µmol/l phenylephrine for 48 h; AA + U, 50 µmol/l AA +
10 µmol/l U0126 for 30 min + 100 µmol/l phenylephrine for 48 h; U,
10 µmol/l U0126 for 30 min + 100 µmol/l phenylephrine for 48 h;
Control, no drug; PCAF, P300/CBP-associated factor; PE,
phenylephrine; H3K9ac, histone 3 acetylation K9; MEF2C, myocyte
enhancer factor 2C; ChIP-qPCR,
chromatin-immunoprecipitation-quantitative PCR; ANP, atrial
natriuretic peptide; β-MHC, β-myosin heavy chain; BNP, brain
natriuretic peptide. |
AA and U0126 reduce the levels of
biomarkers of cardiac hypertrophy and attenuate hypertrophy in
cardiomyocytes
To investigate the effects of MEF2C on
downstream genes associated with cardiac hypertrophy in
cardiomyocytes, the regulatory relationship between MEF2C
and downstream genes associated with cardiac hypertrophy, including
ANP, BNP and β-MHC were investigated. The binding
affinity between MEF2C and the promoters of ANP, BNP
and β-MHC, were detected by PCR following ChIP. The results
showed that MEF2C could bind to the promoters of ANP,
BNP and β-MHC (Fig. 5D).
These results indicate that MEF2C is involved in regulating
cardiac hypertrophy-related ANP, BNP and β-MHC gene
expression. The levels of biomarkers of cardiac hypertrophy (ANP,
BNP and β-MHC) at the protein level in the PE group were
significantly higher than those in the control group. Additionally,
it was found that AA and U0126 could reduce the increase in ANP,
BNP and β-MHC levels in myocardial cells treated with PE (Fig. 6A, B and C). AA and U0126 could also
significantly reduce the surface area of cardiomyocytes treated
with PE (Fig. 6D and E).
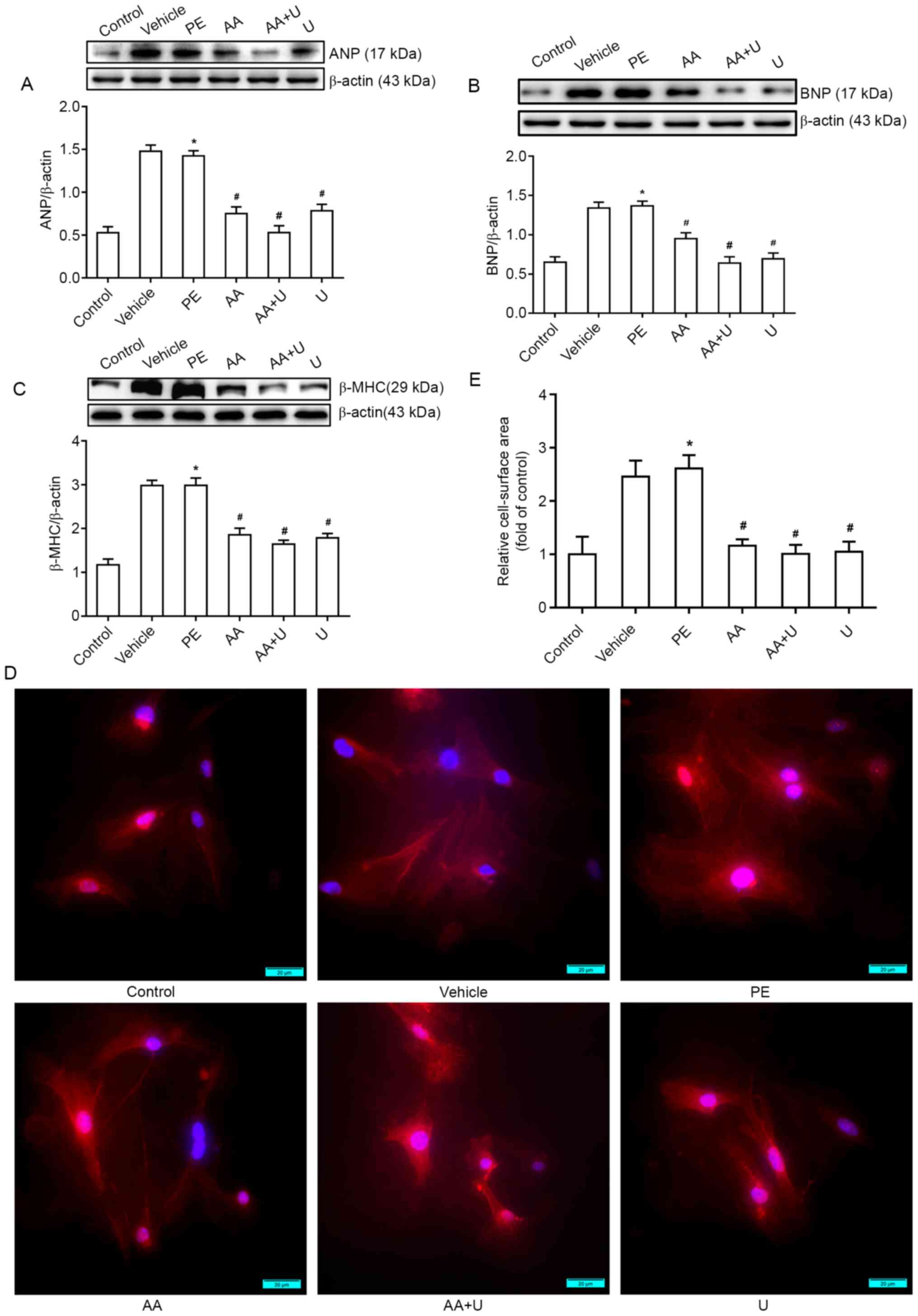 | Figure 6.AA and U0126 alleviates PE-induced
cardiomyocyte hypertrophy. (A-C) Western blotting data for cardiac
hypertrophy biomarkers (ANP, BNP and β-MHC). (D) Cell surface area
was measured by immunofluorescence staining to demonstrate the
hypertrophic responses in cardiomyocytes. (E) Quantification of the
cell surface area of myocardial cells. Scale bar, 20 µm. n=6.
*P<0.05 vs. control group; #P<0.05 vs. PE group.
PCAF, P300/CBP-associated factor; PE, phenylephrine; AA, anacardic
acid; PE, 100 µmol/l phenylephrine for 48 h; Vehicle, 100 µmol/l
phenylephrine + equal volume of DMSO for 48 h; AA, 50 µmol/l AA for
30 min + 100 µmol/l phenylephrine for 48 h; AA + U, 50 µmol/l AA +
10 µmol/l U0126 for 30 min + 100 µmol/l phenylephrine for 48 h; U,
10 µmol/l U0126 for 30 min + 100 µmol/l phenylephrine for 48 h;
Control, no drug; ANP, atrial natriuretic peptide; β-MHC, β-myosin
