Introduction
Diabetic cardiomyopathy (DCM) is a major
cardiovascular complication in diabetes that remains the leading
cause of mortality in diabetic patients (1). Hyperglycemia is the main clinical
manifestation of diabetes, playing a key pathogenic role in the
development of DCM (1,2). However, a clear causative role for
hyperglycemia has not been established. The major biochemical
pathways of hyperglycemic cardiac damage, including chronic
hyperglycemia, result in the activation of the inflammatory
response and the generation of reactive oxygen species (ROS)
(2). Increased inflammatory
response influences upstream regulatory events that impair cardiac
function and result in cardiomyocyte injury. ROS overproduction
subsequently induces injury and an aggressive inflammatory response
(3).
Numerous studies have revealed that nuclear
factor-κB (NF-κB) is a primary regulator of inflammatory responses;
NF-κB is activated in the cardiac myocytes of patients with DCM.
Activated NF-κB induces the expression of proinflammatory
cytokines, including tumor necrosis factor (TNF)-α, interleukin
(IL)-6 and IL-1β, which results in cell infiltration of leucocytes,
including macrophages and neutrophils. The NF-κB-dependent
mechanism plays a significant role in hyperglycemia-induced
myocardial damage and inflammation (2,4). The
aforementioned data suggest that inhibition of the inflammatory
response by preventing NF-κB pathway activation may have
therapeutic potential in DCM.
Cluster of differentiation 36 (CD36), which belongs
to a class B scavenger receptor family, is a glycosylated surface
receptor present in the cell membrane and in the intracellular
compartment of cardiac myocytes (5). High glucose (HG) concentrations can
increase CD36 mRNA translation and subsequently increase
CD36 expression in macrophages, renal tubular epithelial cells and
dendritic cells (6–8). However, the effects of HG on CD36 in
cardiomyocytes have not been demonstrated. Accumulating evidence
has focused on the role of CD36: As a fatty acid transporter, it
mediates fatty acid uptake, leading to cardiac lipotoxicity and
alterations of myocardial energy metabolism (2,5). CD36
is also a signaling molecule that participates in the inflammatory
process (9–12). Diverse animal studies have reported
that CD36 is important for lipid deposition, and also participates
in NF-κB pathway activation and mitochondrial fatty acid oxidation,
which contributes to the regulation of chronic metabolic diseases,
especially in metabolic disease models that include atherogenic
processes and non-alcoholic steatohepatitis (10,13–16).
However, studies investigating the effects of CD36 on inflammatory
stress in DCM are rare.
The present study investigated CD36 expression in HG
conditions, whether CD36 upregulation promotes inflammation stress
via NF-κB in HG-induced H9c2 cells, as well as the relevant
mechanism.
Materials and methods
Reagents and antibodies
Glucose, mannitol, palmitic acid (PA),
N-acetylcysteine (NAC), MitoTEMPO, and MitoSOX were
purchased from Sigma-Aldrich; Merck KGaA. The lentivirus constructs
were provided by Shanghai GeneChem Co., Ltd. 2′,
7′-Dichlorodihydrofluorescein diacetate (DCFDA) was from Invitrogen
(Thermo Fisher Scientific, Inc.). CD36 (cat. no. 14347), NF-κB p65
(cat. no. 8242), inhibitor of κBα (IκBα; cat. no. 4814),
AMP-activated protein kinase (AMPK; cat. no. 2532), and
phosphorylated (p-)AMPK (cat. no. 4184) antibodies were purchased
from Cell Signaling Technology, Inc. Antibodies against β-actin
(cat. no. sc-8432) and lamin B (cat. no. sc-374015) were from Santa
Cruz Biotechnology, Inc. The TNF-α (cat. no. HSTA00E), IL-6 (cat.
no. D6050) and IL-1β (cat. no. DLB50) enzyme-linked immunosorbent
assay (ELISA) kits were from R&D Systems.
Cell culture and transfection
H9c2 cells were obtained from American Type Culture
Collection and cultured in Dulbecco's modified Eagle's medium
(Gibco; Thermo Fisher Scientific, Inc.) with 1 g/l glucose
supplemented with 10% fetal bovine serum (Gibco; Thermo Fisher
Scientific, Inc.), 100 U/ml penicillin and 100 mg/ml streptomycin
at 37°C in an atmosphere of 5% CO2. The cells were
incubated with normal glucose (NG; 5.6 mM), NG plus mannitol (M;
24.4 mM), HG (30 mM), HG plus NAC (HG + N; 5 mM), and/or MitoTEMPO
(HG + T; 10 µM) and HG plus PA (HG + 100 µM PA) for 72 h. The H9c2
cells were infected with LV3 empty vector (LV3-NC), LV3 containing
CD36 (LV3-CD36), CD36 knockout short hairpin (sh)RNA (LV3-shRNA)
(Shanghai GeneChem Co. Ltd.) lentivirus expression vectors (viral
titer: 1×109 transducing U/ml; multiplicity of
infection: 10) for 6 h to knockout or overexpress CD36. Thereafter,
medium was replaced with fully supplemented DMEM for 24 h, the
cells were collected and transduction efficiencies were confirmed
by reverse transcription-quantitative PCR. LV3-NC was used as the
control group. In the experiment to verify the CD36 shRNA knockout
efficiency, scrambled shRNA was used as the control. The sequences
of all the shRNAs were as follows: CD36-RNAi (10899-1),
5′-CCGACGTTAATCTGAAAGGAA−3′; CD36-RNAi (10900-1),
5′-GCCATAATCGACACATATAAA-3′; CD36-RNAi (10902-1),
5′-CCATTGGTGATGAGAAGGCAA−3′; scrambled shRNA,
5′-TTCTCCGAACGTGTCACGT-3′. LV3-CD36: CD36 (64809-1)-p1,
5′-GAACCGTCAGATCCGCTAGCCGCCACCATGGGCTGTGACCGGAACTG-3′; CD36
(64809-1) -p2, 5′-GGAGGGAGAGGGGCGGATCTTATTTTATTGTTTTCGATCTGC-3′.
The H9c2 cells were designated the empty vector control group (NC),
high-CD36 expression group (HC) and CD36 knockout group (KC).
Western blotting
Total protein was extracted from the H9c2 cells
using radioimmunoprecipitation assay buffer (Boster Biological
Technology); nuclear and cytoplasmic proteins were extracted using
a commercial nucleoprotein extraction kit (cat. no. 40010)
referring to the manufacturer's instructions (Active Motif, Inc.).
Western blotting was performed according to a previous publication
(17). The primary antibodies
(CD36, NF-κB p65, IκBα, AMPK, p-AMPK, β-actin, lamin B) were
diluted 1:1,000; goat anti-rabbit (cat. no. SA00001-2; ProteinTech
Group, Inc.) or mouse IgG horseradish peroxidase conjugated
antibodies (cat. no. SA00001-1; ProteinTech Group, Inc.) were
diluted 1:5,000. The results were scanned using the Odyssey Fc
System (LI-COR Biosciences). The intensity of the bands was
measured using LabWorks 4.5 (Analytik Jena AG).
