Introduction
Atrial natriuretic peptide (ANP) is a precursor
protein that is produced by atrial myocytes and then secreted into
the plasma. It is distributed in the heart, pituitary gland, lung,
adrenal gland, and other peripheral tissues and organs, and
possesses numerous bioactivities (1). For example, ANP acts as a diuretic,
natriuretic and vasodilator protein, stimulates vasodilation, fluid
egress, increased glomerular filtration and salt/water excretion
(2), and blocks the release and/or
actions of several hormones, including angiotensin II, aldosterone
and vasopressin (3). There have
been numerous reports demonstrating that ANP is a potential
candidate for the treatment of heart failure (4), nephrosis (5) and other conditions. In addition, ANP
has been reported to exert a protective effect on oxidant injury
(3,6,7).
Humans are frequently exposed to oxidative stress
and detrimental environmental factors, such as
H2O2, endotoxin, UV light and heavy metals.
Under such stressful conditions, oxidative stress is generated,
which disturbs the cellular processes, and as a consequence leads
to cell apoptosis, organ injury and disease (8,9). Cell
apoptosis has often been evaluated by the measurement of the
expression levels of several major apoptosis-associated proteins,
including Bcl-2, Bax and caspase-3. The Bcl-2 and caspase families
are the most common indices used in cell apoptosis research. Bcl-2
and Bax are the most important regulatory genes associated with
apoptosis, and have opposite functions; these genes ultimately
activate the caspase protein family and lead to chondrocyte
apoptosis (10). Bcl-2 inhibits
cell apoptosis and promotes cell survival, whereas Bax and
caspase-3 elicit apoptosis and evoke cell death by promoting the
release of cytochrome c (11). As a result, if the Bax/Bcl-2 ratio
is modulated it can affect activation of the intrinsic apoptotic
pathway. Notably, decreased Bax/Bcl-2 ratio and caspase-3 abrogated
cell apoptosis (12).
Under oxidative stress, cell-protective mechanisms
are activated to fight against oxidant injury and to protect cells.
Single cells and organisms may adapt to harmful oxidative stress
conditions via the activation of stress-related factors. Nuclear
factor erythroid 2-related factor 2 (Nrf2) is an important
transcription factor responsible for regulating the antioxidant
response (13,14). The multifunctional regulator Nrf2 is
regarded as a cell-protective factor that can regulate the
expression of anti-oxidant, anti-inflammatory and detoxifying
proteins. Activation of the Nrf2 pathway composes a cell-protective
system that improves cell viability under harmful conditions
(15,16). To some extent, the main
characteristics of Nrf2 are simulated by Nrf2-dependent genes and
associated proteins, such as heme oxygenase-1 (HO-1), which besides
removing toxic heme, produces biliverdin, iron ions and carbon
monoxide (17). HO-1 and its
products play a beneficial role in protecting against oxidant
injury, inhibiting cell death, modulating inflammation and other
cell toxicities (13,18–20).
Oxidative stress has been reported to be associated
with intervertebral disc degeneration (IDD) (21). The endplate plays a critical role in
biomechanical integrity and disc nutrition (22), and endplate chondrocytes (EPCs) are
vitally important to intervertebral discs under physiological and
pathological conditions (23).
Notably, although ANP has been reported to exert beneficial
effects, including protective effects against oxidant injury, to
the best of our knowledge, the protective effect of ANP on EPCs and
the associated mechanisms involved have not been reported and are
poorly understood. Therefore, on the basis of these reports, it was
hypothesized that ANP may serve an important protective role in
H2O2-induced EPC injury and apoptosis through
activation of the Nrf2/HO-1 signaling pathway.
Materials and methods
Drug and materials
ANP (purity, >99%) was obtained from National
Institutes for Food and Drug Control. Dimethyl sulfoxide (DMSO) was
purchased from Sigma-Aldrich; Merck KGaA. Brusatol (a specific Nrf2
inhibitor) was purchased from MedChemExpress. GAPDH (cat. no.
sc-365062), Bax (cat. no. sc-7480), Bcl-2 (cat. no. sc-7382),
Lamin-B1 (cat. no. sc-374015), Nrf2 (cat. no. sc-365949) and HO-1
(cat. no. sc-136960) antibodies were purchased from Santa Cruz
Biotechnology, Inc. Cleaved (C)-caspase-3 (cat. no. ab2302) was
obtained from Abcam. DAPI (cat. no. C1002) was obtained from
Beyotime Institute of Biotechnology.
Cell culture
EPCs were obtained from The Cell Bank of Type
Culture Collection of Chinese Academy of Sciences [EPCs were
differentiated from ATDC5 cell line (24) in The Cell Bank of Type Culture
Collection of Chinese Academy of Sciences]. It has been shown that
the ATDC5 cell line is a useful in vitro model for examining
the multistep differentiation of chondrocytes. Undifferentiated
ATDC5 cells proliferate rapidly until they reach confluence, at
which point they undergo growth arrest. When treated with insulin,
transferrin and sodium selenite, confluent ATDC5 cells re-enter a
proliferative phase and form cartilaginous matrix nodules (mature
chondrocytes) (24). The EPCs were
seeded into 10-cm culture plates at a density of 1×105
cells/plate. The complete medium was changed every other day, and
the first two and three passages of EPCs were used in subsequent
experiments. Cells were grown as monolayers in DMEM/F12 (Gibco;
Thermo Fisher Scientific, Inc.) supplemented with 10% heat
inactivated fetal bovine serum (Sigma-Aldrich; Merck KGaA) and
antibiotics (1% penicillin/streptomycin). Cells were incubated at
37°C in a humidified atmosphere containing 5% CO2.
Cell treatment
EPCs were treated with different concentrations of
H2O2 (50, 100, 200, 300 and 400 µM), and the
control (0 µM) group was treated with DMSO, for 48 h at 37°C.
Subsequently, ANP (300 nM) was administrated to
H2O2-treated cells for 48 h at 37°C. Blank
control cells were treated with DMSO (model group) and Saline
solution was applied to H2O2-treated cells
serving as a saline group. In addition, brusatol (0.3 µg/ml) was
used to treat H2O2-treated cells for 48 h at
37°C. Finally, brusatol (0.3 µg/ml) was applied in the ANP (300
nM)-treated cells for 48 h at 37°C, which served as the ANP +
Brusatol group.
Cell viability assay
As previously reported (25), the viability of EPCs was
investigated using the Cell Counting Kit-8 (CCK-8) (Dojindo
Molecular Technologies, Inc.) in accordance with the manufacturer's
protocol. Firstly, the EPCs (1×104 cells/well) were
seeded in 96-well plates and incubated at 37°C for 24 h. Cells were
then treated as aforementioned. At the predetermined time, the
cells were washed three times with PBS, and 100 µl DMEM/F12
containing 10% CCK-8 was added and incubated for another 2 h.