heavy chain; BNP, brain natriuretic peptide; PE, phenylephrine. |
Discussion
Myocardial hypertrophy is an adaptive response that
is induced by a wide range of factors, including hemodynamic
overload, myocardial injury and vascular disease. In its early
stages, this disease is characterized by an increased volume of
cardiomyocytes, although the number of these cells remains
constant. Moreover, in an attempt to compensate for these effects,
the myocardial contractility is enhanced; this way, the heart can
maintain a normal ejection fraction (35,36).
These abnormalities further contribute to increased oxygen demand
in the myocardial cells, which leads to insufficiency in terms of
the blood supply and eventually results in myocardial fibrosis,
arrhythmia and heart failure. However, the specific mechanisms
underlying myocardial hypertrophy remain unclear. An increasing
body of research is now investigating the epigenetic regulation of
myocardial hypertrophy in an attempt to identify more effective
treatments. Existing reports indicate that histone modification and
DNA methylation are involved in the occurrence and development of
myocardial hypertrophy (37,38).
The occurrence and development of cardiac hypertrophy is also
accompanied by the abnormal activation of a large number of
important signaling pathways (5,39,40).
Consequently, there is an increased level of interest in how key
signaling pathways participate in the epigenetic regulation of
cardiac hypertrophy (41,42). Our previous studies found that an
imbalance of histone acetylation modification is involved in
PE-induced hypertrophy in cardiomyocytes, and that an HAT
inhibitor, AA, can attenuate PE-induced cardiomyocyte hypertrophy
(4); however, the relevant upstream
signaling pathways have yet to be identified.
The development of cardiac hypertrophy is closely
related to a variety of signaling pathways that interact with each
other, for example, the Wnt signaling transduction pathway, the
mitogen-activated protein kinase (MAPK) signaling transduction
pathway and the microRNA signaling transduction pathway are all
considered to be related to this process (3,6,7,9).
Of these, the MAPK signaling pathway has attracted significant
attention (43,44). In our previous study, it was
confirmed that AA could attenuate pressure-overload cardiac
hypertrophy through inhibition of histone acetylases (31), and the p38/MAPK and JNK/MAPK
signaling pathways were involved in the attenuation of PE-induced
cardiomyocyte hypertrophy in mice treated with AA (20,45).