Reverse transcription-quantitative PCR
(RT-qPCR)
Total RNA and complementary DNA (cDNA) synthesis
from H9c2 cells were prepared using TRIzol® (Invitrogen;
Thermo Fisher Scientific, Inc.) and RevertAid reverse transcription
kits (Invitrogen; Thermo Fisher Scientific, Inc.) according to the
manufacturer's instructions. Real-time PCR was performed on an
Agilent Mx3000P QPCR System using SYBR Premix Ex Taq™ II (Takara
Biotechnology Co., Ltd.) at default thermal cycling conditions: 2
min at 50°C, 10 min at 95°C, followed by 40 cycles of 15 sec at
95°C for denaturation and 1 min at 60°C for annealing and
extension. The expression levels were represented by the
comparative threshold cycle value (2−ΔΔCq) (18) and normalized to β-actin. The primers
used were as follows: CD36 sense, 5′-ATAACTGGATTCACTCTACAGTTTGC-3′
and antisense, 5′-GATCTGCAAGCACAGTATGAAATC−3′; TNF-α sense,
5′-GATCGGTCCCAACAAGGAGG-3′ and antisense,
5′-TTTGCTACGACGTGGGCTAC-3′; IL-6 sense, 5′-CCACCCACAACAGACCAGTA-3′
and antisense, 5′-ACAGTGCATCATCGCTGTTC-3′; IL-1β sense,
5′-GAAGAATCTATACCTGTCCT-3′ and antisense,
5′-CTTTTCCATCTTCTTCTTTG-3′; and β-actin sense,
5′-CCCTAAGGCCAACCGTGAAAAG-3′ and antisense,
5′-TACGTACATGGCTGGGGTGT-3′.
Detection of ROS
Intracellular ROS were measured using DCFDA. H9c2
cells were seeded into 6-well plates at a density of
1.5×106 cells/ml and incubated under different
experimental conditions for 72 h. After culturing, H9c2 cells were
incubated in 10 µM DCFDA at 37°C for 30 min in the dark, and then
washed three times with warm buffer. Mitochondrial ROS (mtROS) were
detected using MitoSOX, a specific mitochondria-targeted superoxide
fluorescent probe. H9c2 cells were incubated with a 5-µM MitoSOX
working solution at 37°C for 30 min before flow cytometry. The
measurement of intracellular and mtROS was performed using flow
cytometry (FACSCalibur; BD Immunocytometry Systems), and the
analysis was performed using FlowJo software (version 10; FlowJo
LLC).
ELISA
After cells were cultured in 6-well plates for 72 h,
the supernatants were collected. TNF-α, IL-6 and IL-1β protein
levels in the culture supernatants were detected using a commercial
ELISA kit according to the manufacturer's instructions (Nanjing
Jiancheng Bioengineering Institute).
Immunofluorescence
H9c2 cells were cultured and stimulated in 6-well
chamber slides under different experimental conditions for 72 h,
then fixed in frozen acetone for 20 min at 4°C, then the cells were
incubated in 0.1% Triton X-100 for 20 min at room temperature to
permeabilize the cell membrane. The cells were blocked with 5%
bovine serum albumin (cat. no. SW3015; Beijing Solarbio Science
& Technology Co., Ltd.) for 30 min, and incubated with
antibodies (anti-NF-κB p65, 1:50, cat. no. 8242; anti-CD36, 1:50,
cat. no. 14347; Cell Signaling Technology, Inc.) overnight at 4°C.
Then, the cells were exposed to FITC-conjugated secondary antibody
(1:200; cat. nos. 31461; Invitrogen, Thermo Fisher Scientific,
Inc.) for 1 h at 37°C. The cells were counterstained with
4′,6-diamidino-2-phenylindole for 5 min at room temperature. Images
were captured using a fluorescence microscope (magnification, ×400;
Olympus BX63; Olympus Corporation) or confocal microscope
(magnification, ×400; Olympus FV 1000 Viewer).
Extracellular flux assay
H9c2 cells were seeded at a density of 50,000-80,000
cells/well in specialized XF96 cell culture microplates (Seahorse
Bioscience; Agilent Technologies, Inc.). The cells were exposed to
different conditions for 72 h at 37°C. Then, the medium was
replaced with Seahorse running medium (XF base medium supplemented
with 10% D-glucose, 100 mM pyruvate and 200 mM glutamine for the
mitochondrial stress test, or with 200 mM glutamine alone for the
glycolysis stress test). Subsequently, incubation was performed in
a non-CO2 incubator for 60 min at 37°C. The basal oxygen
consumption rate and extracellular acidification rate were recorded
for 24 min, followed by performance of the mitochondrial stress
test (1 µM oligomycin, 2 µM trifluoromethoxy carbonylcyanide
phenylhydrazone and 0.5 µM rotenone/antimycin A) and glycolysis
stress test (10 mM glucose, 1 µM oligomycin, and 50 mM 2-DG).
Subsequently, the cells were lysed in RIPA buffer and subjected to
the Bradford protein assay (Bio-Rad Laboratories, Inc.). The oxygen
consumption rate (OCR) and extracellular acidification rate (ECAR)
values were normalized by the protein values in each well.
Quantitative detection of
triglycerides
In total, 4–5 million H9c2 cells were collected,
sonicated by ultrasonication (200 W; 20 KHz, pulse 20%; run for 2
sec, stop 1 sec) for 1 min, then centrifuged at 8,000 × g for 10
min at 4°C, and the supernatant was retained for detection. The
triglyceride content was measured according to the manufacturer's
instructions (cat. no. A110-1-1; Nanjing Jiancheng Bioengineering
Institute).
Statistical analysis
The statistical significance of differences among
groups was analyzed using one-way analysis of variance, followed by
post hoc testing using the Tukey-Kramer method. All experiments
were performed at least three times. P<0.05 was considered to
indicate a statistically significant difference. Data were
expressed as the mean ± standard deviation (SD).
Results
HG-induced CD36 expression is
partially dependent on ROS in H9c2 cells
To determine the effect of HG on CD36 expression
in vitro, H9c2 cells were incubated with high concentrations
of glucose, and the CD36 mRNA and protein levels were examined. HG
induced a time-dependent increase in CD36 expression in the cells
(Fig. 1A and B). CD36 mRNA and
protein expression was increased within 12 h and continued to
increase up to 72 h. CD36 is a fatty acid transport protein; its
function mainly depends on its location in the cell membrane
(12). Thus, the translocation of
CD36 to the cell membrane in HG-treated H9c2 cells was next
evaluated by immunofluorescence staining observed by confocal
microscopy. As shown in Fig. 1C,
fluorescence of CD36 was low at 0 h. HG stimulation for 72 h
resulted in increased fluorescence of CD36. In the 0-h group, the
fluorescence was distributed in the cytoplasm of H9c2 cells;
however, in the 72-h HG-stimulated group, the fluorescence tended
to be distributed around the cell membrane. Flow cytometry analysis
revealed that the levels of CD36 in the cell membrane were higher
at 24 and 72 h compared with 0 h (Fig.
1D).
