Introduction
Atherosclerosis (AS) is a chronic inflammatory
disease as well as a cholesterol storage disease, which is often
asymptomatic at the early stages but can lead to serious ischemic
vascular disease and thrombosis such as coronary heart disease and
carotid artery disease at an advanced stage (1–3).
Risk factors of AS, such as obesity, smoking, stress
and hypertension, are all intimately linked with a higher level of
local or global inflammation (4–8). CXC
chemokine ligand 16 (CXCL16), a member of the CXC chemokine family,
serves an important role in the initiation and maintenance of
inflammation (9,10). Evidence links it with the formation
and progression of AS (11). CXCL16
expresses in aortic smooth muscle cells exclusively within
atherosclerotic plaques (12). Its
two distinct existing isoforms in vivo, transmembrane and
soluble form, give it great influence over inflammation (13). The transmembrane isoform functions
as a selective cell adhesion molecule for CXC receptor
6+ cells, such as monocytes and T lymphocytes, to
facilitate cell-cell interaction (14), while the soluble isoform secreted
into extracellular matrix or plasma exerts a relatively long
distance recruitment of immune cells and regulates a global
inflammation level (15,16). Another role CXCL16 performs is a
scavenger receptor which facilitates internalization of fatty acids
in several central cell types involved in AS, such as macrophages
and T lymphocytes (17). Clinical
researches investigating the role of CXCL16 in AS or carotid artery
disease are controversial, for instance, as to whether CXCL16
promotes or inhibits lesions (18).
Decreased and increased plasma levels of CXCL16 are identified to
be related to cardiovascular or cerebrovascular events, such as
coronary artery disease and atherosclerotic ischemic stroke
(11,19). It is probably due to the
methodological limitation of clinical research that cross-sectional
studies or a relatively short period of prospective study cannot
well determine causal relationship. The present study attempted to
address this problem by introducing CXCL16 gene into C57BL/6J
wild-type mice, a canonical strain for metabolism disorder, to
create a predisposed high expression level of CXCL16 during a
relatively long period of time. The present study also focused on
how this high level of CXCL16 expression affected atherogenicity in
an inflammatory aspect by testing several well-established
atherosclerotic inflammation markers.
Materials and methods
Laboratory animals
Animal experiments were approved by the Laboratory
Animal Welfare and Ethics Review Committee of University of South
China (approval no. SYSK2013-0010). The present study was performed
according to the requirements in the Guide for the Care and Use of
Laboratory Animals (20). Specific
pathogen-free (SPF) C57BL/6J wild-type mice were purchased from
Shanghai Model Organisms Center, Inc. (SMOC) and raised under SPF
conditions. A total of 45 mice (30 females and 15 males; age, 6–8
weeks; weight, 17–20 g) were housed in standard plastic cages at
20–26°C, 40–70% humidity with a 12 h light/dark cycle and had free
access to water and food. The mice were given 2 weeks to acclimate
for breeding before any treatment.
Vector construction
The goal of the present study was to subclone mouse
CXCL16 cDNA into the pGL3-basic mammalian expression vector. PCR
amplification was used to multiply Murine CXCL16 cDNA and
simultaneously as part of the primer a sequence (5′ to 3′) of flag
tag (GATTACAAGGATGACGACGATAAG) was attached to the N terminus.
Later scavenger receptor (SR)-enhancer, SR-promoter and CXCL16-flag
were all respectively inserted into pGL3-basic between restriction
sites KpnI and NheI, XhoI and HindIII,
HindIII and XbaI. A genomic DNA isolation kit (Bio
Basic Inc.) was used to extract and purify DNA (the yield of 5–10
µg) from miniprep according to the manufacturer's protocols.
Verification was performed through Sanger sequencing (21) using an ABI 3730XL automatic
sequencer (Applied Biosystems; Thermo Fisher Scientific, Inc.). A
triple cut site ClaI was set to cut the vector in to three
parts that were respectively 1.8, 2.7 and 0.1 kbp. The 1.8 kbp
fragments including CXCL16 cDNA were collected and purified from
agarose gel electrophoresed in TBE running buffer using the gel
recovery kit (Bio Basic Inc.), according to the manufacturer's
instructions, for further use.
Founder mouse generation
A total of 30 C57BL/6J × CBA F1 hybrid female mice
(age, 2–4 weeks; weight, 7–10 g) supplied by SMOC were housed in
standard plastic cages at 20–26°C and 40–70% humidity with a 12 h
light/dark cycle. These mice were used as their oocytes exhibit
high survival and maturation rates after microinjection (22). At the age of ~6–8 weeks old, 12 h
before dark cycle, they were injected with 10 IU PMSG and ~48 h
afterwards with 10 IU human chorionic gonadotropin i.p. In the same
afternoon they were mated 1:1 with fertile male C57BL/6J mice. For
all mouse experiments, the mice were anesthetized with 50–60 mg/kg
pentobarbital sodium (Sigma-Aldrich; Merck KGaA) and sacrificed by
cervical dislocation. The next day, if a vaginal plug was present,
they were set as donors and fertilized eggs were excised from
oviducts into M2 culture medium (Sigma-Aldrich; Merck KGaA). After
washing, screening and removal of cumulus cells, the zygotes were
transferred into M16 medium (Sigma-Aldrich; Merck KGaA) prior to
microinjection. Linear DNA constructs (1.8 kbp) prepared as
aforementioned were injected into the pronuclei of zygotes using
micromanipulator (Narishige Group). After ~20–30 min rest in M16,
zygotes (20–30 each) were transferred into the oviducts of
pseudopregnant C57BL/6J mice prepared beforehand. Integration of
the transgene was determined by a double check of
transgene-specific PCRs with genomic DNA isolated from tail
biopsies of the progenies. To distinguish genomic CXCL16 from
transgenic CXCL16, special upstream and downstream primers were
located in SR-promoter and CXCL16-flag respectively. The primer
sequences (purchased from Sangon Biotech Co., Ltd.) were given in
Table I.
 | Table I.Primer sequences used. |
Table I.
Primer sequences used.
| Name | Sequence |
|---|
| CXCL16-FP |
5′-TAAAGCGGCCTAAATGGGGTG-3′ |
| CXCL16-RP |
5′-CAAGAAAAGGAAGAACGCAAGAGA-3′ |
| β-Actin-FP |
5′-CATCCTGCGTCTGGACCTGG-3′ |
| β-Actin-RP |
5′-TAATGTCACGCACGATTTCC-3′ |
Transgenic mice strain generation
PCR positive offspring were mated with wild-type
C57BL/6J mice according to female/male ratio (1:1 or 1:2). To
generate homozygous transgenic strain, sib-mating was applied most
of the time and backcrossing when necessary. Genomic DNA was
extracted from the descendants to sequence for the verification of
transgene presence. PCR was used as a screen method. When all
progenies (n>5) of two consecutive generations were all
homozygous, the mouse was considered homozygous. After 10
generations of selective breeding, a
CXCL16+/+ transgenic line was successfully
established. In total, 16 CXCL16+/+ C57BL/6J
mice from F11 combined with 16 wild-type C57BL/6J mice were chosen
as subjects.
Treatment
CXCL16+/+ and wild-type mice
were randomly selected and divided into two treatment groups. One
group was fed with high-fat diet (HFD, 67.5% standard chow, 15%
lard, 10% yolk powder, 3% cholesterol, 4% whole milk powder and
0.5% Sodium cholate; Amresco, LLC) and the other standard chow,
termed low-fat diet (LFD) for 18 weeks (23–25).
Body weight, plasma total cholesterol (TC), triglycerides (TG),
low-density lipoprotein (LDL), high-density lipoprotein (HDL) were
measured at week 18. Mice in each group were intraperitoneally
injected with sodium pentobarbital (50 mg/kg) and were sacrificed
by cervical dislocation followed by decapitation.
ELISA
ELISA kits for Mouse CXCL16 (cat. no. 10030301;
Cusabio Technology LLC), Mouse matrix metalloproteinase-9 (MMP-9;
cat. no. EK1462), monocyte chemoattractant protein-1 (MCP-1; cat.
no. EK1351) and intercellular adhesion molecule-1 (ICAM-1; cat. no.