MAPKs include ERK1/2, p38 and c-Jun N-terminal kinase (JNK)
subfamilies; these pathways can transfer a variety of extracellular
stimuli from the cell membrane to the nucleus and cause a range of
biological effects, including differentiation, hypertrophy and
apoptosis (46,47). With regard to the MAPK signaling
system, research has shown that ERK1/2 is mainly associated with
cell growth, differentiation, development, proliferation, and a
range of other physiological and pathological processes (10,48).
Other studies have shown that p38 and JNK can cause cell
inflammation, apoptosis, growth, differentiation and stress
responses (49,50). Previous studies have also reported
high expression levels of p-ERK in myocardial tissues from rats
with pathological myocardial hypertrophy; furthermore, a reduction
in the levels of p-ERK was effectively shown to improve myocardial
hypertrophy (51). Phosphorylation
of ERK at threonine 188, along with the activation of ERK5, has
also been shown to be related to the pathological process of
cardiomyocyte hypertrophy (41).
Furthermore, as the first angiotensin receptor-enkephalin
inhibitor, LCZ696 has been shown to attenuate cardiac remodeling by
inhibiting the ERK signaling pathway in mice with
pregnancy-associated cardiomyopathy (52). Collectively, these studies suggest
that the ERK1/2 signaling pathway plays a critical role in the
process of cardiomyocyte hypertrophy. Therefore, it was
hypothesized that the ERK1/2 signaling pathway may be involved in
the attenuation of PE-induced cardiomyocyte hypertrophy by AA. The
activation of p-ERK1/2 could phosphorylate downstream nuclear
transcription factors and protein kinase substrates and regulate
the occurrence and development of myocardial hypertrophy. In the
present study, it was confirmed that the expression of p-ERK1/2 in
the PE-treated group was significantly higher than that in the
control group, although there was no change in the expression of
T-ERK1/2; this suggested that phosphorylation of ERK1/2 may play an
important regulatory role in PE-induced cardiomyocyte
hypertrophy.
An increasing body of evidence has suggested that
changes in signaling pathways may bring about epigenetic changes
(53,54). At present, research relating to
histone modification focuses mainly on histone acetylation. As one
of the subtypes closely related to cardiac hypertrophy, PCAF-HAT is
known to acetylate histones and non-histones, activate chromatin
and participate in pathological cardiac hypertrophy (55). However, PCAF-HAT does not bind DNA;
rather PCAF-HAT is attracted to prime subsites by interacting with
sequence-specific activators that mediate transcriptional
activation (56). In the present
study, it was confirmed that p-ERK1/2 can combine with PCAF and
histone H3K9ac to create a complex; this suggests that the p-ERK1/2
signaling pathway may participate in the modification of
PCAF-mediated H3K9ac acetylation to jointly regulate PE-induced
cardiac hypertrophy. Certain studies have found that the ERK1/2
signaling pathway is involved in the overexpression of
transcription factors that are related to cardiac development
induced by alcohol exposure; these factors act by upregulating the
levels of histone H3 acetylation (57). Class I histone deacetylases can
inhibit cardiomyocyte hypertrophy by inhibiting the expression of
genes encoding bispecific phosphatase 5 (a nucleophosphatase that
negatively regulates hypertrophic signals through ERK1/2) (58). Together with the results of the
present study, these data suggest that the regulatory mechanism
underlying cardiac hypertrophy is diverse, and that its occurrence
and development is a complex process.
Transcription factors are a group of protein
molecules that can specifically bind to a specific sequence
upstream of the 5′ terminus of a gene; this ensures that the target
gene is expressed in a specific time and space, and at a specific
intensity (59). The analysis of
transcription factors is a prerequisite and forms the basis for
studying the regulation of gene expression. The overexpression of
several cardiac core transcription factors (GATA4, MEF2A and
MEF2C) is an important factor that can lead to cardiac
hypertrophy; these transcription factors are regulated by HATs
(4,33,60).
In our previous study, it was shown that that the HAT inhibitor,
AA, can downregulate the transcriptional activation of GATA4
by inhibiting P300 and PCAF, and thus attenuate the myocardial
hypertrophy caused by alcohol exposure during pregnancy in fetal
mice (61). Based on this finding,
it was hypothesized that PCAF-mediated changes in the
transcriptional activity of GATA4 were not the only
regulatory factors that lead to cardiac hypertrophy. Indeed,
MEF2C is involved in heart development at various stages and
shows high DNA binding activity in cardiomyocytes (62). Changes in the transcription levels
of transcriptional factors that lie upstream of cardiac hypertrophy
are different to the genes that lie downstream. In the present
study, ChIP-PCR results showed that MEF2C binds to the
promoters of ANP, BNP and β-MHC, whereas an ERK
inhibitor (U0126) and a HAT inhibitor (AA) could inhibit the
transcriptional activity of MEF2C, and therefore reduce the
expression of ANP, BNP and β-MHC in the myocardial cells of mice.