Finally, the absorbance was measured at 470 nm using a microplate
reader and the relative survival rate was calculated as follows:
Cell viability=absorbance of test cells/absorbance of blank control
cells. Each sample was performed in triplicate and the mean was
calculated.
TUNEL assay
The TUNEL method was used to measure cell apoptosis.
Previous studies have used the TUNEL method to determine cell
viability as described previously (26). VECTASHIELD mounting medium
containing DAPI (1.0 µg/ml; Vector Laboratories, Inc.) was used for
visualization of nuclei. EPCs cultured with
H2O2 with or without ANP were harvested after
12 h and transferred to six-well plates. Cells (1×104)
were then fixed in 4% paraformaldehyde (freshly prepared) for 15
min and incubated with 0.1% Triton X-100 for 10 min at 4°C. At each
step, cells were washed three times with aseptic PBS. Finally,
cells were incubated and stained with the TUNEL detection kit (cat
no. G7130; Promega Corporation) for 1 h at 37°C in the dark.
Apoptotic cells were detected using a fluorescence microscope
(Olympus Corporation) and counted in seven random fields.
Western blot analysis
The expression levels of signaling pathway proteins
were detected by western blotting, which has been previously
described (19,26,27).
EPCs were incubated with ANP (300 nM) and/or
H2O2 or the negative control brusatol (0.3
µg/ml) for 48 h at 37°C. The blank control group was incubated with
DMSO. After incubation and aspiration of the culture solution,
cells were washed twice with PBS. Total proteins were extracted
using RIPA buffer [FUJIFILM Wako Pure Chemical Corporation;
containing 50 mM Tris-HCl (pH 7.4), 150 mM NaCl, 2 mM EDTA, 1%
Nonidet P-40 and 0.1% SDS] on ice for 30 min with occasional
swirling to disperse the lysate evenly. The specific nuclear
proteins were extracted using a NE-PER™ nuclear extraction kit
(cat. no. 78835; Thermo Fisher Scientific, Inc.) following the
manufacturer's instructions. Cellular protein lysates were
collected and protein concentration was quantified using a BCA kit
(cat. no. A045-3-2; Nanjing Jiancheng Bioengineering Institute).
Proteins (40 µg/lane) were separated by SDS-PAGE on 10% gels and
transferred onto nitrocellulose membranes. The blots were blocked
with 5% non-fat milk in Tris-buffered saline for 2 h at 25°C and
then probed with primary antibodies against C-caspase-3 (1:500),
Bax (1:1,000), Bcl-2 (1:1,000), GAPDH (1:500), Nrf2 (1:200), HO-1
(1:1,000) and Lamin B1 (1:1,000) at 4°C overnight. Subsequently,
membranes were incubated with the respective secondary antibodies
(cat. no. ab6802; 1:3,000; Abcam) according to the appropriate
protocols. The antibody signals were then detected with Chemistar
High-sig ECL Western Blotting Substrate (Thermo Fisher Scientific,
Inc.). Each sample was analyzed three times. Protein expression
levels were semi-quantified by densitometric analysis using
Quantity One software (version 4.6.6; Bio-Rad Laboratories,
Inc.)
Measurement of oxidative stress
EPCs (3×105 cells/well) were seeded onto
6-well plates and incubated with ANP in the presence/absence of
H2O2 or brusatol. Cells were then harvested
and lysed by ultrasonic disruption (300 W, 15 min) on ice, and then
centrifuged at 12,880 × g for 15 min at 4°C. The supernatant was
collected to detect the levels of superoxide dismutase (SOD) using
a SOD assay kit (cat. no. A001-1-2), nitric oxide (NO) using a NO
assay kit (cat. no. A013-2-1) and malondialdehyde (MDA) using an
MDA assay kit (cat. no. A003-1-2) obtained from Nanjing Jiancheng
Bioengineering Institute according to the manufacturer's
recommendation and as previously described (28).
Statistical analysis
Each experiment was performed independently at least
three times. Statistical analyses were performed using SPSS 18.0
software (SPSS, Inc.). Statistical differences among multiple
groups were assessed by one-way analysis of variance, followed by
Tukey's post hoc tests if there were statistical differences
between two groups. P<0.05 was considered to indicate a
statistically significant difference.
Results
Effect of H2O2
on the viability and apoptosis of EPCs
Before investigating the effect of ANP on EPC
apoptosis and oxidative stress, the cytotoxic effects of
H2O2 on EPCs were evaluated using the CCK-8
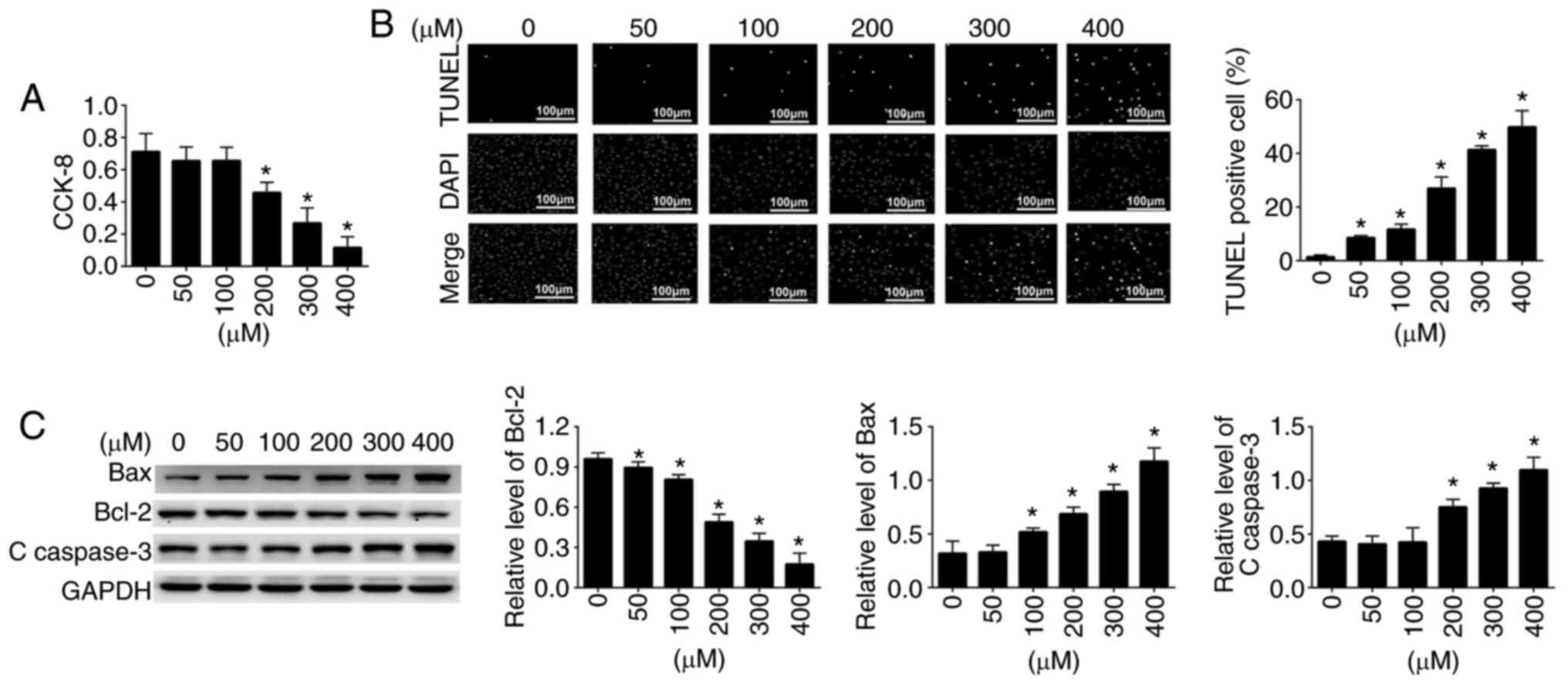
assay. The results are shown in Fig.