EK1726; Wuhan Boster Biological Technology, Ltd.) were used
according to the manufacturer's protocols. Absorbance was measured
at 450 nm using an 800 TS Absorbance Reader (BioTek Instruments,
Inc.). ELISA calc software (BioTNT; V0.1) was used to draw standard
curve and calculate concentration.
Hematoxylin-eosin (HE) staining and
immunohistochemistry
Biopsies were obtained from the aortic root and
artery sinus and were fixed overnight in 4% formaldehyde solution
at room temperature. Aortic root HE staining was performed to
examine the degree of fatty acid infiltration among the groups. The
sections were stained in hematoxylin solution for 10 min and eosin
solution for 1 min at room temperature. Immunohistochemical
staining of the lesions was performed at room temperature for 1 h
using rabbit polyclonal MMP-9 antibody (1:1,000; cat. no. ab38898),
rabbit polyclonal MCP-1 antibody (1:1,000; cat. no. ab73680) (both
Abcam) and rabbit polyclonal ICAM-1 antibody (1:200; cat. no.
PB9081; Wuhan Boster Biological Technology, Ltd.).
Statistical analysis
Each experiment was repeated in triplicate. Data
analysis was performed with SPSS v17.0 (SPSS, Inc.) and presented
as the mean ± standard deviation. Results were determined by
unpaired Student's t test and one-way ANOVA (post hoc test: Tukey's
multiple comparison test) or two-way ANOVA (post hoc test:
Bonferroni). P<0.05 was considered to indicate a statistically
significant difference. All diagrams were generated by GraphPad
Prism7 (GraphPad Software, Inc.).
Results
Transgene construct
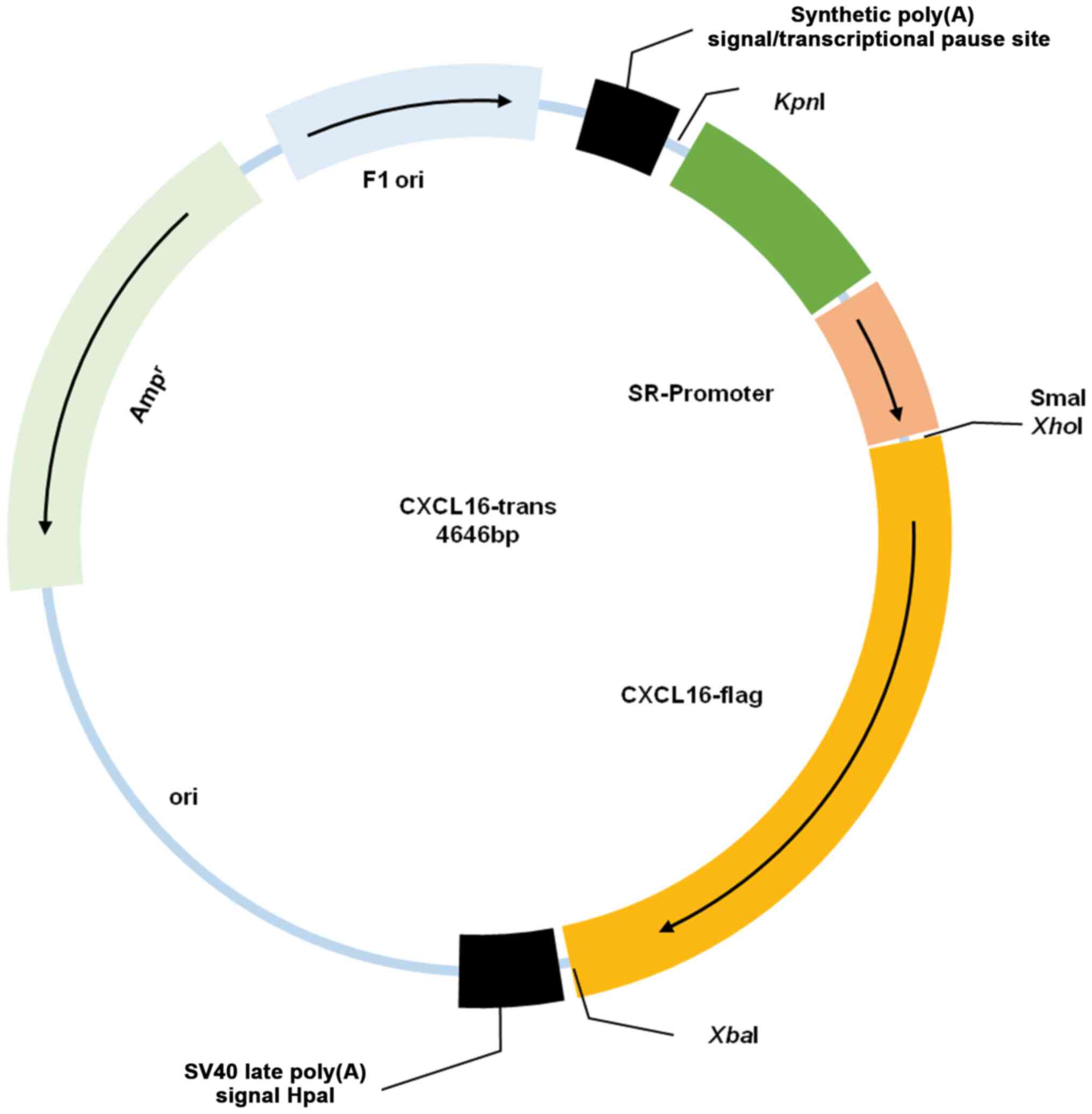
Following several rounds of insertion, a plasmid
with the backbone of pGL3-basic, SR-enhancer, SR-promoter, CXCL16
CDS and flag was successfully constructed (Fig. 1) and confirmed by sequencing.
CXCL16+/+
transgenic mice
A total of 426 microinjected eggs were obtained and
20 eggs as one unit were transferred to oviducts of 20 pseudo
mothers. Among which 12 became pregnant and 171 young were born.
Following PCR screening, 26 were proved to be carriers of the
target gene. After 4 weeks, another double-blind PCR was performed
on 26 transgenic and four wild-type mice, and confirmed the
stability of transgene expression. Founder mice were mated with
wild-type C57BL/6J mice. The PCR-positive offspring were then mated
with positive siblings in order to obtained homozygous transgenic
mice. When all progenies (n>5) of two consecutive generations
were all homozygous, the parent mice were considered homozygous.
F8-1–2 (female) and F9-2–3 (male) were thus proved homozygous and,
when mated with wild type mice, their offspring were all positive.
Backcross F8-1–2 with F9-2-3, 5 descendants were obtained and
termed C10-1–1 to C10-1-5. Sib-mate C10-1 and 19 descendants were
obtained and eight were randomly chosen as
CXCL16+/+ transgenic subject mice. In
addition, eight from the 11th generation of wild-type mice were
randomly selected as subjects.