This further suggests that the involvement of the ERK signaling
pathway in the modification of histone acetylation is strongly
associated with the occurrence and development of pathological
cardiac hypertrophy.
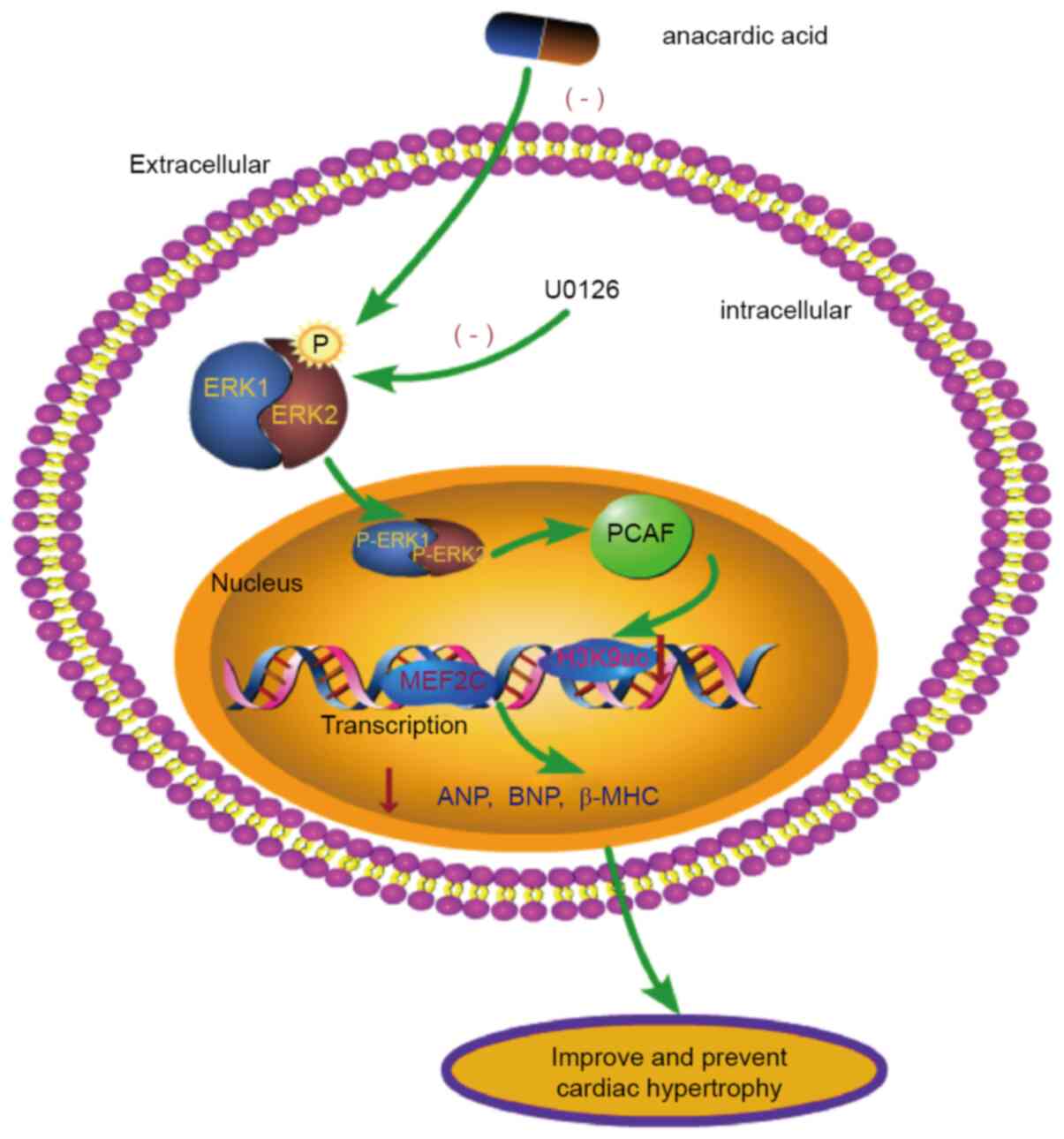
In conclusion, cardiac development is a delicate and
complex process and is closely related to gene transcription and
modification. In the present study, it was shown that the ERK1/2
signaling pathway interacts with PCAF to modify H3K9ac
hyperacetylation and that this process plays a significant role in
the cardiomyocyte hypertrophy induced by PE (Fig. 7). PE induces phosphorylation of
ERK1/2 to generate p-ERK1/2. p-ERK1/2 upregulates expression of
PCAF leading to acetylation of H3K9ac, thereby increasing the
binding of MEF2C in the promoter region. The increased production
of the myocardial hypertrophy factors ANP, BNP and β-MHC cause
cardiomyocyte hypertrophy. These findings may lead to the
development of novel interventional targets and candidate drugs for
the clinical prevention and treatment of myocardial
hypertrophy.