1A. Cell viability was significantly affected and impaired by
exposure to different concentrations of H2O2
in a dose-dependent manner. When EPCs were incubated with
increasing concentrations of H2O2, cell
viability was gradually decreased (Fig.
1A); 200 µM H2O2 was selected for
subsequent experiments. Subsequently, the TUNEL assay was conducted
to investigate the cell death of EPCs. The results are presented in
Fig. 1B. It was observed that the
incidence of cell apoptosis was markedly increased in the
H2O2-treated group in a dose-dependent manner
compared with that in the control group. The TUNEL assay results
were in agreement with the findings of the CCK-8 assay. To further
investigate the apoptosis of EPCs, the expression levels of
apoptosis-associated proteins, Bcl-2, Bax and C-caspase-3, were
detected by western blot analysis. The expression levels of these
three apoptosis-related proteins in control and
H2O2 (50–400 µM)-treated EPCs are shown in
Fig. 1C. Compared with those in the
untreated control cells, increasing concentrations of
H2O2 significantly increased the protein
expression levels of Bax and C-caspase-3 (pro-apoptotic), and
decreased the protein expression levels of Bcl-2 (anti-apoptotic).
These results indicated that H2O2 may induce
EPC death through activation of the apoptosis-related signaling
pathway.
H2O2 induces
oxidative stress in EPCs
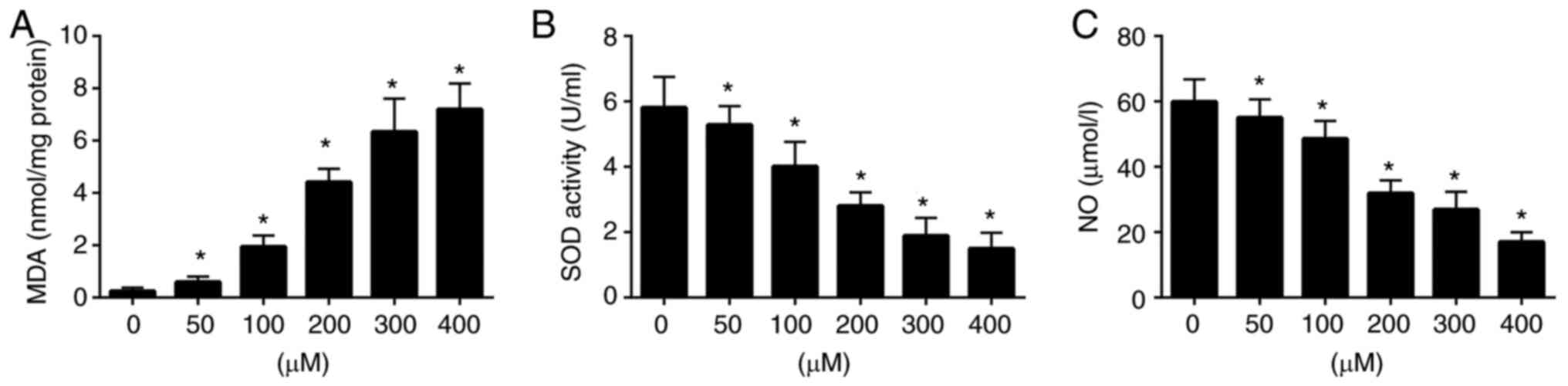
To assess the levels of oxidative stress in
H2O2-induced EPCs, the levels of related
enzymes (MDA) or markers (SOD and NO) were detected using
commercial assay kits. The results revealed that exposure to
H2O2 markedly increased MDA activity, and
decreased SOD and NO levels compared with those in the untreated
group (Fig. 2). These results
revealed that H2O2 induced oxidative stress
in EPCs and affected the levels of associated indices.