Transgene expression in
CXCL16+/+ transgenic mice
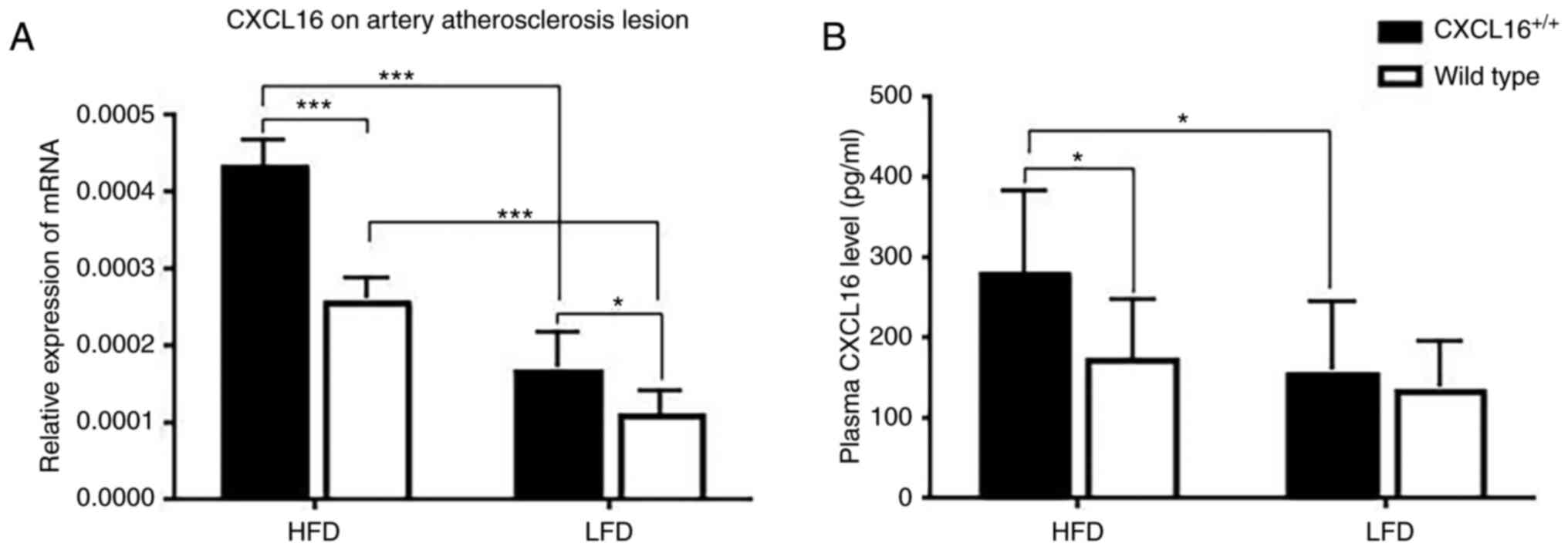
CXCL16 mRNA expression on artery AS lesion and
plasma protein expression was measured at week 18. In the two HFD
groups or the two LFD groups, CXCL16+/+
transgenic mice had a higher CXCL16 mRNA expression (Fig. 2A). CXCL16 mRNA expression of
CXCL16+/+ transgenic mice or wild-type mice
fed with HFD was significantly higher than those fed with LFD
(Fig. 2A). CXCL16 plasma protein
expression was result similar to CXCL16 mRNA expression on artery
AS lesion (Fig. 2B).
Plasma lipid profile and visceral fat
accumulation
Body weight and plasma lipid profile were measured
at the end of the week 18. No significant difference was observed
between the two LFD groups, but the average body weight of
CXCL16+/+ transgenic mice fed with HFD was
significantly highly (Fig. 3A)
compared with that of wild-type mice fed with the same regimen.
Similar results were observed in plasma TC level, LDL-cholesterol
(LDL-C) level, non HDL-C level and TC/HDL-cholesterol (HDL-C) ratio
(Fig. 3B-E). Another significant
change was identified between CXCL16+/+
transgenic mice fed with HFD and LFD. When fed with HFD,
CXCL16+/+ transgenic mice tend to maintain
much higher plasma lipoprotein levels such as LDL-C and HDL-C
compared with the LFD groups (Fig. 3C
and F), even when no significant differences were observed
between the expression levels of the wild-type mice groups. This
suggested that CXCL16 exerted an effect on fatty acid metabolism,
but it was only significant when a higher intake of fatty acid was
present.
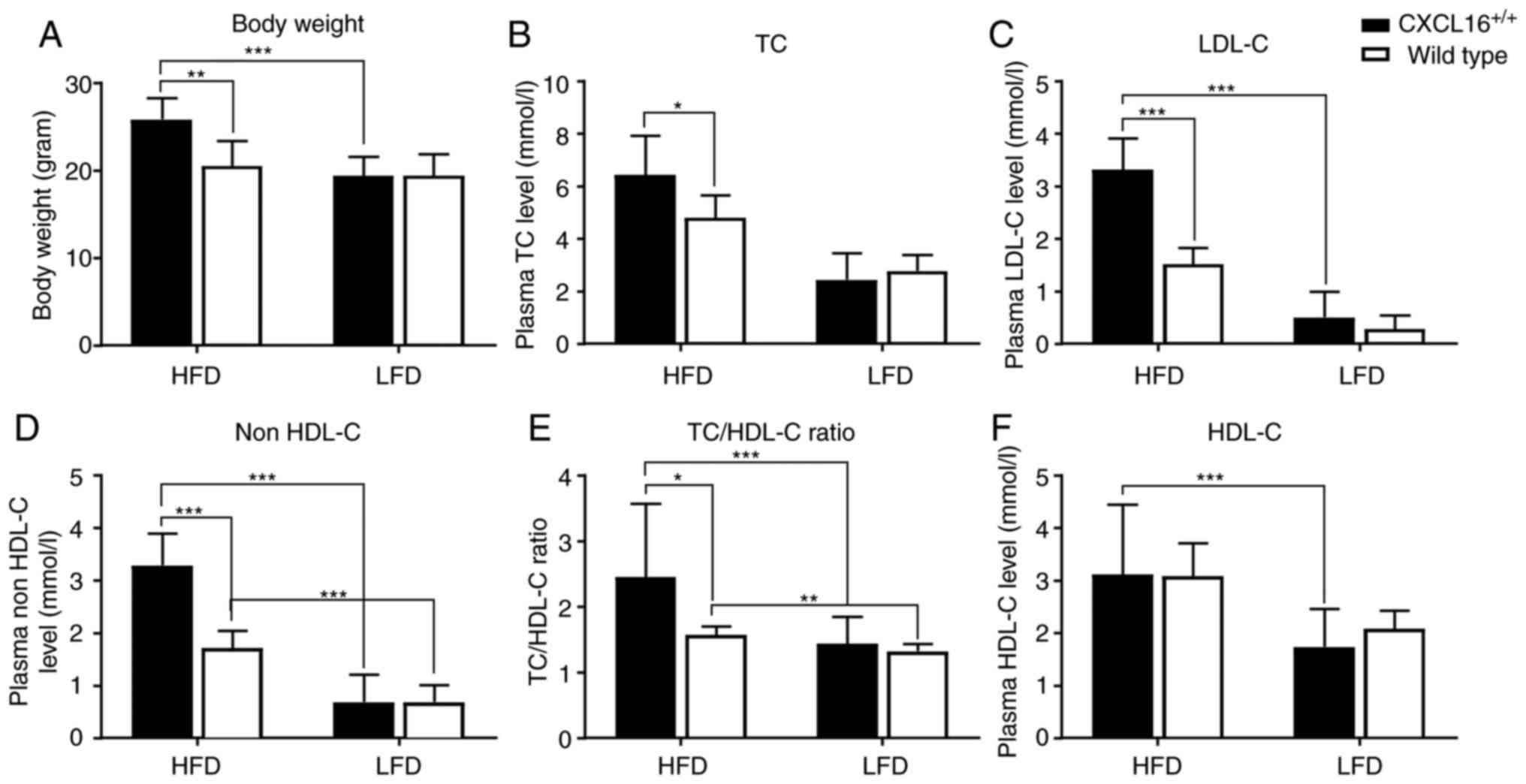 | Figure 3.Body weights and plasma lipid
profiles in different groups. (A) Body weights and plasma level of
(B) TC, (C) LDL-C, (D) non HDL-C, (E) TC/HDL-C ratio and (F) HDL-C.
All data are expressed as the mean ± standard deviation and
analyzed by one-way or two-way analysis of variance (n=8).
*P<0.05, **P<0.01, ***P<0.001. TC, total cholesterol;
LDL-C, low density lipoprotein-cholesterol; HDL-C, high density
lipoprotein-cholesterol; HFD, high-fat diet; LFD, low-fat diet. |
Visceral fat accumulation was recorded in the form
of images (data not shown). Fibrosis status was determined
according to the Ishak staging system (26). Table
II shows the percentage of mice with different degree of liver
disease; after 18 weeks of HFD, 100% of mice with
CXCL16+/+ developed liver diseases, while
only 50% of wild-type mice in HFD did. The degree of fibrosis
status was also relatively less. None of mice in the LFD groups
developed fatty liver disease.
| Table II.Counts of mice with liver disease in
each experimental group (n=8). |
Table II.