Acknowledgements
Not applicable.
Funding
This work was supported by the National Natural
Science Foundation of China (grant nos. 82060046 and 81560040).
Availability of data and materials
All data generated or analyzed during the present
study are included in the published article.
Authors' contributions
QM drafted the manuscript. BHP and CP conceived and
designed the study. SQW and QM performed the experiments and
confirmed the authenticity of all the raw data. XML and LXH
collected the experimental results. HTZ analyzed the data. All
authors have read and approved the final manuscript.
Ethics approval and consent to
participate
All animal experiments were approved by the Animal
Protection and Use Committee of Zunyi Medical University (Zunyi,
China), and complied with Directive 2010/63/EU of the European
Parliament.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
AA
|
anacardic acid
|
|
ANP
|
atrial natriuretic peptide
|
|
BNP
|
brain natriuretic peptide
|
|
β-MHC
|
β-myosin heavy chain
|
|
Co-IP
|
co-immunoprecipitation
|
|
ChIP
|
chromatin immunoprecipitation
|
|
ERK
|
extracellular signal-regulated protein
kinase
|
|
HAT
|
histone acetylase
|
|
H3K9ac
|
histone 3 acetylation K9
|
|
JNK
|
c-Jun N-terminal kinase
|
|
MEF2C
|
myocyte enhancer factor 2C
|
|
MAPK
|
mitogen-activated protein kinases
|
|
PE
|
phenylephrine
|
|
PCAF
|
P300/CBP-associated factor.
|
References
|
1
|
Degoricija V, Trbušić M, Potočnjak I,
Radulović B, Terešak SD, Pregartner G, Berghold A, Tiran B and
Frank S: Acute heart failure developed as worsening of chronic
heart failure is associated with increased mortality compared to de
novo cases. Sci Rep. 8:95872018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Yu B, Zhao Y, Zhang H, Xie D, Nie W and
Shi K: Inhibition of microRNA-143-3p attenuates myocardial
hypertrophy by inhibiting inflammatory response. Cell Biol Int.
42:1584–1593. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Wehbe N, Nasser SA, Pintus G, Badran A,
Eid AH and Baydoun E: MicroRNAs in cardiac hypertrophy. Int J Mol
Sci. 20:47142019. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Peng C, Luo X, Li S and Sun H:
Phenylephrine-induced cardiac hypertrophy is attenuated by a
histone acetylase inhibitor anacardic acid in mice. Mol Biosyst.
13:714–724. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Gao W, Guo N, Zhao S, Chen Z, Zhang W, Yan
F, Liao H and Chi K: Carboxypeptidase A4 promotes cardiomyocyte
hypertrophy through activating PI3K-AKT-mTOR signaling. Biosci Rep.
40:BSR202006692020. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Gai Z, Wang Y, Tian L, Gong G and Zhao J:
Whole genome level analysis of the Wnt and DIX gene families in
mice and their coordination relationship in regulating cardiac
hypertrophy. Front Genet. 12:6089362021. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Siti HN, Jalil J, Asmadi AY and Kamisah Y:
Rutin modulates MAPK pathway differently from Quercetin in
angiotensin II–Induced H9c2 cardiomyocyte hypertrophy. Int J Mol
Sci. 22:50632021. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Bogdanova E, Beresneva O, Galkina O,
Zubina I, Ivanova G, Parastaeva M, Semenova N and Dobronravov V:
Myocardial hypertrophy and fibrosis are associated with
cardiomyocyte Beta-Catenin and TRPC6/Calcineurin/NFAT signaling in
spontaneously hypertensive rats with 5/6 Nephrectomy. Int J Mol
Sci. 22:46452021. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Tamura S, Marunouchi T and Tanonaka K:
Heat-shock protein 90 modulates cardiac ventricular hypertrophy via
activation of MAPK pathway. J Mol Cell Cardiol. 127:134–142. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Hu B, Song JT, Ji XF, Liu ZQ, Cong ML and
Liu DX: Sodium Ferulate protects against angiotensin II–Induced
cardiac hypertrophy in mice by regulating the MAPK/ERK and JNK
Pathways. Biomed Res Int. 2017:37549422017. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Yan ZP, Li JT, Zeng N and Ni GX: Role of
extracellular signal-regulated kinase 1/2 signaling underlying
cardiac hypertrophy. Cardiol J. 28:473–482. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Gu J, Hu W, Song ZP, Chen YG, Zhang DD and
Wang CQ: Rapamycin inhibits cardiac hypertrophy by promoting
autophagy via the MEK/ERK/Beclin-1 pathway. Front Physiol.