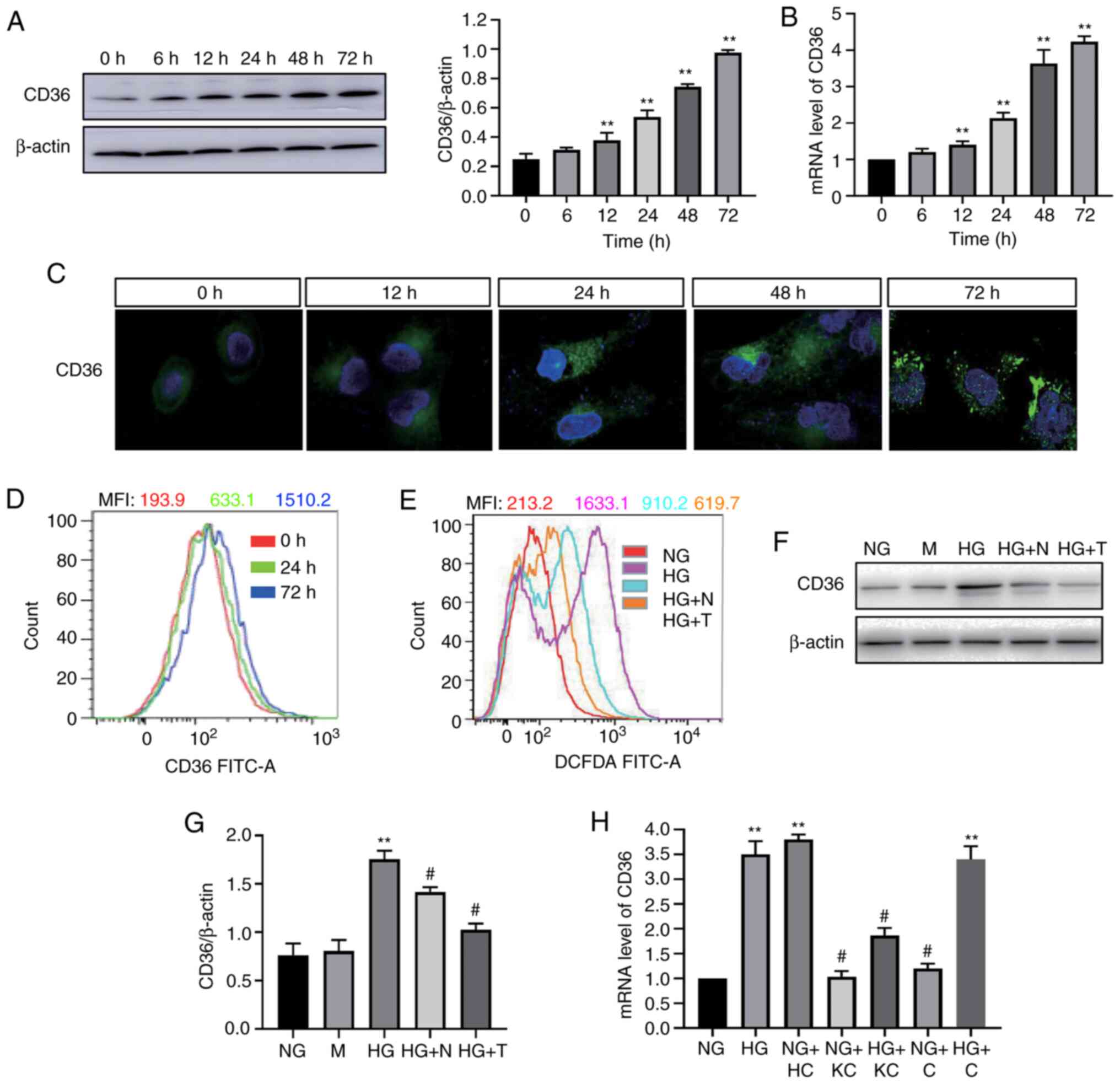 | Figure 1.HG-induced CD36 expression in H9c2
cells is dependent on reactive oxygen species. CD36 expression was
determined by western blotting (A) and reverse
transcription-quantitative PCR (B) in HG-treated cells. (C) CD36
levels in the cell membrane were measured by immunofluorescence
staining in HG-treated cells (magnification, ×400). (D) CD36 levels
in the cell membrane were measured by flow cytometry. (E)
Quantitative analysis of DCFDA fluorescence intensity using flow
cytometry. (F and G) CD36 expression levels were analyzed by
western blotting. (H) mRNA expression levels of CD36 were measured
by reverse transcription-quantitative PCR. H9c2 cells were
incubated with NG (5.6 mM, NG group), NG plus mannitol (24.4 mM, M
group), HG (30 mM, HG group), HG plus N-acetylcysteine (5 mM, HG +
N group) and HG plus MitoTEMPO (10 µM, HG + T group), NG plus LV3
containing CD36 (NG + HC group), NG plus LV3 containing CD36
knockout (NG + KC group), HG plus LV3 containing CD36 knockout (HG
+ KC group), NG plus LV3 empty vector (NG + C group) and HG plus
LV3 empty vector (HG + C group) for 72 h. Data are expressed as the
means ± SD. n=4. **P<0.01 vs. NG or 0 h; #P<0.05
vs. HG. HG, high glucose; NG, normal glucose; DCFDA, 2′,
7′-dichlorodihydrofluorescein diacetate. |
Next, it was assessed whether ROS mediated the
HG-induced CD36 expression in the H9c2 cells. ROS generation was
detected by the cellular ROS indicator DCFDA. Following the
addition of HG, ROS production was increased (Fig. 1E). Mannitol treatment did not affect
CD36 expression (Fig. 1F); NAC (an
intracellular ROS scavenger) and MitoTEMPO (a
mitochondria-targeting antioxidant) significantly decreased
HG-induced generation of ROS (Fig.
1E). The addition of NAC or MitoTEMPO inhibited the HG-induced
CD36 protein overexpression significantly (Fig. 1G).
Knockout of CD36 prevents
NF-κB-mediated inflammatory responses and ROS generation in HG
induced-H9c2 cells
It was investigated whether inhibition of CD36
expression could prevent cellular damage in H9c2 cells by
modulating the inflammatory responses. Lentivirus vectors
(LV3-shRNA) were constructed to knock down CD36 expression, which
was validated by RT-qPCR (Fig. 1H).
Following 72-h exposure to HG or HG + control empty vector
conditions, TNF-α, IL-6 and IL-1β mRNA and protein levels in the
cells were significantly enhanced, whereas knockout of CD36
alleviated these changes (Fig. 2A and
B). Subsequently, the changes in the key transcription factor
NF-κB were assessed. The increased nuclear levels of the p65
subunit of NF-κB and decreased levels of IκBα were used to
represent the activation of NF-κB signaling. Nuclear levels of
NF-κB p65 were increased and cytosolic levels of IκBα were
decreased in H9c2 cells incubated with HG or HG + C for 72 h
compared with that in cells incubated with NG (Fig. 2C-E). CD36 shRNA reversed these
changes. The results of immunofluorescence detection of NF-κB p65
(Fig. 2F) were consistent with the
results showing that knockout of CD36 inhibited NF-κB activation in
HG-treated H9c2 cells.