Counts of mice with liver disease in
each experimental group (n=8).
| Group | n | Cirrhosis | Fatty liver | Healthy liver | Nos. of livers with
pathological changes | Percentage |
|---|
| HDF
CXCL16+/+ | 8 | 4 | 4 | 0 | 8 | 100 |
| HDF wild-type | 8 | 0 | 4 | 4 | 4 | 50 |
| LDF
CXCL16+/+ | 8 | 0 | 0 | 8 | 8 | 0 |
| LDF wild-type | 8 | 0 | 0 | 8 | 0 | 0 |
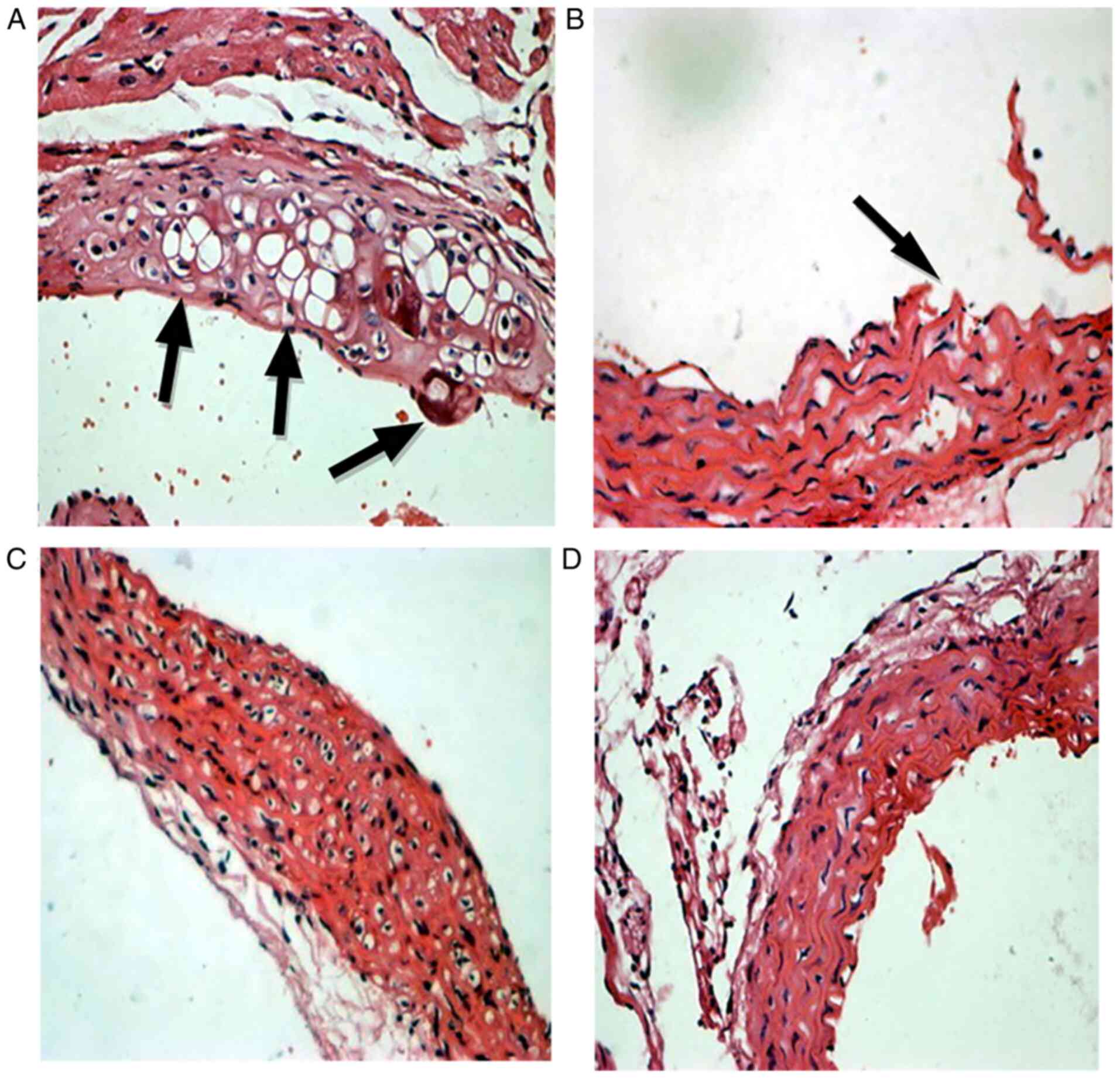
Aortic root HE staining
For aortic root HE staining, CXCL16 double positive
mice in the HFD groups demonstrated the most marked contrast
compared with healthy aorta (Fig.
4).
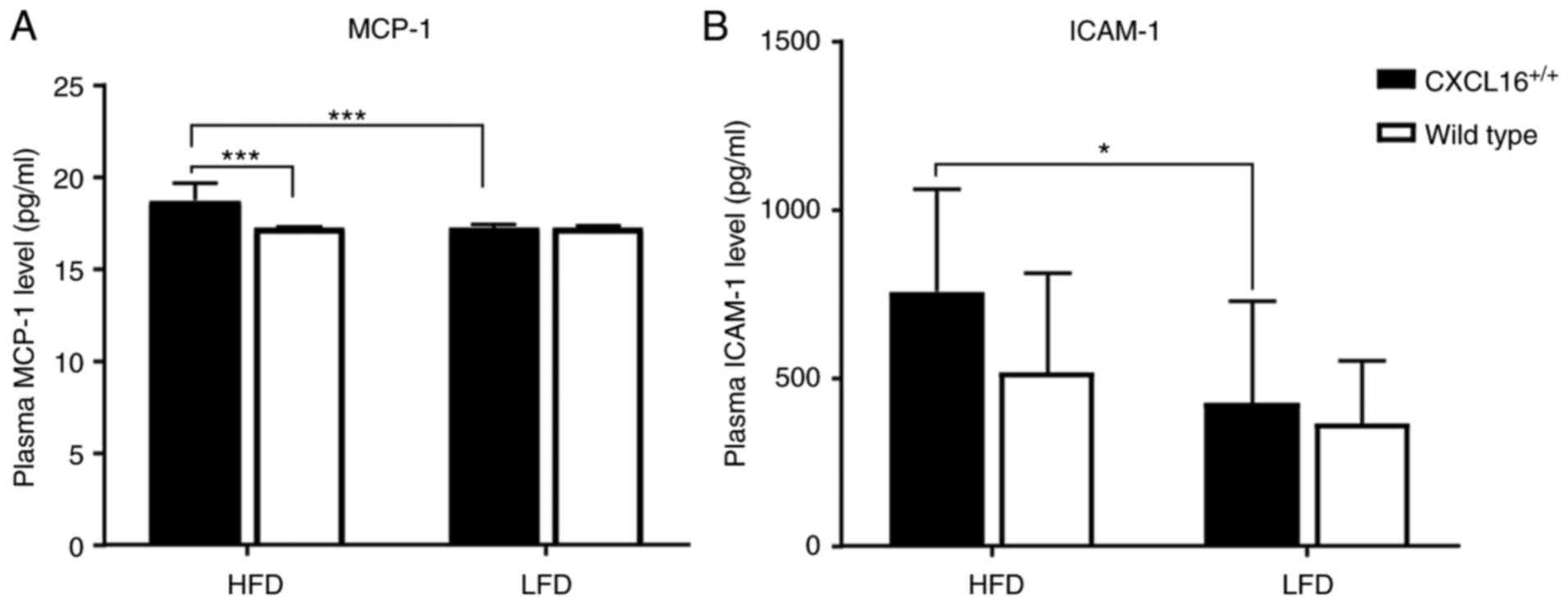
Plasma level of MCP-1 and ICAM-1
Plasma MCP-1 and ICAM-1 expression levels were
determined by ELISA at the end of the week 18. Fig. 5A demonstrates the overall expression
level of MCP-1 in the four groups. MCP-1 expression pattern among
the LFD groups showed no observable discrepancy (Fig. 5A) while among the HFD groups, a
significant difference was observed; with a higher expression of
CXCL16, MCP-1 expression levels were also higher (Fig. 5A).
ICAM-1 expression levels showed an incremental
gradient from LDF wild-type, LDF CXCL16+/+,
HDF wild-type to HDF CXCL16+/+. This had no
statistical significance except for the difference between
CXCL16+/+ transgenic mice fed with HFD and LFD (Fig. 5B).