7:1042016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Huang Y, Wu L, Wu J, Li Y and Hou L:
Cellular FLICE-like inhibitory protein protects against cardiac
hypertrophy by blocking ASK1/p38signaling in mice. Mol Cell
Biochem. 397:87–95. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zhou J, Gao J, Zhang X, Liu Y, Gu S, Zhang
X, An X, Yan J, Xin Y and Su P: MicroRNA-340-5p functions
downstream of cardiotrophin-1 to regulate cardiac eccentric
hypertrophy and heart failure via target gene dystrophin. Int Heart
J. 56:454–458. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Li B, Wang X, Yu M, Yang P and Wang W:
G6PD, bond by miR-24, regulates mitochondrial dysfunction and
oxidative stress in phenylephrine-induced hypertrophic
cardiomyocytes. Life Sci. 260:1183782020. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Melchert RB, Liu H, Granberry MC and
Kennedy RH: Lovastatin inhibits phenylephrine-induced ERK
activation and growth of cardiac. Cardiovasc Toxicol. 1:237–252.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhong L, Chiusa M, Cadar AG, Lin A,
Samaras S, Davidson JM and Lim CC: Targeted inhibition of ANKRD1
disrupts sarcomeric ERK-GATA4 signal transduction and abrogates
phenylephrine-induced cardiomyocyte hypertrophy. Cardiovasc Res.
106:261–271. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Schreckenberg R, Taimor G, Piper HM and
Schlüter KD: Inhibition of Ca2+-dependent PKC isoforms unmasks
ERK-dependent hypertrophic growth evoked by phenylephrine in adult
ventricular cardiomyocytes. Cardiovasc Res. 63:553–560. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
EUR-Lex, . Directive 2010/63/EU of the
European Parliament and of the Council of 22 September 2010 on the
protection of animals used for scientific purposes. Official J
European Union. 53:L 276/33–L 276/79. 2010.
|
|
20
|
Peng BH, Peng C, Huang LX, Luo XM and Han
X: The roles of P38 MAPK in the process of anacardic acid
attenuating mouse cardiomyocyte hypertrophy induced by
phenylephrine. Chin J Pathophys. 36:200–205. 2020.PubMed/NCBI
|
|
21
|
Yin Y, Guan Y, Duan J, Wei G, Zhu Y, Quan
W, Guo C, Zhou D, Wang Y, Xi M and Wen A: Cardioprotective effect
of Danshensu against myocardial ischemia/reperfusion injury and
inhibits apoptosis of H9c2 cardiomyocytes via Akt and ERK1/2
phosphorylation. Eur J Pharmacol. 699:219–226. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhao Q, Zhang X, Cai H, Zhang P, Kong D,
Ge X, Du M, Liang R and Dong W: Anticancer effects of plant derived
anacardic acid on human breast cancer MDA-MB-231 cells. Am J Transl
Res. 10:2424–2434. 2018.PubMed/NCBI
|
|
23
|
Li Q, Li ZM, Sun SY, Wang LP, Wang PX, Guo
Z, Yang HW, Ye JT, Lu J and Liu PQ: PARP1 interacts with HMGB1 and
promotes its nuclear export in pathological myocardial hypertrophy.
Acta Pharmacol Sin. 40:589–598. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Frias MA, Rebsamen MC, Gerber-Wicht C and
Lang U: Prostaglandin E2 activates Stat3 in neonatal rat
ventricular cardiomyocytes: A role in cardiac hypertrophy.
Cardiovasc Res. 73:57–65. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Peng B, Han X, Peng C, Luo X, Deng L and
Huang L: G9α-dependent histone H3K9me3 hypomethylation promotes
overexpression of cardiomyogenesis-related genes in foetal mice. J
Cell Mol Med. 24:1036–1045. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Ren J, Zhang N, Liao H, Chen S, Xu L, Li
J, Yang Z, Deng W and Tang Q: Caffeic acid phenethyl ester
attenuates pathological cardiac hypertrophy by regulation of
MEK/ERK signaling pathway in vivo and vitro. Life Sci. 181:53–61.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Saleem N, Prasad A and Goswami SK:
Apocynin prevents isoproterenol-induced cardiac hypertrophy in rat.
Mol Cell Biochem. 445:79–88. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Önal B, Özen D, Demir B, Gezen Ak D,
Dursun E, Demir C, Akkan AG and Özyazgan S: The anti-inflammatory
effects of Anacardic acid on a TNF-α-Induced human saphenous vein
endothelial cell culture model. Curr Pharm Biotechnol. 21:710–719.