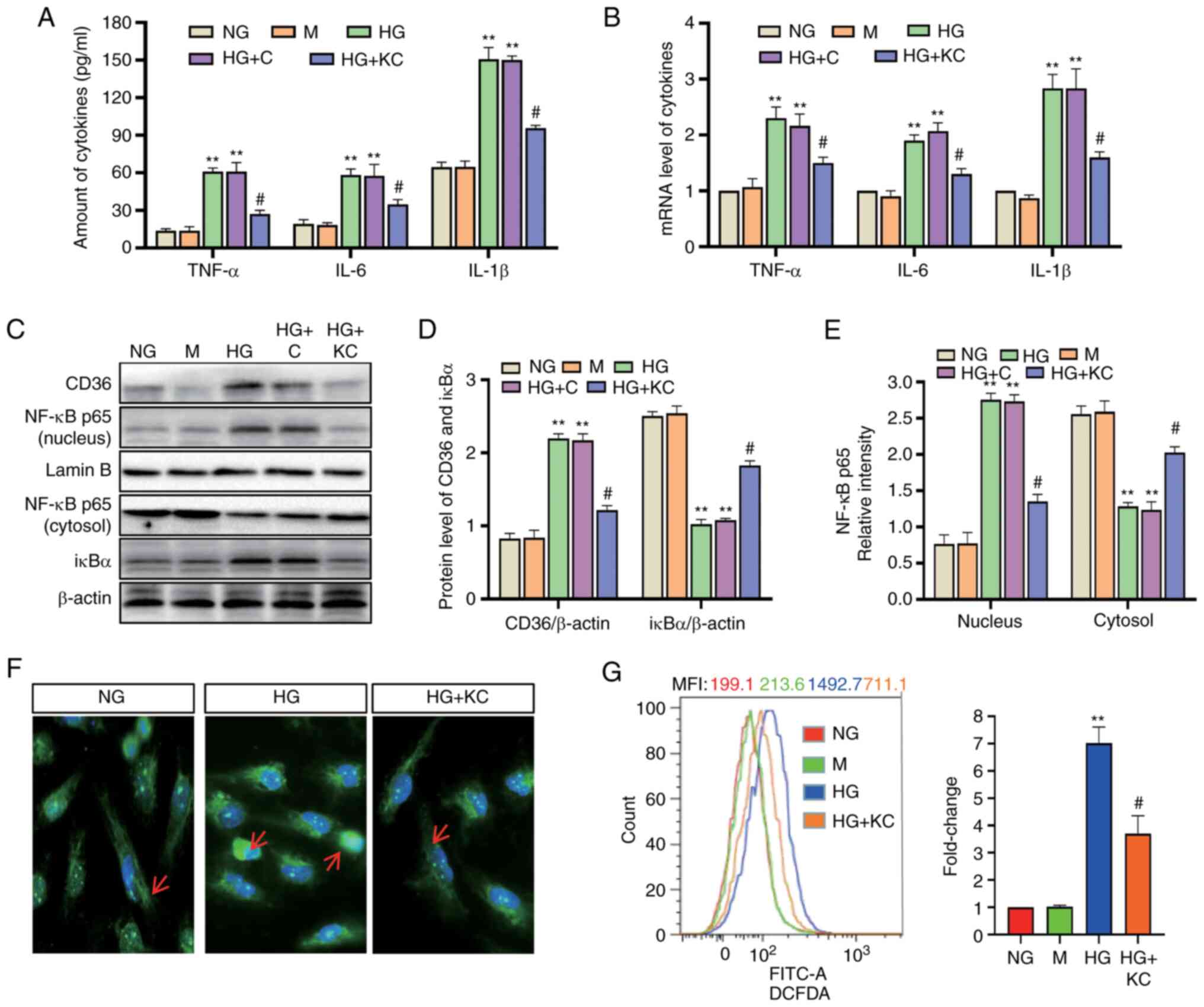 | Figure 2.Knockout of CD36 prevents
NF-κB-mediated inflammatory responses and ROS generation in
HG-induced H9c2 cells. H9c2 cells were incubated with NG (5.6 mM,
NG group), NG plus mannitol (24.4 mM, M group), HG (30 mM, HG
group), HG plus LV3 empty vector (HG + C group) or HG plus LV3
containing CD36 mutant (HG + KC group) for 72 h. (A) The TNF-α,
IL-6 and IL-1β levels in the culture supernatant were measured by
enzyme-linked immunosorbent assay. (B) The mRNA levels of TNF-α,
IL-6 and IL-1β were measured by reverse transcription-quantitative
PCR and normalized to β-actin. (C and D) CD36 in the nucleus and
cytosol and nuclear translocation of NF-κB p65 protein were
detected by western blotting. (C and E) IκBα protein in cytosolic
extracts was measured by western blotting. (F) Immunofluorescence
labeling with anti–NF-κB p65 (magnification, ×400). (G)
Intracellular ROS were detected by flow cytometry. Data are
expressed as the means ± SD. n=4. **P<0.01 vs. NG;
#P<0.05 vs. HG. NF-κB, nuclear factor-κB; ROS,
reactive oxygen species; HG, high glucose; NG, normal glucose; TNF,
tumor necrosis factor; IL, interleukin. |
Next, the effect of CD36 on HG-induced ROS
production was determined. Intracellular ROS levels were examined
using DCFDA. Flow cytometry showed increased fluorescence in the HG
group compared with the NG group, and CD36 knockout significantly
inhibited HG-induced ROS (Fig.
2G).
Knockout of CD36 prevents NF-κB by
inhibiting ROS, and mitochondrial ROS plays a major role
ROS is one of the important activators of NF-κB
(4). Next, it was determined
whether CD36 activates NF-κB through ROS. H9c2 cells were
transfected by lentivirus vector to overexpress CD36 before HG
stimulation. The CD36-overexpressing cells had increased nuclear
levels of NF-κB p65 and decreased cytosolic levels of IκBα, which
were blocked by MitoTEMPO (Fig.
3A-C). The results of immunofluorescence detection of NF-κB p65
(Fig. 3D) were consistent with the
western blotting results. To further demonstrate the role of mtROS
in this pathway, the mtROS level was detected using the specific
mitochondria-targeted superoxide fluorescent probe MitoSOX. As
shown in Fig. 3E, CD36 knockout
significantly inhibited HG-induced mtROS, by ~50% at 72 h. These
data support for a critical role of mtROS in CD36-induced NF-κB
activation in H9c2 cells.