Artery sinus expression of MCP-1 and
ICAM-1
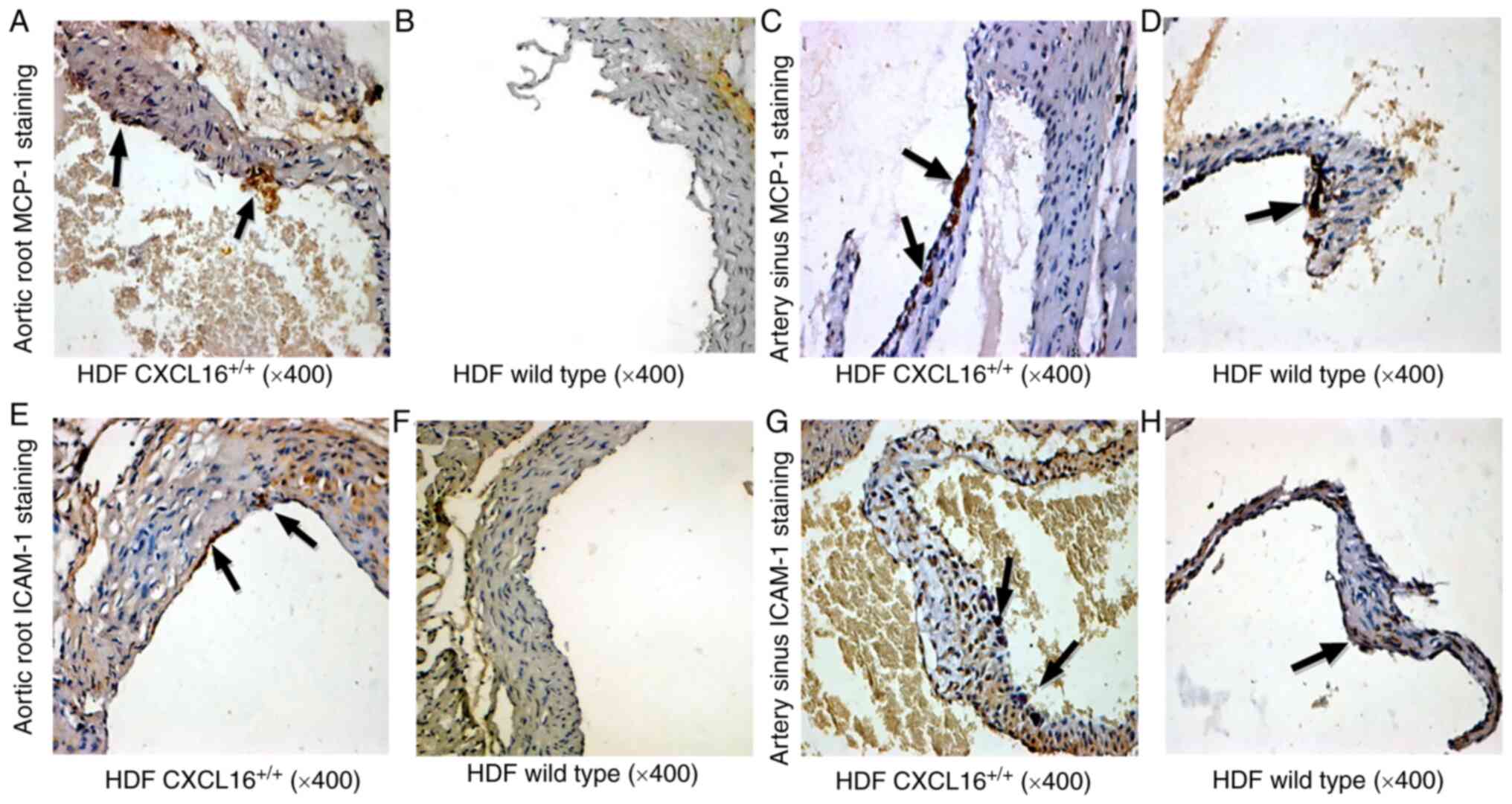
Immunohistochemistry staining also indicated a
higher local expression of MCP-1 and ICAM-1 in transgenic mice at
the aortic root and artery sinus (Fig.
6). Positive MCP-1 signals were seen within the AS lesions,
while accumulation of ICAM-1 was mainly located on the aortic
intima surface (Fig. 6E-H). An
overall higher expression of MCP-1 and ICAM-1 was observed at the
artery sinus compared with the aortic root (Fig. 6).
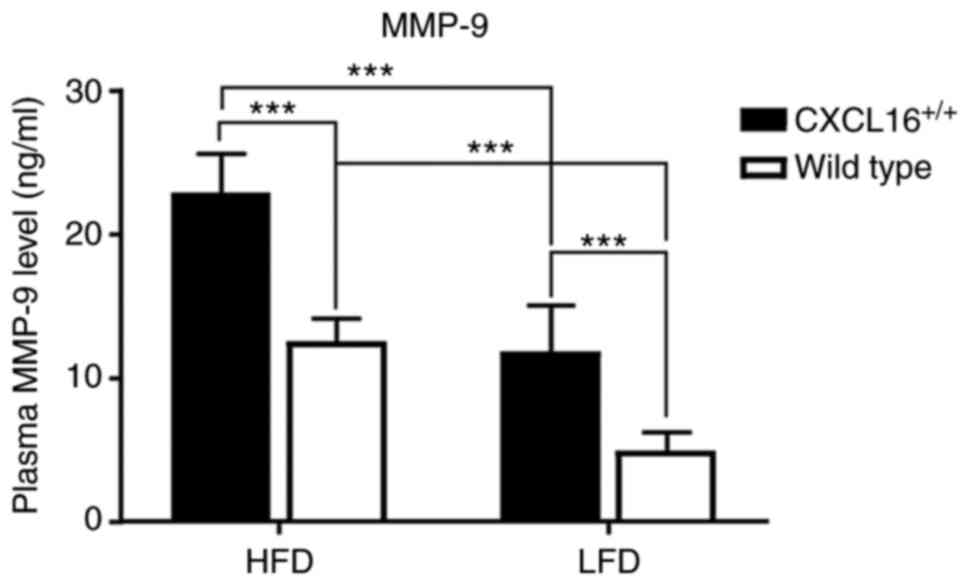
Plasma level of MMP-9
Plasma MMP-9 level was tested at the end of the week
18. It showed high specificity among the four groups. HDF
CXCL16+/+ mice had the highest plasma level
of all, and it was statistically significant when compared with HDF
wild-type or LDF CXCL16+/+ mice (Fig. 7). Additionally, LDF
CXCL16+/+ mice also expressed a higher level
of plasma MMP-9 than LDF wild-type mice (Fig. 7).