H2O2 inhibits
the expression of proteins associated with the Nrf2/HO-1 signaling
pathway in EPCs
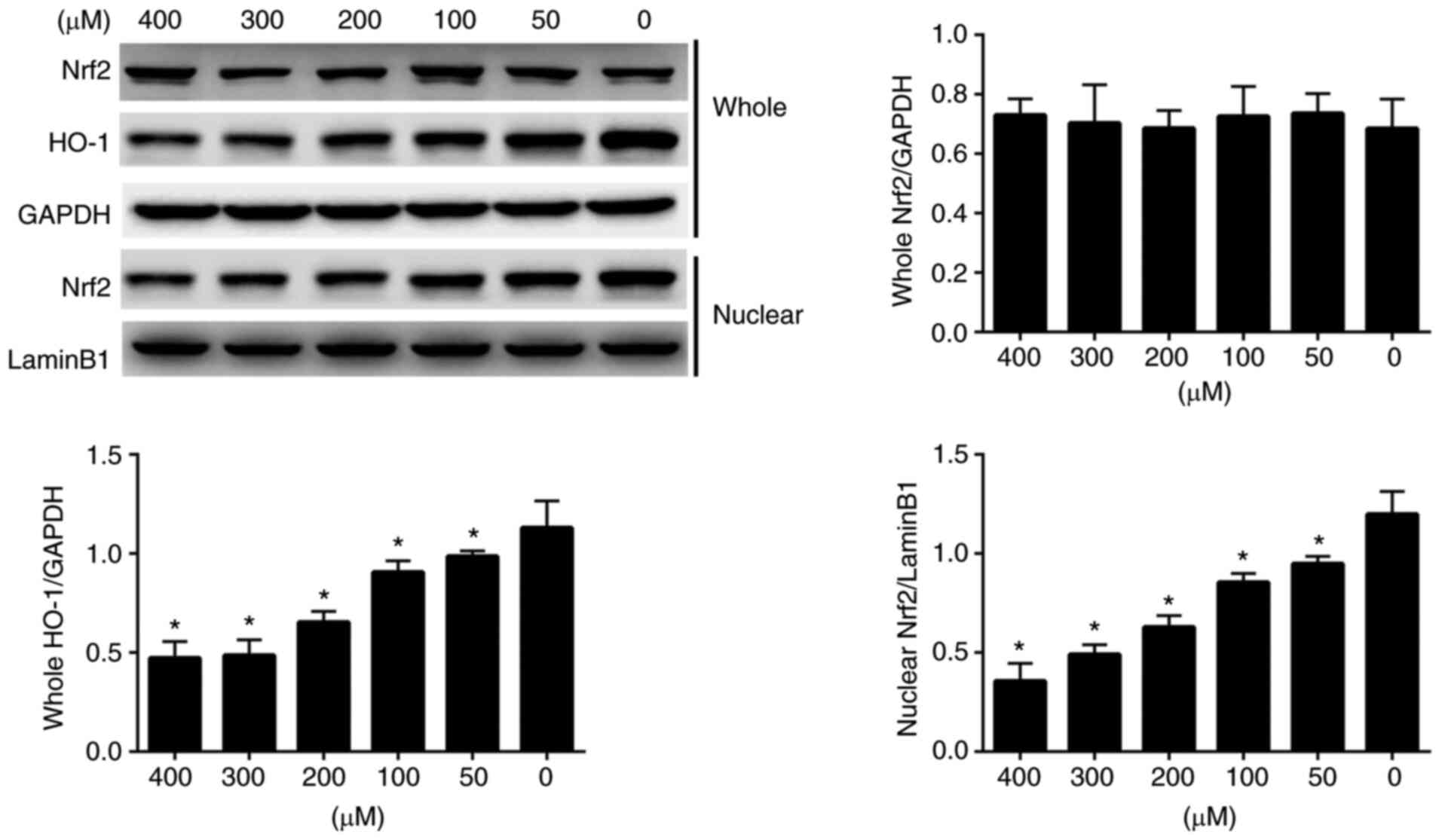
The expression levels of Nrf2 and HO-1 were detected
in H2O2-induced EPCs by western blotting
(Fig. 3). With regard to the
protein expression levels of Nrf2 in whole cells, there was a
slight difference among the different groups; however, this was not
statistically significant. By contrast, the protein expression
levels of HO-1 in whole cells were markedly lower in the
H2O2-treated groups compared with those in
the control group; a moderate inhibition of ~60% was achieved in
response to 200 µM H2O2 compared with 0 µM
H2O2. The protein expression levels of Nrf2
in the nucleus were also detected; the results indicated that the
protein expression levels of Nrf2 in the nucleus were markedly
decreased with increasing H2O2 dose. These
findings suggested that a large proportion of Nrf2 protein had
moved from the nucleus and the expression levels of the related
protein HO-1 were also decreased after treatment with
H2O2. These results indicated that
H2O2 treatment may block the nuclear
translocation of Nrf2 signaling pathway components and may inhibit
HO-1 expression in whole EPCs.
ANP activates the NrF2/HO-1 signaling
pathway
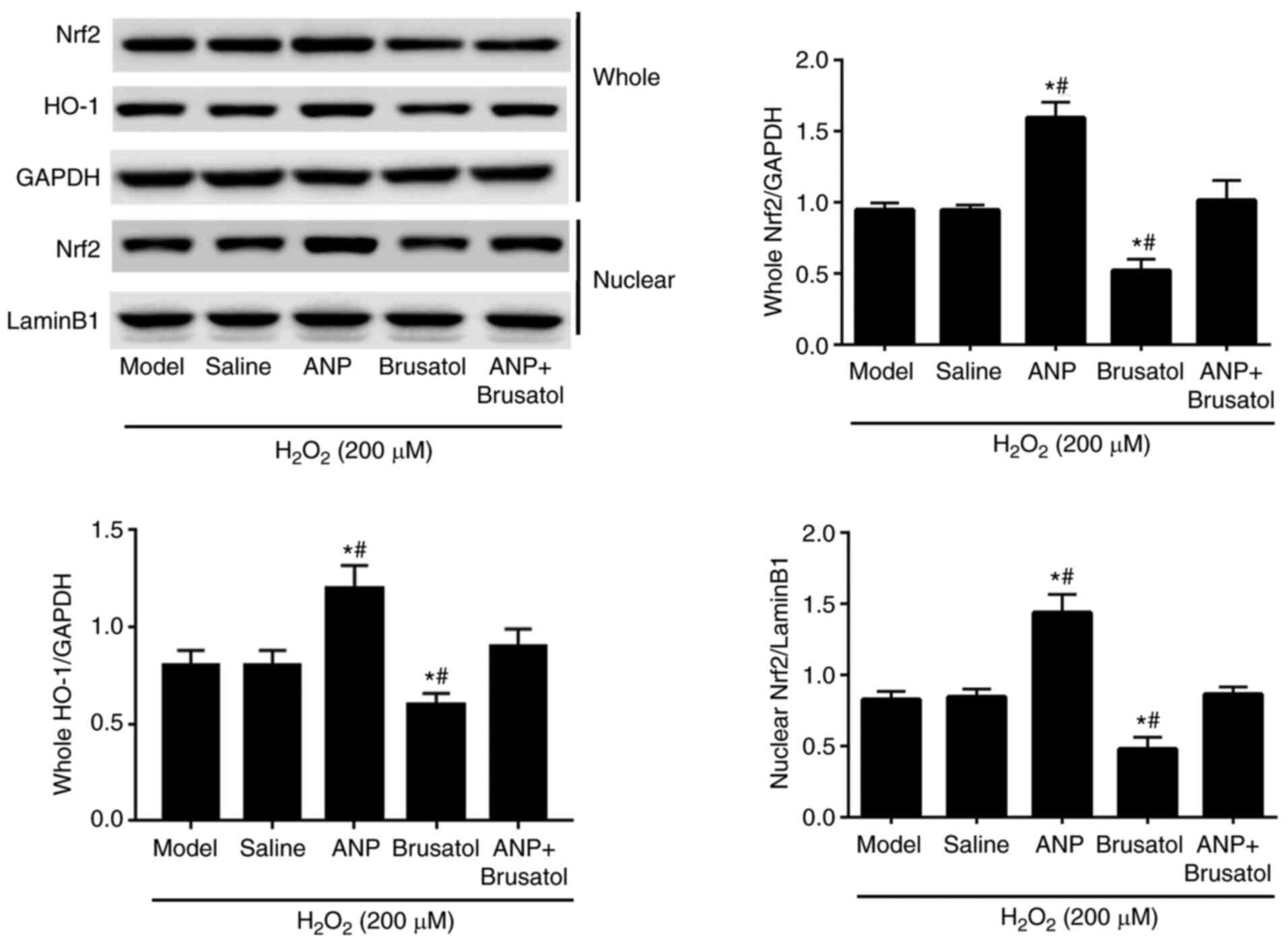
Western blotting was conducted to measure the
protein expression levels of Nrf2 and HO-1 in EPCs treated with
H2O2, ANP and brusatol, alone or together
(Fig. 4). When compared with those
in the model and saline groups, the whole Nrf2, nuclear Nrf2 and
whole HO-1 protein expression levels were increased in the ANP
treatment group. Conversely, the ANP-induced increase in whole
Nrf2, nuclear Nrf2 and whole HO-1 protein expression levels was
offset by brusatol. Collectively, ANP treatment increased Nrf2 and
HO-1 expression in H2O2-induced EPCs, and may
exert protective effects against oxidant stress.