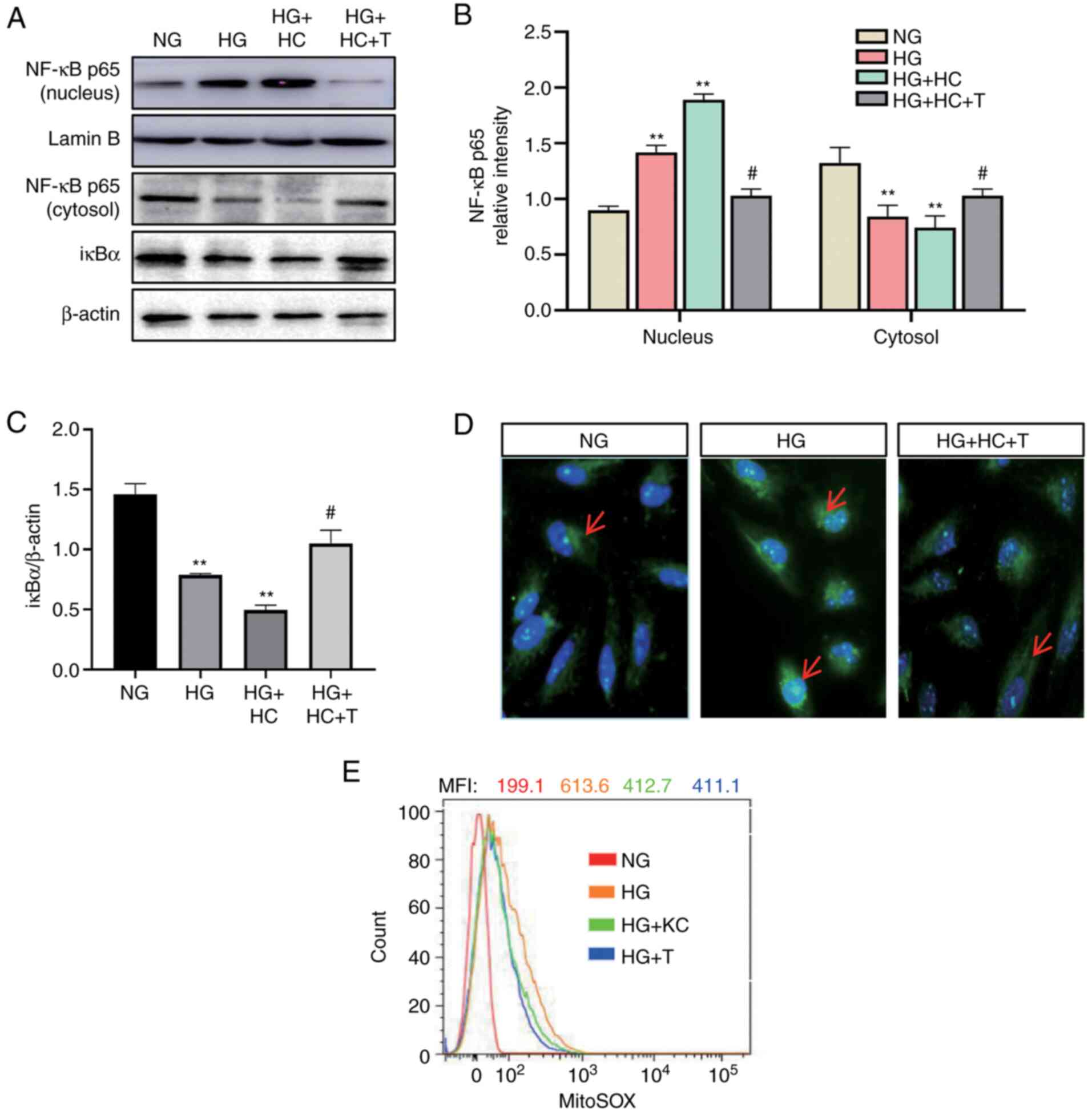 | Figure 3.Knockout of CD36 prevents NF-κB by
inhibiting ROS in H9c2 cells. H9c2 cells were incubated with NG
(5.6 mM, NG group), HG (30 mM, HG group), HG plus LV3 empty vector
(HG + C group), HG plus LV3 containing CD36 (HG + HC group), HG +
HC plus MitoTEMPO (100 nM, HG + HC + T group), HG plus LV3
containing the CD36 short hairpin RNA (HG + KC group), and HG plus
MitoTEMPO (10 µM, HG + T group) for 72 h. (A and B) The nuclear
translocation of NF-κB p65 protein in the nucleus and cytosol was
detected by western blotting. (A and C) The IκBα protein expression
in cytosolic extracts was measured by western blotting. (D)
Immunofluorescence labeling with anti-NF-κB p65 (magnification,
×400). (E) Expression levels of mitochondrial ROS were detected by
FACS using MitoSOX reagent. Data are expressed as the mean ± SD.
n=4. **P<0.01 vs. NG; #P<0.05 vs. HG. NF-κB,
nuclear factor-κB; ROS, reactive oxygen species; HG, high glucose;
NG, normal glucose. |
Knockout of CD36 ameliorates
metabolism reprogramming and lipid accumulation caused by HG in
H9c2 cells
It has previously been demonstrated that mtROS
production is associated with altered bioenergetics (3). Therefore, the present study
investigated the effects of HG on metabolism reprogramming by
treating H9c2 cells with HG for 72 h, then subjecting them to
extracellular flux analysis, which measures the OCR and ECAR
(Fig. 4A). Basal respiration was
unchanged in the HG or HG + C groups when compared with the NG
group, but ATP production and maximal respiration were
significantly decreased after HG treatment (Fig. 4B). Non-mitochondrial respiration and
proton leak were increased in response to HG treatment. Knockout of
CD36 attenuated the OCR changes caused by HG.
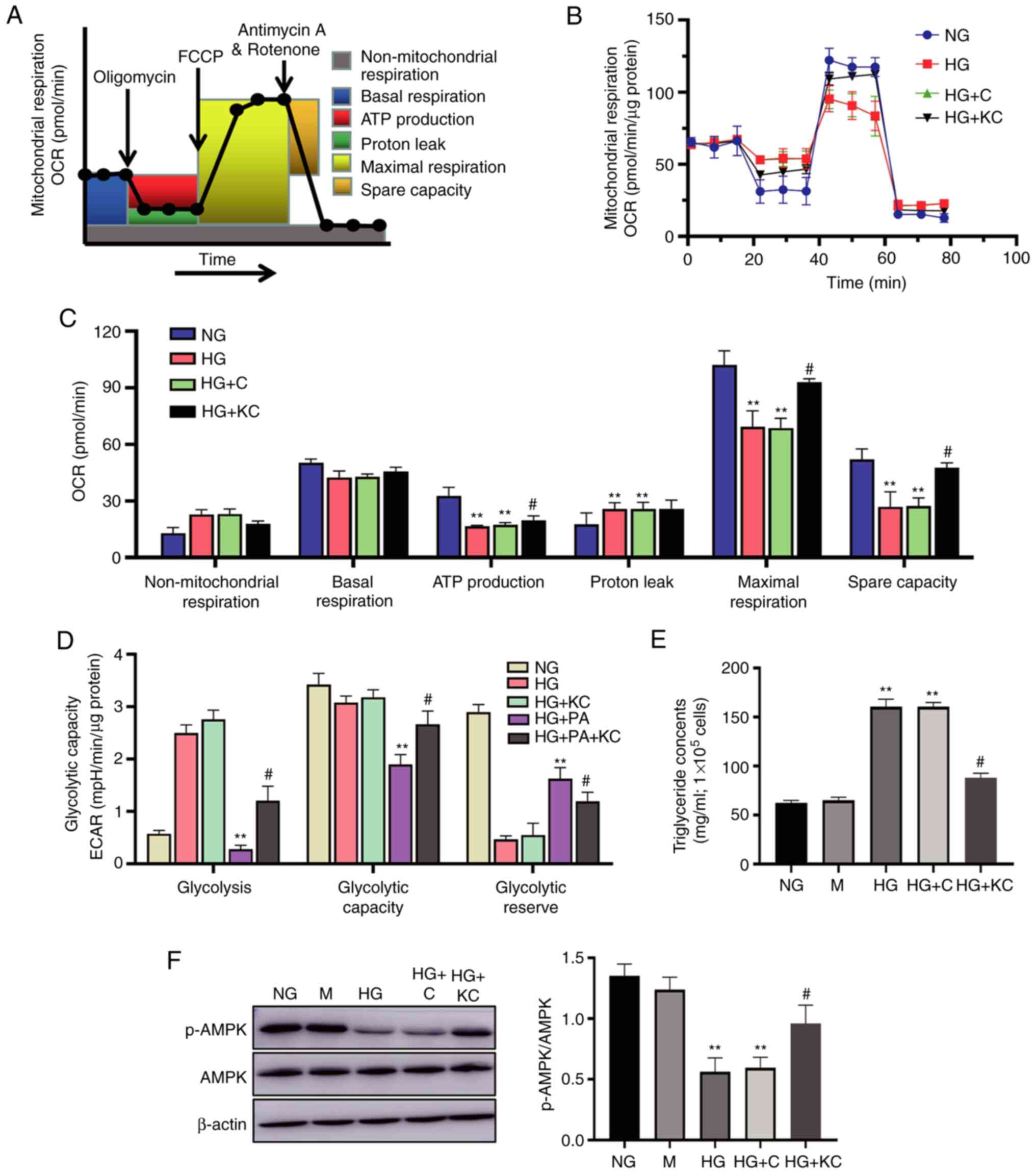 | Figure 4.Knockout of CD36 ameliorates
metabolism reprogramming and lipid accumulation caused by HG in
H9c2 cells. H9c2 cells were incubated with NG (5.6 mM, NG group),
HG (30 mM, HG group), HG plus LV3 containing CD36 mutant (HG + KC
group), HG plus PA (100 µM HG + PA group), or HG + PA plus LV3
containing CD36 mutant (HG + PA + KC group) for 72 h. (A) Protocol
used in data collection and calculations for evaluating
mitochondrial respiration. (B) Representative OCR curve. (C)
Quantitative analysis of non-mitochondrial respiration, basal
respiration, ATP production, proton leak, maximal respiration and
spare respiratory capacity. (D) Quantitative analysis of
glycolysis, glycolytic capacity and glycolytic reserve. All
measured OCR and ECAR were normalized by total cellular protein.