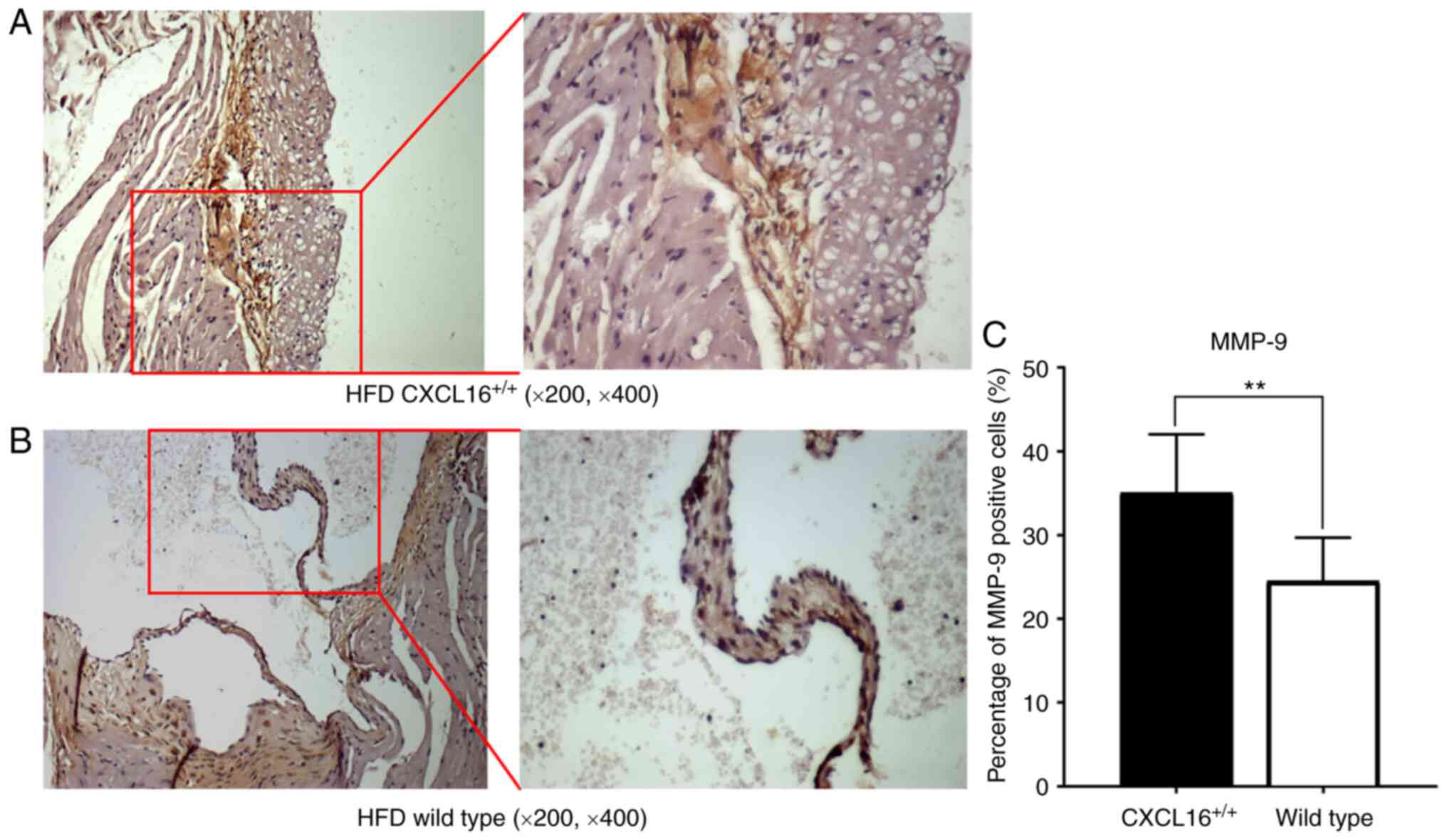
Artery sinus MMP-9 expression
Local expression of MMP-9 in artery sinus was also
tested among the HFD groups (Fig. 8A
and B). Atherosclerotic plaques of
CXCL16+/+ mice possessed a higher percentage
of cells that were MMP-9 positive (Fig.
8C).
Discussion
Growing attention has been paid to chemokines
involved in the pathogenesis of AS (27). CXCL16 is one of those chemokines,
although a large-scale review of overexpression of CXCL16 in
transgenic mice is still required. CXCL16 is specifically expressed
in arteries with AS and is mainly undetectable in normal arteries
(14). The present study was the
first, to the best of the authors' knowledge, to examine how
overexpression of CXCL16 directs the progress of AS under LFD and
HFD. Although apolipoprotein E (ApoE) knockout mice are used by
numerous research groups for AS changes, they possess some
limitations (28,29). ApoE is a multifunctional protein
that has an effect on inflammation, oxidation, lipid transport and
AS pathogenesis (30). These
functions might affect atherosclerotic plaque development in
ApoE−/− mice, independent of plasma lipid
levels (29). It has been reported
that HFD for 10–20 weeks can cause typical AS lesions in the aortic
sinus valve of mice, which are mainly filled with foam cells and
lipid streaks (23,31). The present study identified that a
high expression level of CXCL16 combined with an HFD presented a
strong pro-atherogenesis ability. Blood lipid levels have a
regulatory effect on the expression of CXCL16. A higher level of
lipid profile was identified in CXCL16+/+
transgenic mice fed with HFD and greater fatty acid accumulation
was observed in the liver and the artery wall. Higher plasma levels
of AS markers and denser foci expression of risk indicators were
detected, while the levels in transgenic mice fed with LFD showed
no significant difference with its corresponding control. It was a
limitation to the present study that oil Red O staining was not
performed to check the formation of lipid plaques and to observe
the changes in foam cells. The observation of changes in plaques
and fibrous caps will be added in future studies, which may further
illustrate this part of the results.
HDF CXCL16+/+ mice expressed a
significant high level of pro-inflammatory or pro-AS markers while
the other three groups retained a relatively similar low level of
such markers, suggesting that both HFD and high CXCL16 level were
prerequisite for a severer degree of AS to occur. Overexpression of
the CXCL16 gene in transgenic mice did have a pro-inflammatory
effect, but not as great as the HFD. When an HFD was combined with
high CXCL16 expression, it showed a predisposing effect greater
than either of these factors (HFD or CXCL 16 overexpression) alone.
As adipose tissue is a potential origin for inflammation, this may
suggest that CXCL16 alone is not enough to initiate inflammation.
However, CXCL16 when initiated is a potent booster of inflammation
and, furthermore, AS.
MCP-1 and ICAM-1 are involved mainly in the
initiation of atherosclerotic plaques (32). MCP-1 has a definite
pro-atherogenesis effect via its potent chemotactic ability for
monocytes and also helps them through endothelial cells by
diapedesis (33). MCP-1 is
predominantly expressed in macrophages, smooth muscle cells and
endothelial cells (34). It induces
transendothelial migration of monocytes so as to facilitate
atherosclerotic lesion formation (35). Absence of MCP-1 results in a milder
extent of AS. Modified LDL, local fluid stress or short-term of
high glucose exposure may all be stimulants to upregulate MCP-1
expression (36–38). The present study proposed MCP-1 as
an early stage in indicator of AS.
ICAM-1 by contrast, is an adhesion molecule.
Adhesion molecules represent another group of factors closely
linked with inflammation and AS (39). They are upregulated in the regions
predisposed to atherosclerotic lesion. Deficiency of certain
adhesion molecules exert a protective effect from atheroma
formation (40,41). Among adhesion molecules, ICAM-1 and
vascular cell adhesion protein 1 (VCAM-1) are upregulated in
atherosclerotic plaques (32,43),
while ICAM-1 is a more responsive marker to diet cholesterol
compared with VCAM-1, which remains relatively stable during or
after a high cholesterol diet (44). Thus, the present study proposed
ICAM-1 as a marker representing adhesion molecules in AS.
Under the combined action of elevated blood lipids
and CXCL16 overexpression, the expression levels of MCP-1 and
ICAM-1 in the aortic root and artery sinus of transgenic mice with
HFD were significantly higher compared with those of wild-type mice
with HFD, suggesting that they may have a common regulatory pathway
under the influence of CXCL16 overexpression. The distribution of
inflammatory factors on the tissue surface is consistent with the
location of the initial stage of AS. Overexpression of CXCL16 in
ApoE knockout mice can also promote the expression of MCP-1 mRNA in
arterial plaques, but its mechanism or signal pathway remain to be
elucidated (45). These results of
the present study can also indirectly support the hypothesis that
CXCL16 has the effect of promoting AS lesions, which supports the
views of Wuttge et al (46).
In a future study, additional markers in CXCL16
inflammation-related pathways need to be investigated to further
explain the process of promoting AS lesions.
MMP-9 is one of the zinc-dependent endopeptidases
and has a broad spectrum of substrates including a full length
interstitial collagens, certain cytokine and adhesion molecules
(47–49). MMP-9 is closely involved in AS in
that it can regulate inflammatory processes by proteolyzing
chemokines (50,51) or releasing chemokines from their
functional location (50). MMP-9
may also serve a role in macrophage migration (50). Another function, which is also the
main role it serves in AS, is that MMP-9 affects plaque stability
(52). Clinical and cell
experimental studies have shown that a high level of MMP-9
expression, active form foci or total plasma MMP-9 are correlated
with plaque vulnerability to rupture (53–56).