2020. View Article : Google Scholar
|
|
30
|
Lee MJ, Tsai YJ, Lin MY, You HL, Kalyanam
N, Ho CT and Pan MH: Calebin-A induced death of malignant
peripheral nerve sheath tumor cells by activation of histone
acetyltransferase. Phytomedicine. 57:377–384. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Li S, Peng B, Luo X, Sun H and Peng C:
Anacardic acid attenuates pressure-overload cardiac hypertrophy
through inhibiting histone acetylases. J Cell Mol Med.
23:2744–2752. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Luo XM, Che JL, Liu SR, Long S, Zhao PX,
Xu P, Wei Y and Peng C: The effects of histone acetylation
modification on cardiac hypertrophy induced by two different
modeling methods in mice. J Clin Cardiol. 33:1106–1110. 2017.
|
|
33
|
Zhang G and Ni X: Knockdown of TUG1
rescues cardiomyocyte hypertrophy through targeting the
miR-497/MEF2C axis. Open Life Sci. 16:242–251. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Khajehlandi M, Bolboli L, Siahkuhian M,
Rami M, Tabandeh M, Khoramipour K and Suzuki K: Endurance training
regulates expression of some angiogenesis-related genes in cardiac
tissue of experimentally induced diabetic rats. Biomolecules.
11:4982021. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Wang HN, Li JL, Xu T, Yao HQ, Chen GH and
Hu J: Effects of Sirt3 autophagy and resveratrol activation on
myocardial hypertrophy and energy metabolism. Mol Med Rep.
22:1342–1350. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ding J, Liu S, Qian W, Wang J, Chu C, Wang
J, Li K, Yu Y, Xu G, Mao Z, et al: Swietenine extracted from
Swietenia relieves myocardial hypertrophy induced by isoprenaline
in mice. Environ Toxicol. 35:1343–1351. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Zhang LX, Du J, Zhao YT, Wang J, Zhang S,
Dubielecka PM, Wei L, Zhuang S, Qin G, Chin YE and Zhao TC:
Transgenic overexpression of active HDAC4 in the heart attenuates
cardiac function and exacerbates remodeling in infarcted
myocardium. J Appl Physiol. 125:1968–1978. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Stenzig J, Schneeberger Y, Löser A, Peters
BS, Schaefer A, Zhao RR, Ng SL, Höppner G, Geertz B, Hirt MN, et
al: Pharmacological inhibition of DNA methylation attenuates
pressure overload-induced cardiac hypertrophy in rats. J Mol Cell
Cardiol. 120:53–63. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Lin L, Xu W, Li Y, Zhu P, Yuan W, Liu M,
Shi Y, Chen Y, Liang J, Chen J, et al: Pygo1 regulates pathological
cardiac hypertrophy via a β-catenin-dependent mechanism. Am J
Physiol Heart Circ Physiol. 320:H1634–H1645. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Kuwabara Y, Horie T, Baba O, Watanabe S,
Nishiga M, Usami S, Izuhara M, Nakao T, Nishino T, Otsu K, et al:
MicroRNA-451 exacerbates lipotoxicity in cardiac myocytes and
high-fat diet-induced cardiac hypertrophy in mice through
suppression of the LKB1/AMPK pathway. Circ Res. 116:279–288. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Gallo S, Vitacolonna A, Bonzano A,
Comoglio P and Crepaldi T: ERK: A key player in the pathophysiology
of cardiac hypertrophy. Int J Mol Sci. 20:21642019. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Breitenbach T, Lorenz K and Dandekar T:
How to steer and control ERK and the ERK signaling cascade
exemplified by looking at cardiac insufficiency. Int J Mol Sci.
20:21792019. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Liu R and Molkentin JD: Regulation of
cardiac hypertrophy and remodeling through the dual-specificity
MAPK phosphatases (DUSPs). J Mol Cell Cardio. 101:44–49. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Zhang Y, Cui Y, Dai S, Deng W, Wang H, Qin
W, Yang H, Liu H, Yue J, Wu D, et al: Isorhynchophylline enhances
Nrf2 and inhibits MAPK pathway in cardiac hypertrophy. Naunyn
Schmiedebergs Arch Pharmacol. 393:203–212. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Peng BH: Role of JNK/MAPK
signaling-dependent regulation of histone acetylation on the
attenuation of anacardic acid for cardiomyocyte hypertrophy induced
by phenylephrine (unpublished PhD thesis). Zunyi Medical
University; 2020
|
|
46
|
Ba L, Gao J, Chen Y, Qi H, Dong C, Pan H,
Zhang Q, Shi P, Song C, Guan X, et al: Allicin attenuates
pathological cardiac hypertrophy by inhibiting autophagy via
activation of PI3K/Akt/mTOR and MAPK/ERK/mTOR signaling pathways.