(E) The triglyceride content in the cells. (F) The p-AMPK and AMPK
expression levels were analyzed by western blotting. Data are
expressed as the mean ± SD. n=4. **P<0.01 vs. NG,
#P<0.05 vs. HG. HG, high glucose; OCR, oxygen
consumption rate; ECAR, extracellular acidification rate; p-,
phosphorylated-. |
The H9c2 cells in the NG group had a low ECAR,
consistent with low glycolytic metabolism. HG or HG + C treatment
for 72 h induced elevation in the ECAR, consistent with an increase
in glycolysis (Fig. 4C). Given the
existence of glucose and lipid metabolism disorder in DCM, ECAR
changes were also measured in H9c2 cells treated with HG plus PA
(100 µM; HG + PA). HG + PA treatment induced a decreased ECAR,
consistent with a metabolic switch to fatty acid oxidation
(Fig. 4D). Knockout of CD36
reversed the ECAR changes caused by HG (Fig. 4D), but similar responses were not
observed when transfected with empty vector.
To elucidate the mechanisms by which CD36 regulates
fatty acid oxidation metabolism, the levels of lipid accumulation
and fatty acid oxidation-related pathway were examined in the H9c2
cells. The cellular triglyceride content in the HG or HG + C group,
which was significantly higher than that in the NG group, could be
partially reversed by knockout of CD36 (Fig. 4E). Further, the level of AMPK
phosphorylation was measured, which was a key regulator in the
maintenance of cellular fatty acid homeostasis and controls fatty
acid β-oxidation in mitochondria (19), with western blotting. HG treatment
for 72 h induced a decline in p-AMPK protein in the H9c2 cells
(Fig. 4F). Knockout of CD36
prevented these alterations, as compared with the HG group, but
similar effects were not observed in the HG + C group.
Discussion
The present study demonstrated that HG can
upregulate CD36 at mRNA and protein levels in H9c2 cells, which was
partially dependent on ROS generation. Knockout of CD36 attenuated
HG-induced activation of NF-κB signaling. It was further
demonstrated that the underlying molecular mechanisms may be due to
inhibited mitochondrial ROS production, changes in metabolism and
decreased lipid deposition.
CD36, a membrane glycoprotein, serves an important
role in DCM. The main mechanism is that, as a fatty acid
transporter, CD36 mediates fatty acid transport, promoting fatty
acid oxidation and leading to cardiac lipotoxicity and altered
myocardial energy metabolism (2).
Two major signaling pathways for regulating CD36 in DCM have been
explored: Insulin levels and the energy demand of the heart
(20). However, some studies have
also found that sarcolemmal CD36 levels in cardiac muscle are
increased in type 1 DCM, in which plasma insulin levels are greatly
decreased or absent, suggesting that in addition to insulin levels
(20–22), hyperglycemia may have a regulatory
effect on CD36 expression. Hyperglycemia can upregulate CD36
protein and mRNA expression in macrophages (6). In HG-treated HK-2 cells, the levels of
CD36 mRNA transcripts and subsequent protein translation
were increased in a time-dependent manner (7). Consistently, it was found that
exposure to HG induced significantly increased CD36 expression in
H9c2 cells. However, CD36 is present not only at the cell surface
but is also found in endosomes, the endoplasmic reticulum and the
mitochondria. CD36 can migrate between these locations via
vesicular transport along exocytotic and endocytic pathways to
control lipid homeostasis and energy reprogramming (12). There is subcellular localization of
CD36 in cardiomyocytes (23). CD36
is a fatty acid transporter, and its main role depends on the
amount localized in the cell membrane (12). Therefore, in the present study, the
total protein expression of CD36 was first detected in H9c2 cells
induced by HG, and the localization of CD36 was then observed in
cells with a confocal microscope. The results show that CD36
fluorescence was distributed in the cytoplasm of H9c2 cells in the
0-h groups. However, CD36 fluorescence tended to be distributed
around the cell membrane in the 72-h groups. To quantify CD36
expression in the cell membrane, flow cytometry was used (which
mainly detects cell surface proteins). The flow cytometry results
are consistent with that of the western blotting. It was also found
that at early-stage stimulation by HG for 24 or 48 h, CD36
localization in the cell membrane of H9c2 cells was increased, but
not significantly, while it was increased at 72 h. H9c2 cell
responses to a HG environment first involve an increased ATP/AMP
ratio, which results in AMPK inactivation. AMPK inactivation may
inhibit the membrane translocation of CD36 (16,24).
However, persistent HG can contribute to cardiomyocyte damage, such
as oxidative stress, mitochondrial dysfunction and the activation
of several signaling molecules (2).
All of these may have the effect of increasing CD36 expression and
membrane translocation in H9c2 cells. ROS generation induces CD36
expression in other cell types (7,25).
Further, the present data show that MitoTEMPO or NAC can decrease
CD36 overexpression in HG-induced H9c2 cells, suggesting that there
may be crosstalk between ROS and the CD36 pathway in HG-treated
H9c2 cells.
CD36 is not only a fatty acid transporter, but also
a signaling molecule involved in inflammatory responses in a
variety of disease states (7–16). In
atherogenic processes, CD36 is important for activating the NLRP3
(NLR family pyrin domain containing 3) inflammasome and the NF-κB
pathway (9,10). In non-alcoholic steatohepatitis,
knockout of CD36 decreased inflammation via the NF-κB pathway
inactivation (16). NF-κB is a
primary regulator of inflammatory responses, and serves an
important role in the development of DCM. Activated NF-κB can
induce the expression of proinflammatory cytokines, including TNFα,
IL-6 and IL-1β. These cytokines perform an important stress
response in myocardial injury (2).
In the present study, knockout of CD36 prevented inflammatory
responses in HG-induced H9c2 cells. The mechanism that affected
this involved the restoration of abnormal activation of the NF-κB
system. These results certainly warrant further study of the
mechanism of activation of NF-κB by CD36. Previous studies have
confirmed that ROS is an important activator of NF-κB (26–28).
The results of the present study indicate that when MitoTEMPO was
used to inhibit ROS production, NF-κB activation by CD36
overexpression was significantly suppressed. These data provide
clear evidence that ROS may play an important role in CD36
activation of the NF-κB pathway in HG-induced injury and
inflammation in H9c2 cells. As a membrane fatty acid transporter,
CD36 mediates fatty acid transport, leading to increased
intracellular lipid deposition in a variety of disease states,
causing mitochondrial damage via increased ROS production and
cellular inflammation. Additionally, CD36 is a cell transduction
protein that activates multiple signaling pathways (10,12,23,29).