The present study proposed MMP-9 level as a marker for advanced AS
and its corresponding myocardial infarction. Following an HFD, all
mice had elevated peripheral blood MMP-9. Immunohistochemistry
showed that the expression of MMP-9 in the aortic sinus AS lesions
of transgenic mice was significantly increased. Unfortunately,
there was the lack of protein expression analysis of MCP-1, ICAM-1
and MMP-9 using western blot analysis in the present study.
According to a pooled analysis conducted by NCD Risk
Factor Collaboration, the global body mass index has been
constantly increasing since 1975 (57). This adds an extra burden to the task
of artery disease control, although the pathological conditions in
the arterial wall are reversible before certain point of the
disease (58). More studies similar
to the present study could lead the way to a significant
elucidation of the mechanism of AS and may show the way to
prevention or cure.
In conclusion, the present study was the first, to
the best of the authors' knowledge, to establish a CXCL16
homozygous transgenic mice model to study how overexpressed CXCL16
is associated with AS. It is hypothesized that overexpression of
CXCL16 combined with HFD may promote the AS lesions by upregulating
the aforementioned inflammatory-related genes at a protein level.
Overexpression of CXCL16 may lead to high blood lipids and strong
inflammatory response. Whether blocking the expression of CXCL16
could reduce the local inflammatory response in AS, thereby
intervening in the occurrence and development of AS, remains to be
elucidated.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
JZ and JW conceived and designed the experiments.
JZ, MY and JW performed the experiments. JZ and JW confirm the
authenticity of all the raw data. JZ and JW analyzed the data. JW
wrote the manuscript. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
All animal experiments were performed according to
the guidelines of the Chinese Experimental Animals Administration
Legislation and were approved by the Ethics Committee for Animal
Care and Use of Shandong Academy of Medical Sciences.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Wang HH, Garruti G, Liu M, Portincasa P
and Wang DQ: Cholesterol and lipoprotein metabolism and
atherosclerosis: Recent advances in reverse cholesterol transport.
Ann Hepatol. 16 (Suppl 1):S27–S42. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Galkina E and Ley K: Immune and
inflammatory mechanisms of atherosclerosis (*). Annu Rev Immunol.
27:165–197. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Toth PP: Subclinical atherosclerosis: What
it is, what it means and what we can do about it. Int J Clin Pract.
62:1246–1254. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ambrose JA and Barua RS: The
pathophysiology of cigarette smoking and cardiovascular disease: An
update. J Am Coll Cardiol. 43:1731–1737. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Balistreri CR, Caruso C and Candore G: The
role of adipose tissue and adipokines in obesity-related
inflammatory diseases. Mediators Inflamm. 2010:8020782010.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Gustafson B: Adipose tissue, inflammation
and atherosclerosis. J Atheroscler Thromb. 17:332–341. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wannamethee SG, Lowe GDO, Shaper AG,
Rumley A, Lennon L and Whincup PH: Associations between cigarette
smoking, pipe/cigar smoking, and smoking cessation, and haemostatic
and inflammatory markers for cardiovascular disease. Eur Heart J.
26:1765–1773. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Mazurek T, Zhang L, Zalewski A, Mannion
JD, Diehl JT, Arafat H, Sarov-Blat L, O'Brien S, Keiper EA, Johnson
AG, et al: Human epicardial adipose tissue is a source of
inflammatory mediators. Circulation. 108:2460–2466. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Andersen T, Ueland T, Ghukasyan Lakic T,
Åkerblom A, Bertilsson M, Aukrust P, Michelsen AE, James SK, Becker
RC, Storey RF, et al: C-X-C ligand 16 is an independent predictor
of cardiovascular death and morbidity in acute coronary syndromes.
Arterioscler Thromb Vasc Biol. 39:2402–2410. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Bakogiannis C, Sachse M, Stamatelopoulos K
and Stellos K: Platelet-derived chemokines in inflammation and
atherosclerosis. Cytokine. 122:1541572019. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ma A, Pan X, Xing Y, Wu M, Wang Y and Ma
C: Elevation of serum CXCL16 level correlates well with
atherosclerotic ischemic stroke. Arch Med Sci. 10:47–52. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Cybulsky MI, Iiyama K, Li H, Zhu S, Chen
M, Iiyama M, Davis V, Gutierrez-Ramos JC, Connelly PW and Milstone
DS: A major role for VCAM-1, but not ICAM-1, in early
atherosclerosis. J Clin Invest. 107:1255–1262. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ludwig A, Schulte A, Schnack C, Hundhausen
C, Reiss K, Brodway N, Held-Feindt J and Mentlein R: Enhanced
expression and shedding of the transmembrane chemokine CXCL16 by
reactive astrocytes and glioma cells. J Neurochem. 93:1293–1303.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Hofnagel O, Engel T, Severs NJ, Robenek H
and Buers I: SR-PSOX at sites predisposed to atherosclerotic lesion
formation mediates monocyte-endothelial cell adhesion.
Atherosclerosis. 217:371–378. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Lehrke M, Millington SC, Lefterova M,
Cumaranatunge RG, Szapary P, Wilensky R, Rader DJ, Lazar MA and
Reilly MP: CXCL16 is a marker of inflammation, atherosclerosis, and
acute coronary syndromes in humans. J Am Coll Cardiol. 49:442–449.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Holmøy T, Løken-Amsrud KI, Bakke SJ,
Beiske AG, Bjerve KS, Hovdal H, Lilleås F, Midgard R, Pedersen T,
Šaltytė Benth J, et al: Inflammation markers in multiple sclerosis:
CXCL16 reflects and may also predict disease activity. PLoS One.
8:e750212013. View Article : Google Scholar
|
|
17
|
Canton J, Neculai D and Grinstein S:
Scavenger receptors in homeostasis and immunity. Nat Rev Immunol.
13:6212013. View
Article : Google Scholar : PubMed/NCBI
|
|
18
|
Sheikine Y and Sirsjö A: CXCL16/SR-PSOX-a
friend or a foe in atherosclerosis? Atherosclerosis. 197:487–495.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Sheikine Y, Bang CS, Nilsson L, Samnegård
A, Hamsten A, Jonasson L, Eriksson P and Sirsjö A: Decreased plasma
CXCL16/SR-PSOX concentration is associated with coronary artery
disease. Atherosclerosis. 188:462–466. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
National Research Council (US) Committee
for the Update of the Guide for the Care and Use of Laboratory
Animals, . Guide for the Care and Use of Laboratory Animals. 8th
edition. National Academies Press (US); Washington, DC: 2011
|
|
21
|
Crossley BM, Bai J, Glaser A, Maes R,
Porter E, Killian ML, Clement T and Toohey-Kurth K: Guidelines for
Sanger sequencing and molecular assay monitoring. J Vet Diagn
Invest. 32:767–775. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Thouas GA, Trounson AO and Jones GM:
Effect of female age on mouse oocyte developmental competence
following mitochondrial injury. Biol Repord. 73:366–373. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Stewart-Phillips JL, Lough J and Skamene
E: Genetically determined susceptibility and resistance to
diet-induced atherosclerosis in inbred strains of mice. J Lab Clin
Med. 112:36–42. 1988.PubMed/NCBI
|
|
24
|
Zhou YG, Lan XH, Li X and Xu R:
Establishment and evaluation of an atherosclerosis model in rats.
Pharm J Chin PLA. 27:399–403. 2011.
|
|
25
|
Wang W, Zhang ZZ, Wu Y, Wang RQ, Chen JW,
Chen J, Zhang Y, Chen YJ, Geng M, Xu ZD, et al:
(−)-Epigallocatechin-3-gallate ameliorates atherosclerosis and
modulates hepatic lipid metabolic gene expression in apolipoprotein
E knockout mice: Involvement of TTC39B. Front Pharmacol. 9:1952018.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ishak K, Baptista A, Bianchi L, Callea F,
De Groote J, Gudat F, Denk H, Desmet V, Korb G, MacSween RN, et al:
Histological grading and staging of chronic hepatitis. J Hepatol.