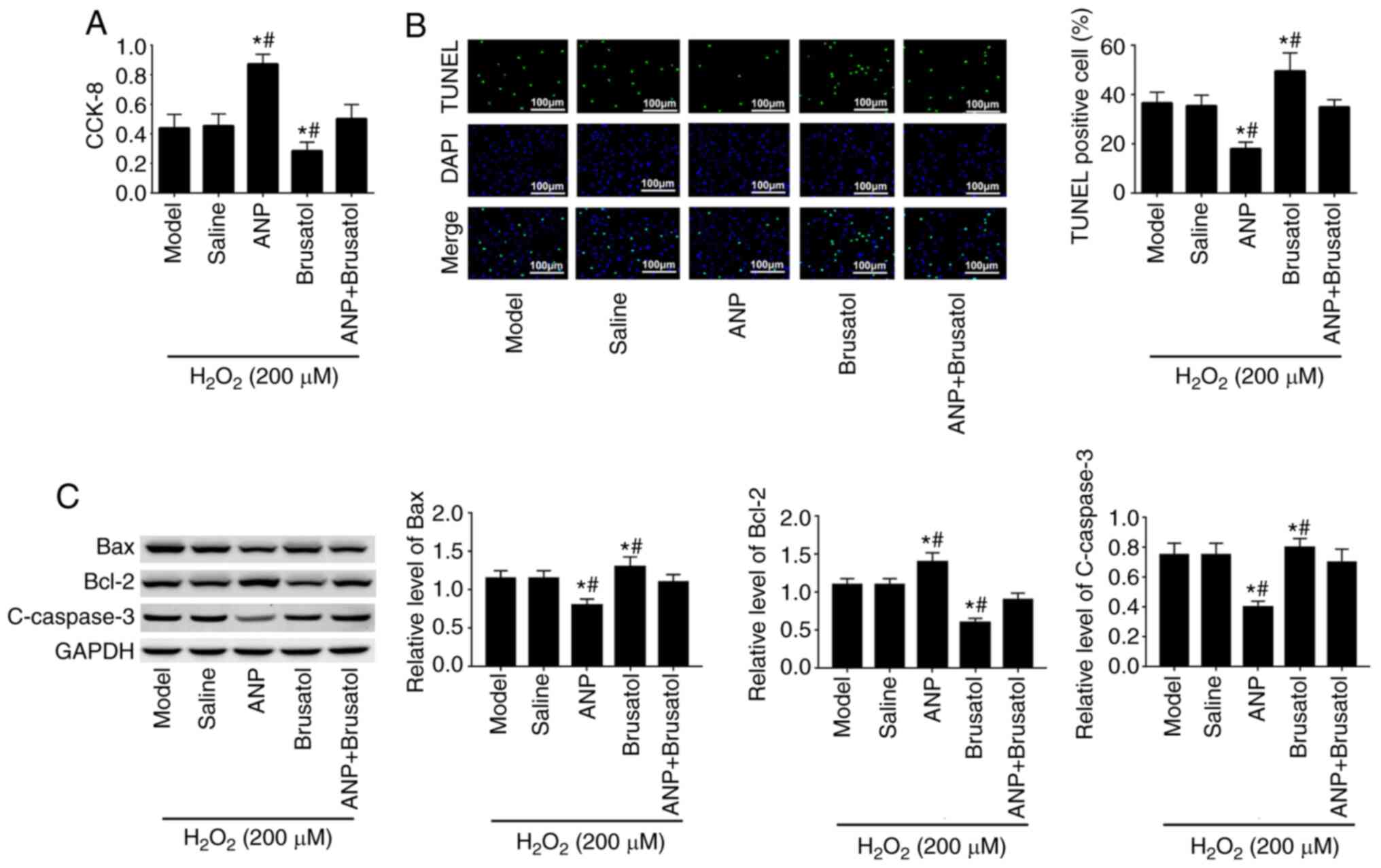
ANP inhibits the
H2O2-induced apoptosis of EPCs
The cell viability and apoptosis of EPCs were
measured to determine whether ANP may confer protection against
H2O2-induced cell damage. As presented in
Fig. 5A, cell viability was
inhibited in brusatol-treated cells compared with that in the
saline and ANP + brusatol groups, whereas this phenomenon was
reversed by ANP treatment. The cell viability in the ANP-pretreated
group was higher than that in cells without ANP. The results of the
TUNEL assay (Fig. 5B) also revealed
that ANP pretreatment decreased H2O2-induced
cell apoptosis when compared with that in the saline group.
Subsequently, the results of western blotting revealed that in
brusatol-treated cells, the protein expression levels of Bax and
C-caspase-3 were increased, whereas those of Bcl-2 were decreased;
these effects were reversed by ANP treatment. Notably, when ANP +
brusatol were present, the expression levels of these proteins
almost returned to the same levels as the model group. Taken
together, these results suggested that ANP protected EPCs against
H2O2-induced apoptosis.