Thus, it is believed that, in cardiomyocytes, the increased CD36
expression induced by HG also acts both ways. Therefore, both
CD36-induced lipid deposition and NF-κB inflammatory signaling
pathway activation were detected. The results indicated that
CD36-induced intracellular lipid deposition and inflammatory
signaling pathway activation plays a role in DCM.
ROS production is closely associated with
mitochondrial metabolism. Numerous studies have found that
metabolic remodeling, increased lipid utilization and decreased
glucose metabolism play a role in the development of DCM 1–3. The
present study examined the metabolic changes in cultured H9c2 cells
under HG stimulation. HG decreased the levels of oxidative
phosphorylation and induced the elevation of glycolysis in H9c2
cells, as demonstrated by extracellular flux analysis. The base
ECAR represents the non-glycolytic acid production of the cell; the
ECAR after the addition of glucose represents the glycolytic
capacity of the cells at the time. With the addition of the
oligomycin, oxidative phosphorylation was inhibited, and the cells
were supplied with oxygen by glycolysis. At this point, acid
production increases, and the increased ECAR represents the cell's
remaining glycolytic capacity, or potential; the total value
represents the maximum glycolytic capacity of the cell. Finally, a
glycolytic inhibitor is added, and the ECAR after the addition of
this drug represent acid production beyond glycolysis; this can be
due to a lack of mitochondrial respiration (30). The 72-h HG treatment induced ECAR
elevation; knockout of CD36 reversed the ECAR, promoting
mitochondrial respiration. In addition, there are complex metabolic
disorders of hyperglycemia and hyperlipidemia on a whole-body
level. It was also found that in HG and high PA conditions, H9c2
cells begin to rely on fatty acid oxidation for energy production,
similar to the metabolic remodeling in vivo. Knockout of
CD36 protected H9c2 cells from the imbalance of energy homeostasis
and lipid accumulation caused by HG, indicating that the metabolic
disturbance caused by HG may partly be due to CD36.
AMPK regulates fatty acid oxidation, and has emerged
as a potential therapeutic target for various metabolic disorders,
including diabetes mellitus (31).
In non-alcoholic steatohepatitis, CD36 regulates AMPK activation
via the phosphorylation of LKB1 (serine/threonine kinase 11)
(16). In skeletal muscle, exercise
regulates AMPK activation partly through CD36 (32). AMPK activation was decreased in
diabetic nephropathy, neuropathy and retinopathy (33). Nevertheless, in the present study,
AMPK activation was significantly decreased in H9c2 cells that had
been stimulated by HG for 72 h. CD36 is a well-known cell membrane
protein and has been studied extensively in cardiomyocytes;
however, current studies have focused on the role of CD36 as a
fatty acid transporter (34). The
association between CD36 and fatty acid oxidation, as well as
CD36-induced cellular inflammation and cell metabolism, has not
been clarified. Previous studies have found that CD36 can inhibit
AMPK activation, which has been confirmed in liver cells and
macrophages (16,32,35).
Therefore, the association between CD36 and AMPK in cardiomyocytes
was further investigated. The knockout of CD36 can improve AMPK
inactivation by HG. The results suggest an association between CD36
and AMPK in H9c2 cells. However, the relevant mechanism still
requires further research and confirmation.
However, a few limitations of the present study
should be addressed. Firstly, HG-stimulated H9c2 cells were used to
simulate a diabetic renal tubular injury model in vitro.
This cell model was used based on previous research (36–38).
However, there have been several studies on H9c2 cells as a model
in DCM (36–39). H9c2 cells are derived from rat
cardiomyocytes and differ from primary cardiomyocytes; human
cardiomyocytes or primary cardiomyocytes would represent a better
model. Relevant in vivo studies will be conducted later,
which should be more appropriate. Secondly, previous studies have
reported that CD36 mediates ROS production (7,25,40);
inhibiting intracellular ROS decreased CD36 expression (7,25).
However, this is unclear in cardiomyocytes. The present study only
proves that the association between CD36 and ROS and HG-induced
CD36 expression in H9c2 cells relies partially on ROS.
In conclusion, HG can increase CD36 expression,
which leads to the activation of NF-κB signaling. This effect is
mediated by metabolism reprogramming and lipid accumulation,
resulting in enhanced ROS generation.
Acknowledgements
Not applicable.
Funding
The present study was supported by the grant from
Shanxi Cardiovascular Hospital (grant no. XYS20200101).
Availability of data and materials
All data generated or analyzed during the present
study are included in this article.
Authors' contributions
BH and JL designed the study. JWa supervised the
entire project and conducted analyses of western blotting images.
BH wrote the manuscript. BH and JL performed most of the
experiments and data analysis. JWu analyzed the western blot
images. FY contributed to data analysis. YW assisted with the
experiments and manuscript preparation. BH and JL confirm the
authenticity of all the raw data. All authors reviewed the data and
read and approved the final version of the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Falcão-Pires I and Leite Moreira AF:
Diabetic cardiomyopathy: Understanding the molecular and cellular
basis to progress in diagnosis and treatment. Heart Fail Rev.
17:325–344. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Jia G, Hill MA and Sowers JR: Diabetic
cardiomyopathy: An update of mechanisms contributing to this
clinical entity. Circ Res. 122:624–638. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kaludercic N and Di Lisa F: Mitochondrial
ROS formation in the pathogenesis of diabetic cardiomyopathy. Front
Cardiovasc Med. 7:122020. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Jia G, DeMarco VG and Sowers JR: Insulin
resistance and hyperinsulinaemia in diabetic cardiomyopathy. Nat
Rev Endocrinol. 12:144–153. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Brinkmann JF, Abumrad NA, Ibrahimi A,
vander Vusse GJ and Glatz JF: New insights into long-chain fatty
acid uptake by heart muscle: A crucial role for fatty acid
translocase/CD36. Biochem J. 367:561–570. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Griffin E, Re A, Hamel N, Fu C, Bush H,
McCaffrey T and Asch AS: A link between diabetes and
atherosclerosis: Glucose regulates expression of CD36 at the level
of translation. Nat Med. 7:840–846. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Hou Y, Wu M, Wei J, Ren Y, Du C, Wu H, Li
Y and Shi Y: CD36 is involved in high glucose-induced epithelial to
mesenchymal transition in renal tubular epithelial cells. Biochem
Biophys Res Commun. 468:281–286. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lu H, Yao K, Huang D, Sun A, Zou Y, Qian J
and Ge J: High glucose induces upregulation of scavenger receptors
and promotes maturation of dendritic cells. Cardiovasc Diabetol.
12:802013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Sheedy FJ, Grebe A, RaynerK J, Kalantari
P, Ramkhelawon B, Carpenter SB, Becker CE, Ediriweera HN, Mullick
AE, Golenbock DT, et al: CD36 coordinates NLRP3 inflammasome
activation by facilitating intracellular nucleation of soluble
ligands into particulate ligands in sterile inflammation. Nat
Immunol. 14:812–820. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Chen Y, Yang M, Huang W, Chen W, Zhao Y,
Schulte ML, Volberding P, Gerbec Z, Zimmermann MT, Zeighami A, et
al: Mitochondrial metabolic reprogramming by CD36 signaling drives
macrophage inflammatory responses. Circ Res. 125:1087–1102. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Bhat A, Das S, Yadav G, Chaudhary S, Vyas
A, Islam M, Gupta AC, Bajpai M, Maiwall R, Maras JS and Sarin SK:
Hyperoxidized albumin modulates platelets and promotes inflammation
through CD36 receptor in severe alcoholic hepatitis. Hepatol
Commun. 4:50–65. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Yang X, Okamura DM, Lu X, Chen Y, Moorhead
J, Varghese Z and Ruan XZ: CD36 in chronic kidney disease: Novel
insights and therapeutic opportunities. Nat Rev Nephrol.