22:696–699. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Munjal A and Khandia R: Atherosclerosis:
Orchestrating cells and bimolecules involved in its activation and
inhibition. Adv Protein Chem Struct Biol. 120:85–122. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Emini Veseli B, Perrotta P, De Meyer GRA,
Roth L, Van der Donckt C, Martinet W and De Meyer GRY: Animal
models of atherosclerosis. Eur J Pharmacol. 816:3–13. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Getz GS and Reardon CA: Apoprotein E as a
lipid transport and signaling protein in the blood, liver, and
artery wall. J Lipid Res. 50 (Suppl):S156–S161. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Zhang HL, Wu LM and Wu J: Cross-talk
between apolipoprotein E and cytokines. Mediators Inflamm.
2011:9490722011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Yang XY, Yang YZ, Tan JM, Yuan ZH and Wang
ZY: The estbablishment of an Atheroscleroti murine model and
pathologieal observation of plaques by laser seaning confoeal
mieroseopy. Chin J Arterioseler. 4:54–57. 1996.
|
|
32
|
Roebuck KA: Oxidant stress regulation of
IL-8 and ICAM-1 gene expression: Differential activation and
binding of the transcription factors AP-1 and NF-kappaB (Review).
Int J Mol Med. 4:223–230. 1999.PubMed/NCBI
|
|
33
|
Gu L, Okada Y, Clinton SK, Gerard C,
Sukhova GK, Libby P and Rollins BJ: Absence of monocyte
chemoattractant protein-1 reduces atherosclerosis in low density
lipoprotein receptor-deficient mice. Mol Cell. 2:275–281. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Yadav A, Saini V and Arora S: MCP-1:
Chemoattractant with a role beyond immunity: A review. Clin Chim
Acta. 411:1570–1579. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Kanda H, Tateya S, Tamori Y, Kotani K,
Hiasa K, Kitazawa R, Kitazawa S, Miyachi H, Maeda S, Egashira K and
Kasuga M: MCP-1 contributes to macrophage infiltration into adipose
tissue, insulin resistance, and hepatic steatosis in obesity. J
Clin Invest. 116:1494–1505. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Cushing SD, Berliner JA, Valente AJ,
Territo MC, Navab M, Parhami F, Gerrity R, Schwartz CJ and Fogelman
AM: Minimally modified low density lipoprotein induces monocyte
chemotactic protein 1 in human endothelial cells and smooth muscle
cells. Proc Natl Acad Sci USA. 87:5134–5138. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Shyy YJ, Hsieh HJ, Usami S and Chien S:
Fluid shear stress induces a biphasic response of human monocyte
chemotactic protein 1 gene expression in vascular endothelium. Proc
Natl Acad Sci USA. 91:4678–4682. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Piga R, Naito Y, Kokura S, Handa O and
Yoshikawa T: Short-term high glucose exposure induces
monocyte-endothelial cells adhesion and transmigration by
increasing VCAM-1 and MCP-1 expression in human aortic endothelial
cells. Atherosclerosis. 193:328–334. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Zhu Y, Xian X, Wang Z, Bi Y, Chen Q, Han
X, Tang D and Chen R: Research progress on the relationship between
atherosclerosis and inflammation. Biomolecules. 8:802018.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Preiss DJ and Sattar N: Vascular cell
adhesion molecule-1: A viable therapeutic target for
atherosclerosis? Int J Clin Pract. 61:697–701. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Marzolla V, Armani A, Mammi C, Moss ME,
Pagliarini V, Pontecorvo L, Antelmi A, Rosano G, Jaffe IZ and
Caprio M: Essential role of ICAM-1 in aldosterone-induced
atherosclerosis. Int J Cardiol. 232:233–242. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Nakashima Y, Raines EW, Plump AS, Breslow
JL and Ross R: Upregulation of VCAM-1 and ICAM-1 at
atherosclerosis-prone sites on the endothelium in the
ApoE-deficient mouse. Arterioscler Thromb Vasc Biol. 18:842–851.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Iiyama K, Hajra L, Iiyama M, Li H,
DiChiara M, Medoff BD and Cybulsky MI: Patterns of vascular cell
adhesion molecule-1 and intercellular adhesion molecule-1
expression in rabbit and mouse atherosclerotic lesions and at sites
predisposed to lesion formation. Circ Res. 85:199–207. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Fotis L, Agrogiannis G, Vlachos IS,
Pantopoulou A, Margoni A, Kostaki M, Verikokos C, Tzivras D,
Mikhailidis DP and Perrea D: Intercellular adhesion molecule
(ICAM)-1 and vascular cell adhesion molecule (VCAM)-1 at the early
stages of atherosclerosis in a rat model. In Vivo. 26:243–250.
2012.PubMed/NCBI
|
|
45
|
Yi GW, Zeng QT, Mao XB, Cheng M, Yang XF,
Liu HT, Mao Y, Guo M, Ji QW and Zhong YC: Overexpression of CXCL16
promotes a vulnerable plaque phenotype in apolipoprotein E-knockout
mice. Cytokine. 53:320–326. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Wuttge DM, Zhou X, Sheikine Y, Wågsäter D,
Stemme V, Hedin U, Stemme S, Hansson GK and Sirsjö A:
CXCL16/SR-PSOX is an interferon-gamma-regulated chemokine and
scavenger receptor expressed in atherosclerotic lesions.
Arterioscler Thromb Vasc Biol. 24:750–755. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Vandooren J, Van den Steen PE and
Opdenakker G: Biochemistry and molecular biology of gelatinase B or
matrix metalloproteinase-9 (MMP-9): The next decade. Crit Rev
Biochem Mol Biol. 48:222–272. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Patterson J and Hubbell JA: Enhanced
proteolytic degradation of molecularly engineered PEG hydrogels in
response to MMP-1 and MMP-2. Biomaterials. 31:7836–7845. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Lindsey ML and Zamilpa R: Temporal and
spatial expression of matrix metalloproteinases and tissue
inhibitors of metalloproteinases following myocardial infarction.
Cardiovasc Ther. 30:31–41. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Van Lint P and Libert C: Chemokine and
cytokine processing by matrix metalloproteinases and its effect on
leukocyte migration and inflammation. J Leukoc Biol. 82:1375–1381.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Gong Y, Hart E, Shchurin A and Hoover-Plow
J: Inflammatory macrophage migration requires MMP-9 activation by
plasminogen in mice. J Clin Invest. 118:3012–3024. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Li F, Sun XJ, Xie H, Wang Y and Zhang YK:
The relationship between chronic periodontitis and the instability
of carotid atherosclerotic plaque by serum level of MMP-9, MCP-1
and MMP-7. Shanghai Kou Qiang Yi Xue. 24:589–593. 2015.(In
Chinese). PubMed/NCBI
|
|
53
|
Loftus IM, Naylor AR, Goodall S, Crowther
M, Jones L, Bell PR and Thompson MM: Increased matrix
metalloproteinase-9 activity in unstable carotid plaques. A
potential role in acute plaque disruption. Stroke. 31:40–47. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Rao VH, Kansal V, Stoupa S and Agrawal DK:
MMP-1 and MMP-9 regulate epidermal growth factor-dependent collagen
loss in human carotid plaque smooth muscle cells. Physiol Rep.
2:e002242014. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Huang Z, Wang L, Meng S, Wang Y, Chen T
and Wang C: Berberine reduces both MMP-9 and EMMPRIN expression
through prevention of p38 pathway activation in PMA-induced
macrophages. Int J Cardiol. 146:153–158. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Gostiljac D, Dorđević PB, Djurić D,
Peruničić J, Lasica R, Colak E, Canovic F, Srećković VD, Ilić M and
Obrenović R: The importance of defining serum MMP-9 concentration
in diabetics as an early marker of the rupture of atheromatous
plaque in acute coronary syndrome. Acta Physiol Hung. 98:91–97.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
NCD Risk Factor Collaboration (NCD-RisC),
. Trends in adult body-mass index in 200 countries from 1975 to
2014: A pooled analysis of 1698 population-based measurement
studies with 19·2 million participants. Lancet. 387:1377–1396.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Öörni K, Lehti S, Sjövall P and Kovanen
PT: Triglyceride-rich lipoproteins as a source of proinflammatory
lipids in the arterial wall. Curr Med Chem. 26:1701–1710. 2019.
View Article : Google Scholar
|