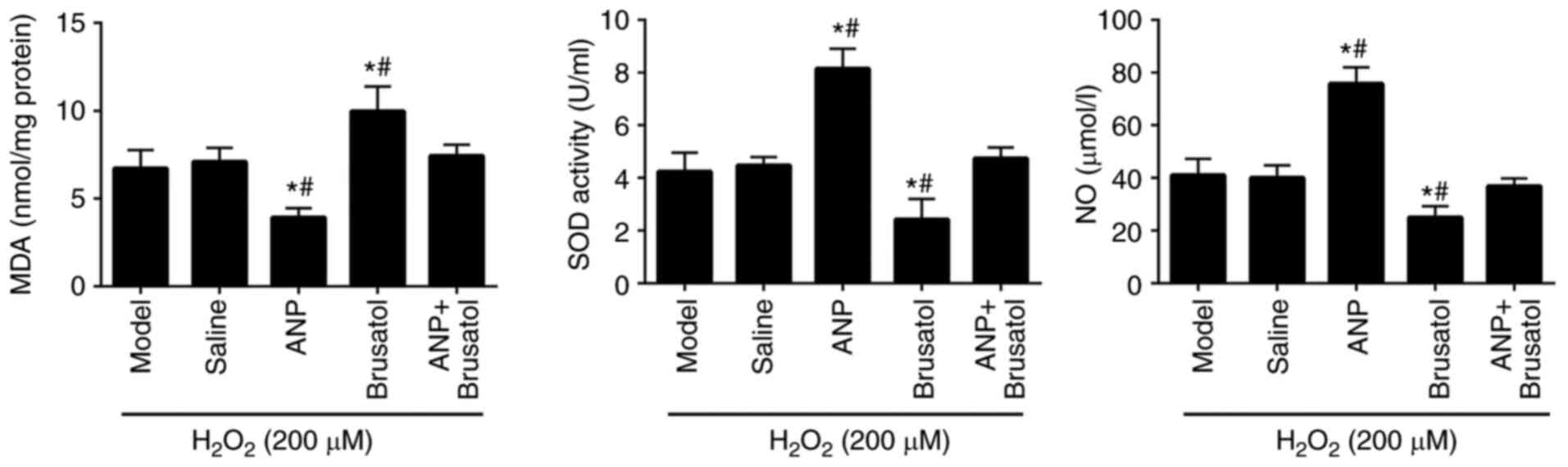
ANP regulates the levels of MDA, SOD
and NO in EPCs treated with H2O2
The levels of MDA, SOD and NO were examined in
ANP-treated EPCs. The results revealed that pretreatment with ANP
increased the levels of SOD, which is able to eliminate
H2O2, and NO, and reduced MDA content when
compared with those in the model group (Fig. 6). By contrast, the Nrf2 specific
inhibitor, brusatol reversed the reduced MDA, and increased SOD and
NO levels compared with the ANP group. These findings suggested
that ANP blocked the adverse effects of H2O2
on EPCs and may have potential antioxidant effects. These results
coincided with the increased expression and nuclear translocation
of Nrf2 and Nrf2/HO-1, whereas no obvious differences in enzymes
were found between the saline group and ANP + brusatol group.
Discussion
ANP has been proven to exert beneficial effects on
intervertebral discs under physiological and pathological
conditions (3), and has also been
shown to exert a protective effect on oxidant injury (3,6,7).
However, its effect and associated mechanism on EPCs is poorly
understood and, to the best of our knowledge, has never been
reported. Based on the previous reports regarding the beneficial
and protective effects of ANP during oxidant injury, it was
hypothesized that ANP may help to protect EPCs against
H2O2-induced oxidant injury; therefore, a
series of experiments were performed to confirm and explore its
underlying mechanism.
In the present study, the oxidative stress model was
induced by H2O2, and EPCs were pre-incubated
in the presence or absence of ANP. The inhibitory effects of ANP on
cell death were evaluated by detecting cell viability and the
expression levels of apoptosis-related proteins (C-caspase-3, Bax
and Bcl-2) in EPCs. The results indicated that
H2O2-treated cells exhibited a higher rate of
apoptosis, whereas ANP pre-incubation could significantly inhibit
cell death. Furthermore, H2O2-induced
C-caspase-3 and Bax activation, as well as Bcl-2 suppression, was
also significantly reversed when EPCs were treated with ANP,
whereas the effect of ANP was attenuated in the presence of
brusatol. In consistent with this finding, previous studies have
found similar results (29–31). Furthermore, it was reported that
cell apoptosis was suppressed by increased Bcl-2, reduced Bax and
caspase-3 activity (12). Taken
together, along with the findings of other studies, the present
results suggested that ANP could inhibit
H2O2-induced cell apoptosis and regulate the
levels of apoptosis-related proteins.
Previous studies have revealed that oxidative stress
can induce cell apoptosis (32,33)
and since the present results revealed that ANP could reduce
H2O2-induced cell apoptosis, it was
hypothesized that ANP may have a role in the regulation of several
enzymes/proteins that can protect cells against oxidant injury. To
explore the underlying relationship between the prevention of
oxidant injury and inhibition of cell death, the levels of three
essential oxidative stress-related enzymes (SOD-1, MDA and NO) were
assessed (34,35). The results indicated that
H2O2 induced changes in the levels of SOD,
MDA and NO, and these changes were reversed by ANP.
It has been reported that under normal conditions,
Nrf2 is sequestered in the cytoplasm by Keap1, whereas under
oxidative stress, Keap1 is modified by -SH groups and Nrf2 is
phosphorylated. This facilitates Nrf2 dissociation from Keap1 and
its translocation into the nucleus (36). The antioxidant response element can
be activated by Nrf2 and the transcription of Nrf2-regulated genes
(such as HO-1 and glutathione S-transferase) can be increased after
binding with Maf protein. Among these genes, HO-1 enzyme activity
may remove the prooxidant molecule heme, and lead to the generation
of iron ions, biliverdin and carbon monoxide (37). Biliverdin is reduced to bilirubin by
the action of biliverdin reductase, and bilirubin is a potent
antioxidant (38); consequently,
HO-1 is generally considered as a cellular defense against
oxidative stress. Notably, the expression of Nrf2-facilitated HO-1
shows an antioxidant effect (18).
These findings indicated that the Nrf2/HO-1 signaling pathway may
serve a significant role in oxidative stress. Based on these
reports, it was hypothesized that ANP may prevent oxidative stress
through regulation of the Nrf2/HO-1 signaling pathway. Western
blotting was conducted to investigate the expression levels of
proteins in the Nrf2/HO-1 pathway, and the specific inhibitor of
the Nrf2 pathway, brusatol (39),
was used in the present study as a negative control. The results
revealed that ANP activated Nrf2 expression and nuclear
translocation, and reversed H2O2-induced
inhibition of the Nrf2/HO-1 pathway, whereas brusatol treatment
blocked the inhibitory effects of ANP on cell apoptosis and
oxidative stress. According to these results, it was suggested that
the decreased expression of Nrf2/HO-1 in the
H2O2-treated group was related to cell death
and oxidative stress, and that ANP prevented oxidative stress
through activating the Nrf2/HO-1 signaling pathway. Specifically,
ANP promoted Nrf2 nuclear translocation and upregulated HO-1, which
suggested that it may directly activate the Nrf2/HO-1 signaling
pathway. As a consequence of activating the Nrf2/HO-1 signaling
pathway, ANP could suppress the expression of apoptosis-related
proteins, such as Bax and C-caspase 3, promote expression of the
anti-apoptotic protein Bcl-2, and inhibit apoptosis and oxidant
injury in EPCs.