13:769–781. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kunz A, Abe T, Hochrainer K, Shimamura M,
Anrather J, Racchumi G, Zhou P and Iadecola C: Nuclear
factor-kappaB activation and postischemic inflammation are
suppressed in CD36-null mice after middle cerebral artery
occlusion. J Neurosci. 28:1649–1658. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Cao D, Luo J, Zang W, Chen D, Xu H, Shi H
and Jing X: Gamma-linolenic acid suppresses NF-κΒ signaling via
CD36 in the lipopolysaccharide-induced inflammatory response in
primary goat mammary gland epithelial cells. Inflammation.
39:1225–1237. 2016.PubMed/NCBI
|
|
15
|
Sp N, Kang DK, Kim DH, Park JH, Lee HG,
Kim HJ, Darvin P, Park YM and Yang YM: Inhibits CD36-dependent
tumor angiogenesis, migration, invasion, and sphere formation
through the Cd36/Stat3/Nf-Κb signaling axis. Nutrients. 10:7722018.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zhao L, Zhang C, Luo X, Wang P, Zhou W,
Zhong S, Xie Y, Jiang Y, Yang P, Tang R, et al: CD36 palmitoylation
disrupts free fatty acid metabolism and promotes tissue
inflammation in non-alcoholic steatohepatitis. J Hepatol.
69:705–717. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Hou Y, Shi Y, Han B, Liu X, Qiao X, Qi Y
and Wang L: The antioxidant peptide SS31 prevents oxidative stress,
downregulates CD36 and improves renal function in diabetic
nephropathy. Nephrol Dial Transplant. 33:1908–1918. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PcR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ruderman N, Carling D, Prentki M and
Cacicedo J: AMPK, insulin resistance, and the metabolic syndrome. J
Clin Invest. 123:2764–2772. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ramírez E, Picatoste B, González-Bris A,
Oteo M, Cruz F, Caro-Vadillo A, Egido J, Tuñón J, Morcillo MA and
Lorenzo Ó: Sitagliptin improved glucose assimilation in detriment
of fatty-acid utilization in experimental type-II diabetes: Role of
GLP-1 isoforms in Glut4 receptor trafficking. Cardiovasc Diabetol.
17:122018. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Palomer X, Salvadó L, Barroso E and
Vázquez-Carrera M: An overview of the crosstalk between
inflammatory processes and metabolic dysregulation during diabetic
cardiomyopathy. Int J Cardiol. 168:3160–3172. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Koonen DP, Sung MM, Kao CK, Dolinsky VW,
Koves TR, Ilkayeva O, Jacobs RL, Vance DE, Light PE, Muoio DM, et
al: Alterations in skeletal muscle fatty acid handling predisposes
middle-aged mice to diet-induced insulin resistance. Diabetes.
59:1366–1375. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Abumrad NA and Goldberg IJ: CD36 actions
in the heart: Lipids, calcium, inflammation, repair and more?
Biochim Biophys Acta. 1861:1442–1449. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Han L, Yang Q, Li J, Cheng F, Zhang Y, Li
Y and Wang M: Protocatechuic acid-ameliorated endothelial oxidative
stress through regulating acetylation level via CD36/AMPK pathway.
J Agric Food Chem. 67:7060–7072. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Li W, Febbraio M, Reddy SP, Yu DY,
Yamamoto M and Silverstein RL: CD36 participates in a signaling
pathway that regulates ROS formation in murine VSMCs. J Clin
Invest. 120:3996–4006. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
26
|
Liang E, Liu X, Du Z, Yang R and Zhao Y:
Andrographolide ameliorates diabetic cardiomyopathy in mice by
blockage of oxidative damage and NF-κB-mediated inflammation. Oxid
Med Cell Longev. 2018:90867472018. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Morgan M and Liu Z: Crosstalk of reactive
oxygen species and NF-κB signaling. Cell Res. 21:103–115. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Frati G, Schirone L, Chimenti I, Yee D,
Biondi-Zoccai G, Volpe M and Sciarretta S: An overview of the
inflammatory signalling mechanisms in the myocardium underlying the
development of diabetic cardiomyopathy. Cardiovasc Res.
113:378–388. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Febbraio M, Hajjar DP and Silverstein RL:
CD36: A class B scavenger receptor involved in angiogenesis,
atherosclerosis, inflammation and lipid metabolism. J Clin Invest.
108:785–791. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
30
|
Lisa S, Winer P and Wu M: Rapid analysis
of glycolytic and oxidative substrate flux of cancer cells in a
microplate. PLoS One. 9:e1099162014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Day EA, Ford RJ and Steinberg GR: AMPK as
a therapeutic target for treating metabolic diseases. Trends
Endocrinol Metab. 28:545–560. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Samovski D, Sun J, Pietka T, Gross RM,
Eckel RH, Su X, Stahl PD and Abumrad NA: Regulation of AMPK
activation by CD36 links fatty acid uptake to β-oxidation.
Diabetes. 64:353–359. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Shrikanth CB and Nandini CD: AMPK in
microvascular complications of diabetes and the beneficial effects
of AMPK activators from plants. Phytomedicine.
24:1528082018.PubMed/NCBI
|
|
34
|
Glatz JFC, Luiken JJFP and Nabben M: CD36
(SR-B2) as a target to treat lipid overload-induced cardiac
dysfunction. J Lipid Atheroscler. 9:66–78. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Li Y, Yang P, Zhao L, Chen Y, Zhang X,
Zeng S, Wei L, Varghese Z, Moorhead JF, Chen Y and Ruan XZ: CD36
plays a negative role in the regulation of lipophagy in hepatocytes
through an AMPK-dependent pathway. J Lipid Res. 60:844–855. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Zhu Y, Qian X, Li J, Lin X, Luo J, Huang J
and Jin Z: Astragaloside-IV protects H9C2(2-1) cardiomyocytes from
high glucose-induced injury via miR-34a-mediated autophagy pathway.
Artif Cells Nanomed Biotechnol. 47:4172–4181. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Ma L, Cao Y, Zhang L, Li K, Pan Y and Zhu
J: Celastrol mitigates high glucose-induced inflammation and
apoptosis in rat H9c2 cardiomyocytes via miR-345-5p/growth
arrest-specific 6. J Gene Med. 22:e32012020. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Huang Z, Dong X, Zhuang X, Hu X, Wang L
and Liao X: Exogenous hydrogen sulfide protects against high
glucose-induced inflammation and cytotoxicity in H9c2 cardiac
cells. Mol Med Rep. 14:4911–4917. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Zhao MX, Zhou B, Ling L, Xiong XQ, Zhang
F, Chen Q, Li YH, Kang YM and Zhu GQ: Salusin-β contributes to
oxidative stress and inflammation in diabetic cardiomyopathy. Cell
Death Dis. 8:e26902017. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Kotla S and Rao GN: Reactive oxygen
species (ROS) mediate p300-dependent STAT1 protein interaction with
peroxisome proliferator-activated receptor (PPAR)-γ in CD36 protein
expression and foam cell formation. J Biol Chem. 290:30306–30320.
2015. View Article : Google Scholar : PubMed/NCBI
|