Phytomedicine. 58:1527652019. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Kim MJ, Im MA, Lee JS, Mun JY, Kim DH, Gu
A and Kim IS: Effect of S100A8 and S100A9 on expressions of
cytokine and skin barrier protein in human keratinocytes. Mol Med
Rep. 20:2476–2483. 2019.PubMed/NCBI
|
|
48
|
Sun Y, Liu WZ, Liu T, Feng X, Yang N and
Zhou HF: Signaling pathway of MAPK/ERK in cell proliferation,
differentiation, migration, senescence and apoptosis. J Recept
Signal Transduct Res. 35:600–604. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Gentile MT, Russo R, Pastorino O, Cioffi
S, Barbieri F, Illingworth EA, Grieco M, Chambery A and
Colucci-D'Amato L: Ruta graveolens water extract inhibits cell-cell
network formation in human umbilical endothelial cells via
MEK-ERK1/2 pathway. Exp Cell Res. 64:50–58. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Dai X, Song R and Xiong Y: The expression
of ERK and JNK in patients with an endemic osteochondropathy,
Kashin-Beck disease. Exp Cell Res. 359:337–341. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Cipolletta E, Rusciano MR, Maione AS,
Santulli G, Sorriento D, Del Giudice C, Ciccarelli M, Franco A,
Crola C, Campiglia P, et al: Targeting the CaMKII/ERK interaction
in the heart prevents cardiac hypertrophy. PLoS One.
10:e01304772015. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Wang Y, Guo Z, Gao Y, Liang P, Shan Y and
He J: Angiotensin II receptor blocker LCZ696 attenuates cardiac
remodeling through the inhibition of the ERK signaling pathway in
mice with pregnancy-associated cardiomyopathy. Cell Biosci.
9:862019. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Thienpont B, Aronsen JM, Robinson EL,
Okkenhaug H, Loche E, Ferrini A, Brien P, Alkass K, Tomasso A,
Agrawal A, et al: The H3K9 dimethyltransferases EHMT1/2 protect
against pathological cardiac hypertrophy. J Clin Invest.
127:335–348. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Xing S, Tian JZ, Yang SH, Huang XT, Ding
YF, Lu QY, Yang JS and Yang WJ: Setd4 controlled quiescent
c-Kit+ cells contribute to cardiac neovascularization of
capillaries beyond activation. Sci Rep. 11:116032021. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Wei J, Joshi S, Speransky S, Crowley C,
Jayathilaka N, Lei X, Wu Y, Gai D, Jain S, Hoosien M, et al:
Reversal of pathological cardiac hypertrophy via the
MEF2-coregulator interface. JCI Insight. 2:e910682017. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Marmorstein R and Zhou MM: Writers and
readers of histone acetylation: Structure, mechanism, and
inhibition. Cold Spring Harb Perspect Biol. 6:a0187622014.
View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Gao W, Pan B, Liu L, Huang X, Liu Z and
Tian J: Alcohol exposure increases the expression of cardiac
transcription factors through ERK1/2-mediated histone3
hyperacetylation in H9c2 cells. Biochem Biophys Res Commun.
466:670–675. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Ferguson BS, Harrison BC, Jeong MY, Reid
BG, Wempe MF, Wagner FF, Holson EB and McKinsey TA:
Signal-dependent repression of DUSP5 by class I HDACs controls
nuclear ERK activity and cardiomyocyte hypertrophy. Proc Natl Acad
Sci USA. 110:9806–9811. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Lambert M, Jambon S, Depauw S and
David-Cordonnier MH: Targeting transcription factors for cancer
treatment. Molecules. 23:14792018. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Peng C, Zhang W, Zhao W, Zhu J, Huang X
and Tian J: Alcohol-induced histone H3K9 hyperacetylation and
cardiac hypertrophy are reversed by a histone acetylases inhibitor
anacardic acid in developing murine hearts. Biochimie. 113:1–9.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Peng C, Zhu J, Sun HC, Huang XP, Zhao WA,
Zheng M, Liu LJ and Tian J: Inhibition of histone H3K9 acetylation
by anacardic acid can correct the over-expression of Gata4 in the
hearts of fetal mice exposed to alcohol during pregnancy. PLoS One.
9:e1041352014. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Cardoso AC, Pereira AHM, Ambrosio ALB,
Consonni SR, Rocha de Oliveira R, Bajgelman MC, Dias SMG and
Franchini KG: FAK forms a complex with MEF2 to couple biomechanical
signaling to transcription in cardiomyocytes. Structure.
24:1301–1310. 2016. View Article : Google Scholar : PubMed/NCBI
|