The present study, in combination with previous
reports (40,41), suggested that oxidative stress may
be a crucial contributor towards cell apoptosis, and oxidative
stress was related to the elevated levels of oxidative stress
markers, which were affected by an intracellular oxidative and
antioxidant defense mechanism in H2O2-treated
EPCs. In the present study, the H2O2-induced
cell apoptosis was accompanied by oxidative stress and inhibition
of the Nrf2/HO-1 signaling pathway in EPCs, whereas these changes
were reversed by ANP treatment. Therefore, it was suggested that
the associated potential mechanisms of protection were possibly
linked to activation of the Nrf2/HO-1 signaling pathway in EPCs
under oxidative stress conditions, and that the Nrf2/HO-1 signaling
pathway may serve a conservative role in cell viability. As a
result, the protective effects of ANP on EPCs were revealed to be
due to direct activation of the Nrf2/HO-1 signaling pathway.
There were some limitations in the present study.
First, only in vitro experiments were performed. In
addition, only the Nrf2/HO-1 signaling pathway was assessed with
regard to the potential mechanism of ANP in this study. It is
essential to evaluate related regulators or other signaling
pathways, such as the PI3K/Akt and TLR pathways, which may be
detected by reverse transcription-quantitative PCR.
In the future, a rat model of IDD may be established
via percutaneous needle puncture of the rat tail to further explain
the association between ANP and IDD. In addition, future studies
should focus on the role of the PI3K/Akt and TLRs signaling
pathways in the inhibition of oxidant stress in EPCs.
The present study revealed that in EPCs, ANP may
activate Nrf2 nuclear translocation and expression of Nrf2/HO-1,
which may inhibit cell apoptosis and oxidant injury induced by
H2O2 under oxidative stress conditions. In
conclusion, it was suggested that the Nrf2/HO-1 signaling pathway
may represent a target for protection against
H2O2-induced cell apoptosis and oxidant
stress. ANP may represent a potential treatment for various
diseases related to oxidant injury through its activation of the
Nrf2/HO-1 signaling pathway, including IDD.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
FH and QF conceived and designed the study, and
reviewed and revised the manuscript. JG and JW performed the
experiments and collected the experimental data. LT and YL
interpreted the results and contributed to the manuscript
preparation. FH and QF confirm the authenticity of all the raw
data. All authors agreed to be accountable for the content of the
work, and read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
ANP
|
atrial natriuretic peptide
|
|
EPCs
|
endplate chondrocytes
|
|
NO
|
nitric oxide
|
|
MDA
|
malondialdehyde
|
|
SOD
|
superoxide dismutase
|
|
Nrf2
|
nuclear factor erythroid 2-related
factor 2
|
|
HO-1
|
heme oxygenase-1
|
|
DMSO
|
dimethyl sulfoxide
|
References
|
1
|
Hu RM, Levin ER, Pedram A and Frank HJL:
Atrial natriuretic peptide inhibits the production and secretion of
endothelin from cultured endothelial cells: Mediation through the C
receptor. J Biol Chem. 267:17384–9. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Smith S, Anderson S, Ballermann BJ and
Brenner BM: Role of atrial natriuretic peptide in adaptation of
sodium excretion with reduced renal mass. J Clin Invest.
77:1395–1398. 1986. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Baxter JD, Lewicki JA and Gardner DG:
Atrial Natriuretic Peptide. Nat Biotechnol. 6:529–546. 1988.
View Article : Google Scholar
|
|
4
|
Abraham WT, Hensen J, Kim JK, Dürr J and
Schrier RW: Atrial natriuretic peptide and urinary cyclic guanosine
monophosphate in patients with chronic heart failure. J Am Soc
Nephrol. 2:1697–1703. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Rahman SN, Kim GE, Mathew AS, Goldberg CA,
Allgren R, Schrier RW and Conger JD: Effects of atrial natriuretic
peptide in clinical acute renal failure. Kidney Int. 45:1731–1738.
1994. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Bilzer M, Jaeschke H, Vollmar AM,
Paumgartner G and Gerbes AL: Prevention of Kupffer cell-induced
oxidant injury in rat liver by atrial natriuretic peptide. Am J
Physiol. 276:G1137–G1144. 1999.PubMed/NCBI
|
|
7
|
Bilzer M, Jaeschke H, Vollmar AM,
Paumgartner G and Gerbes AL: Reduction of Kupffer cell-induced
oxidant injury of the rat liver by atrial natriuretic peptide and
cyclo-GMP receptor proteins. J Hepatol. 28:471998. View Article : Google Scholar
|
|
8
|
Fahn S and Cohen G: The oxidant stress
hypothesis in Parkinson's disease: Evidence supporting it. Ann
Neurol. 32:804–812. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Jie B, Fan Y, Li D and Yi Z: Ghrelin
protects human lens epithelial cells against oxidative
stress-induced damage. Oxid Med Cell Longev.
2017:19104502017.PubMed/NCBI
|
|
10
|
Martinou JC and Youle RJ: Mitochondria in
apoptosis: Bcl-2 family members and mitochondrial dynamics. Dev
Cell. 21:92–101. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kluck RM, Bossy-Wetzel E, Green DR and
Newmeyer DD: The release of cytochrome c from mitochondria:
A primary site for Bcl-2 regulation of apoptosis. Science.
275:1132–1136. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Shi L, Chen J, Yang J, Pan T, Zhang S and
Wang Z: miR-21 protected human glioblastoma U87MG cells from
chemotherapeutic drug temozolomide induced apoptosis by decreasing
Bax/Bcl-2 ratio and caspase-3 activity. Brain Res. 1352:255–264.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kensler TW, Wakabayashi N and Biswal S:
Cell Survival responses to environmental stresses via the
Keap1-Nrf2-ARE Pathway. Annu Rev Pharmacol Toxicol. 47:89–116.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Mukaigasa K, Nguyen LTP, Li L, Nakajima H,
Yamamoto M and Kobayashi M: Genetic evidence of an evolutionarily
conserved role for Nrf2 in the protection against oxidative stress.
Mol Cell Biol. 32:4455–4461. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Strom J: A critical role of Nrf2 in
protecting cardiomyocytes against oxidative stress and ischemic
injury. PhD dissertation, The University of Arizona, . 2014.
|
|
16
|
Buendia I, Michalska P, Navarro E, Gameiro
I and León R: Nrf2-ARE pathway: An emerging target against
oxidative stress and neuroinflammation in neurodegenerative
diseases. Pharmacol Ther. 157:84–104. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Yagishita Y, Fukutomi T, Sugawara A,
Kawamura H, Takahashi T, Pi J, Uruno A and Yamamoto M: Nrf2
protects pancreatic beta-cells from oxidative and nitrosative
stress in diabetic model mice. Diabetes. 63:605–618. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Loboda A, Damulewicz M, Pyza E, Jozkowicz
A and Dulak J: Role of Nrf2/HO-1 system in development, oxidative
stress response and diseases: An evolutionarily conserved
mechanism. Cell Mol Life Sci. 73:3221–3247. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Jian Z, Li K, Liu L, Zhang Y, Zhou Z, Li C
and Gao T: Heme Oxygenase-1 protects human melanocytes from
H2O2-induced oxidative stress via the
Nrf2-ARE pathway. J Invest Dermatol. 131:1420–1427. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Sahin K, Tuzcu M, Sahin N, Ali S and Kucuk
O: Nrf2/HO-1 signaling pathway may be the prime target for
chemoprevention of cisplatin-induced nephrotoxicity by lycopene.
Food Chem Toxicol. 48:2670–2674. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Han Y, Li X, Yan M, Yang M, Wang S, Pan J,
Li L and Tan J: Oxidative damage induces apoptosis and promotes
calcification in disc cartilage endplate cell through
ROS/MAPK/NF-κB pathway: Implications for disc degeneration. Biochem
Biophys Res Commun. 516:1026–1032. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Giers MB, Munter BT, Eyster KJ, Ide GD,
Newcomb AGUS, Lehrman JN, Belykh E, Byvaltsev VA, Kelly BP, Preul
MC and Theodore N: Biomechanical and endplate effects on nutrient
transport in the intervertebral disc. World Neurosurg. 99:395–402.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ariga K, Yonenobu K, Nakase T, Hosono N,
Okuda S, Meng W, Tamura Y and Yoshikawa H: Mechanical
stress-induced apoptosis of endplate chondrocytes in organ-cultured
mouse intervertebral discs: An ex vivo study. Spine (Phila Pa
1976). 28:1528–1533. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Otero M, Lago R, Lago F, Reino JJG and
Gualillo O: Signalling pathway involved in nitric oxide synthase
type II activation in chondrocytes: Synergistic effect of leptin
with interleukin-1. Arthritis Res Ther. 7:R581–R591. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zhang JX, Han YP, Bai C and Li Q: Notch1/3
and p53/p21 are a potential therapeutic target for APS-induced
apoptosis in non-small cell lung carcinoma cell lines. Int J Clin
Exp Med. 8:12539–12547. 2015.PubMed/NCBI
|
|
26
|
Zhang Z, Lin J, Tian N, Wu Y, Zhou Y, Wang
C, Wang Q, Jin H, Chen T, Nisar M, et al: Melatonin protects
vertebral endplate chondrocytes against apoptosis and calcification
via the Sirt1-autophagy pathway. J Cell Mol Med. 23:177–193. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhou YF, Guo B, Ye MJ, Liao RF and Li SL:
Protective effect of rutin against
H2O2-induced oxidative stress and apoptosis
in human lens epithelial cells. Curr Eye Res. 41:933–942. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Liang B, Wei W, Wang J, Zhang M, Xu R, Wu
F, Xiao H and Tang L: Protective effects of Semiaquilegia adoxoides
n-butanol extract against hydrogen peroxide-induced oxidative
stress in human lens epithelial cells. Pharm Biol. 54:1656–1663.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Salakou S, Kardamakis D, Tsamandas AC,
Zolota V, Apostolakis E, Tzelepi V, Papathanasopoulos P, Bonikos
DS, Papapetropoulos T, Petsas T and Dougenis D: Increased Bax/Bcl-2
ratio up-regulates caspase-3 and increases apoptosis in the thymus
of patients with myasthenia gravis. In Vivo. 21:123–132.
2007.PubMed/NCBI
|
|
30
|
Ghate NB, Hazra B, Sarkar R, Chaudhuri D
and Mandal N: Alteration of Bax/Bcl-2 ratio contributes to
Terminalia Belerica-induced apoptosis in human lung and breast
carcinoma. In Vitro Cell Dev Biol Anim. 50:527–537. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Chen HM, Lai ZQ, Liao HJ, Xie JH, Xian YF,
Chen YL, Ip SP, Lin ZX and Su ZR: Synergistic antitumor effect of
brusatol combined with cisplatin on colorectal cancer cells. Int J
Mol Med. 41:1447–1454. 2018.PubMed/NCBI
|
|
32
|
Hart BA: Characterization of
cadmium-induced apoptosis in rat lung epithelial cells: Evidence
for the participation of oxidant stress. Toxicology. 133:43–58.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Olayanju A, Copple IM, Bryan HK, Edge GT,
Sison RL, Wong MW, Lai ZQ, Lin ZX, Dunn K, Sanderson CM, et al:
Brusatol provokes a rapid and transient inhibition of Nrf2
signaling and sensitizes mammalian cells to chemical
toxicity-implications for therapeutic targeting of Nrf2. Free Radic
Biol Med. 78:202–212. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
El-Beltagi HS and Mohamed HI: Reactive
oxygen species, lipid peroxidation and antioxidative defense
mechanism. Notulae Botanicae Horti Agrobotanici Cluj-Napoca.
41:44–57. 2013. View Article : Google Scholar
|
|
35
|
Zheng Y, Liu Y, Ge J, Wang X and Liu P:
Resveratrol protects human lens epithelial cells against
H2O2-induced oxidative stress by increasing
catalase, SOD-1, and HO-1 expression. Mol Vis. 16:1467–1474.
2010.PubMed/NCBI
|
|
36
|
Velichkova M and Hasson T: Keap1 regulates
the oxidation-sensitive shuttling of Nrf2 into and out of the
nucleus via a Crm1-dependent nuclear export mechanism. Mol Cell
Biol. 25:4501–4513. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Camara NO and Soares MP: Heme oxygenase-1
(HO-1), a protective gene that prevents chronic graft dysfunction.
Free Radic Biol Med. 38:426–435. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Abraham NG and Kappas A: Heme oxygenase
and the cardiovascular-renal system. Free Radic Biol Med. 39:1–25.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Ren D, Villeneuve NF, Jiang T, Wu T, Lau
A, Toppin HA and Zhang DD: Brusatol enhances the efficacy of
chemotherapy by inhibiting the Nrf2-mediated defense mechanism.
Proc Natl Acad Sci USA. 108:1433–1438. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Tang XQ, Feng JQ, Chen J, Chen PX, Zhi JL,
Cui Y, Guo RX and Yu HM: Protection of oxidative preconditioning
against apoptosis induced by H2O2 in PC12
cells: Mechanisms via MMP, ROS, and Bcl-2. Brain Res. 1057:57–64.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Buttke TM and Sandstrom P: Oxidative
stress as a mediator of apoptosis. Immunol Today. 15:7–10. 1994.
View Article : Google Scholar : PubMed/NCBI
|